

Abstract
Disturbances in redox equilibrium in tissue can lead to inflammatory state, which is a mediatory factor in many human diseases. The mechanism(s) by which exogenous oxidants may activate an inflammatory response is not fully understood. Emerging evidence suggest that oxidant-induced Toll-like receptor 4 (TLR4) activation play a major role in “sterile” inflammation. In the present study, we used murine macrophage RAW-Blue cells, which are chromosomally integrated with secreted embryonic alkaline phosphatase (SEAP) inducible by NF-κB. We confirmed the expression of TLR4 mRNA and protein in RAW-Blue cells by RT-PCR and Western blot, respectively. We showed that oxidants increased intracellular reactive oxygen species production and lipid peroxidation, which resulted in decreased intracellular total antioxidant capacity. Consistent with the actions of TLR4-specific agonist LPS-EK, exogenous oxidants increased transcriptional activity of NF-κB p65 with subsequent release of NF-κB reporter gene SEAP. These effects were blocked by pretreatment with TLR4 neutralizing pAb and TLR4 signaling inhibitor CLI-095. In addition, oxidants decreased the expression of IκBα with enhanced phosphorylation at the Tyr42 residue. Finally, oxidants and LPS-EK increased TNFα production, but did not affect IL-10 production, which may cause imbalance between pro- and anti-inflammatory processes, which CLI-095 inhibited. For biological relevance, we confirmed that oxidants increased release of TNFα and IL-6 in primary macrophages derived from TLR4-WT and TLR4-KO mice. Our results support the involvement of TLR4 mediated oxidant-induced inflammatory phenotype through NF-κB activation in macrophages. Thus exogenous oxidants may play a role in activating inflammatory phenotypes that propagate and maintain chronic disease states.
Keywords: Exogenous oxidants, Oxidative stress, Sterile inflammation, Macrophages, nuclear factor kappa B
Graphical abstract
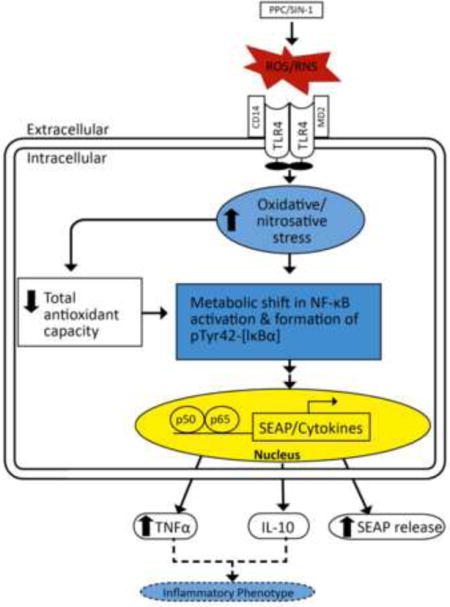
1. Introduction
Reactive oxygen species (ROS) [a term used here to also encompass reactive nitrogen species (RNS)] can cause damage to lipid, DNA, and other cellular macromolecules. Oxidative stress, induced by ROS from exogenous and endogenous sources, can initiate, propagate and maintain cellular inflammatory states. The mechanism(s) by which exogenous oxidants can activate inflammatory responses that can potentially initiate, propagate and maintain many human diseases has not been elucidated. However, accumulating evidence suggests that pattern recognition receptors (PRRs) of the innate immune system such as toll-like receptors (TLRs) may be involved in integrating this response [1].
TLRs, the first identified member of PRR, play a key role in the host defense system. TLRs in innate immune cells detect danger signals, which can be pathogen associated molecular patterns (PAMPs) in pathogenic infection, or damage associated molecular patterns (DAMPs) in “sterile” inflammation due to tissue injury. Particularly, TLR4- or TLR2-dependent signaling pathway can be triggered by various environmental factors, including ozone, atmospheric particulate matter, pentachlorophenol ionizing radiation [2] and cigarette smoke extract [3]. Exogenous oxidants can stimulate TLR4 by TLR4/ROS coupling that in turn activates intracellular signaling pathways such as NF-κB, which increases the production of ROS/RNS (RONS) and pro-inflammatory cytokines. Thus, the activation of inflammatory pathways derived from extracellular factors may be associated with the onset and potentially the maintenance of inflammatory diseases such as chronic fatigue syndrome, cardiovascular disorders, chronic pain, and neurodegeneration diseases [2].
TLR4 activation can initiate inflammatory responses by recruiting intracellular adaptor proteins such as myeloid differentiating primary response gene 88 (MyD88), TIR adaptor protein (TIRAP), TIR-domain-containing adapter-inducing interferon-β (TRIF), and TRIF-related adaptor molecule (TRAM) [1]. Association of TLR4 with MyD88 further recruits other adaptor proteins leading to the activation of transforming growth factor-β-activated protein kinase 1 (TAK-1), which in turn results in phosphorylation of inhibitory NF-κB protein (IκB) and activation of NF-κB. NF-κB is considered a crucial regulator in the immune system, and its activation leads to increased transcription of genes related to innate immunity and inflammatory response [4].
TLR4, an integral part of the innate immune system, is mainly expressed on immune cells, such as neutrophil and macrophages. We hypothesize that TLR4 expressed on immune effector cells such as macrophages and microglia can be stimulated directly by exogenous oxidants [1]. Functioning as one of the primary sensors of danger in the host, macrophages retain remarkable plasticity that allows them to efficiently respond to environmental cues and change their phenotypes [5]. Generally, classically activated macrophages (M1 phenotype) are pro-inflammatory and can induce tissue damage, whereas alternatively activated macrophages (M2 phenotype) are anti-inflammatory that can initiate tissue repair. Macrophage activation is responsible for acute and chronic inflammation by releasing a battery of inflammatory mediators [6]. Indeed, uncontrolled production of inflammatory cytokines by activated macrophages directly mediates the immuno-pathogenesis in several autoimmune diseases including rheumatoid arthritis, and inflammatory bowel disease [7].
Our previous study has demonstrated that TLR4 is involved in exogenous oxidant-induced NF-κB activation in human embryonic kidney (HEK) cells transfected with mouse TLR4 [8, 9]. In the present study, we further determined the role of TLR4 in oxidant-induced NF-κB activation in a well established and characterized murine macrophage cell line, a biologically relevant model with respect to immune response.
We challenged murine macrophages RAW-Blue cells with two distinct oxidants namely potassium peroxychromate (PPC) and SIN-1. PPC decomposition can release several oxygen-centered free radicals such as superoxide anion and hydroxyl radicals under physiological conditions as produced by activated phagocytes [10]. SIN-1 produces cell permeable peroxynitrite anions that can react with lipids, DNA and proteins by direct oxidative reaction or by indirect radical-induced mechanisms [11, 12]. Macrophages, microglia, neutrophils, monocytes and endothelial cells can also generate peroxynitrite in their normal cellular processes. We validated some of the data with primary peritoneal macrophages (pM).
In the present set of experiments, we provide further evidence for molecular mechanism(s) of the role of TLR4/RONS-coupled NF-κB activation in macrophages that may be related to the function of innate immune system.
2. Materials and Methods
2.1 Chemicals and Materials
RAW-Blue selection antibiotic Zeocin, Quanti-Blue detection reagent (alkaline phosphatase detection medium), LPS-EK from E. coli K12 (LPS-EK Ultrapure), and TLR4 signaling inhibitor CLI-095 were obtained from InvivoGen (San Diego, CA, USA). Linsidomine chloride (SIN-1) was obtained from AdipoGen (San Diego, CA, USA). TRI Reagent for RNA extraction was obtained from Molecular Research Center (Cincinnati, OH, USA). EUK-134 and the total antioxidant assay kit were purchased from Cayman Chemical (Ann Arbor, MI, USA). Low endotoxin, azide-free affinity purified rat IgG2a, κ-isotype anti-mouse TLR4/MD-2 complex polyclonal antibody (pAb)(clone MT510) for neutralization of TLR4 and enzyme-linked immunosorbent assay (ELISA) kit for mouse-specific TNF-α, IL-6 and IL-10 were purchased from BioLegend (San Diego, CA, USA). High-capacity cDNA reverse transcription assay kit, CellROX™ deep red reagent and NucBlue Live ReadyProbes reagent were obtained from ThermoFisher Scientific (Grand Island, NY, USA). Parameter thiobarbituric acid-reactive substances (TBARS) kit was purchased from R &D Systems (Minneapolis, MN, USA). TransAM® NF-κB p65 transcription factor assay kit and Nuclear Extract kit were obtained from Active Motif (Carlsbad, CA, USA). OxiSelect 4-hydroxynonenal (4-HNE) Adduct Competitive ELISA kit and anti-Nitrotyrosine polyclonal antibody were obtained from Cell Biolabs, Inc. (San Diego, CA, USA). Mammalian cell-PE-LB and Protease Arrest™ for protein lysates were purchased from G-Biosciences (St. Louis, MO. USA) supplemented with appropriate protease inhibitors (G-Biosciences, St. Louis, MO. USA). Anti-NF-κB p65 monoclonal antibody (mAb) was purchased from Cell Signaling (Danvers, MA, USA). Protein A/G plus-agarose and anti-lamin B antibody (Ab) was purchased from Santa Cruz Biotechnology (Dallas, Texas, USA). Anti-IκBα (c-terminal) and anti-I IκBα (Tyr-42) phosphor-specific Ab were purchased from ECM Biosciences (Versailles, KY, USA). Anti-β-actin and α-tubulin Ab were purchased from Proteintech (Chicago, IL, USA).
2.2 Preparation of potassium peroxychromate (PPC)
PPC, used in the study as a primary exogenous source of ROS, is not available commercially, but was synthesized in the laboratory according to a published protocol [10]. It was characterized by elemental and infrared analyses with a purity of > 98%.
2.3 Cell lines and culture
RAW-Blue cells, purchased from InvivoGen, are derived from the murine macrophages RAW 246.7 cell line. They are chromosomally integrated with secreted embryonic alkaline phosphatase (SEAP) reporter construct inducible by NF-κB and AP-1. We chose to use this cell model to quantify NF-κB activation directly in real time with a robust read-out as well. We assessed the levels of SEAP released into the culture medium to quantify the extent of NF-κB transcriptional activity. Cells were grown in a 37°C, 100% humidified incubator in Dulbecco’s modified Eagle’s medium (DMEM) (4.5 g of glucose/L) without pyruvate, but supplemented with 2 mM L-glutamine, 10 % (v/v) FBS, 50 units/ml penicillin and 50 ug/ml streptomycin. RAW-Blue cells were maintained in growth medium supplemented with selective antibiotic reagent Zeocin (50 μg/ml). The supplier of the RAW-Blue cells authenticated these cells to be of purely mouse macrophage lineage without cell contaminants from another species.
We also used primary peritoneal macrophages (pM) isolated from TLR4-WT and TLR-4 KO mice to validate some of the data obtained with RAW-Blue cells. We prepared and characterized pM according to an established method [13]. The purity of the pM (≥ 95.5 %) as determined by the level of cell surface expression of CD11b antigen was confimed by both immunofluorescence and flow cytometry.
2.4 Determination of optimal concentrations of PPC and SIN-1 by LDH cytotoxicity assay and live cell counting
LDH cytotoxicity assay and counting of live cell number were used to provide a rationale for the concentrations we used in all subsequent studies. Cells were plated in tissue culture dishes and grown overnight. After stimulation with PPC (100, 500 μM), SIN-1 (1, 5 mM) or LPS-EK (10 ng/ml) for 2 h, the culture medium was replaced with fresh growth medium in the absence of LPS-EK or oxidants. Cells were then incubated overnight for ~16 h. Samples (cells and culture supernatants) were collected after either 2 h stimulation with oxidants (or LPS-EK) or 16 h incubation with fresh growth medium.
For the LDH cytotoxicity assay, cells (1×105/well) were plated in 24-well plates and grown overnight to 70% confluence. LDH levels in culture supernatant after either 2 h stimulation or 16 h incubation were quantified according to the manufacturer’s instructions. LDH catalyzes the formation of red colored formazan and its absorbance is read at 490 nm. The amount of formazan produced is proportional to the activity of LDH. The content of LDH released into culture medium was determined from standard curves.
To determine live cell numbers, cells (5×105/well) were plated in 6-well plates and grown overnight to 70% confluence. Cells were scrapped and counted either after 2 h stimulation or 16 h incubation in the absence of the oxidant or LPS-EK using the Countess Automated Cell counter (Life technologies, CA, USA). Live and dead cells were distinguished using 0.2% trypan blue dye. Live cells would exclude the dye while dead cells will take up the dye into their cytoplasm.
2.5 Expression of TLR4 in RAW-Blue cells, RNA extraction and real time PCR
RAW 264.7 cell line expresses TLR4 and its co-receptors MD-2 and CD14 [14]. We confirmed the expression of TLR4 in RAW-Blue cells by real time quantitative PCR (RT-qPCR) and Western blot.
RAW-Blue cells were collected in TRI Reagent. Total RNA was extracted by chloroform, and precipitated by isopropanol. Single-stranded cDNA was synthesized from total RNA with the reverse transcription kit using StepOnePlus™ instrument (Life technologies, PA, USA). The PCR product was resolved on polyacrylamide gel, stained with ethidium bromide, and visualized under UV light. The primer sequences that were used for TLR4 (NCBI Reference Sequence: NM_021297.3) are: forward primer (FP): 5′-AAC CAG CTG TAT TCC CTC AGC ACT-3′; reverse primer (RP): 5′-ACT GCT TCT GTT CCT TGA CCC ACT-3′. The primer sequences for ß-actin that were used for normalization of gene expression (NCBI Reference Sequence: NM_007393.5) are: FP: 5′-GTTGGAGCAAACATCCCCCA-3′; RP: 5′-ACGCGACCATCCTCCTCTTA-3′. We followed the guidelines for the minimum information for publication of quantitative real time PCR experiments (MIQE) [15,16], which included uniformity in cell treatment with incubation conditions, RNA extraction protocols, and its essential purity across samples, quantification, storage and uniformity of the # of cycles with the linearity of samples.
2.6 Quantification of lipid peroxidation by measurement of thiobarbituric acid reactive substances (TBARS) and 4-hydroxynonenal (4-HNE)
Exposure of plasma membrane to ROS leads to lipid peroxidation during which malondialdehyde (MDA) and 4-HNE are generated as the end products [17]. We quantified levels of MDA and 4-HNE to determine lipid peroxidation caused by ROS released from PPC or SIN-1. We used EUK-134, a synthetic serum-stable scavenger for oxidative species to confirm the action of oxidants. Cells were preincubated with EUK-134 (4 μM) for 30 min before stimulation with oxidants for 2 h. Culture supernatant at the end of 2 h stimulation was collected for lipid peroxides quantification. MDA can be quantified as TBARS according to the instructions in the Parameter TBARS assay kit.
Culture medium was centrifuged to remove debris, and clarified by trichloroacetic acid treatment. The precipitated interfering proteins and other substances were removed by centrifugation at 12, 000 × g for 4 min. In the presence of heat and acid, free MDA in the sample reacts with thiobarbituric acid (TBA) to produce MDA-TBA adduct which was quantified colorimetrically using a Bio-Tek microplate reader at 532 nm. The content of MDA in cell lysate was determined from TBARS standard curves.
The levels of 4-HNE adduct in the cell culture medium was quantified by Oxiselect ™ HNE Adduct Competitive ELISA kit according to the manufacturer’s instructions.
2.7 Measurement of intracellular ROS (iROS) production by fluorescent imaging microscopy and flow cytometry
Cells were incubated with oxidants or LPS-EK for 2 hr. The iROS production was determined using the CellROX deep red reagent by fluorescent imaging microscopy and flow cytometry as per manufacturer’s instructions and as previously described [18]. Images were acquired using a fluorescence microscope (Axiovert 200M; Zeiss) at excitation and emission wavelengths of ~644/655 nm for CellROX Deep Red reagent and 405/410-550 nm for NucBlue. Fluorescent density was quantitatively analyzed by Image J software (Version 1.44, http://imagej.nih.gov/ij/).
The flow cytometric data acquisition and analysis were conducted on FACSCanto II® flow cytometer (BD Biosciences, San Jose, CA, USA). Electronic gating was used to exclude doublets and subcellular debris with forward scatter threshold set at 500. The fluorescence intensity corresponding to iROS was determined using Allophycocyanin (APC) filter at excitation and emission wavelengths of 645/660 nm (which corresponds the excitation/emission λ of CellROX® Deep Red reagent) with unstained cells used as negative controls for iROS. For each parameter investigated, at least 104 events (cells) were analyzed per sample. The fluorescence intensities as logarithmically amplified data were compared between different treatments.
2.8 Measurement of intracellular total antioxidant capacity (iTAOC)
Cells (5×105/well) were plated in 6- or 12-well plates and grown to 70% confluence overnight before treatment, whereas for the primary culture, cells were selected and grown overnight before use. After simulation with oxidants or LPS-EK for 2 h, cells were lysed for analysis of iTAOC using the antioxidant assay kit as per manufacturer’s instructions. The assay relies on the ability of antioxidants in the sample to inhibit the formation of oxidized ABTS® + [2,2-Azino-di-(3-ethylbenzthiazoline) sulphonate] by met-myoglobin. The amount of ABTS® + produced was monitored by reading the absorbance at 405 nm with a Bio-Tek microplate reader (Burlington, VT, USA). The capacity of the antioxidants in cell lysate was calculated from Trolox (a water-soluble tocopherol analogue) standard curves.
2.9 Quanti-Blue SEAP reporter assay
Quanti-Blue assay was used to quantify SEAP levels in cell supernatants, which is a sensitive indicator of NF-κB activation. Cells were plated in 96-well plates and grown to 70% confluence overnight. Cells were preincubated for 30 min with 4 μM EUK-134 (an antioxidant), or for 3 h with anti-TLR4/MD2 pAb or CLI-095, prior to incubation with either oxidants or LPS-EK for 2 h. Culture medium was replaced with fresh growth medium, and cells were then incubated overnight (~16 h). Aliquots of cell culture medium (5 μl) were removed and transferred to new 96-well plates containing 195 μl of pre-warmed Quanti-Blue detection reagent per well as per the manufacturer’s instructions. Color was allowed to develop for 30 min, and absorbance was read at 650 nm in a Bio-Tek microplate reader.
2.10 Preparation of nuclear and cytoplasmic extract from cells
Cells (3×106) were plated in 100 mm dishes, grown to 70% confluence overnight and then treated with oxidants or LPS-EK for 0, 15, 30, and 120 min. Nuclear fraction (NF) and cytoplasmic fraction [19] were prepared using nuclear extract kit as per manufacturer’s instructions. Briefly, the cells were first collected in ice-cold PBS supplemented with phosphatase inhibitors. Cells were then resuspended in hypotonic buffer to swell the cell membrane and make them fragile. Addition of a detergent caused leakage of the cytoplasmic proteins into lysis buffer. After collection of the CF by centrifugation at 14, 000×g for 10 min at 4°C, the nuclei were lysed and the nuclear proteins were solubilized in detergent-free lysis buffer supplemented with protease inhibitor cocktail. NF was used to determine p65 DNA binding activity using the TransAM® assay kit. NF and CF were subjected to immunoblot assay.
2.11 Preparation of whole-cell extract
RAW-Blue cells (5 × 105 cells/well) were plated in 6-well plates and grown to 70% confluence overnight. They were treated with oxidant or LPS-EK for the indicated time, and then lysed in whole cell extraction lysis buffer supplemented with appropriate protease inhibitors. Cell lysates were centrifuged at 12,000g at 4°C for 20 min to remove cell debris. Aliquots of cell extracts containing 20–30 μg of total protein were mixed with loading buffer in a total volume of 20 μl and denatured at 100 °C for 10 min.
2.12 Western blot
Equal amount (30 μg) of denatured total protein was loaded per lane, fractionated on a 4–12% Bis-Tris electrophoresis gel, and transferred onto PVDF membranes. After blocking with 5% non-fat milk in TBS-T, the membranes were probed with primary antibody for overnight. After three washes in TBS-T, the membranes were incubated for 2 h with secondary antibodies. Finally, the membranes were developed using ECL, and signals were visualized with the Fujifilm LAS-400 imaging system.
2.13 TransAM® assay
Nuclear extract was prepared for analysis of p65 DNA binding activities using TransAM® assay as per manufacturer’s instructions. Briefly, 10 ng nuclear extract was added into the 96-well plate to which an oligonucleotide containing the NF-κB consensus site (5′-GGGACTTTCC-3′) was immobilized. After 3 × washes, the primary antibodies was added for 1 h at room temperature. Then, HRP-conjugated secondary antibody was added to provide a sensitive colorimetric readout that was quantified by reading absorbance at 450 nm.
2.14 Quantification of TNF-α and IL-10 levels in RAW-Blue cells
Cells (5 × 105 cells/well) were plated in 6-well plates, grown to 70% confluence overnight and then preincubated with CLI-095 for 3 h followed by incubation with either oxidants or LPS-EK for 2 h. Culture medium was replaced with fresh growth medium in the absence of oxidants or LPS-EK. Cells were then incubated overnight for ~16 h. Culture supernatant were collected and centrifuged to remove cell debris. The contents of TNF-α and IL-10 released into the medium were quantified by using TNF-α and IL-10 ELISA kits according to the manufacturer’s instructions.
2.15 Quantification of the levels of oxidant-induced release of TNF-α and IL-6 in primary peritoneal murine macrophages (pM) derived from TLR4-WT and TLR-KO mice
To validate that oxidant stimulation of TLR4 would enhance only pro-inflammatory phenotypes, we quantified both TNF-α and IL-6 expression whose genes are confirmed to be targets of the NF-B transcriptional activity. We used pM derived from TLR4-WT and TLR4-KO mice. We quantified the levels of TNF-α and IL-6 released into the culture medium following stimulation with PPC or SIN-1 by using TNF-α and IL-6 ELISA kits according to the manufacturer’s instructions. We also stimulated TLR4 with LPS-EK as positive control. We further confirmed the role of NF-κB activation in oxidant-mediated TNF-α and IL-6 release by pretreatment of pM with CLI-095 prior to overnight incubation with either PPC or SIN-1 and quantification of the proinflammatory cytokines.
2.16 Immunoprecipitation (IP)
The supernatant of whole-cell extract was precleared by adding rabbit IgG together with protein A/G plus-agarose for 30 min at 4°C followed by centrifugation at 1,000×g for 5 min at 4°C. The lysate containing 500 μg of total protein in extraction buffer was incubated with 2.5 μg/ml IκBα (c-terminal) pAb together with protein A/G plus-agarose for overnight at 4°C. Beads with the immunoprecipitates were collected by centrifugation, washed 4 × in lysis buffer, resuspended in 1× loading buffer, then heated at 100 °C for 10 min, and subjected to immunoblot assays with anti-p-IκBα (Tyr 42) Ab.
2.17 Statistical analysis
The IBM Statistical Package for the Social Sciences (SPSS) version 22 was used to perform data analysis in all experiments. Data are presented as the mean ± SEM from at least 3 to 6 independent experiments carried out in duplicates or triplicates, where applicable, and analyzed by 1-or 2-way analysis of variance (ANONA) followed by Tukey’s post hoc tests. Significance was assigned at ≤ 0.05 %
3. Results
3.1 Expression of TLR4 in RAW-Blue cells
RAW-Blue® cells (InvivoGen®) are derived from murine macrophage cell line RAW 264.7, which expresses TLR4 complex and respond to TLR4 agonist stimulation [14]. Before determining the role of TLR4 in exogenous oxidant-induced NF-κB activation, we confirmed the expression of TLR4 at both the messenger RNA (mRNA) and protein levels using RT-PCR and Western blot, respectively. Our results affirmed that RAW-Blue cells express TLR4 at the mRNA (Fig. 1A & 1B) and protein (Fig. 1C) levels. RNA extracted from peritoneal macrophages isolated from TLR4-wild type (TLR4-WT) and TLR4 homozygous knockout (KO) mice were used as positive and negative controls, respectively.
Fig. 1. Confirmation of transcriptional and translational expression of TLR4 in RAW-Blue cells.
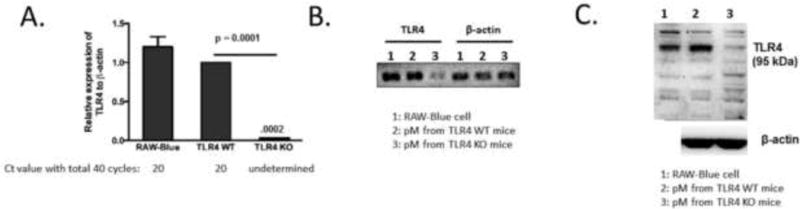
(A) Transcriptional expression of TLR4 mRNA in RAW-Blue cells by RT-qPCR. Primary peritoneal macrophages (pM) isolated from TLR4-WT and TLR4-KO mice were used as positive and negative control, respectively. B) Comparative TEB gel electrophoresis of TLR4 real time PCR products. (C) TLR4 expression in RAW-Blue cells and pM from TLR4-KO mice following subjection of cell lysates to Western blot analysis.
3.2 Determination of optimal concentrations of oxidants used
Previous kinetic studies revealed that 2 h was sufficient for PPC [20] and SIN-1[21] to completely generate their potential free radical components under physiological conditions. Therefore, we incubated cells for 2 h with oxidants (or LPS-EK) followed by one rinse with fresh media. Cells were then incubated with fresh growth medium in the absence of oxidants (or LPS-EK) for 16 h. We used this exposure protocol in all subsequent experiments. Initially, we determined the optimal effective concentrations for each oxidant using the LDH release assay. PPC (100, 500 μM), SIN-1 (1, 5 mM), and LPS-EK (10 ng/ml) did not cause significant increase in LDH release either at 2 h (Fig. 2A) or 16 h (Fig. 2B). Then, we confirmed the effects of oxidants on cell viabilities by live cell counting stained with trypan blue. At 2 h incubation (Fig. 2C), PPC (100, 500 μM), and SIN-1 (1, 5 mM) did not affect the live cell number compared with untreated cells. However at 16 h (Fig. 2D) PPC (500 μM) and SIN-1 (5 mM) significantly reduced cell numbers compared to untreated control cells, the cell number were still higher than initial seeding number (5×105 cells/well).
Fig. 2. Effects of oxidants on RAW-Blue cell viabilities.
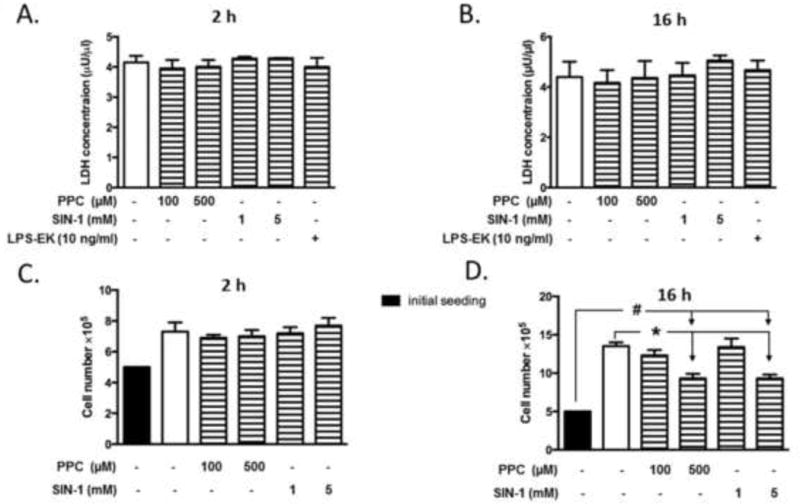
RAW-Blue cells were plated at 5 × 105 cells/well in 24-well plates. Cells were treated with oxidants or LPS-EK at the indicated concentrations for 2 h. Media was collected and cells rinsed in fresh media followed by re-incubation with fresh medium in the absence of oxidants or LPS-EK for the following 16 h. The cytotoxicity resulting from treatment with PPC, SIN-1 or LPS-EK at indicated concentrations were determined by quantification of LDH released into culture medium at the end of 2 h (A) and 16 h incubation (B). Cell viabilities upon different treatments were also determined by live cell counting with trypan blue staining at 2 h (C) and 16 h (D) for cell proliferation assay. Cell # was counted at the end of 16 h treatment. The white (blank) bars represent vehicle control, while the black bars represent the initial cell seeding number of 5 × 105 cells. The data represent 3 independent experiments conducted in duplicate at different times. # p ≤ 0.01 vs initial seeding number; * p ≤ 0.01 vs untreated cells. 1-way ANOVA followed Tukey’s post hoc tests.
These results suggested that short term and high exposure to PPC (100, 500 μM) or SIN-1 (1, 5 mM) did not cause significant cell death. To obviate effects resulting from oxidant cytotoxicity, the maximal concentrations of PPC and SIN-1 used in all subsequent experiments were 500 μM and 5 mM, respectively, with incubation for 2 h.
3.3 Oxidant stimulation of RAW-Blue cells increased formation of lipid peroxides
The peroxychromate anion, CrO83−, decomposes readily in aqueous systems to release several ROS capable of causing lipid peroxidation [20]. To confirm ROS release from PPC decomposition, the level of MDA in cell culture supernatant, indicative of treatment-induced lipid peroxidation product, was quantified as TBARS. Treatment with PPC significantly increased the thiobarbituric acid reactive substances (TBARS) concentration in a dose-dependent manner (Fig. 3A). Increased TBARS levels were reduced by pretreatment with the anti-oxidant reagent EUK-134 (4 μM) (Fig. 3A), a superoxide dismutase/catalase (SOD/CAT) mimetic [22].
Fig. 3. Effects of anti-oxidants on oxidant-induced lipid peroxidation, and confirmation of protein tyrosine nitration in RAW-Blue cells.
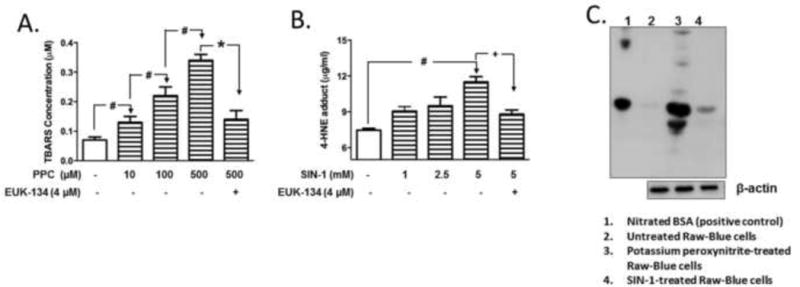
Cells were preincubated with antioxidant EUK-134 (4 μM) for 30 min, followed by stimulation with PPC (A) or SIN-1 (B) at various concentrations for 2 h. Cell culture medium was used to quantify the end product of MDA-TBARs as for PPC treatment, and 4-HNE for SIN-1 treatment according to the manufacturer’s instructions. The data represent 3 independent experiments carried out in duplicates. # p ≤ 0.01, * ≤ 0.01,+p ≤ 0.05. (C) Representative immunoblots of nitrated protein. Cells were treated with equimolar concentration (1 mM) of either potassium peroxynitrite (PN) or SIN-1 for 2 h and cell lysates were subjected to immunoblot using anti-nitrotyrosine. Nitrated BSA was used as positive marker for protein nitration.
Due to interference of SIN-1 in the TBARS assay, levels of 4-hydroxynonenal (4-HNE), another major end product of lipid peroxidase, were measured in cell culture supernatant. SIN-1 (5 mM) significantly increased the concentration of 4-HNE, which was also reduced by preincubation with EUK-134 (4 μM) (Fig. 3B).
To examine the generation of PN from SIN-1, we confirmed the extent of protein tyrosine nitration following SIN-1 treatment by Western blot. Treatment with SIN-1 produced a single band of nitrated protein confirming its effectiveness in generating nitrated proteins (Fig. 3C). Potassium PN, which releases PN directly in situ, was used as comparative positive control. We used nitrated bovine serum albumin was used as positive marker of nitrated proteins.
3.4 Stimulation of RAW-Blue cells increased iROS production
Using fluorescent microscopy imaging with Image J analysis, we showed that 2 h treatment with PPC (500 μM) or SIN-1 (5 mM) increased iROS production by a mean of 4.2- and 3.6-fold over control cells, respectively, under the same conditions (Fig. 4A and 4B). Enhanced iROS following treatment with PPC and SIN-1 was further confirmed by flow cytometry. Incubation of cells with PPC (500 μM), or SIN-1 (5 mM) for 2 h increased iROS production by a mean of 25%, and 30%, over control cells, respectively (Fig. 4 C and 4D). The positive control treatment with TLR4 agonist LPS-EK (10 ng/ml) also increased iROS production.
Fig. 4. Effects of oxidants on levels of intracellular ROS (iROS) and total antioxidant capacity (iTAOC) following stimulation of RAW-Blue cells.
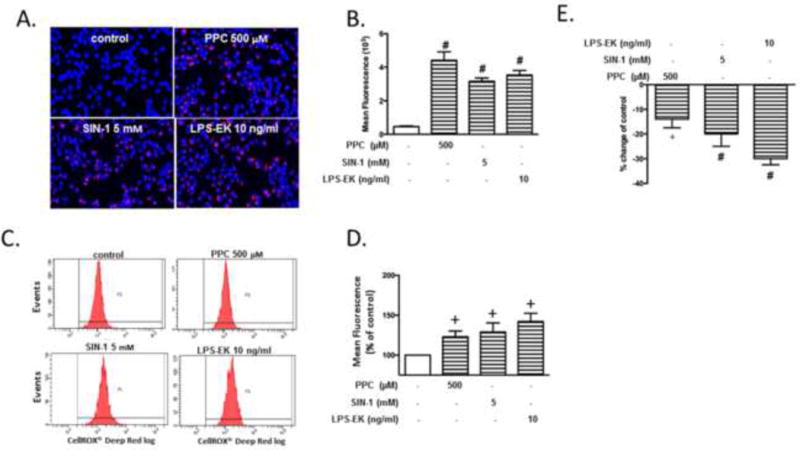
Cells were treated with oxidants or LPS-EK for 2 h followed by determination of iROS production and iTAOC. (A) Cells were incubated with CellROX™ or together with NucBlue live cell stain followed by image acquisition using a fluorescence microscope. Merged representative pictures of fluorescent images (B) Semi-quantitative histograms of (A) generated by using image J software. The data represent 4 independent experiments. #p ≤ 0.01 vs control (C) The fluorescence intensity following CellROX incubation was also analyzed by flow cytometer with representative images of the flow cytometry data shown in (D) Quantitative histograms of the fluorescence intensity. The data represent 3 independent experiments. +p ≤ 0.05 vs control (E) Cell lysates were subjected to total antioxidants capacity according to the manufacturer’s instructions. Intracellular total antioxidant capacity (iTAOC) was quantified as mM Trolox equivalents (TE). % Change of control was calculated as [(TEtreatment –TEcontrol) ⁄ TEcontrol ×100%] to represent the effects of oxidants or LPS-EK on iTAOC over control cells. The data represent 3 independent experiments carried out in triplicate. # p ≤ 0.01 vs control (0 %), +p ≤ 0.05 vs control (0%), 1-way ANOVA in all cases followed by Tukey’s post-hoc tests.
3.5 Stimulation of RAW-Blue cells decreased intracellular TAOC
The burden of ROS production is largely counteracted by an intricate antioxidant defense system [23]. After we determined that oxidants increased iROS production in RAW Blue cells, we also examined the effects of PPC or SIN-1 on iTAOC. Treatment of cells with PPC (500 μM) or SIN-1 (5 mM) for 2 h decreased iTAOC by mean values of 12 % and 19 %, respectively, compared with control (Fig. 4E). LPS-EK (10 ng/ml) decreased iTAOC by 30% in comparison with control cells. These results affirmed that oxidants induced intra-cellular oxidative stress.
3.6 TLR4 mediates oxidant-stimulated NF-κB activation
We had earlier demonstrated that TLR4 mediates exogenous oxidant-induced NF-κB activation in an artificial cell system HEK-Blue mTLR4 cells [8]. In the preset study, we confirmed the role of TLR4 in a more biologically relevant macrophage cell line.
First, to determine the effects of oxidants on the transcriptional activity of NF-κB, we quantified the levels of SEAP, a reporter gene widely used to study promoter activity or gene expression of NF-κB, in the RAW-Blue cell culture medium. Both PPC and SIN-1 induced SEAP release in a dose-dependent manner in this cell model (Fig. 5A and 5B). PPC (500 μM) (Fig. 5A) and SIN-1 (5 mM) (Fig. 5B) treatment induced SEAP release by 3.2- and 2.7-fold over the same cells in the absence of either of the oxidants, respectively. To validate and confirm the experimental system, we incubated cells with LPS-EK, as a positive control of TLR4 activation, which produced a robust SEAP release.
Fig. 5. The role of TLR4 in oxidants induced secreted embryonic alkaline phosphatase (SEAP) release in RAW-Blue cells.
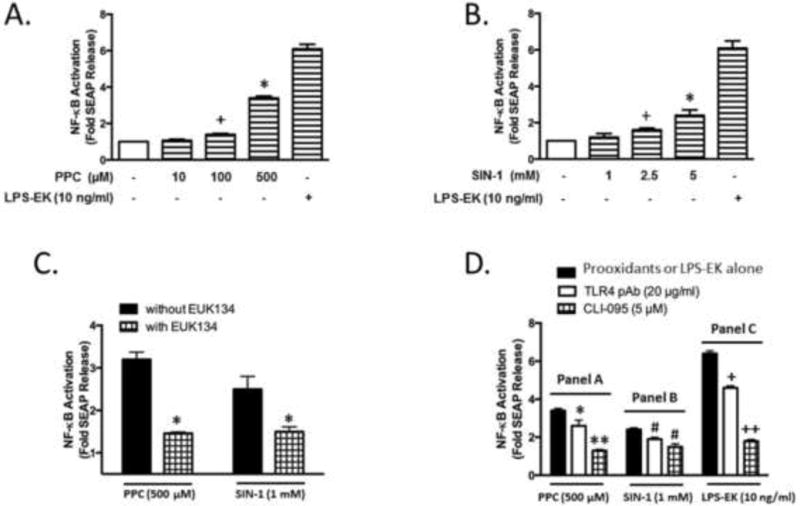
Cells were exposed to oxidants or LPS-EK at the indicated concentrations for 2 h followed by incubation with complete growth medium (oxidants or LPS-EK free) for next 16 h. Anti-TLR4/MD2 pAb (20 μg/ml) or CLI-095 (5 μM) was added 3 h before oxidants or LPS-EK stimulation. SEAP released in the culture medium at the end of 16 h incubation was determined using Quanti-Blue assay as per the manufacturer’s instructions, and the absorbance was read at 650 nm. (A) PPC concentration-dependent SEAP release. The data represent 6 independent experiments carried out in triplicate. *p ≤ 0.005 vs control; +p ≤ 0.05 vs control. (B) SIN-1 stimulated SEAP release in a dose-dependent manner. The data represent 6 independent experiments carried out in triplicate. +p ≤ 0.05 vs control, *p ≤ 0.005 vs control. (C) Pretreatment with anti-oxidant reagent EUK-134 (4 μM) inhibited PPC or SIN-1 induced SEAP release. The data represent 4 independent experiments carried out in triplicate. *p ≤ 0.005 vs treatment with PPC or SIN-1 alone. (D) Pretreatment with Anti-TLR4 pAb or CLI-095 reduced oxidants or LPS-EK induced SEAP release in RAW-Blue cells. The data represent 4 independent experiments carried out in triplicate. *p ≤ 0.05, **p ≤ 0.005 vs treatment with PPC alone; # p ≤ 0.05 vs SIN-1 treatment alone; +p ≤ 0.05 vs LPS-EK treatment alone, ++p ≤ 0.005 vs LPS-EK treatment alone, 1-way ANOVA in all cases followed by Tukey’s post-hoc tests.
To further clarify the role of the oxidants in activating NF-κB, we pretreated the cells with ROS scavenger EUK-134 (4 μM) for 30 min prior to stimulation with oxidants. Preincubation with EUK-134 decreased PPC- or SIN-1-induced SEAP release by 53.1% and 44.4% compared with cells stimulated with PPC or SIN-1 alone, respectively (Fig. 5C).
To confirm the role of TLR4 in oxidant-mediated NF-κB activation, RAW-Blue cells were pretreated with anti-TLR4/MD2 pAb or a specific TLR4 signaling inhibitor CLI-095 for 3 h before oxidants or LPS-EK treatments. Pre-incubation of the cells with anti-TLR4 pAb inhibited PPC- or SIN-1-mediated SEAP release by 23.5% or 20.8%, respectively (Fig. 5D). Similarly, pre-treatment with CLI-095 inhibited PPC- or SIN-1-mediated SEAP release by 61.8% or 37.5%, respectively (Fig. 5D). Pre-incubation with anti-TLR4 pAb or CLI-095 reduced SEAP release induced by LPS-EK by 28.1% and 71.9%, respectively. These results suggest that TLR4 is involved in mediating oxidant-stimulated NF-κB activation measured by quantification of the reporter gene SEAP expression in macrophages.
3.7 TLR4 mediates oxidant-induced NF-κB p65 DNA binding activities
To further confirm the role of TLR4 in oxidants mediated NF-κB activation, DNA binding capacity of p65 in nuclear extract was quantified by TransAM® assay. PPC (500 μM) increased the DNA binding activity of p65 by 22.5%, 83.5% and 68.0%, at 15, 30 and 120 min, respectively, compared with untreated controls (with time 0 min) (Fig. 6). Whereas, SIN-1(1 mM) initially decreased the DNA binding activity of p65 by 13% at 15 min, but then it increased it by 32% at 30 min, which returned to the resting state or base line levels at 120 min.
Fig. 6. The role of TLR4 in oxidants stimulated p65 DNA binding activity in cellular nuclear fraction.
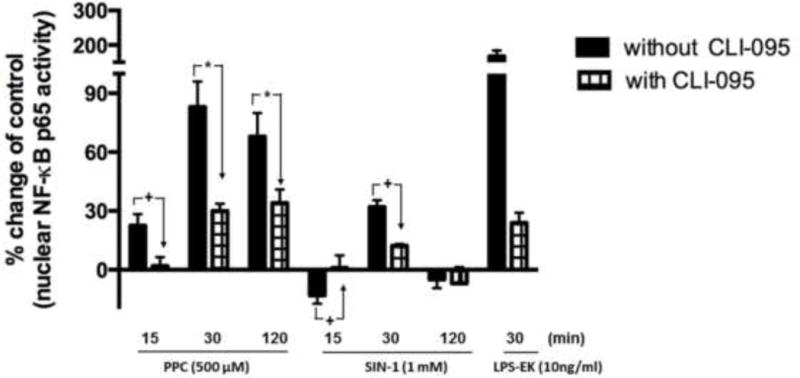
Cells were preincubated with CLI-095 for 3 h followed by stimulation with either oxidants for 0, 15, 30 and 120 min, or LPS-EK for 30 min. Nuclear faction (NF) was prepared with a kit according to the manufacturer’s. p65 DNA binding activity in NF was determine by TransAM® assay as per manufacturer’s instruction, and the absorbance was read at 450 nm. % Change of control was calculated as [(ODtreatment –ODcontrol) ⁄ ODcontrol ×100%] to represent the effects of oxidants or LPS-EK on p65 DNA binding activity over control untreated cells. The data represent 3 independent experiments carried out in duplicate. +p ≤ 0.05, *p ≤ 0.001, 2-way ANOVA followed by Tukey’s post-hoc tests.
The increased transcriptional activities of p65 caused by PPC were decreased by 20%, 53%, and 34% in the presence of CLI-095 (Fig. 6). Similarly, pretreatment with CLI-095 decreased the stimulated DNA binding activities of p65 caused by SIN-1 treatment at 30 min. LPS-EK, as a positive activator of TLR4, increased DNA binding activity by 124% at 30 min which also was blocked by pretreatment with CLI-095. These results affirmed that TLR4 activation was required in oxidants stimulated NF-κB activation.
3.8 Oxidants stimulate NF-κB nuclear translocation
To further confirm NF-κB activation by oxidants, we determined the level of p65 nuclear translocation. Complete fractionation of cytoplasmic fraction (CF) [19] and nuclear fraction (NF) was confirmed by Western blot (Fig. 7). α-Tubulin, a marker for CF was undetectable in the NF, and lamin B, a marker for NF, was undetectable in the CF. β-Actin present in both NF and CF was used as a housekeeping gene for semi-quantitative normalization because the expression of lamin B was affected by PPC (Fig. 7A) and SIN-1 (Fig. 7B) treatment.
Fig. 7. The role of TLR4 in oxidants stimulated p65 nuclear translocation.
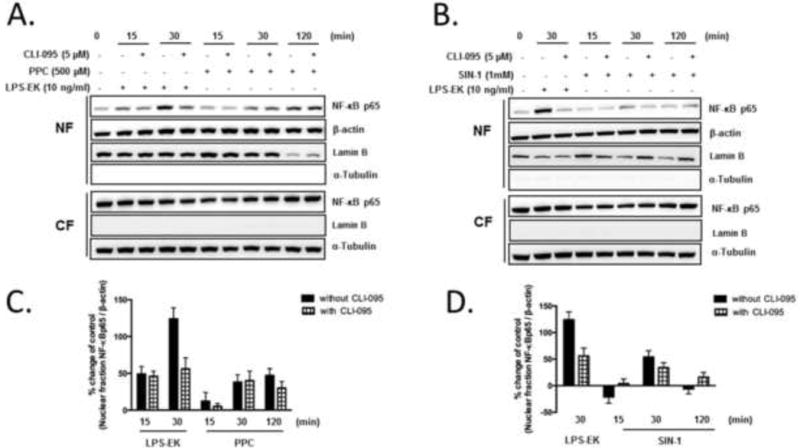
Cells were preincubated with CLI-095 for 3 h followed by stimulation with either PPC (A) or SIN-1 (B) for 0, 15, 30 and 120 min. LPS-EK stimulation for 30 min was used as a positive control of TLR4 activation. Nuclear fraction and cytoplasmic fraction were separated. The amount of NF-κB p65 in NF and CF were analyzed by Western blot. Lamin B and α-Tubulin were used as marker for NF and CF, respectively. β-Actin in NF was used as a housekeeping gene to semi-quantify the amount of p65 in NF. (C and D) The histograms represent the OD ratios of p65 immunoblot signals in NF normalized to those of β-actin in NF from (A) and (B).
At the resting state (time = 0 min), p65 was mainly sequestered in the CF with a negligible amount located in NF (Fig. 7A). PPC treatment increased the levels of p65 in NF in a time-dependent manner with maximal effect at 120 min, but with no significant effect on levels of p65 in CF (Fig. 7A). On the other hand, SIN-1 treatment maximally enhanced levels of p65 in NF at 30 min without affecting p65 levels in CF (Fig. 7B). LPS-EK increased nuclear levels of p65 at 30 min, which was blocked by TLR4 signal inhibitor CLI-095 (Fig. 7D). These results suggested that oxidants stimulated NF-κB p65 translocation to the nucleus, which was consistent with results of the SEAP release and TransAM® assays. The effects of CLI-095 on oxidants stimulated p65 nuclear translocation were not detected by Western blot.
3.9 Oxidants stimulated degradation and phosphorylation of IκBα
Following confirmation that oxidants stimulated NF-κB activation, we determined the mechanisms involved in this process. TLR4 agonist LPS-EK induced NF-κB activation by inducing IκBα degradation [24]. We investigated whether NF-κB activation induced by oxidants is mediated through the same mechanisms. The effects of oxidants on IκBα degradation at different time points were determined by Western blot. PPC induced IκBα degradation as early as 15 min, which lasted for up to 120 min (Fig. 8). To investigate the role of TLR4 stimulation in the process, we preincubated cells with the TLR4 signal inhibitor CLI-095 before stimulation with PPC. The results showed that PPC-mediated IκBα degradation was blocked by pretreatment with CLI-095 suggesting that TLR4 activation is required in the process (Fig. 8). LPS-EK also induced robust IκBα degradation at 30 min, which was inhibited by CLI-095 (Fig. 8).
Fig. 8. The role of TLR4 in oxidants mediated IκBα degradation in RAW-Blue cells.
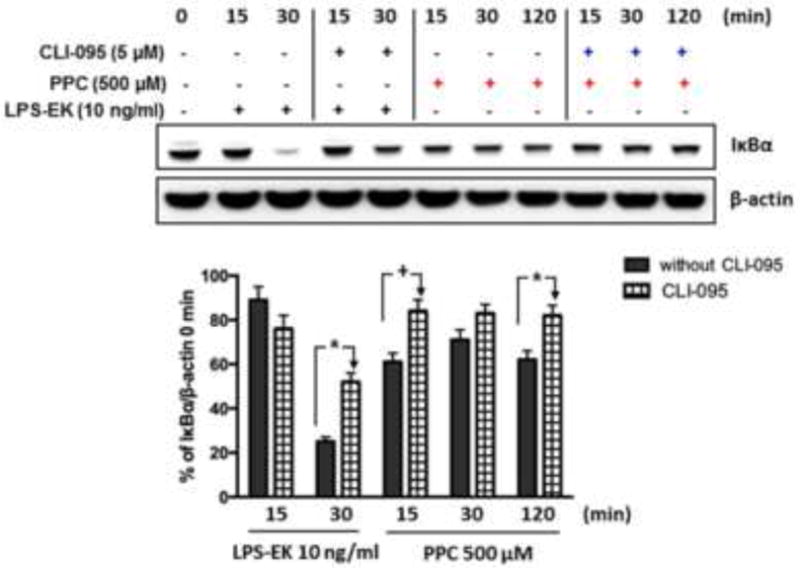
Cells were preincubated with CLI-095 for 3 h followed by stimulation with either LPS-EK or PPC for 0, 15, 30 and 120 min. IκBα degradation was determined by Western blot. % Change of OD ratios (IκBα to β-actin) compared with time = 0 min was used to represent the effects of oxidants on IκBα expression. Representative immunoblot results are shown here. The data represent 3 independent experiments conducted in duplicate (+ p ≤ 0.05, *p ≤ 0.01). 2-way ANOVA followed by Tukey’s post-hoc tests.
LPS-EK induces canonical phosphorylation of serine (Ser) 32/36 residues in IκBα leading to a rapid degradation of IκBα [24]. So we investigated whether PPC induced phosphorylation of IκBα at the same residues. Consistent with previous studies, LPS-EK appeared to stimulate phosphorylation of IκBα on Ser32 and 36 residues at 15, 30 and 120 min, whereas PPC had no effect on the Ser residues of IκBα (Figs. 9A and 9B). Phosphorylated IκBα (Tyr 42) was determined by immuno-precipitation (IP) assay with anti-IκBα Ab (C-terminal) followed by immunoblot probed with anti-p-IκBα (Tyr 42) Ab. Cells following PPC treatment exhibited enhanced Tyr 42 phosphorylation of IκBα in a time-dependent manner (Fig. 9C and 9D). PPC stimulated Tyr 42 phosphorylation in 30 min, which continued for up to 120 min. We confirmed the specificity of immunoprecipitation by stripping the membrane and probing it with anti-IκBα Ab (C-terminal). A similar level of β-actin demonstrated equal amounts of total protein was used for the IP analysis.
Fig. 9. Effects of LPS-EK and PPC on IκBα phosphorylation in RAW-Blue cells.
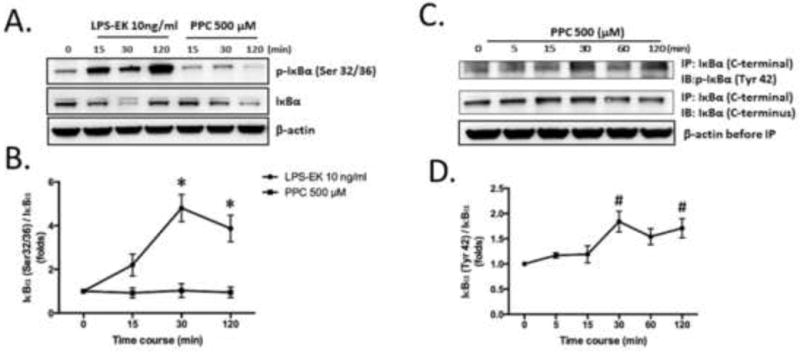
Cells were stimulated with LPS-EK or PPC for the indicated time points. IκBα phosphorylation at Ser32/36 residues was determined by Western blot (A). IκBα phosphorylation at Tyr42 was determined after immunoprecipitation (IP) followed by Western blot (B). Total IκBα was immunoprecipitated by anti-IκBα antibody (C-terminal). Anti-p-IκBα (Tyr 42) was used to probe the membrane (C and D). The time-course graphs represent the OD ratio of p-IκBα immunoblot signal from (A) and (B) after normalization with total IκBα. Representative immunoblot results are shown here. The data represent 3 independent experiments with #p ≤ 0.05 vs time 0 min, *p ≤ 0.005 vs time 0 min, 1-way ANOVA in all cases followed by Tukey’s post-hoc tests.
3.10 TLR4 mediates oxidant-induced imbalance between pro- and anti-inflammatory cytokines
Finally, we investigated the biological significance of oxidant-mediated NF-κB activation in macrophages. NF-κB is the central transcriptional regulator of a myriad of pro- and anti-inflammation mediators. We only investigated the effects of oxidants on the production of TNFα as a pro- and IL-10 as a counter balance anti-inflammatory cytokines in RAW-Blue cells specifically. The rationale for our choice was based on the observation that TNFα appears to drive the proinflammatory processes in immune cells of macrophage lineage. Exposure of these cells to PPC significantly induced TNFα production, but surprisingly, SIN-1 treatment had no measurable effect (Fig. 10A). As a positive control, we also stimulated these cells with LPS-EK (Fig. 10A). The role of TLR4 in oxidant-mediated TNFα production was further confirmed with CLI-095 (Fig. 10A), a potent TLR4 signaling inhibitor. For the anti-inflammatory mediator IL-10, both oxidants and LPS-EK had no significant effect on the levels of its production (Fig. 9B). We calculated the ratios of TNFα to IL-10 following stimulation with oxidants and LPS-EK, which represent the parameters for TLR4/ROS-coupled effects as a gauge for a potential dominant proinflammatory phenotype. Both PPC and LPS-EK treatment increased the ratio of TNFα to IL-10 whereas SIN-1 had no effect on the ratio. However, cell incubation with a direct peroxynitrite (PN)-generating agent produced a robust TNFα to IL-10 ratio (data not shown).
Fig. 10. Role of TLR4 in oxidant-induced TNFα and IL-10 production.
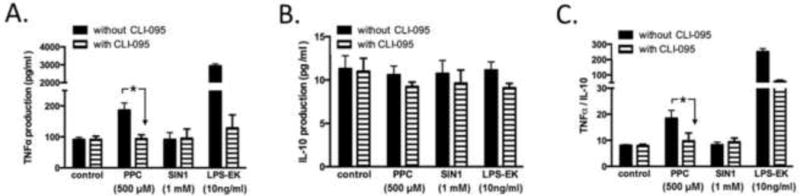
Cells were exposed to oxidants or LPS-EK for 2 h following which the media was removed. Attached cells were rinsed and then replaced with complete media without oxidants or LPS-EK for the next 16 h. CLI-095 (5 μM) was added 3 h before oxidants or LPS-EK stimulation. TNFα (A) and IL-10 (B) levels in the conditioned media were determined using their respective ELISA kits according to the manufacturer’s instructions. (C) The ratios of TNFα and IL-10 after various treatments with oxidants or LPS-EK were calculated. The data represent 3 independent experiments with *p ≤ 0.01, 2-way ANOVA in all cases followed by Tukey’s post-hoc tests.
3.11 Prooxidants increase proinflammatory cytokines TNF-α and IL-6 release in pM derived from wild-type TLR4-WT, but not in pM from TLR4-KO mice
To validate the physiological relevance of RONS-coupled stimulation of TLR4 as demonstrated in RAW-Blue cells, we measured the effect of TLR4 stimulation on TNF-α and IL-6 release. We used pM derived from TLR4-WT and TLR4-KO mice. We quantified the levels of TNFα (Fig. 11A) and IL-6 (Fig. 11B) as proinflammatory mediators, produced by the interactions of prooxidants through TLR4 stimulation. Treatment with PPC or SIN-1 significantly increased both TNFα (by 3 – 6X) and IL-6 (by 2.5 – 4X) in pM derived from TLR4-WT compared to those from TLR4-KO. We also treated pM with LPS-EK, a TLR4-specific agonist, to confirm and delineate the fidelity of the pM derived from both TLR4-WT and TLR4-KO mice. As is clearly evident (Figs. 11A & 11B), LPS-EK produced significant increases in TNFα (by 7X) and IL-6 (by 20X) only in TLR4-WT pM compared with TLR4-KO-derived cells. Overall the data indicate that increased release of both TNFα and IL-6 was due to an intact and functional TLR4 expression. There were no significant differences in basal release of TNFα and IL-6 in macrophages derived from both TLR4-WT and TLR4-KO animals in the absence of prooxidants.
Fig. 11. Effect of prooxidants on TNF-α and IL-6 release in primary murine macrophages derived from TLR4-WT and TLR4-KO mice.
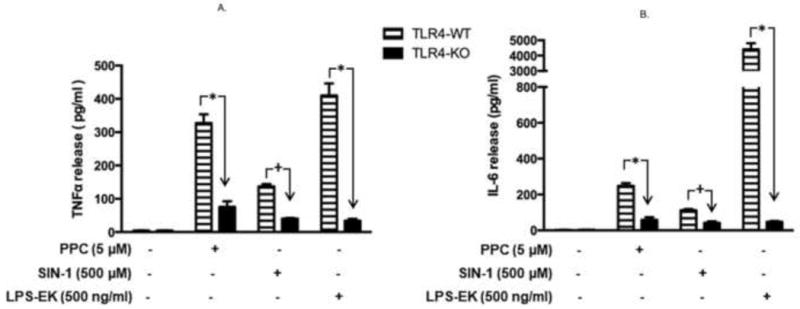
Primary macrophages derived from TLR4-WT and TLR4-KO mice were incubated overnight with PPC or SIN-1 alone. The levels of TNF-α (Fig. 11A) and IL-6 (Fig. 11B) released into the culture media were quantified using the ELISA kits of the relevant murine cytokine. The effect of LPS-EK on TNF-α and IL-6 release was used as positive control. The data represent 3 – 6 independent experiments conducted in duplicate with *p < 0.0001; +p ≤ 0.01.
We also showed that prior incubation of pM with CLI-095 followed by overnight stimulation of the cells with oxidants significantly decreased oxidant-induced TNF-α and IL-6 release only in pM derived from TLR4-WT, but not in pM from TLR4-KO (data not shown). CLI-095 suppresses both ligand and ligand-independent TLR4 signaling, and potently inhibits NF-κB activation induced by ultra-pure LPS [25]. This is further evidence that the increased proinflammatory cytokine release could only be due to a direct oxidant stimulation of TLR4 specifically through NFκB activation.
4. Discussion
In the present study, we had hypothesized that TLR4 mediates exogenous oxidant-induced NF-κB activation in macrophages. To test this hypothesis, we used murine macrophage RAW-Blue cells, which are stably transfected with NF-κB reporter gene SEAP. We confirmed the expression of TLR4 at both the mRNA and protein levels. We treated cells with ROS/RNS-producing agents (PPC or SIN-1) followed by measurement of SEAP release, DNA binding activity and nuclear translocation of p65 as a readout for NF-κB activation, and TNFα and IL-10 production as biological outcomes of NF-κB activation. We used TLR4 neutralizing antibody and a specific TLR4 signal transduction inhibitor CLI-095 to define the role of TLR4 activation. A major finding of this study is that TLR4 is involved in oxidant-mediated NF-κB activation in macrophages. The data that support these conclusions include the following findings: (i) anti-TLR4/MD2 pAb and CLI-095 significantly attenuated oxidant-induced SEAP release, and (ii) CLI-095 significantly inhibited oxidant-induced p65 DNA biding activity, IκBα degradation and imbalance between TNFα and IL-10 production. Thus, our findings have further defined the role of TLR4 in exogenous (environmental) oxidant-mediated NF-κB activation in innate immune cells with expansion and confirmation of our previous work [18].
On one hand, we used the physiological decomposition of PPC in the extracellular milieu as a source of ROS to study their activation of NF-κB through coupled stimulation of TLR4 based on their effects on cellular and biochemical function [26, 27]. Peroxychromate anion, CrO83−, decomposes readily in aqueous systems to release several ROS, including hydrogen peroxide, hydroxyl radical, singlet oxygen, and possibly superoxide. Singlet oxygen and hydroxyl radicals have been suggested as the primary lipid oxidants. Thus, we confirmed ROS release from PPC decomposition by quantifying the end-products of lipid peroxidation, MDA and 4-HNE. Consistent with previous studies [20, 26, 28], in vitro exposure of cells to PPC resulted in the release of lipid peroxidation products into the culture medium, which was attenuated by EUK-134 (Fig. 3A). EUK-134, a synthetic serum stable ROS scavenger reduced the propagation rate of ROS-induced lipid peroxidation [29]. These results suggest that singlet oxygen and hydroxyl radical were released following PPC decomposition.
On the other hand however, SIN-1 under physiological condition produces both nitric oxide (NO) and superoxide anion, which combine to form peroxynitrite (PN) anion (OONO−) [11]. PN can further decompose to nitrogen dioxide and hydroxyl radicals [30], to induce membrane lipid peroxidation [31]. In the present experiments, incubation of cells with SIN-1 increased levels of lipid peroxide 4-HNE in the culture medium, confirming the production of free radical following SIN-1 decomposition in our experimental model. We determined the levels of 4-HNE instead of MDA upon treatment with SIN-1 because SIN-1 interfered in the TABRS assay. This could perhaps be due to the morpholinyl structure within SIN-1 that may interfere with the pyrimidine cycle within TBA. In addition, PN has the capacity to nitrate phenolic compounds including free and protein-bound Tyr residues resulting in specific formation of nitrotyrosine adducts [32]. In the present study, we confirmed the formation of cellular nitrotyrosine upon treatment with SIN-1, suggesting the formation of OONO−, which has been reported in cell culture system [11]. Thus, both lipid peroxidation and protein nitration assays confirmed that PN and its decomposition product hydroxyl radical were produced during SIN-1 decomposition in the culture media.
With a combination of fluorescence microscopy (FM) and flow cytometry (FC) [33], we determined the effects of oxidants on the levels of intracellular ROS (iROS). As an indicator of oxidative stress, iROS was increased upon treatment with PPC, SIN-1 or LPS-EK, which implies that TLR4 stimulation is necessary for oxidant-induced stress that can emanate from environmental stressors.
Interestingly, we showed that FM yielded a greater magnitude of iROS than those obtained by FC for the same treatments. This could be due to loss of fluorescent positive cells inherent in sample preparation prior to FC analysis. Possibly, FM could encompass an approximation of ROS in both extra- and intra-cellular compartments, whereas FC quantified only intra-cellular fluorescence [34]. Nevertheless, accurate ROS quantification poses technical challenges. Thus, we used a combination of two approaches to quantify total iROS levels. However, it is realized that iROS production in an inappropriate place or time, for too long, at too high a level or inappropriate forms can cause disturbance of ROS homeostasis. Thus total levels of iROS ought to be used as the final or a sole indicator of disturbances of ROS homeostasis. However, we still need new strategies for a more accurate subcellular localization or quantification of iROS in free radical biology.
Using CellRox deep red reagent to detect cellular oxidative stress has some limitations as well because it mainly identifies superoxide in live cells [35]. Distinguishing one type of ROS from each other by specific assays presents technical challenges as well [36]. For example, cellular oxidative stress caused by hydrogen peroxide cannot be detected. To overcome this limitation, we also quantified another marker of oxidative stress, iTAOC, which has been used to monitor oxidative status in cancer patients [37] and in infants exposed to passive smoking [38]. Cells inherently possess antioxidant defense system to counteract both exogenous and endogenous ROS and reduce their damaging effects. The cellular catabolism of ROS includes (i) enzymes such as superoxide dismutase (SOD), GSH (glutathione) reductase, catalase, peroxidase etc., and (ii) an array of small molecules reacting with ROS non-enzymatically, including ascorbic acid, pyruvate, α-ketoglutarate, and oxaloacetate [36]. In the present study, we quantified iTAOC in cell lysate, which reflects a more comprehensive biological information of reduction-oxidation status inside of cells than measurement of any one individual component. Decrease by oxidants as well as LPS-EK in iTAOC (Fig. 4E), suggests that impaired iTAOC may fail to buffer ROS production resulting in oxidative stress. It could also be due to the fact that the ROS produced following oxidant treatment exceeded and depleted cellular antioxidants. Alternatively, it could be due to tyrosine nitration of mitochondrial Mn superoxide dismutase (SOD), which has been demonstrated to diminish the ability of cells to cope with oxidative stress [39]. The mechanism (s) of decreased iTAOC following TLR4 stimulation would require more experiments.
RAW-Blue cells are chromosomally integrated with NF-κB reporter gene SEAP and, its levels in culture medium were used as a reliable quantitative and sensitive marker for NF-κB activation. RAW-Blue cells express TLR4 complex with the myeloid differentiation factor 2 (MD-2) and CD14 co-receptors [40]. To further define the role of TLR4 in oxidant-stimulated NF-κB activation, we used anti-mouse TLR4/MD2 clone MT510, which is reported to bind to an epitope of TLR4/MD2 complex to block interaction with its ligands [41]. Soluble MD-2 is specifically physically associated with the extracellular domain of TLR4 on the cell surface, and confers responsiveness to LPS, with TLR4 being the only TLR that has been confirmed to form an active heterodimer with MD-2 [42,43]. However, our results clearly show that preincubation with anti-TLR4/MD2 pAb at the concentration we used only sub-optimally attenuated oxidant-induced SEAP release (Fig. 5D) suggesting that oxidants are able to stimulate TLR4 with its co-receptor. This suggests that TLR4 with its co-receptor MD2 mediated oxidant-induced NF-κB activation in macrophages. Because of the high cost of the antibody, we could not use a higher concentration.
Activation of intracellular signaling requires interaction between TLR4 and adaptor molecules containing Toll/IL-1 receptor (TIR) domain such as TIRAP and TRAM [44]. TIR domain of TLR4 is critical for signal transduction, because a single point mutation can abolish the response to TLR4 activation [45]. CLI-095, an intracellular TLR4 antagonist, covalently binds to the Cys747 within the TIR domain of TLR4 among 10 human TLRs [46], thus interfering with interactions between TLR4 and its adaptor molecules TIRAP and TRAM [47]. Thus, CLI-095 selectively inhibits TLR4 signaling [48,49], and showed beneficial effects in a mouse endotoxin model [49]. In agreement with other studies, our results clearly showed that pre-treatment with CLI-095 inhibited oxidant-induced SEAP release (Fig. 5D), further confirming that TIR domain of TLR4 is both required and necessary in oxidant-induced NF-κB activation. Although TIR domain of TLR4 shares high homology with that of type I interleukin I (IL-1) receptor, type I IL-1 receptor is predominantly expressed on T cells and fibroblasts. There is no report on the interaction between CLI-095 and IL-1 receptor. Furthermore, the mediatory role of TLR4 in oxidant-induced NF-κB activation has been demonstrated in peritoneal macrophages derived from tlr4−/− mice in a previous report [18].
NF-κB family comprises of five members: RelA (p65), ReIB, C-Rel, p105 and p100, which form homo-or hetero-dimers. The most frequently activated form of NF-κB in TLR4-dependent signaling is a heterodimer composed of p65 and p50 [50]. At rest (i.e., in the inactive state), NF-κB is retained in cytosol by direct interaction with inhibitory kappa B (iκB) proteins. Upon stimulation, NF-κB translocates into nucleus and binds to promoter region of target genes. Thus, in the present study, we demonstrated the oxidant-induced stimulation of NF-κB by measuring p65 DNA binding activities and nuclear translocation by TransAM® assay and Western blot, respectively. Consistent with the results in SEAP release assay, our results clearly showed increased p65 DNA binding activity (Fig. 6) and its nuclear translocation (Fig. 7) upon oxidant treatment. However, we observed a discrepancy regarding the effects of CLI-095: its inhibitory effects on p65 DNA binding activity was not detected in p65 nuclear translocation by Western blot. Compared with semi-quantification of p65 in nuclear fraction by western blot, TransAM® assay is a more accurate assay to quantify transcriptional factor activation. First, it is an ELISA based assay. Second, the primary antibody used in TransAM® assay specifically recognizes transcriptional active form of p65 when it binds to DNA. Therefore, Trans AM assay quantifies transcriptionally active form of p65 while Western blot is semi-quantitative measurement of total amount p65, which may produce less reliable results.
Next, we determined the mechanisms of NF-κB activation in response to oxidants. In quiescent cells, NF-κB remains sequestered in the cytoplasm bound to IκBα proteins. With “sterile” inflammation or oxidant stress, IκBα is phosphorylated, causing dissociation and unmasking of the nuclear localization sequence of NF-κB. Classically, phosphorylation of IκBα occurs on Ser residues at positions 32/36, with subsequent rapid degradation through the proteosomal pathway, which leads to NF-κB activation [4]. Alternatively, phosphorylation of IκBα on Tyr 42 residue is an atypical pathway of NF-κB activation, which occurs after stimulation with pervanadate, nerve growth factor, H2O2, ischemia-reperfusion, and hypoxia [24, 51, 52]. Previously, we have shown that oxidants can activate NF-κB through Tyr phosphorylation [9, 24]. Our present study clearly suggests different post-TLR4 mechanisms of NF-κB activation. First, oxidant-induced degradation of IκBα occurred as early as 15 min and continued up to 120 min (Fig. 8 and 9A) suggesting both rapid and delayed effects, whereas levels of IκBα went up back to basal level after LPS-EK treatment for 120 min (Fig. 9A) suggesting a possible resynthesis of IκBα [24]. Second, PPC induced robust phosphorylation at the Tyr 42 residue but not Ser residue 32/36, which was observed following LPS-EK treatment (the native ligand for TLR4) (Fig. 9A and 9C). This suggests that oxidants can stimulate NF-κB activation mainly by phosphorylation at the Tyr 42 residue with subsequent degradation of IκBα. Our study is in agreement with the suggested mechanism of NF-κB activation by pervanadate or UV radiation, in which phosphorylation of IκBα at Tyr42 residue was necessary for NF-κB activation [52,53].
Differences in TLR4 signaling in response to damage-associated molecular patterns (DAMPs) released by oxidative stress compared to pathogen-associated molecular patterns (PAMPs) released by “infective” stress are beginning to emerge. For example, high mobility group box 1 (HMGB1), a DAMP molecule, activates both IKKα and IKKβ, but LPS stimulate the activity of IKKβ only in cultured neutrophils and macrophages [54].
A major outcome of NF-κB activation in macrophages is the production of myriad of cytokines including TNFα and chemokines [55]. The correlation between NF-κB activity and the severity of inflammation has been established, but their association is difficult to interpret. This is because the production of pro- and anti-inflammatory cytokines during inflammation orchestrates the inflammatory response [6]. TNFα appears to generate a feed-forward mechanism to amplify the inflammatory response by regulating the expression of other pro-inflammatory cytokines including IL-1, IL-6, PDGF, and TGF-β and enhancing chemotaxis of macrophages and neutrophils at the site of inflammation [56]. However, IL-10 showed multifaceted anti-inflammatory effects, such as inhibition of NF-κB activation, resulting in suppression of cytokines production [57].
Thus, we determined the production of pro-inflammatory mediator TNFα and anti-inflammatory mediator IL-10 following different oxidant treatment. We also calculated their ratios to represent the balance of biological effects between the two cytokines. Our results clearly showed that stimulation of TLR4 signaling pathway by PPC significantly induced the production of TNFα but not IL-10, thus increasing the ratio between them (Fig. 9). This suggests that exogenous oxidants may trigger pro-inflammatory response by releasing TNFα in the absence of counteracting anti-inflammatory effects from IL-10. Interestingly, SIN-1 had little effect on the expression of TNFα. This may be related to the treatment condition or/and cell model system we used. Our previous studies clearly showed that treatment with SIN-1 (1 mM) overnight significantly stimulated the production of TNF-α and IL-6 in primary murine peritoneal macrophages [18].
The detailed molecular mechanisms by which oxidants may activate TLR4 are not yet fully appreciated or understood. Speculatively, this could be because ROS can alter plasma membrane fluidity resulting in the ligand-independent clustering of membrane receptors [29]. Alternatively, exogenous oxidants may directly activate TLR4 through oxidization of its cysteine residue specifically to change the receptor structural configuration. ROS can oxidize a subset of sulfur atoms in the side chains of cysteine or methionine residues to form sulfenic acid moieties, which are unstable, but are able to form disulfide bridges with one another, thereby resulting in a change in structural configuration and activity [58]. This provides a foundation for our hypothesis, which may require more biophysical experiments to further clarify it.
Because of the association of the contribution of TLRs dysfunction in many disease states, developing highly specific small-molecule inhibitors for these innate immune receptors is a dynamic research area in drug discovery/development. Previously used strategies suffered from significant shortcomings, e.g., LPS-RS, which is a potent LPS-mimicking antagonist of TLR4 that targets MD-2 co-receptor [59], or Eritoran, an LPS mimetic [60], both showed poor pharmacological profiles [61]. Even CLI-095, the most potent/active TLR4 binding small-molecule antagonist, failed to suppress cytokine levels in patients with sepsis and shock [62]. Therefore, novel strategies, e.g., specific inhibition of the TLR4/MD-2 association, might eventually lead to selective drug candidates that target TLR4 signaling pathway [63]. Overall, the modulation of iROS levels is crucial for cellular micro-homeostasis, and different ROS levels can induce different biological responses.
In conclusion, we show that environmental and intrinsic oxidants are capable of promoting macrophages toward the pro-inflammatory phenotype by a TLR4-dependent pathway. We have confirmed the functional significance of oxidant-induced TLR4-signaling through NFB activation with the pM derived from TLR4-WT and TLR4-KO mice. Our present findings suggest that oxidant-induced activation of TLR4 signaling pathway is potentially associated with the initiation, propagation and maintenance of “sterile” inflammation of innate immunity that may act as the nucleus of chronic disease processes [64, 65]. Thus our data lend support to a still emerging hypothesis that all disease processes may have a common mechanistic origin in the cumulative effects over time of oxidant non-lethal stress [66].
Acknowledgments
The authors will like to thank Dr. Michael Wacker for use of his immunofluorescence microscope and Dr. Mingui Fu for use of his RT-PCR equipment. The project was supported by NIH-NIDCR 021888 (OJI). This work is part of the doctoral thesis for YZ.
Abbreviations
- ONS
oxidative/nitrosative stress
- TLR
toll-like receptor
- MD
myeloid of differentiation
- CD
cluster of differentiation
- pAb
polyclonal antibody
- LPS-EK (Ultrapure)
lipopolysaccharide from E. coli K12
- TLR4-KO macrophages
macrophages derived from complete TLR4 knock-out mice
- iTAOC
intracellular total antioxidant capacity
- LDH
lactate dehydrogenase
- TLR4-WT macrophages
macrophages derived from wild-type mice
- pM
primary peritoneal macrophages
- PPC
potassium peroxychromate
- SEAP
secreted embryonic alkaline phosphatase
- FBS
fetal bovine serum
- DMEM
Dulbecco’s modified Eagle’s medium
- ANOVA
analysis of variance
- MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- TBARS
thiobarbituric acid reacting substances
- MDA
malonyldialdehyde
- NF-κB
nuclear factor kappa B
- iROS
intracellular reactive oxygen species
- TNF-α
tumor necrosis factor alpha
- ELISA
enzyme-linked immuno-sorbent assay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest:
The authors declare no conflicts of interest.
References
- 1.Gill R, Tsung A, Billiar T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med. 2010;48:1121–32. doi: 10.1016/j.freeradbiomed.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lucas K, Maes M. Role of the Toll Like Receptor (TLR) Radical Cycle in Chronic Inflammation: Possible Treatments Targeting the TLR4 Pathway. Mol Neurobiol. 2013:190–204. doi: 10.1007/s12035-013-8425-8427. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paul-Clark MJ, McMaster SK, Sorrentino R, Sriskandan S, Bailey LK, Moreno L, Ryffel B, Quesniaux VF, Mitchell JA. Toll-like receptor 2 is essential for the sensing of oxidants during inflammation. Am J Respir Crit Care Med. 2009;179:299–306. doi: 10.1164/rccm.200707-1019OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janssen-Heininger YM, Poynter ME, Baeuerle PA. Recent advances towards understanding redox mechanisms in the activation of nuclear factor kappaB. Free Radic Biol Med. 2000;28:1317–1327. doi: 10.1016/s0891-5849(00)00218-5. [DOI] [PubMed] [Google Scholar]
- 5.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawrence T, Gilroy DW. Chronic inflammation: a failure of resolution? Int J Exp Pathol. 2007;88:85–94. doi: 10.1111/j.1365-2613.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. J Pathol. 2008;214:161–178. doi: 10.1002/path.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karki R, Igwe OJ. Toll-like receptor 4-mediated nuclear factor kappa B activation is essential for sensing exogenous oxidants to propagate and maintain oxidative/nitrosative cellular stress. PLoS One. 2013;8:e73840. doi: 10.1371/journal.pone.0073840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karki R, Zhang Y, Igwe OJ. Activation of c-Src: a hub for exogenous pro-oxidant-mediated activation of Toll-like receptor 4 signaling. Free Radic Biol Med. 2014;71:256–69. doi: 10.1016/j.freeradbiomed.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miesel R, Kroger H, Kurpisz M, Weser U. Induction of arthritis in mice and rats by potassium peroxochromate and assessment of disease activity by whole blood chemiluminescence and 99mpertechnetate-imaging. Free Radic Res. 1995;23:213–227. doi: 10.3109/10715769509064035. [DOI] [PubMed] [Google Scholar]
- 11.Trackey JL, Uliasz TF, Hewett SJ. SIN-1-induced cytotoxicity in mixed cortical cell culture: peroxynitrite-dependent and -independent induction of excitotoxic cell death. J Neurochem. 2001;79:445–455. doi: 10.1046/j.1471-4159.2001.00584.x. [DOI] [PubMed] [Google Scholar]
- 12.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008 doi: 10.1002/0471142735.im1401s83. Chapter 14, Unit 14 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Cui Y, Cao F, Qin Y, Li W, Zhang J. Ganglioside GD1a suppresses LPS-induced pro-inflammatory cytokines in RAW264.7 macrophages by reducing MAPKs and NF-kappaB signaling pathways through TLR4. Int Immunopharmacol. 2015;28:136–145. doi: 10.1016/j.intimp.2015.05.044. [DOI] [PubMed] [Google Scholar]
- 15.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 16.Johnson G, Nour AA, Nolan T, Huggett J, Bustin S. Minimum information necessary for quantitative real-time PCR experiments. Methods Mol Biol. 2014;1160:5–17. doi: 10.1007/978-1-4939-0733-5_2. [DOI] [PubMed] [Google Scholar]
- 17.Zhong H, Yin H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox biology. 2015;4:193–199. doi: 10.1016/j.redox.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Karki R, Igwe OJ. Toll-like receptor 4 signaling: A common pathway for interactions between prooxidants and extracellular disulfide high mobility group box 1 (HMGB1) protein-coupled activation. Biochem Pharmacol. 2015;98:132–143. doi: 10.1016/j.bcp.2015.08.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002;22:644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards JC, Quinn PJ. Decomposing potassium peroxychromate produces hydroxyl radical (.OH) that can peroxidize the unsaturated fatty acids of phospholipid dispersions. J Lipid Res. 1982;23:994–1000. [PubMed] [Google Scholar]
- 21.Thome U, Lazrak A, Chen L, Kirk MC, Thomas MJ, Forman HJ, Matalon S. Novel SIN-1 reactive intermediates modulate chloride secretion across murine airway cells. Free Radic Biol Med. 2003;35:662–675. doi: 10.1016/s0891-5849(03)00392-7. [DOI] [PubMed] [Google Scholar]
- 22.Rong Y, Doctrow SR, Tocco G, Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc Natl Acad Sci U S A. 1999;96(17):9897–9902. doi: 10.1073/pnas.96.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 24.Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J Biol Chem. 2003;278:24233–24241. doi: 10.1074/jbc.M212389200. [DOI] [PubMed] [Google Scholar]
- 25.Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. 2008;584(1):40–48. doi: 10.1016/j.ejphar.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 26.Baird MB, Massie HR, Piekielniak MJ. Formation of lipid peroxides in isolated rat liver microsomes by singlet molecular oxygen. Chem Biol Interact. 1977;16:145–53. doi: 10.1016/0009-2797(77)90124-7. [DOI] [PubMed] [Google Scholar]
- 27.Hodgson EK, Fridovich I. The production of superoxide radical during the decomposition of potassium peroxochromate(V) Biochemistry. 1974;13:3811–3815. doi: 10.1021/bi00715a030. [DOI] [PubMed] [Google Scholar]
- 28.Sharma SK, Ebadi M. Metallothionein attenuates 3-morpholinosydnonimine (SIN-1)-induced oxidative stress in dopaminergic neurons. Antioxid Redox Signal. 2003;5:251–264. doi: 10.1089/152308603322110832. [DOI] [PubMed] [Google Scholar]
- 29.Decraene D, Smaers K, Gan D, Mammone T, Matsui M, Maes D, Declercq L, Garmyn M. A synthetic superoxide dismutase/catalase mimetic (EUK-134) inhibits membrane-damage-induced activation of mitogen-activated protein kinase pathways and reduces p53 accumulation in ultraviolet B-exposed primary human keratinocytes. J Invest Dermatol. 2004;122:484–91. doi: 10.1046/j.0022-202X.2004.22215.x. [DOI] [PubMed] [Google Scholar]
- 30.Hogg N, Darley-Usmar VM, Wilson MT, Moncada S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochem J. 1992;281(Pt 2):419–424. doi: 10.1042/bj2810419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys. 1991;288:481–487. doi: 10.1016/0003-9861(91)90224-7. [DOI] [PubMed] [Google Scholar]
- 32.Beckman JS, Ischiropoulos H, Zhu L, van der Woerd M, Smith C, Chen J, Harrison J, Martin JC, Tsai M. Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-v. [DOI] [PubMed] [Google Scholar]
- 33.Moretto MB, Arteni NS, Lavinsky D, Netto CA, Rocha JB, Souza DO. WofchukS Hypoxic-ischemic insult decreases glutamate uptake by hippocampal slices from neonatal rats: prevention by guanosine. Exp Neurol. 2005;195:400–406. doi: 10.1016/j.expneurol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Muratori M, Forti G, Baldi E. Comparing flow cytometry and fluorescence microscopy for analyzing human sperm DNA fragmentation by TUNEL labeling. Cytometry A. 2008;73:785–787. doi: 10.1002/cyto.a.20615. [DOI] [PubMed] [Google Scholar]
- 35.Plaza Davila M, Martin Munoz P, Tapia JA, Ortega Ferrusola C, Balao da Silva CC, Pena FJ. Inhibition of Mitochondrial Complex I Leads to Decreased Motility and Membrane Integrity Related to Increased Hydrogen Peroxide and Reduced ATP Production, while the Inhibition of Glycolysis Has Less Impact on Sperm Motility. PLoS One. 2015;10:e0138777. doi: 10.1371/journal.pone.0138777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13:349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel BP, Rawal UM, Dave TK, Rawal RM, Shukla SN, Shah PM, Patel PS. Lipid peroxidation, total antioxidant status, and total thiol levels predict overall survival in patients with oral squamous cell carcinoma. Integr Cancer Ther. 2007;6:365–372. doi: 10.1177/1534735407309760. [DOI] [PubMed] [Google Scholar]
- 38.Aycicek A, Erel O, Kocyigit A. Decreased total antioxidant capacity and increased oxidative stress in passive smoker infants and their mothers. Pediatr Int. 2005;47:635–639. doi: 10.1111/j.1442-200x.2005.02137.x. [DOI] [PubMed] [Google Scholar]
- 39.MacMillan-Crow LA, Crow JP, Kerby JD, Beckman JS, Thompson JA. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci U S A. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 41.Laplante P, Amireault P, Subang R, Dieude M, Levine JS, Rauch J. Interaction of beta2-glycoprotein I with lipopolysaccharide leads to Toll-like receptor 4 (TLR4)-dependent activation of macrophages. J Biol Chem. 2011;286:42494–42503. doi: 10.1074/jbc.M111.230383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dziarski R, Gupta D. Role of MD-2 in TLR2- and TLR4-mediated recognition of Gram-negative and Gram-positive bacteria and activation of chemokine genes. J Endotoxin Res. 2000;6:401–405. doi: 10.1179/096805100101532243. [DOI] [PubMed] [Google Scholar]
- 43.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 45.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 46.Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol. 2011;79:34–41. doi: 10.1124/mol.110.068064. [DOI] [PubMed] [Google Scholar]
- 47.Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y, Ii M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol. 2009;157:1250–1262. doi: 10.1111/j.1476-5381.2009.00297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, Hazeki O, Kitazaki T, Iizawa Y. A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-Chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol Pharmacol. 2006;69:1288–1295. doi: 10.1124/mol.105.019695. [DOI] [PubMed] [Google Scholar]
- 49.Sha T, Sunamoto M, Kitazaki T, Sato J, Ii M, Iizawa Y. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur J Pharmacol. 2007;571:231–239. doi: 10.1016/j.ejphar.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 50.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 51.Wright CJ, Zhuang T, La P, Yang G, Dennery PA. Hyperoxia-induced NF-kappaB activation occurs via a maturationally sensitive atypical pathway. Am J Physiol Lung Cell Mol Physiol. 2009;296:L296–L306. doi: 10.1152/ajplung.90499.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mukhopadhyay A, Manna SK, Aggarwal BB. Pervanadate-induced nuclear factor-kappaB activation requires tyrosine phosphorylation and degradation of IkappaBalpha. Comparison with tumor necrosis factor-alpha. J Biol Chem. 2000;275:8549–8555. doi: 10.1074/jbc.275.12.8549. [DOI] [PubMed] [Google Scholar]
- 53.Li N, Karin M. Ionizing radiation and short wavelength UV activate NF-kappaB through two distinct mechanisms. Proc Natl Acad Sci U S A. 1998;95:13012–13017. doi: 10.1073/pnas.95.22.13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 55.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Larrick JW, Wright SC. Cytotoxic mechanism of tumor necrosis factor-alpha. FASEB J. 1990;4:3215–3223. doi: 10.1096/fasebj.4.14.2172061. [DOI] [PubMed] [Google Scholar]
- 57.Wang P, Wu P, Siegel MI, Egan RW, Billah MM. Interleukin (IL)-10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem. 1995;270:9558–9563. doi: 10.1074/jbc.270.16.9558. [DOI] [PubMed] [Google Scholar]
- 58.Kim JR, Yoon HW, Kwon KS, Lee SR, Rhee SG. Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Anal Biochem. 2000;283:214–221. doi: 10.1006/abio.2000.4623. [DOI] [PubMed] [Google Scholar]
- 59.Coats SR, Pham TT, Bainbridge BW, Reife RA, Darveau RP. MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J Immunol. 2005;175(7):4490–4498. doi: 10.4049/jimmunol.175.7.4490. [DOI] [PubMed] [Google Scholar]
- 60.Barochia A, Solomon S, Cui X, Natanson C, Eichacker PQ. Eritoran tetrasodium (E5564) treatment for sepsis: review of preclinical and clinical studies. Expert Opin Drug Metab Toxicol. 2011;7(4):479–494. doi: 10.1517/17425255.2011.558190. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR, Yin H. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci U S A. 2012;109:6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38:1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, Wu J, Ying S, Chen G, Wu B, Xu T, Liu Z, Liu X, Huang L, Shan X, Dai Y, Liang G. Discovery of new MD2 inhibitor from chalcone derivatives with anti-inflammatory effects in LPS-induced acute lung injury. Sci Rep. 2016;6:25130. doi: 10.1038/srep25130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 65.Rider P, Voronov E, Dinarello CA, Apte RN, Cohen I. Alarmins: Feel the Stress. J Immunol. 2017;198(4):1395–1402. doi: 10.4049/jimmunol.1601342. [DOI] [PubMed] [Google Scholar]
- 66.De Araujo RFF, Martins DBG, Borba MACSM. Oxidative Stress and Disease. In: Morales-Gonzalez JA, Moralez-Gonzalez A, Madrigal-Santillon, editors. A Master Regulator of Oxidative stress - The Transcription Factor Nrf2, Croatia, European Union. Publisher: InTech Open Access; 2016. pp. 185–199. Chapter 10. [DOI] [Google Scholar]