

Abstract
Human islet amyloid polypeptide (hIAPP) is a 37-residue peptide hormone, which upon misfolding changes from the physiologically active monomer into pathological amyloid fibril aggregates in the pancreas of type 2 diabetes mellitus patients. During this process, the insulin-producing pancreatic β-cells are damaged; however, the underlying mechanism of this mode of cytotoxicity remains elusive. It is known that anionic lipids accelerate amyloid fibril formation, implicating the importance of the cellular membrane in the process, and that a pH close to the level in the β-cell secretory granules (pH = 5.5) inhibits the amyloid fibril formation. Using all-atom molecular dynamics simulations, we have investigated the membrane-associated monomer state of α-helical hIAPP and analyzed specific interactions of hIAPP with a mixed anionic-zwitterionic lipid membrane, examined the influence of pH on the structure and dynamics of hIAPP and its interaction with the membrane. We find that hIAPP primarily interacts with the membrane by forming favorable interactions between anionic lipids and the positively charged residues in the N-terminal part of the peptide. Rationalizing experimental findings, the simulations show that the N-terminal part of the peptide interacts with the membrane in the lipid head-group region. At neutral pH the C-terminal part of the peptide, which contains the residues that initiate fibril formation, displays a highly dynamic, unfolded state, which interacts significantly less with the membrane than the N-terminal part. Such an unfolded form can be proposed to contribute to the acceleration of fibril formation. At low pH, protonation of His18 mediates a stronger interaction of the C-terminal part with the membrane, resulting in the immobilization of the C-terminal part on the membrane surface which may constitute a mechanism by which low pH inhibits fibril formation.
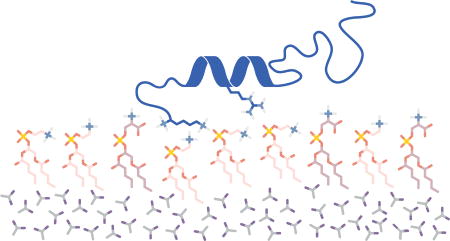
Graphical abstract
INTRODUCTION
Type 2 diabetes mellitus (T2DM) represents a major pathological condition, estimated to affect 285 million adults in 2010 and to increase to 439 million by 2030.1 40–100% of T2DM patients are reported to have pancreatic amyloid deposits of human islet amyloid polypeptide (hIAPP),2–5 also called amylin. Amyloid deposits are insoluble proteinaceous assemblies containing mainly amyloid fibrils and are the hallmark of the amyloid diseases, of which more than 40 have been identified, including neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases. Misfolding and aggregation of a particular protein or peptide into insoluble amyloid fibrils characterize the pathology of this class of diseases. The amyloid fibrils of hIAPP are not considered to be the main cause of T2DM, however, they are clearly a contributing factor to the pathology and responsible for the failure of islet grafts.6,7
hIAPP is a 37-residue peptide hormone secreted along with insulin by the pancreatic β-cells in response to elevated blood glucose.8 Its physiological function has not yet been fully determined, however, hIAPP has been implicated in blood glucose homeostasis, gastric emptying, suppression of glucagon release, and controlling satiety.9
The amino acid sequence of hIAPP is shown in Figure 1a. It contains a disulphide bridge between Cys2 and Cys7 and a C-terminal amidation. Based on nuclear magnetic resonance (NMR) and circular dichroism (CD) measurements, the hIAPP monomer in solution can be characterized as a mostly random coil structure with some α-helical content.10,11 However, the peptide adopts a mainly α-helical structure when it is either bound to an anionic phospholipid membrane, dissolved in hydrophobic solvents such as trifluoroethanol, or bound to detergent micelles.12–14 If a solution of hIAPP is left for an extended period of time, the peptides self-assemble into larger aggregates, the secondary structure changes from random coil to mainly β-sheet, and insoluble amyloid fibrils are formed as revealed by CD spectroscopy, Fourier transform infrared spectroscopy (FTIR), and electron microscopy (EM).15 Insoluble and kinetically stable amyloid fibrils are composed of extended β-sheet structures with inter-peptide hydrogen bonds running parallel to the fibril elongation axis (Figure 1b).16
Figure 1.

a) Amino acid sequence of hIAPP. b) Schematic representation of a cross-β structure composed of extended β-sheets with the peptide strands in light and dark grey positioned perpendicular to the fibril elongation axis. The inter-strand hydrogen bonds running along the fibril length are shown schematically by dotted lines. c) Structure of hIAPP monomer (PDB ID: 2L86). Side chains are shown in licorice and Lys1, Arg11, and His18 are shown in a lighter blue color.
Two structures of monomeric hIAPP bound to an SDS-micelle have been determined by NMR.17,18 One was determined at pH 4.6 and did not contain the physiologically important C-terminal amidation,17 while the other structure was determined at pH 7.3 and did contain the C-terminal amidation (Figure 1c).18 The resulting structures are rather similar, both being mainly α-helical and showing a sharp kink at His18; however, the structural ensemble of the peptides containing the C-terminal amidation was more uniform than the one without. An atomic resolution structure of mature amyloid fibrils has been difficult to obtain using traditional structure determination methods, such as X-ray crystallography, due to the fast aggregation of the monomer and the insoluble nature of the aggregates. The structure of the mature fibrils has been studied using a variety of techniques such as EM, atomic force microscopy (AFM), X-ray fiber diffraction, and solid-state NMR (ss-NMR),19,20 and two atomic-resolution structures of hIAPP fibrils have been published.21,22 These structures reveal long, regular, and parallel β-sheets with elongated grooves on the surface along the long axis of the fibril. The grooves are created by the repetition of identical side chains on neighboring β-strands.
Physiologically, hIAPP is stored in secretory granules in pancreatic β-cells along with insulin. The pH in the secretory granules is around 5.5.23 CD and FTIR experiments have shown that low pH (5.5) inhibits the aggregation of hIAPP, both in the presence and absence of a lipid bilayer in the form of large unilamellar vesicles (LUVs).24,25 Furthermore, it was shown by Thioflavin T fluorescence, which probes the formation of amyloid fibrils, and by calcein release from LUVs, revealing membrane disruption, that fibril formation coincides with membrane leakage at both pH 5.5 and 7.4, indicating a link between fibril formation and cytotoxicity.24 Moreover, monolayer experiments monitoring the change in surface pressure of the membrane have shown almost identical membrane insertion of hIAPP at low and neutral pH.24
The hIAPP species responsible for cytotoxicity to the islet β-cells was initially thought to be the mature amyloid fibrils; however, this hypothesis has been revised in recent years.26 Currently, several hypotheses exist about the cause of the cytotoxicity of hIAPP. These can be roughly divided into two groups; some propose the involvement of a specific cytotoxic species often referred to as a toxic oligomer, while others suggest that a cytotoxic event occurs during the fibril formation, which does not necessarily exclude the toxic oligomer hypothesis.27 Pore-like assemblies composed of five domains have been observed with AFM.28 Leakage of the contents of phospholipid vesicles in the presence of soluble hIAPP has been reported.29–33 Patch-clamp experiments have also shown that hIAPP can form pores (or defects) in the membrane which exhibited open-close transitions.34
It is generally accepted that hIAPP interacts with the cell membrane, and characterizing the details of interaction of hIAPP with different membrane systems has been the subject of several studies.35–40 The presence of membranes, especially those composed of anionic lipids, accelerates the formation of hIAPP fibrils,35 possibly through the formation of a membrane-induced conformation of the peptide prone to aggregation. Increase in the anionic lipid content of the islet β-cells has been shown to be associated with an elevated glucose content of the cell growth environment.36 Therefore, failure to produce required amount of insulin could initiate a vicious circle of anionic lipids accelerating the formation of hIAPP fibrils, leading to further β-cell cytotoxicity. Maximum acceleration of aggregation is caused by an anionic lipid content of 25–30 mol%.35,37 Several studies have shown that hIAPP binds to the membrane as a monomer, undergoing conformational changes initially from random coil to an α-helical structure upon binding, and after a lag-phase to β-sheet.38–40 Using infrared reflection absorption spectroscopy, Lopes et al. showed that the N-terminal part of the peptide interacts strongly with negatively charged lipids.39 Furthermore, from surface tension measurements, Engel et al. showed that hIAPP20–29 (a truncated version of the peptide including only residues 20–29) does not interact with anionic membranes at all, while hIAPP1–19 interacts stronger than the full-length hIAPP1–37.38 Both studies clearly support the involvement of the N-terminal part in membrane interaction of the peptide.
Due to the rapid aggregation of hIAPP, it is difficult experimentally to study the interaction of the peptide with membranes. Molecular dynamics (MD) simulation provides a useful method to study the atomic details of the interaction, and several computational studies investigating the interactions between hIAPP and membranes have been reported. Zhang et al. used an SDS-bound NMR structure of monomeric hIAPP to investigate the dynamics of hIAPP monomers and dimers with the N-terminus pre-inserted into 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) bilayers using MD simulations.41 They reported that the electrostatic interactions between the positively charged residues and negatively charged lipids are important for the peptide-membrane association. Furthermore, they also found that the C-terminal residues mostly mediate the inter-peptide interactions in the dimer.41 It is, however, not known if and to what extent the peptide is inserted into the membrane, and the assumption that the N-terminal part of the peptide is the membrane-inserting element will of course influence the resulting model. Zhao et al. have constructed membrane pores from ss-NMR structures of the hIAPP fibril21 to determine the number of monomers per pore by comparing the size of the pores with AFM images.42 Using MD simulations they showed that the peptides partitioned into 5 domains, the number of domains also observed by AFM;28 however, the relevance of the study can be questioned, because the inserted monomers were assumed to be composed of the hairpin-like peptide monomers observed in the mature fibrils. Additionally, Duan et al. investigated the monomer conformational dynamics of the fast aggregating mutant, hIAPP S20G, initially built in an α-helical conformation with a mixed 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS) membrane using MD simulations.43 They speculated that a kink at His18 present in the conformational ensemble could prime the peptide for adopting the hairpin motif observed in the mature amyloid fibrils. However, in that study, the peptide was also placed at a certain position in the membrane prior to the simulation. To our knowledge, only one study has sought to investigate the process of binding of the α-helical monomer peptide to the membrane to study the relative orientation. Jia et al. positioned an hIAPP monomer above a POPG bilayer and investigated its insertion into the membrane using MD simulations.44 They observed that the binding event is driven by electrostatic attraction between the positively charged residues of the N-terminal part of the peptide and the anionic lipids. The membrane-bound structure that was investigated in that study most likely resembles an aggregation inhibited structure, because an anionic lipid content of 100% (POPG) was used, which has been shown to inhibit the aggregation of hIAPP.37
Capturing the process of membrane binding of an intrinsically disordered peptide such as hIAPP in its entirety using conventional lipid bilayers in atomistic simulations, poses a challenging sampling problem due to the slow diffusion of lipids that leads to only a few Å translation during 10 ns of MD simulation.45 Insertion of peptides and proteins into the membrane requires significant displacement (and mixing) of the lipids; therefore, investigating how hIAPP interacts with the membrane, once membrane and peptide have adapted to each other, using a conventional membrane may only be achieved by prohibitively long simulations. To partly overcome this challenge, the highly mobile membrane mimetic (HMMM) model, in which lipid diffusion is accelerated, has been developed.45 This model substitutes the hydrophobic interior of a phospholipid bilayer by the organic solvent 1,1-dichloroethane (DCLE), and models the head-group region by short-tailed lipids. The model is able to reproduce closely the interfacial energetic profiles of a conventional membrane,46 but with the advantage of a significantly faster diffusion of the lipids. Given the increased lipid mobility, the model accelerates various membrane-associated phenomena while preserving atomic accuracy of the membrane surface. HMMM membranes have thus been used to successfully capture membrane interaction and insertion of a variety of peripheral proteins, e.g., the GLA domain of the human coagulation factor VII45, cytochrome P45047, talin48, α-synuclein (using a micelle-bound starting structure)49, and synaptotagmin.50 Insertion of phospholipids into the membrane51, as well as determining the optimal tilt angle of transmembrane helices,52 have also been successfully accomplished with this model. Taking advantage of the HMMM model, we herein present all-atom MD simulations investigating the orientation and insertion of hIAPP into a mixed anionic-zwitterionic phospholipid bilayer starting from the SDS-micelle bound conformation, because such a conformation is likely to be very similar to the membrane bound conformation.53–55 The simulations are designed to capture the most probable orientation of the peptide on the membrane, and the dynamical properties and lipid interactions of the monomeric hIAPP peptide, as well as the influence of pH on these features.
COMPUTATIONAL METHODS
Peptide Models
hIAPP was modeled using the ss-NMR structure which contains all 37 residues (PDB ID: 2L86).18 It was determined at pH 7.3 allowing for His18 to be found in all three tautomer/protonation states. The disulphide bridge between Cys2 and Cys7 and the amidated C-terminus were included in the model, hereby representing the physiological important variant of hIAPP.7 Since it is not expected that atomistic simulations will be able to capture folding of the peptide, the starting structure of the peptide when bound to an SDS micelle was chosen, instead of a water-equilibrated peptide. It is known from the literature that a micelle-bound conformation is closer to the membrane bound conformation, because SDS micelles have been shown to be effective mimics of membrane environments, especially for monomeric peptides as multimeric peptides may be dissociated.55 Since monomeric hIAPP is most likely surface-bound,40 SDS micelles are thus a reasonable membrane mimic for this peptide. Different initial orientations of the peptide were used when placing it at least 12 Å above the membrane in bulk solution in order not to bias the orientation and insertion of the peptide in the membrane. In the A-orientation the side chain of His18 is pointing away from the membrane (Figure 2a), while in the B-orientation the protein is rotated 180° around an axis parallel to the membrane plane (the y-axis here) leaving the side chain of His18 pointing toward the membrane (Figure 2b). To model two different pH levels, three forms of the peptide were prepared, one for each tautomer and protonation state of His18: the two neutral tautomers, with the hydrogen placed respectively at Nδ (HSD) or Nε (HSE) positions of the imidazole ring, as well as the positively charged histidinium (HSP) form. In the following, a particular tautomer form will be referred to as HSD, HSE, or HSP. At times, the two neutral His18 peptides will be referred to collectively as HSD/E. The overall charge of the peptide is +3 for HSD/E, and +4 for HSP. To equally sample the neutral and charged His18 forms, two (1×A and 1×B), two (1×A and 1×B), and four (2×A and 2×B) systems were prepared for the HSD, HSE, and HSP forms, respectively (Table 1). Reference to a specific trajectory such as HSP1–37(A2) refers to the second full-length peptide with a positive His18 (HSP) with the peptide in the A-orientation, while a group of simulations such as the hIAPP1–37 simulations with a protonated histidine will be referred to as HSP1–37.
Figure 2.

Two different initial orientations of hIAPP with respect to the membrane, where the short-tailed lipids of the nearest leaflet is shown in pink using a space-filling model, were used: a) the A-orientation with His18 pointing away from the membrane, and b) the B-orientation with His18 pointing towards the membrane. c) The membrane was composed of two type of lipids: 30% DVPS and 70% DVPC. d) A representative initial starting structure for the simulations. The simulation setup is composed of one hIAPP peptide shown in dark blue placed at least 12 Å above the HMMM membrane. The lipid bilayer is composed of a central hydrophobic core of DCLE shown in purple and grey, between two layers of DVPC (light-pink) and DVPS lipids (dark-pink) at the interface between DCLE and water. The system is neutralized with Na+ ions shown in dark grey.
Table 1.
Summary of MD simulations of hIAPP and its fragments.
| System | Peptide | His18 | No. of simulations |
Membrane (lipids/leaflet) |
Time per simulation (ns) |
|---|---|---|---|---|---|
| HSD/E1–19 | hIAPP1–19 | His(δ)/(ε) | 4 (2/2)1 | HMMM (36) | 50 |
| HSP1–19 | hIAPP1–19 | His(+) | 4 | HMMM (36) | 50 |
| hIAPP20–37 | hIAPP20–37 | - | 4 | HMMM (36) | 50 |
| HSD/E1–37 | hIAPP1–37 | His(δ)/(ε) | 4 (2/2) | HMMM (49) | 250 |
| HSP1–37 | hIAPP1–37 | His(+) | 4 | HMMM (49) | 250 |
| HSD/E1–37 (no membrane) | hIAPP1–37 | His(δ)/(ε) | 6 (3/3) | - | 50 |
| HSP1–37 (no membrane) | hIAPP1–37 | His(+) | 4 | - | 50 |
| HSD/E1–37 (DOPC/PS) | hIAPP1–37 | His(δ)/(ε) | 2 (1/1) | DOPC/DOPS2 (49) | 100 |
| HSP1–37 (DOPC/PS) | hIAPP1–37 | His(+) | 2 | DOPC/DOPS (49) | 100 |
The number of simulations with (δ/ε) protonation of His18.
The systems with DOPC/DOPS membrane were extended from HMMM simulations by growing the tails of the short-tailed lipids as detailed in Methods.
Simulations were performed with the full-length peptide (hIAPP1–37) and two truncated peptides, hIAPP1–19 and hIAPP20–37, containing residues 1–19 and 20–37, respectively (Table 1). hIAPP1–19 was included in the study since it has previously been shown to induce liposome leakage to the same extent as hIAPP1–37.56 hIAPP20–37 was included as a negative control, because hIAPP20–29 has been shown not to interact with monolayers.38 Eight simulations (two HSD, two HSE, and four HSP) were performed with hIAPP1–19, since it contains His18, while only four simulations were performed with hIAPP20–37. hIAPP1–19 was amidated at the C-terminus as is often the case in experiments with hIAPP1–19,38, 56 and hIAPP20–37 was capped with an acetyl group at the N-terminus to mimic the peptide linkage which is also comparable to experiments.56
In order to compare the behavior of the membrane-bound peptide with the behavior of the hIAPP1–37 monomer in solution, 3 simulations of each of the neutral His18 peptides (HSD and HSE), and 4 simulations of the HSP peptide were similarly performed in water.
Membrane Model
The lipid bilayer is modeled using the HMMM and is composed of a central hydrophobic phase of DCLE which is separated from water (on each side) by a 70:30 mixture of two types of short-tailed lipids, zwitterionic divalerylphosphocholine (DVPC) and anionic divalerylphosphoserine (DVPS), i.e., lipids including five carbons in each tail (Figure 2c).45 This ratio was chosen to match the lipid composition of most hIAPP-membrane experiments, the lipid composition of the islet β-cell membrane after prolonged exposure to high glucose concentration, and the optimum lipid composition for acceleration of hIAPP aggregation.35,36 The upper and lower leaflets are each composed of 49 lipids for the systems with hIAPP1–37, and 36 lipids for the systems with hIAPP1–19 and hIAPP20–37. To minimize overlap, the lipids were initially placed on a 7×7 (6×6 for the smaller membranes) grid in each leaflet, with random rotation around the z-axis. The initial positions of the DVPC and DVPS lipids were chosen randomly. The separation of the phosphates in the lipid head-groups of the two leaflets can be tuned by the amount of DCLE included between the leaflets. The separation was set to 38 Å to mimic the average bilayer thickness of DOPC and DOPS membranes (we note that given the expected peripheral interaction of the peptide with only one leaflet of the membrane, membrane thickness is not a critical parameter here).57 533 and 734 DCLE molecules were included between the short-tailed lipids of the small and large membranes, respectively, using PACKMOL58 with a DCLE density of 0.0073 molecules/Å3. The carbonyls of the short-tailed lipids were restrained along the z-direction using a weak harmonic potential with a force constant of 0.05 kcal/mol/Å2 to prevent excessively large fluctuations perpendicular to the membrane. This procedure has previously been employed to bring the transmembrane atomic density distribution of the HMMM membrane even closer to that of a conventional membrane.47 The systems were solvated with TIPS3P59 water and neutralized with Na+ ions.
Prior to incorporation of the peptide in the system, the membrane was minimized for 1000 steps and equilibrated for 1 ns using the same simulation protocol as for the production runs described below. The peptide was then incorporated into the system by placing it in the water phase at least 12 Å above the equilibrated membrane and removing overlapping water molecules (within 1.0 Å). Three or four Na+ ions, depending on the protonation state of His18, were randomly removed to neutralize the additional charge introduced by the protein, resulting in a final Na+ ion concentration of 0.10–0.12 M. A representative simulation system is displayed in Figure 2d.
Simulation Conditions and Protocols
All systems were simulated with NAMD 2.9 using the “helix-coil”-balanced CHARMM22* force field60 for the protein, while CHARMM3661 parameters were used for the lipids. The CHARMM22* force field has been shown to be advantageous in describing the conformational distribution and energetics of small peptides.62 Initially, the systems were minimized for 10,000 steps. Then, a short 10-ps equilibration of water was performed in the NVT ensemble while keeping everything but water and ions fixed. Finally, production runs were performed keeping the xy area fixed to ensure an area per lipid approximately 8% larger than the average 68 Å2 for DOPC and DOPS lipids.57 This procedure has been employed previously to allow protein insertion into the membrane without creating too much surface compression.47 The sides (x and y dimensions) of the simulation box were 51.4 Å for the 36 lipids/leaflet systems and 60.0 Å for the 49 lipids/leaflet systems, leaving a final area per lipid of ~73.5 Å2. A time-step of 2 fs was used while constraining all bonds to hydrogen atoms using the SHAKE algorithm.63,64 The temperature was kept constant at 310 K using the Langevin thermostat with a damping coefficient of 0.5 ps−1. A Nosé-Hoover Langevin piston65,66 was used to keep the pressure constant at 1 atm with an oscillation period of 200 fs and a damping coefficient of 200 fs. Periodic boundary conditions were employed to remove boundary effects, and PME with a grid-size of 1 Å was used for electrostatic interactions.67–69 The vdW interactions were cut off at 12 Å with a switching function starting at 10 Å. The pair list contained pairs of atoms within 13.5 Å and was updated every 40 fs. The non-bonded interactions were calculated every 2 fs, and the full-electrostatics every 4 fs. The simulations of the two peptide fragments were performed for 50 ns each, while the simulations with full-length hIAPP1–37 were performed for 250 ns each to obtain additional sampling, given the larger system size and the greater physiological relevance of this set-up (Table 1). The simulations of the peptide in water without a membrane were each performed for 50 ns.
Full-Membrane Simulations
To examine the stability of the resulting membrane-bound peptides from HMMM simulations, full-tailed lipid, conventional membrane systems (70:30 DOPC:DOPS) were generated for four of the HMMM-hIAPP1–37 simulations after 100 ns of simulation (Table 1) by growing the short lipid tails of the HMMM model to the full length. To preserve the existing lipid-protein interactions captured during the HMMM simulations, the coordinates of the atoms of the short-tailed lipids were retained. A random DOPS or DOPC lipid from a pre-equilibrated full membrane system was chosen, and the head-group and the first five atoms of the lipid tail were aligned with an existing short-tailed lipid. The missing atom coordinates were then adopted from the pre-equilibrated full-tailed lipid. This procedure was repeated for each lipid in the system. The system was then minimized for 10,000 steps, which was followed by an equilibration of the newly added atoms along with water while keeping the protein as well as the lipid head-groups, the carbonyl, and the first four CH2 groups in the tails restrained using a harmonic potential with a force constant of 0.10 kcal/mol/Å2 for 100 ps. The systems were then simulated without restraints in the NPT ensemble for 100 ns each.
Analysis
The torsional root mean square fluctuation (RMSF) was determined as the mean fluctuation from the average torsional angle of the backbone dihedral angles. The average dihedral angle was determined by converting the angles to Cartesian coordinates and determining the arithmetic average of the angles in a two-dimensional coordinate system; the angle of the vector from the origin to the geometrical average was then the mean angle used to determine the torsional RMSF.70
The analysis for figures 4b, 7, 8, 9, the histograms in figures 3 and 6, as well as table S1 in the Supporting Information have been performed excluding the first 20 ns of simulation.
Figure 4.

a) The z-coordinate of the center of mass of each side chain for two hIAPP1–37 simulations, HSE1–37(B) and HSP1–37(A1). Plots for all the simulations can be found in the Supporting Information. The center of the membrane is at z = 0 Å and the average position of the phosphates is at z = 18 Å. Data points with z > 40 Å are the same color as z = 40 Å (white). b) Average z-coordinate of the center of mass of each side chain during the hIAPP1–37 simulations excluding the first 20 ns. The horizontal dashed line represents the average position of the phosphates, and the vertical dashed lines mark the position of the positive residues.
Figure 7.

a) Torsional RMSF of the hIAPP1–37 HMMM backbone Φ and Ψ angles. b) Torsional RMSF differences. If the difference is positive, the top peptide is more flexible, whereas a negative difference indicates more flexibility in the bottom peptide. The difference between HMMM-bound HSD/E1–37 and HMMM-bound HSP1–37 shows the effect of protonating His18. The difference between HMMM-bound HSP1–37 and solution HSP1–37 shows the effect of the membrane on the HSP1–37 peptide. The difference between HMMM-bound HSD/E1–37 and solution HSD/E1–37 shows the effect of the membrane on the HSD/E1–37 peptide. The difference between solution HSD/E1–37 and solution HSP1–37 shows the effect of low pH on the free peptide. The first 20 ns of simulation have not been included in the analysis. The vertical dashed lines in all panels indicate the divide between the 19 N-terminal residues and the 18 C-terminal residues.
Figure 8.

a) Helical and b) β structure propensity of the hIAPP1–37 peptide with and without HMMM membrane in the simulation. A value of 100% indicates that the residue is in a helical or β-sheet conformation 100% of the simulation time. The first 20 ns of simulation have not been included in the analysis. The secondary structure was determined using STRIDE.71 The vertical dashed lines indicate the divide between the 19 N-terminal residues and the 18 C-terminal residues.
Figure 9.

a) Average number of heavy atom contacts (within 5 Å) between the peptide and each lipid type (excluding the first 20 ns of the simulations). The average numbers have been normalized by the number of lipids for each type (70% PC and 30% PS). b) Average number of hydrogen bonds and salt bridges between the two types of lipids, and the peptide residues and N-terminus for the HSD/E1–37 and HSP1–37 peptides (excluding the first 20 ns of the simulations), normalized by the number of lipids for each type (70% PC and 30% PS). Hydrogen bonds were counted when donor and acceptor heavy atoms were within 3.5 Å and within a maximum deviation of 30° from a linear donor-H-acceptor configuration. Salt-bridges were counted when heavy atoms of oppositely charged groups were within 4.0 Å.
Figure 3.

Minimum z-coordinate of the protein heavy atoms for a) the hIAPP1–19 peptides, b) the hIAPP1–37 peptides, and c) the hIAPP20–37 peptides. The HSD/E peptides are shown in green, and the HSP peptides are shown in blue. The black horizontal line represents the average position of the membrane phosphate layer. Histograms do not include the first 20 ns of simulation.
Figure 6.

The Cα RMSD to the minimized structure of the hIAPP1–37 peptide bound to the HMMM membrane was calculated for a) residues 1–19, b) residues 20–37, and c) the entire hIAPP1–37 peptide. The Cα RMSD to the minimized structure of the hIAPP1–37 peptide in solvent was calculated for d) the entire hIAPP1–37 peptide, e) residues 1–19, and f) residues 20–37. Histograms do not include the first 20 ns of simulation.
RESULTS AND DISCUSSION
The overall aim of the present study is to characterize the membrane-bound state of hIAPP and the impact of lipids on its structure and dynamics. A list of the simulations performed on the full-length peptide and its N- and C-terminal parts is provided in Table 1, detailing solvated peptides as well as peptides interacting with a membrane (both HMMM and conventional membranes), and including the three possible forms of His18.
hIAPP Dynamics in Water
The hIAPP peptide was simulated in water for 50 ns. Three simulations of each of the two tautomer forms at neutral pH and four simulations of the peptide with a positively charged histidine were performed. These simulations were included for comparison of the dynamic properties of the membrane-bound form with a solution form. An alignment of snapshots from the simulations taken at an interval of 1 ns (Figure S1 in Supporting Information) shows that the peptide is quite dynamic, and has changed from the micelle-bound structure, which can be seen in Figure 1c, which is expected for an intrinsically disordered peptide in water. To study the membrane surface mediated dynamics and subsequent change of the α-helical conformation to an aggregation prone conformation, we used knowledge from the literature revealing that micelle-bound peptide structures are good structural models of the membrane-bound conformations of the peptides,55 especially for surface-bound and monomeric peptides such as IAPP; therefore, the micelle-bound structure of the peptide was chosen as the starting structure for the HMMM simulations. This choice is ideal for studying aspects of the proposed mechanism for fibril formation; firstly, the conformation of a monomeric α-helical peptide can be found by following the adaption of the micelle-conformation to the lipid bilayer, and secondly the dynamics can be followed which may result in an aggregation prone form of the peptide, as suggested by Nanga et al.18
Full-Length hIAPP and hIAPP1–19 Binds to the Membrane
During the HMMM simulations, spontaneous association and insertion of the peptides into the membranes were observed for hIAPP1–37 and hIAPP1–19. Binding to the membrane occurs within the first 20 ns (Figure 3) irrespective of the protonation/tautomeric state of His18. As the aim of the simulations is to describe membrane-associated properties of the peptide, the analyses of average properties presented in this paper have been performed excluding the first 20 ns of the simulations. Four hIAPP1–37 peptides have His18 in a neutral configuration (HSD/E1–37), and four have a positively charged His18 (HSP1–37). The HSP1–37 peptides exhibit deeper penetration into the membrane than the HSD/E1–37 peptides (Figure 3b). In contrast to the hIAPP1–19 and hIAPP1–37 peptides, the hIAPP20–37 peptides only contact the membrane superficially and occasionally (Figure 3c). It is clear that hIAPP20–37 does not have a strong affinity for the membrane and the interaction between the peptide and the membrane is a diffusional encounter facilitated by the confinement within the simulation box. The differential behavior of the simulated peptides rationalizes the results of experimental surface pressure measurements indicating that both hIAPP1–37 and hIAPP1–19 insert into the head-group region of a mixed 70:30 DOPC:DOPS phospholipid monolayer, while hIAPP20–29 does not.38
N-terminal Residues Dominates Binding of Full-Length hIAPP
The N-terminal part of hIAPP1–37 binds to the membrane first (Figure 4a), a behavior that can be attributed to the positive charges present in this region of the peptide. Lys1 carries two positive charges, one N-terminal and one side chain amino group, while Arg11 also carries a positive charge. Initially, the C-terminal part of HSD/E1–37 exhibits only transient interactions with the membrane, which makes it very flexible and dynamic. Closer interaction of the C-terminus with the membrane is present for the HSP1–37 peptides, however the HSD/E1–37 peptides also exhibit interaction of the C-terminus with the membrane albeit much later and to a lesser extent than the HSP1–37 peptides.
Protonation State of His18 Governs Position and Orientation of Full-Length hIAPP in a Membrane
Generally, the HSD/E1–37 peptides have most of the peptide above the membrane, although HSE1–37(A) is moving deeper into the membrane with the N-terminal part than the other peptides. For HSP1–37, a rapid and close interaction of the C-terminus with the membrane is generally observed, which is most likely a result of the additional positive charge at His18 which interacts favorably with the anionic lipids thus bringing the C-terminal region closer to the membrane. The peptide in simulation HSP1–37(B2) does not interact as closely with the membrane as the other HSP1–37 peptides (Figure S2).
Monitoring the position of the individual residues in the hIAPP1–37 simulations provides insight into the differences in peptide insertion and orientation relative to the membrane (Figure 4b). All hIAPP1–37 peptides adopt an orientation of the N-terminal part that is approximately parallel to the membrane plane, with HSD/E adopting a slightly larger angle with the membrane than HSP. This orientation cannot be due to the initial placement of the peptide, because in 7 out of 8 hIAPP1–37 simulations the peptide approaches the membrane with the N-terminus first, i.e., adopting an almost vertical orientation of the N-terminal part, before assuming the final horizontal orientation in its membrane-bound form. As shown in Figure 4b, the HSP1–37 peptides (except HSP1–37(B2); see above), establish a more intimate interaction with the membrane upon binding than the HSD/E1–37 peptides. Furthermore, the rotation around the N-terminal axis is reversed in the two different protonation states (HSD/E vs. HSP). Therefore, the N-terminal region of the HSD/E1–37 peptides (except HSE1–37(A)) is positioned above the lipid phosphate groups and has the positively charged residues pointing downward, enabling interaction with the anionic lipids (Figure 4b and Figure 5a). In contrast, the rotation of the HSP1–37 N-terminal region is opposite because it is positioned in the lipid head-group region and therefore has the positively charged residues pointing upward to enable interaction with the anionic head-groups (Figure 4b and Figure 5b). A saw-toothed pattern displaying 3–4 residues between each maximum is observed in Figure 4b, indicative of a helix with a parallel orientation with respect to the bilayer. The horizontal orientation of all hIAPP1–37 peptides and the rotation around the N-terminal α-helix of the HSP1–37 peptides are consistent with accessibility measurements derived from electron paramagnetic resonance (EPR) spectroscopy of hIAPP bound to an 80% anionic membrane.40 It is surprising that the experimental rotation around the N-terminal part matches the HSP1–37 peptide, since the experiments were performed at neutral pH; thus, we would expect the rotation of the HSD/E1–37 peptides to match the experiments. However, the lipid membrane in the accessibility experiments had a very high content of anionic lipids, which can strongly affect the pKa of His18. The free form of histidine has a pKa of 6, making the protonation state highly sensitive to changes in the immediate environment. The highly anionic lipid content used in the experiments can thus significantly favor a charged form of His18 by shifting its pKa upwards.
Figure 5.

Representative structures of a) HSE1–37(B) and b) HSP1–37(A1). The structures are from the largest and second largest cluster, for HSE1–37(B) and HSP1–37(A1), respectively, from a clustering analysis performed using VMD (Figure S3). Both clusters contain the final structure of the simulation. The lipids are shown in red colors, DCLE is shown in grey and purple, and the peptide is shown in dark blue. The final frames of all hIAPP1–37 peptides bound to the HMMM membrane are provided in Supporting Information.
To illustrate the differences in the membrane-bound form of the peptides at neutral and low pH, representative structures determined through a clustering analysis of the HSE1–37(B) and HSP1–37(A1) simulations are shown in Figure 5 (representatives of the remaining hIAPP1–37 peptides can be found in Figure S4 in the Supporting Information). HSP1–37(A1) has displaced the short-tailed lipids and is reaching the DCLE level. In contrast, only the positively charged residues and a few other side-chains of HSE1–37(B) have inserted between the lipid head groups, while most of the peptide is positioned above the surface of the membrane. The C-terminal part of HSP1–37(A1) is associated closely with the lipids (only residues 29–37 are above the membrane), while the C-terminal part of HSE1–37(B) is solvated and not interacting with the membrane.
Dynamic Properties of Membrane-Associated hIAPP
The conformation of the peptide changes significantly during the membrane-binding simulations (Figure 6), which reflects the relaxation of the peptide from the experimentally determined, micelle-bound conformation and adaption to a more planar bilayer. The Cα RMSD values of the solvated and HMMM-bound hIAPP1–37 peptides plateau around 8 Å (Figure 6c and d), which suggests that both structures are different from the micelle-bound structure. The effect is mostly arising from the C-terminal part of the HMMM-bound hIAPP1–37 (Figure 6b), as the first 19 residues have a much lower RMSD of about 3–4 Å (Figure 6a). The membrane appears to stabilize the helical structure in the N-terminal part of hIAPP1–37, since it is found that the RMSD of the first 19 residues is higher in solution (Figure 6e) than when the peptide is bound to the membrane (Figure 6a). This is consistent with EPR experiments showing stabilization of residues 9–20 upon membrane binding, experiments that also showed a higher mobility of the C-terminal than the N-terminal region.40 The torsional RMSF demonstrates this higher flexibility in the C-terminal region in the simulations (Figure 7a and Figure S5 including standard deviations between simulations). The difference in torsional RMSF between the two protonation states of the membrane-bound hIAPP1–37 peptide reveals that HSP1–37 is not as dynamic as HSD/E1–37 (Figure 7b top panel). This is not the case when the peptide is not bound to the membrane (Figure 7b lower panel). The effect of the membrane on peptide properties is found to vary significantly depending on the protonation state of His18 (Figure 7b central panels). It is seen that the membrane serves to reduce the flexibility of the HSP1–37 peptide, where both the N- and C-terminal regions bind to the membrane, and are thus not as flexible as in solution (Figure 7b second panel). However, residues 32–37 do seem to increase in flexibility upon membrane binding, as these residues are primarily hydrophilic and do not insert easily into the membrane region. The effect of the membrane on the N- and C-termini of the HSD/E1–37 peptide are opposite; the membrane stabilizes the N-terminus with respect to the fluctuation in the solvent, while the C-terminus becomes more dynamic when the peptide is bound to the membrane (Figure 7b third panel). This is a consequence of the affinity of the N-terminus for the membrane and the lack of affinity of the C-terminus for the membrane. When the N-terminus binds to the membrane, it releases the C-terminus from its interaction with the N-terminus, which consequently makes it even more flexible than seen from the simulations of the peptide in water.
Secondary Structure Analysis Correlates with Experimental Measurements
The secondary structure of the peptides during the simulations was evaluated using STRIDE71 and reported in Table S1. The helical content is less pronounced in HMMM simulations mimicking neutral pH (HSD/E1–37) (27%) compared to low pH (HSP1–37) (42%), which is consistent with CD experiments of hIAPP1–37 in the presence of 70:30 DOPC:DOPS LUVs.24 The peptide is, as expected, found to be less structured in solution compared to its membrane-bound state, which is in agreement with CD experiments showing a low helical content in buffer (~10%)13, and a higher helical content when bound to a negatively charged membrane (39–50%).13,37 Estimation of helical structure from CD experiments assumes that the signal in the far-UV region of the CD spectrum only originates from the amide bonds; however, both aromatic side chains and disulphide bonds can contribute to the far-UV region thereby leading to an even higher apparent helical content.72 This may account for the observed small discrepancies between the experimentally and computationally determined helical propensities.
The N-terminal region of the peptide has a high helical content (Figure 8a), especially when bound to the membrane. The region around His18 has a very low degree of helical structure, while the C-terminus has some helical structure with a reduced helical propensity around Ser29. Low pH (as modeled through the protonated His18) increases the helical content, particularly for the C-terminal part of the membrane-bound peptide. It is evident from Figure 8a that the membrane has a preserving effect on the helical structure of HSP1–37 when compared with its solution form. In addition, the tendency for HSD/E1–37 to form helical structure is increased in the N-terminal region and decreased in the C-terminal region by the membrane compared to its solution form. The presence of helical structure in the N-terminal region is in line with EPR experiments suggesting an amphipathic nature of the helix between residues 9 and 20–22 when hIAPP1–37 is interacting with 20:80 POPC:POPS unilamellar vesicles.40 It was not possible experimentally to determine the structure of the C-terminal region. The N-terminal helix was determined based on mobility and accessibility measurements of individual residues in contact with the membrane. The simulations show that the C-terminal region is not buried as deep in the membrane as the N-terminal region, which therefore suggests that it would not be possible to use the same experiment to probe the structure of the C-terminus. Only very little β-strand structure is present in the C-terminal part of the membrane-bound HSD/E1–37 peptide, which has been suggested to contain the residues responsible for initiating fibril formation (Figure 8b).73
hIAPP-Lipid Interactions
The average numbers of contacts between each type of lipid (normalized by the fraction of each lipid type) and the peptides are compiled in Figure 9a. The C-terminal of the HSD/E1–37 peptides has a lower number of contacts with the lipids than the HSP1–37 peptides, especially for residues 12–27. Two hydrophobic stretches are present in this region, 12–17 and 23–27, which do not interact favorably with the highly charged head-group region of the membrane, thereby keeping the C-terminal region away from the membrane. Residues 27–37, however, contain several hydrophilic residues, which are able to form favorable contacts with the lipid head groups. The normalized numbers of contacts for both tautomer forms to the positively charged residues and the residues immediately beside them are higher for the anionic PS lipids than the PC lipids (Figure 9a). Lipid contacts of HSP1–37 peak around Lys1, Arg11, His18, and Phe23, and are more evenly distributed along the length of the polypeptide chain than for HSD/E1–37 (Figure 9a). HSP1–37 inserts in the interface between the lipid head-groups and the hydrophobic core region of the membrane, which allows its hydrophobic residues to interact with the hydrophobic tails of the lipids as well as with DCLE (Figure S6), while the hydrophilic and charged residues interact with the lipid head-groups. The doubly charged Lys1 clearly achieves the highest number of contacts, while a second interaction peak is discernible at Arg11 (Figure 9a). Thus, the positively charged residues are anchoring the peptide to the membrane. This is in line with the high affinity of hIAPP for anionic membranes, and with the accelerating effect of anionic lipids on the rate of fibril formation where this monomer binding may be of importance.35,37
The number of hydrogen bonds and salt bridges between the peptide and the lipids can be used to quantify specific interactions between the peptide and the membrane. Generally, a higher number of specific interactions are present with the PS lipids (Figure 9b), likely due to the net negative charge of PS while PC is zwitterionic. Furthermore, PS lipids can form additional hydrogen bonds compared to PC. The hydrophilic residues 18–22 of HSD/E1–37 form very few contacts with the lipids (Figure 9b). This is surprising but may be rationalized by the fact that the surrounding hydrophobic residues (12–17 and 23–27) may be preventing residues 18–22 from interacting with the membrane. For HSP1–37 the number of hydrogen bonds from His18 is highest to PS, and residues 19–22 form hydrogen bonds with both types of lipids.
Full-Membrane Simulations Support HMMM-Findings
To ensure that the observed membrane-bound conformations are not biased by the short-tailed lipid of HMMM, full-tailed lipid membrane control simulations were performed after 100 ns of HMMM simulation. The HMMM membrane was converted to conventional full-tail DOPC/DOPS membranes in four of the systems by growing the full lipid tails on the existing short-tailed lipids. HSD1–37(A) and HSE1–37(A) were selected based on the structure of the peptide with the N-terminal region bound to the membrane and the C-terminal region solvated, i.e., representing the predominant binding structure observed during the simulations. Similarly, two HSP1–37 simulations were selected: HSP1–37(A1) because the N-terminal region of the structure is buried between the head-groups and the C-terminal region is associated closely with the N-terminal part and the membrane, and HSP1–37(B2) representing the outlier system. These four simulations were extended by 100 ns after the conversion of short-tailed lipids to full PC and PS lipids.
As can be seen in Figure 10a, the peptides all remain bound to the membrane. The minimum z-coordinate of the peptide fluctuates around the level of the phosphates similar to the HMMM simulations (Figure 3). From the Cα RMSD calculated relative to the structure after 100 ns of HMMM simulation, it is apparent that the structure of the HSD/E1–37 peptides changes the most (Figure 10b). Similar to the HMMM simulations, it is the C-terminal part of the peptide which is most flexible during the simulation, thereby causing the RMSD to rise. From the alignment of the peptide structures to the structures obtained after 100 ns of HMMM simulation (Figure 10c) it is evident that the HSP1–37(A1) peptide, which is embedded most deeply in the membrane, has the least dynamic structure, characterized by a lower RMSD value. Although at around 65 ns, a jump in the RMSD of HSP1–37(A1) is found from around 1 Å to around 4 Å due to a change in the conformation of the C-terminal part of the peptide (Figure 10b). The HSP1–37(B2) peptide does not insert in the DOPC/DOPS membrane, however, it is still attached to the membrane via the positively charged N-terminus and the side chains of Lys1, Arg11, and His18. The RMSD increases quickly to around 3 Å due to the flexibility of the peptide when it is not fully inserted into the membrane, and levels out at around 5 Å after 100 ns. The HSD1–37(A) and HSE1–37(A) peptides show much more flexibility than the HSP1–37 peptides, mainly in the C-terminal region, while the N-terminus also here is stable due to the interaction with the membrane. The influence of the HMMM on the binding and structure of hIAPP compared to a conventional DOPC/DOPS membrane is thus minimal based on the tests performed here.
Figure 10.

a) Minimum z-coordinate and b) RMSD for the DOPC/DOPS simulations to the final snapshot of the HMMM simulations. The black horizontal line indicates the position of the lipid phosphates. c) Alignment of the peptides using Ca atoms of residues 1–19 from the DOPC/DOPS simulations that were extended from the HSD1–37(A), HSE1–37(A), HSP1–37(A1), and HSP1–37(B2) HMMM simulations. The peptides are colored by residue number going from the N-terminus colored blue to the C-terminus colored red. Each alignment contains one structure per 0.1 ns.
CONCLUSION
The results obtained in this study suggest that the effect of the membrane on hIAPP strongly depends on the protonation state of His18, which is an arginine residue in many other species.74 At neutral pH (HSD/E1–37 simulations) upon binding of the peptide to the membrane, the N-terminal part is stabilized and anchored to the membrane, while the C-terminal part is disordered and found in the solution. It has been suggested previously that the acceleration of hIAPP fibril formation by membranes is caused by an increased local concentration of hIAPP.40 The present simulations suggest an additional accelerating factor; namely the unstructured C-terminal region. It has been shown that residues 20–29 are important for fibril formation, and that fibril formation is initiated in this region.73,75 An unstructured, water solvated C-terminal region exposes the backbone and primes it for interaction with another peptide, an event which is recognized from other amyloid peptides to be important in the fibril formation pathway.16 The unstructured peptide chain is more favorable entropically than a structured fibril, however, the enthalpic gain of the formation of regular β-sheets combined with the entropic gain of water-release from the solvation shells or the interacting surfaces will most likely be able to overcome the entropically favored unstructured state.16 A situation such as this, where the equilibrium of the pre-aggregation structures is altered, is likely to affect the aggregation rate, causing an acceleration of the fibril formation.76 However, when the C-terminal part of the peptide is associated closely with and restrained by the membrane, as was observed in the HSP1–37 simulations, it is not free to interact with other peptides, consequently leading to a reduction in aggregation rate. Electrostatic repulsion between the peptides most likely also plays a role in the inhibition of aggregation, however, the effect is probably smaller when the peptide is bound to an anionic membrane than in solution, since the anionic lipids serve to screen the charges of the peptides. This may also provide a mechanism for the known inhibitory effect of highly anionic lipid bilayers on the fibril formation37 because the higher content of anionic lipids will induce an increase in pKa of His18, which consequently changes the structure of membrane bound hIAPP from an exposed and dynamic C-terminus to a deeper buried and rigid structure less prone to aggregation. The acceleration of fibril formation by an anionic membrane with a lower content of anionic lipids is most likely due to an increased local concentration of hIAPP without increasing the pKa of His18 significantly.
Supplementary Material
Acknowledgments
K.K.S., O.J.A. and B.S. are supported by the Danish National Research Foundation (inSPIN, DNRF59) and by the Danish Council for Independent Research | Technology and Production Sciences (FTP). Computations were made possible through grants from the Danish Center for Scientific Computing. T.V.P. is grateful for the support from the Illinois Campus Research Board and the Extreme Science and Engineering Discovery Environment (XSEDE, grant TG-MCB130112), which is supported by National Science Foundation grant number ACI-1053575. T.V.P. was a Faculty Fellow of the National Center for Supercomputing Applications when this work was completed. E.T. acknowledges receiving support from the National Institutes of Health, grant numbers R01-GM101048, R01-GM086749, U54-GM087519, and P41-GM104601, and from XSEDE resources (grant number MCA06N060).
ABBREVIATIONS
- Aβ
amyloid-β
- AFM
atomic force microscopy
- CD
circular dichroism
- DCLE
dichloroethane
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPS
1,2-dioleoyl-sn-glycero-3-phosphoserine
- DVPC
1,2-divaleryl-sn-glycero-3-phosphocholine
- DVPS
1,2-divaleryl-sn-glycero-3-phosphoserine
- EM
electron microscopy
- EPR
electron paramagnetic resonance
- FTIR
Fourier transform infrared spectroscopy
- hIAPP
human islet amyloid polypeptide (amylin)
- HMMM
highly mobile membrane mimetic
- LUVs
large unilamellar vesicles
- NMR
nuclear magnetic resonance
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-(phospho-L-serine)
- RMSD
root mean squared deviation
- RMSF
root mean squared fluctuation
- ss-NMR
solid-state nuclear magnetic resonance
- T2DM
type 2 diabetes mellitus.
Footnotes
ASSOCIATED CONTENT
Supporting Information. The Supporting Information contains additional figures and a table not included in the main manuscript. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Bell ET. Hyalinization of the islets of langerhans in diabetes mellitus. Diabetes. 1952;1:341–344. doi: 10.2337/diab.1.5.341. [DOI] [PubMed] [Google Scholar]
- 3.Maloy AL, Longnecker DS, Robert Greenberg E. The relation of islet amyloid to the clinical type of diabetes. Hum. Pathol. 1981;12:917–922. doi: 10.1016/s0046-8177(81)80197-9. [DOI] [PubMed] [Google Scholar]
- 4.Westermark P. Islet Amyloid Polypeptide and Amyloid in the Islets of Langerhans. In: Leslie RDG, Robbins D, editors. Diabetes: Clinical Science in Practice. Cambridge Univ. Press; Cambridge, UK: 1995. pp. 189–199. [Google Scholar]
- 5.Zhao HL, Lai FMM, Tong PCY, Zhong DR, Yang D, Tomlinson B, Chan JCN. Prevalence and clinicopathological characteristics of islet amyloid in chinese patients with type 2 diabetes. Diabetes. 2003;52:2759–2766. doi: 10.2337/diabetes.52.11.2759. [DOI] [PubMed] [Google Scholar]
- 6.Stumvoll M, Goldstein BJ, van Haeften TW. Pathogenesis of type 2 diabetes. Endocr. Res. 2007;32:19–37. doi: 10.1080/07435800701743810. [DOI] [PubMed] [Google Scholar]
- 7.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 8.Lukinius A, Wilander E, Westermark GT, Engstrom U, Westermark P. Co-localization of islet amyloid polypeptide and insulin in the B cell secretory granules of the human pancreatic-islets. Diabetologia. 1989;32:240–244. doi: 10.1007/BF00285291. [DOI] [PubMed] [Google Scholar]
- 9.Lutz TA. Control of energy homeostasis by amylin. Cell. Mol. Life Sci. 2012;69:1947–1965. doi: 10.1007/s00018-011-0905-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williamson JA, Loria JP, Miranker AD. Helix stabilization precedes aqueous and bilayer-catalyzed fiber formation in islet amyloid polypeptide. J. Mol. Biol. 2009;393:383–396. doi: 10.1016/j.jmb.2009.07.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williamson JA, Miranker AD. Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caillon L, Lequin O, Khemtémourian L. Evaluation of membrane models and their composition for islet amyloid polypeptide-membrane aggregation. Biochim. Biophys. Acta, Biomembr. 2013;1828:2091–2098. doi: 10.1016/j.bbamem.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 14.McLean LR, Balasubramaniam A. Promotion of β-structure by interaction of diabetes associated polypeptide (amylin) with phosphatidylcholine. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1992;1122:317–320. doi: 10.1016/0167-4838(92)90411-6. [DOI] [PubMed] [Google Scholar]
- 15.Goldsbury C, Goldie K, Pellaud J, Seelig J, Frey P, Muller SA, Kistler J, Cooper GJS, Aebi U. Amyloid fibril formation from full-length and fragments of amylin. J. Struct. Biol. 2000;130:352–362. doi: 10.1006/jsbi.2000.4268. [DOI] [PubMed] [Google Scholar]
- 16.Harrison RS, Sharpe PC, Singh Y, Fairlie DP. Amyloid peptides and proteins in review. Rev. Physiol. Biochem. Pharmacol. 2007;159:1–77. doi: 10.1007/112_2007_0701. [DOI] [PubMed] [Google Scholar]
- 17.Patil SM, Xu S, Sheftic SR, Alexandrescu AT. Dynamic α-helix structure of micelle-bound human amylin. J. Biol. Chem. 2009;284:11982–11991. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nanga RPR, Brender JR, Vivekanandan S, Ramamoorthy A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta, Biomembr. 2011;1808:2337–2342. doi: 10.1016/j.bbamem.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tycko R. Solid state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pillay K, Govender P. Amylin uncovered: A review on the polypeptide responsible for type II diabetes. BioMed Res. Int. 2013;2013:826706–826706. doi: 10.1155/2013/826706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luca S, Yau W, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: Constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bedrood S, Li Y, Isas JM, Hegde BG, Baxa U, Haworth IS, Langen R. Fibril structure of human islet amyloid polypeptide. J. Biol. Chem. 2012;287:5235–5241. doi: 10.1074/jbc.M111.327817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutton JC. The internal ph and membrane-potential of the insulin-secretory granule. Biochem. J. 1982;204:171–178. doi: 10.1042/bj2040171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khemtémourian L, Doménech E, Doux JPF, Koorengevel MC, Killian JA. Low pH acts as inhibitor of membrane damage induced by human islet amyloid polypeptide. J. Am. Chem. Soc. 2011;133:15598–15604. doi: 10.1021/ja205007j. [DOI] [PubMed] [Google Scholar]
- 25.Chargé SBP, de Koning EJP, Clark A. Effect of pH and insulin on fibrillogenesis of islet amyloid polypeptide In vitro. Biochemistry. 1995;34:14588–14593. doi: 10.1021/bi00044a038. [DOI] [PubMed] [Google Scholar]
- 26.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 27.Zraika S, Hull RL, Verchere CB, Clark A, Potter KJ, Fraser PE, Raleigh DP, Kahn SE. Toxic oligomers and islet beta cell death: Guilty by association or convicted by circumstantial evidence? Diabetologia. 2010;53:1046–1056. doi: 10.1007/s00125-010-1671-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quist A, Doudevski L, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U. S. A. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Last NB, Rhoades E, Miranker AD. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane. Proc. Natl. Acad. Sci. U. S. A. 2011;108:9460–9465. doi: 10.1073/pnas.1102356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee C, Sun Y, Huang HW. How type II diabetes-related islet amyloid polypeptide damages lipid bilayers. Biophys. J. 2012;102:1059–1068. doi: 10.1016/j.bpj.2012.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anguiano M, Nowak RJ, Lansbury PT. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry. 2002;41:11338–11343. doi: 10.1021/bi020314u. [DOI] [PubMed] [Google Scholar]
- 32.Scalisi S, Sciacca MFM, Zhavnerko G, Grasso DM, Marletta G, La Rosa C. Self-assembling pathway of HiApp fibrils within lipid bilayers. Chem Bio Chem. 2010;11:1856–1859. doi: 10.1002/cbic.201000090. [DOI] [PubMed] [Google Scholar]
- 33.Engel MFM, Khemtémourian L, Kleijer CC, Meeldijk HJD, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, Höppener JWM. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mirzabekov T, Lin M, Kagan B. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 1996;271:1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 35.Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 36.Rustenbeck I, Matthies A, Lenzen S. Lipid-composition of glucose-stimulated pancreatic-islets and insulin-secreting tumor-cells. Lipids. 1994;29:685–692. doi: 10.1007/BF02538912. [DOI] [PubMed] [Google Scholar]
- 37.Jayasinghe S, Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 38.Engel MFM, Yigittop H, Elgersma RC, Rijkers DTS, Liskamp RMJ, de Kruijff B, Höppener JWM, Killian JA. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol. 2006;356:783–789. doi: 10.1016/j.jmb.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 39.Lopes DHJ, Meister A, Gohlke A, Hauser A, Blume A, Winter R. Mechanism of islet amyloid polypeptide fibrillation at lipid interfaces studied by infrared reflection absorption spectroscopy. Biophys. J. 2007;93:3132–3141. doi: 10.1529/biophysj.107.110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Apostolidou M, Jayasinghe SA, Langen R. Structure of α-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem. 2008;283:17205–17210. doi: 10.1074/jbc.M801383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Luo Y, Deng Y, Mu Y, Wei G. Lipid interaction and membrane perturbation of human islet amyloid polypeptide monomer and dimer by molecular dynamics simulations. PLoS One. 2012;7:e38191. doi: 10.1371/journal.pone.0038191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao J, Luo Y, Jang H, Yu X, Wei G, Nussinov R, Zheng J. Probing ion channel activity of human islet amyloid polypeptide (amylin) Biochim. Biophys. Acta, Biomembr. 2012;1818:3121–3130. doi: 10.1016/j.bbamem.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duan M, Fan J, Huo S. Conformations of islet amyloid polypeptide monomers in a membrane environment: Implications for fibril formation. PLoS One. 2012;7:e47150. doi: 10.1371/journal.pone.0047150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jia Y, Qian Z, Zhang Y, Wei G. Adsorption and orientation of human islet amyloid polypeptide (hIAPP) monomer at anionic lipid bilayers: Implications for membrane-mediated aggregation. Int. J. Mol. Sci. 2013;14:6241–6258. doi: 10.3390/ijms14036241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohkubo YZ, Pogorelov TV, Arcario MJ, Christensen GA, Tajkhorshid E. Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys. J. 2012;102:2130–2139. doi: 10.1016/j.bpj.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pogorelov TV, Vermaas JV, Arcario MJ, Tajkhorshid E. Partitioning of amino acids into a model membrane: Capturing the interface. J. Phys. Chem. B. 2014;118:1481–1492. doi: 10.1021/jp4089113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. Characterizing the membrane-bound state of cytochrome P450 3A4: Structure, depth of insertion, and orientation. J. Am. Chem. Soc. 2013;135:8542–8551. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arcario M, Tajkhorshid E. Membrane-induced structural rearrangement and identification of a novel membrane anchor in talin F2F3. Biophys. J. 2014;107:2059–2069. doi: 10.1016/j.bpj.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vermaas JV, Tajkhorshid E. Conformational heterogeneity of α-synuclein in membrane. Biochim. Biophys. Acta, Biomembr. 2014;1838:3107–3117. doi: 10.1016/j.bbamem.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Z, Schulten K. Synaptotagmin’s role in neurotransmitter release likely involves Ca2+-induced conformational transition. Biophys. J. 2014;107:1156–1166. doi: 10.1016/j.bpj.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vermaas JV, Tajkhorshid E. A microscopic view of phospholipid insertion into biological membranes. J. Phys. Chem. B. 2014;118:1754–1764. doi: 10.1021/jp409854w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blanchard A, Arcario M, Schulten K, Tajkhorshid E. A highly tilted membrane configuration for the prefusion state of synaptobrevin. Biophys. J. 2014;107:2112–2121. doi: 10.1016/j.bpj.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valiyaveetil FI, Zhou Y, MacKinnon R. Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry (N. Y.) 2002;41:10771–10777. doi: 10.1021/bi026215y. [DOI] [PubMed] [Google Scholar]
- 54.Chill JH, Louis JM, Miller C, Bax A. NMR study of the tetrameric KcsA potassium channel in detergent micelles. Protein Science. 2006;15:684–698. doi: 10.1110/ps.051954706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guangshun Wang. NMR of membrane-associated peptides and proteins. 2008 doi: 10.2174/138920308783565714. [DOI] [PubMed] [Google Scholar]
- 56.Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J. Am. Chem. Soc. 2008;130:6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petrache HI, Tristram-Nagle S, Gawrisch K, Harries D, Parsegian VA, Nagle JF. Structure and fluctuations of charged phosphatidylserine bilayers in the absence of salt. Biophys. J. 2004;86:1574–1586. doi: 10.1016/S0006-3495(04)74225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez L, Andrade R, Birgin EG, Martinez JM. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009;30:2157–2164. doi: 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- 59.Neria E, Fischer S, Karplus M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996;105:1902–1921. [Google Scholar]
- 60.Piana S, Lindorff-Larsen K, Shaw D. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011;100:L47–L49. doi: 10.1016/j.bpj.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD., Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-Chain χ1 and χ2 Dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindorff-Larsen K, Maragakis P, Piana S, Eastwood MP, Dror RO, Shaw DE. Systematic validation of protein force fields against experimental data. PLoS One. 2012;7:e32131. doi: 10.1371/journal.pone.0032131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 64.Weinbach Y, Elber R. Revisiting and parallelizing SHAKE. J. Comput. Phys. 2005;209:193–206. [Google Scholar]
- 65.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 66.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 67.Ewald PP. Die berechnung optischer und elektrostatischer gitterpotentiale. Ann. Phys. 1921;64:253–287. [Google Scholar]
- 68.Darden T, York D, Pedersen L. Particle mesh ewald: An N·log(N) method for ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 69.York DM, Wlodawer A, Pedersen LG, Darden TA. Atomic-level accuracy in simulations of large protein crystals. Proc. Natl. Acad. Sci. U. S. A. 1994;91:8715–8718. doi: 10.1073/pnas.91.18.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bishop CM. Pattern Recognition and Machine Learning. 1. Springer Science+Business Media, LLC; New York: 2006. Periodic Variables. [Google Scholar]
- 71.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins: Struct., Funct., Genet. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 72.Kelly SM, Price NC. The application of circular dichroism to studies of protein folding and unfolding. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1997;1338:161–185. doi: 10.1016/s0167-4838(96)00190-2. [DOI] [PubMed] [Google Scholar]
- 73.Shim S, Gupta R, Ling YL, Strasfeld DB, Raleigh DP, Zanni MT. Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. Proc. Natl. Acad. Sci. U. S. A. 2009;106:6614–6619. doi: 10.1073/pnas.0805957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cao P, Marek P, Noor H, Patsalo V, Tu L, Wang H, Abedini A, Raleigh DP. Islet amyloid: From fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 2013;587:1106–1118. doi: 10.1016/j.febslet.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Westermark P, Engström U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. U. S. A. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roberts CJ. Non-native protein aggregation kinetics. Biotechnol. Bioeng. 2007;98:927–938. doi: 10.1002/bit.21627. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.