

Abstract
Within the last decade, the Bromodomain and Extra-Terminal domain family (BET) of proteins have emerged as promising drug targets in diverse clinical indications including oncology, auto-immune disease, heart failure, and male contraception. The BET family consists of four isoforms (BRD2, BRD3, BRD4, and BRDT/BRDT6) which are distinguished by the presence of two tandem bromodomains (BD1 and BD2) that independently recognize acetylated-lysine (KAc) residues and appear to have distinct biological roles. BET BD1 and BD2 bromodomains differ at five positions near the substrate binding pocket: the variation in the ZA channel induces different water networks nearby. We designed a set of congeneric 2- and 3-heteroaryl substituted tetrahydroquinolines (THQ) to differentially engage bound waters in the ZA channel with the goal of achieving bromodomain selectivity. SJ830599 (9) showed modest, but consistent, selectivity for BRD2-BD2. Using isothermal titration calorimetry, we showed that the binding of all THQ analogs in our study to either of the two bromodomains was enthalpy driven. Remarkably, the binding of 9 to BRD2-BD2 was marked by negative entropy and was entirely driven by enthalpy, consistent with significant restriction of conformational flexibility and/or engagement with bound waters. Co-crystallography studies confirmed that 9 did indeed stabilize a water-mediated hydrogen bond network. Finally, we report that 9 retained cytotoxicity against several pediatric cancer cell lines with EC50 values comparable to BET inhibitor (BETi) clinical candidates.
1. Introduction
Transcriptional regulation in eukaryotes is tightly regulated by post-translational chemical modification of histones, specifically in the tail region.1,2 One critical modification is ξ-acetylation of lysine residues—a hallmark for transcriptional activation.3,4 The lysine acetylation (KAc) state of histone tails is governed by histone acetyltransferases (HATs) and histone deacetylases (HDACs).5 Upon acetylation, activated histone tails can be read by bromodomain containing proteins (BRDs),6,7 which act as adaptors to transcriptional factors and/or other non-histone partner(s). These complexes are integral to gene transcription and regulation.8
All BRDs contain a left-handed bundle of four highly conserved alpha-helices (named αZ, αA, αB and αC) that are tethered together by two loop regions (ZA and BC) of variable length and sequence. The BET family (Bromodomain and Extra-Terminal domain), consisting of BRD2, BRD3, BRD4 and BRDT, are distinguished from other BRDs by the presence of tandem N- and C- terminal bromodomains (BDs), hereby designated ‘BD1’ and ‘BD2’, respectively.9
While BRD2, 3, and 4 are ubiquitously expressed, BRDT (BRD6) is exclusively expressed in the testes.10 Under normal conditions, BET proteins assist in gene transcription by interacting with KAc moieties on either histone tails or transcription factors. BRD2 is involved in hematopoiesis,11 obesity,12,13 and acute inflammatory response.14 BRD2-histone binding has been studied using BRD2-BD115 and BRD2-BD216,17 and has been found to bind to the modified lysine marks H4K5Ac, H4K12Ac and H3K9Ac.18 BRD2 has also been reported to interact with STAT5, a transcription factor that is constitutively activated in hematologic malignancies.19 BRD3-BD1 interacts with the acetylated transcription factor GATA1,20 which regulates the expression of erythroid and megakaryocyte-specific genes.21 BRD4-BD1, like BRD2-BD1, has higher affinity for di-and tetra-acetylated H4-marks while BRD4-BD2 has minimal affinity for any histone modifications.22 BRD4-BD2 recruits the acetylated transcription factor, TWIST, to the chromatin bound complex, which is anchored by BRD4-BD1.23 As a consequence, disruption of the TWIST-BRD4 complex has been suggested as a potential drug target for basal-like breast cancer.24 BRD4-BD2 is also known to regulate transcriptional elongation by recruiting the positive transcription elongation factor B (P-TEFb) complex, ultimately resulting in the activation of RNA polymerase II.25,26,27 The testis-specific BRDT is essential for spermatogenesis. Mutagenesis studies with this protein have revealed that the BD1 domain is required for this process, while BD2 is not.28 This presents a double edged sword, as small molecule inhibition of BRDT-BD1 could either serve as potential target for male contraception,29 or result in undesired reproductive effects.
While pan-BETi have been useful therapeutic leads and have facilitated greater understanding of BET function, the poor selectivity within the BET family limits studies of the function of individual BET family members and their respective BDs. Poor intra-BET family selectivity may result in off-target effects, and/or safety concerns, as these compounds advance in clinical studies (e.g., undesired reproductive side-effects due to BRDT inhibition). A similar situation has occurred in the development of HDAC inhibitors; while initial leads have shown promising anti-tumor responses in early-phase clinical trials, undesired side effects attributed to poor selectivity between the HDAC isoforms halted these studies.30
BET BD1 and BD2 bromodomains show 100% sequence conservation in the substrate binding pocket, and differ at only five positions when including residues immediately adjacent to those in the binding pocket (V1–V5, Table 1). Position V4, which resides in the ZA channel, is unique: in BRD2-4, this residue is a glutamine (BRD2 Q101, BRD3 Q61, and BRD4 Q85), while in BRD2-4 BD2, the homologous residue is a lysine (BRD2 K374, BRD3 K336, and BRD4 K378). The physicochemical properties of the residue at this position are reversed in BRDT, which has an arginine in BD1 and an asparagine in BD2. Crystal structures of both bromodomains suggest that the glutamine in BD1 prefers an “in” conformation that establishes hydrogen bonds with both a water molecule (‘W5’) and the tryptophan backbone carbonyl (W97 in BRD2 numbering) in the ‘WPF’ motif (residues W97, P98, and F99). The lysine residue in BD2 (K374), on the other hand, adopts an “out” conformation and extends away from the ZA pocket into solvent. Consequently, the ZA channel in BRD2-4 BD2 domains is more solvent exposed, and a stable water (‘W6’) mimics the role of the glutamine side-chain and forms hydrogen bonds to W5 and the tryptophan backbone carbonyl.
Table 1.
Sequence variation near the substrate binding pockets of BET BD1 and BD2 bromodomains
| Position | BRD2 (BD1/BD2) | BRD3 (BD1/BD2) | BRD4 (BD1/BD2) | BRDT (BD1/BD2) |
|---|---|---|---|---|
| V1 | I162/V435 | I122/V397 | I146/V439 | I115/V357 |
| V2 | D160/H433 | D120/H395 | D144/H437 | D113/H355 |
| V3 | K107/A380 | K67/A342 | K91/A384 | K60/A302 |
| V4 | Q101/K374 | Q61/K336 | Q85/K378 | R54/N296 |
| V5 | R100/Y373 | Y60/Y335 | Q84/Y377 | Q53/Y295 |
We hypothesized that BD-selective inhibitors could be designed by exploiting differences in these water-mediated interactions. As proof-of-concept, we synthesized a congeneric series of 2- and 3-heteroaryl substituted tetrahydroquinolines (THQ) that differed by only one atom in the vicinity of the ZA channel. Overall, bromodomain selectivity was modest. The 2- and 3-furan 9 and 12 show consistent BRD2 BD2-selectivity in TR-FRET and SPR assays. Using ITC, we showed that the binding of all THQ analogs in our study to either bromodomain is enthalpy driven. Remarkably, the binding of 9 to BRD2-BD2 was marked by negative entropy and was entirely driven by enthalpy, consistent with significant restriction of conformational flexibility and/or engagement with bound waters. To validate this hypothesis, we determined the co-crystal structure of 9 bound to BRD2-BD2 and confirmed that the compound did indeed stabilize a water-mediated hydrogen bond network in the ZA channel, albeit weakly. Finally, we showed that 9 potently suppressed c-MYC protein levels in tumor cells and demonstrated cytotoxicity against several pediatric cancer cell lines with EC50 values comparable to two BETi clinical candidates, GSK525762A (2) and RVX-208 (3).
2. Results
2.1 Design and synthesis of compounds
After searching the literature for established synthetically-tractable BETi scaffolds that would permit targeting the V4 Q/K variance between BD1/BD2, we selected the N-acyl THQ scaffold as our starting point.31,32 This scaffold has been reported as a fragment that blocks p53-CREB association.33 BETi based on this scaffold were first reported in several GSK patents.34–38 Co-crystal structures of the THQ GSK1324726A39 (4) bound to BRD2-BD1 (PDB ID: 4UYF) and BRD2-BD2 (PDB ID: 4UYG) showed that the p-benzoic acid moiety projected into the ZA channel. Based on synthetic tractability at the time these studies were performed, we developed a modified version of the reported route37,39 that would enable facile replacement of the p-benzoic acid moiety with 2- and 3-heteroaryl substitutions (9–14), and synthesized intermediate 8 in four steps (Figure 1) with 90% ee (see SI for details). Bromide 8 was used to generate aryl derivatives at the 4-position using Suzuki conditions.
Figure 1. Exemplar BETi and synthetic scheme for THQ analogs 9–14.

Reagents and conditions: a) crotonoyl chloride, LiHMDS, THF −78 °C; b) 4-bromo-aniline, [(R)-(+)-2,2′-bis(diphenylphosphino)-1,1′binaphthyl]-diaquo-Pd(II) bis(triflate), toluene, RT; c) NaBH4, MgCl2, EtOH −10 °C; d) AcCl, DIEPA, CH2Cl2 0 °C; e) R-B(OH)2, K3PO4, BrettPhos Palladacycle Gen. 3, BrettPhos, 2-methyl-2-butanol, 100 °C, 20–89% yield range. See Supporting Information (SI) for specific structures and details.
Docking studies of 9–14 into BRD2-BD1 and BRD2-BD2 confirm that the heteroatoms at the 2- and 3-positions are adjacent to different sets of bound waters in the ZA channel (Figure 2A–B, respectively). The 2-substitued THQ analogs were designed to interact with W5 in BD1 and W5 and W6 in BD2 (orange circles), whereas the 3-substitued analogs could potentially interact with waters near V103/V376, K107/A380, and W97/W370 (purple circles). In this proof-of-concept study, we sought to minimize confounding effects by adopting a minimalistic approach and only modifying the type (oxygen, sulfur, and nitrogen) and position (2 or 3) of a single atom in the 5-membered aryl ring. The thiophenes are bio-isosteres of phenyl rings and were expected to have limited polar interaction. The nitrogen in pyrrole is a strong hydrogen bond donor and was expected to make significant interactions with water in both bromodomains. However, strong hydrogen bond donors and acceptors also have strong desolvation penalties, and these groups can negatively impact binding affinity unless they are reasonably satisfied with new bonds to the target. Thus, the utility of this group to exploit specific water interactions was debatable. In contrast, furan oxygen is a weak hydrogen bond acceptor with a substantially lower desolvation penalty. We expected this group to be able to weakly stabilize or destabilize bound water without inducing significant rearrangement of the water network.
Figure 2. Potential water-mediated interactions with 2- and 3-substituted THQ analogs.

Docked pose of 9–14 in BRD2-BD1 (A) and BRD2-BD2 (B). W5 and W6 waters are highlighted in orange. Water molecules present in the ZA channel but distal to the V4 residue (Q101/K374) are highlighted in purple. Compound 4 (yellow sticks) is shown for reference.
2.2 Evaluation of the in vitro activity of compounds 9–14
Inhibitor potency was determined using a competitive TR-FRET assay measuring interaction of the respective BET-BD with an acetylated peptide ligand (Table 2). The widely studied BET inhibitor (+)-JQ1 (1) and the exemplar THQ 4 were included for reference. Interestingly, all three 3-substitued heteroaryl THQs possessed some degree of BD2-selectivity, with 13 and 14 exceeding that observed for 9. Although the 3-furanyl analog (12) showed selectivity comparable to 9, it was also approximately 3-fold less potent on both BD1 and BD2. Much to our surprise, none of the isopropyl carbamate-based THQs matched the BD2 selectivity observed for 4, which has an aniline that interacts with the hydrophobic pocket created by the V2 histidine and WPF shelf.
Table 2.
TR-FRET competitive binding data for exemplar BETi and THQ analogs 9–14.
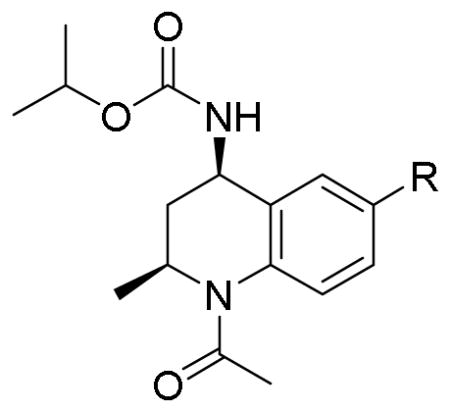
| ||||
|---|---|---|---|---|
| Comp. | R | TR-FRET, IC50 [μM]* | ||
| BRD2-BD1 | BRD2-BD2 | Selectivity [BD1/BD2] | ||
| 1 | -- | 0.37 (0.26 – 0.52) | 0.17 (0.11 – 0.28) | 2.1 |
| 4 | -- | 0.72 (0.39 – 1.33) | 0.03 (0.01 – 0.08) | 21.3 |
| 9 | 2-furan | 2.02 (1.18 – 3.44) | 0.61 (0.46 – 0.81) | 3.3 |
| 10 | 2-thiophene | 1.77 (1.42 – 2.20) | 0.83 (0.61 – 1.15) | 2.1 |
| 11 | 2-pyrrole | 1.40 (0.85 – 2.31) | 1.40 (0.52 – 3.81) | 1.0 |
| 12 | 3-furan | 5.80 (3.46 – 9.74) | 1.91 (1.24 – 2.94) | 3.0 |
| 13 | 3-thiophene | 5.21 (2.72 – 9.96) | 1.08 (0.92 – 1.28) | 4.8 |
| 14 | 3-pyrrole | 2.48 (1.38 – 4.45) | 0.41 (0.23 – 0.74) | 6.0 |
Average of 3–9 replicates per compound. 95% confidence interval reported in parenthesis.
Binding affinity (Kd) of each compound to both BRD2 bromodomains was determined using surface plasmon resonance (‘SPR’, Table 3). SPR confirmed moderately tight binding for all compounds to either bromodomain with Kd values less than 1 μM. Selectivity was lower across all samples compared to the TR-FRET experiment. Compounds 9 and 12 showed a high level of BD2-selectivity, consistent with the competitive binding assay, whereas the selectivity for 13 and 14 was lower.
Table 3.
SPR results for 1 and THQ analogs 9–14.
| Comp. | R | SPR, KD [nM]* | ||
|---|---|---|---|---|
| BRD2-BD1 | BRD2-BD2 | Selectivity [BD1/BD2] | ||
| 1 | -- | 104 | 116 | 0.9 |
| 9 | 2-furan | 80 | 47 | 1.7 |
| 10 | 2-thiophene | 66 | 61 | 1.1 |
| 11 | 2-pyrrole | 99 | 74 | 1.3 |
| 12 | 3-furan | 245 | 148 | 1.7 |
| 13 | 3-thiophene | 105 | 72 | 1.5 |
| 14 | 3-pyrrole | 104 | 77 | 1.4 |
Average of 3 experiments per compound
Next, we explored the thermodynamics of inhibitor binding to individual BRD2 domains using isothermal calorimetry (‘ITC’, Table 4). Interestingly, the binding of all compounds in this series to either BD1 or BD2 appeared to be driven primarily by enthalpy (ΔH), whereas 1 was more dependent on the entropic component (ΔS). Compound 9 showed an exceptionally large negative change in ΔH on BD2 binding compared to other compounds, and this was countered by a large negative change in ΔS. This phenomenon was consistent with the hypothesis that binding of the 2-furan THQ induces one or more specific water-mediated ligand-protein interactions that result in favorable enthalpy. However, this effect was partially compensated by a negative ΔS, potentially stemming from a decrease in rotational and translational freedom when solvent water is bound and oriented. When the ring oxygen in the furan was moved from the 2-position (9) to the 3-position (12), the large negative changes in enthalpy and entropy for BD2 were lost. The thermodynamics of binding of 12 change slightly between BD2 and BD1, with binding to BD1 almost completely driven by enthalpy, while BD2 binding showed more conventional contributions from ΔH and ΔS to total binding energy.
Table 4.
ITC results for 1 and THQ analogs 9–14.
| BRD2 | Measurement | Compound | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 9 | 10 | 11 | 12 | 13 | 14 | ||
| BD1 | average Kd, nM* | 88.99 | 2188.61 | 61.26 | 131.22 | 162.47 | 71.65 | 91.53 |
| ΔG, kcal/mol | −9.29 | −7.47 | −9.52 | −9.08 | −8.96 | −9.43 | −9.28 | |
| ΔH, kcal/mol | −4.9 | −7.27 | −8.27 | −6.9 | −9.9 | −7.69 | −7.33 | |
| −TΔS, kcal/mol | −4.39 | −0.2 | −1.25 | −2.18 | 0.94 | −1.74 | −1.95 | |
| BD2 | average Kd, nM* | 112.14 | 280.44 | 20.87 | 35.86 | 299.51 | 17.12 | 19.94 |
| ΔG, kcal/mol | −9.3 | −8.62 | −10.12 | −9.82 | −8.61 | −10.25 | −10.15 | |
| ΔH, kcal/mol | −1.47 | −15.27 | −7.18 | −6.89 | −6.31 | −6.92 | −7.01 | |
| −TΔS, kcal/mol | −7.83 | 6.65 | −2.94 | −2.92 | −2.29 | −3.33 | −3.32 | |
| Selectivity [BD1/BD2] | 0.8 | 7.8 | 2.9 | 3.7 | 0.5 | 4.2 | 4.6 | |
Average of 3 experiments per compound; N values ranged from 0.95 to 1.08
The more lipophilic thiophene analogs (10, 13) exhibited a moderate selectivity towards BD2, and their respective binding affinities were guided by changes in ΔS. This is expected, as the nature of their interactions is primarily hydrophobic with each protein. Interestingly, the BD2-selectivity of the pyrrole analogs (11, 14) was also driven primarily by ΔS. In contrast to the weak hydrogen bond acceptor potential of the furan oxygen, the pyrrole nitrogen is a strong hydrogen bond donor40 that carries a significant desolvation penalty upon binding. If the polar interactions formed at the binding site are sub-optimal, then the net change in enthalpy for compounds such as the pyrroles might be negligible or even unfavorable. Consistent with this hypothesis, pyrrole 11 had the lowest enthalpy among 2-substituted heteroaryls and pyrrole 14 had the lowest enthalpy among the 3-substituted compounds in BD1. In BD2, enthalpies for all compounds fell within a narrow range (−6.31 to −7.18), with 9 behaving as an outlier. The relatively flat SAR for enthalpy and entropy in BD2 might be the result of the greater extent of solvent accessibility in this domain. As noted above, the exceptional thermodynamic behavior of 9 might be the result of the formation of specific, geometrically-constrained enthalpic interactions.
It should be noted that the rank ordering of Kd values as determined by the ITC assay were inconsistent with those reported from both the TR-FRET and SPR assays. In particular, 9 and 12 are approximately ten-fold less potent than the other analogs on BRD2-BD2. We attribute this discrepancy to the lack of DMSO in the ITC buffer, which may have adversely affected compound solubility. Therefore, the magnitude of the Kd values between compounds in the ITC assay should be interpreted with caution; however, comparison for the same compound between bromodomains is still valid. Taking this observation into account, 9 consistently showed BD2 selectivity in all binding assays tested.
2.3 Crystallographic study of 9 bound to BRD2-BD2
To rationalize the binding observed in TR-FRET, SPR, and ITC, we performed crystallography studies with 9 to understand how it interacted with BRD2-BD1 and BRD2-BD2 domains. While the lack of potency on BD1 hindered structure determination, we were able to solve the 9:BRD2-BD2 co-crystal structure at 2 Å resolution (PDB ID: 5EK9). The asymmetric unit of the crystal contained two monomers which appeared to reflect two opposing low energy binding modes for 9. In chain A, 9 was bound with the isopropyl carbamate moiety extended into a hydrophobic pocket on the WPF shelf, but the 2-furan was pulled out of the ZA channel (Figure 3A). In chain B, the isopropyl carbamate group extended away from the WPF shelf, but the 2-furan moiety engaged the ZA channel (Figure 3B) and the W5 water molecule formed a highly structured water network that involved three protein-bound waters, the backbone amides of D376 and K374, and the backbone carbonyls of P375 and P371.
Figure 3. Hydrogen bond analysis of the water network near V4 and 9 from 5EK9.

(A) Co-crystal structure of 9 bound to BRD2-BD2 from chain A, with the missing W5 water highlighted in red; (B) Co-crystal structure of 9 bound to BRD2-BD2 from chain B. Hydrogen bonds involving water are depicted as broken lines.
The orientation of the water marked at position W6 had not been observed in our earlier analysis, as the normal W6 water orientation involves hydrogen bonding to the K374 amide and the W370 carbonyl. In all BD1 and BD2 structures analyzed, the P371 carbonyl stabilized a water molecule (defined as W4); in this structure, it formed an additional hydrogen bond with the W6 water. Consequently, the other hydrogen atom on W6 was able to form a hydrogen-bond with the W5 water, such that both lone pairs on W5 were occupied. This orientation reduced electrostatic repulsion with the negative electron density near the 2-furan oxygen and allowed 9 to settle more deeply into the ZA channel. The formation of a well-defined hydrogen bond network between water molecules and surrounding residues is consistent with the negative change in the entropy of binding observed in ITC. As noted earlier, the furan oxygen is a relatively weak hydrogen bond acceptor due to its low electronegative potential.40 Hence, very few crystal structures comprising furan molecules have shown hydrogen bond interactions with a furan oxygen. However, the negative dipole of the furan oxygen can still assert a directionality with respect to the orientation of neighboring water. A slight change in the direction of the dipole, as reflected in the 3-furan (12), appears to be enough to prevent the stabilization of the water network, resulting in contributions from ΔH and ΔS that are similar to the other THQ analogs. However, the fact that the water network was not observed in one of the two monomers suggests that this interaction is weak. Indeed, observed BD2 selectivity for 9 was modest and significantly less than the exemplar THQ 4.
2.4 Evaluation of cell-based activity of select BETi and 9
To confirm cell-based activity, we assessed the cytotoxicity of compound 9 in several cancer cell lines and compared EC50 values to other reported BETi (Table 5). We were pleased to see that the EC50 values of 9 were comparable to the clinical compound GSK525762 (2) and exemplar THQ 4, although slightly less active than 1 in general. Compound 9 exhibited greater cellular cytotoxicity than the reported BD2-selective compound RVX-208 (3), but did not exhibit cytotoxicity at the highest concentrations tested in three control cell lines used to assess non-specific cytotoxicity: BJ, an immortalized fibroblast cell line; HEK293, an embryonic kidney cell line; and HEPG2, a hepatocarcinoma cell line. Finally, the furan moiety can be metabolized in cells to generate toxic species. To confirm the reported on-target behavior of BETi in tumor cells41, we found that 9 reduced levels of MYC protein in the human Group 3 medulloblastoma cell line, HD-MB0342, with an EC50 value of 480 nM [dose range 140–1600nM] (Figure 4), which is consistent with the potency of the compound in the biochemical and cytotoxicity assays.
Table 5.
Cytotoxicity of select compounds in cancer cell lines.
| Cell Line | Cancer Type | Compound, EC50 [μM]* | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 9 | ||
| HD-MB03 | MB | 0.16 | 3.50 | >5 | 0.20 | 0.17 |
| NALM-6 | ALL | 0.06 | 0.39 | >5 | NA | 0.55 |
| NALM-16 | ALL | 0.03 | 0.27 | >5 | 0.31 | 0.46 |
| 697 | ALL | 0.09 | 1.17 | >5 | NA | 1.08 |
| Loucy | ETP-ALL | 0.09 | >5 | >5 | 0.7 | 2.27 |
| BJ** | Control | ND | ND | ND | ND | ND |
| HEK293** | Control | ND | ND | ND | NA | ND |
| HEPG2** | Control | ND | ND | ND | ND | ND |
5 days exposure,
3 days exposure
Figure 4. Quantification of c-MYC protein levels.

HD-MB03 medulloblastoma cells (nuclei in blue) were treated with either DMSO, or increasing concentrations of compound 9, then fixed and stained for c-MYC expression after 72 hours. Immunofluorescence with an antibody against c-MYC shows decreased levels of the protein (green) with increasing concentration of compound when compared to the DMSO control. Images acquired at 10x.
3. Discussion
In our efforts to generate a BET bromodomain selective compound, we designed and synthesized a congeneric series 2- and 3-heteroaryl substituted THQs in order to probe the different water networks present in the ZA channel of BD1 and BD2. Several compounds from our small library inhibit histone peptide:BRD2-BD2 interactions with IC50 values <1 μM. Remarkably, analysis of the thermodynamics of binding between BRD2-BD2 and 9 revealed that the interaction is entirely driven by enthalpy that is partially compensated by significant negative entropy. This phenomenon was not observed in the binding of 9 and BRD2-BD1, nor was it observed in any other compounds within this library set, including regioisomer 12. Together, these findings suggest that 9 is involved in highly specific, geometrically constrained interactions. Indeed, co-crystallographic studies with 9 bound to BRD2-BD2 support the hypothesis that the 2-furan induces a well-defined water-mediated hydrogen bond network.
It should be emphasized that the selectivity observed for 9 was modest and does not meet the current guideline for an effective chemical probe43. While there was disagreement among the assays regarding the magnitude and direction of selectivity for some of the THQ analogs in this study, 9 showed consistent selectivity. The observation that one—but not both—monomers from the co-crystallographic studies showed the presence of a water network also supports the conclusion of weak, but observable, selectivity. Furthermore, the interpretation of thermodynamic signatures is complicated by a number of factors, including solute effects, conformational flexibility, and cooperativity44, and therefore, other factors driving selectivity cannot be ruled out. Despite these caveats, we believe that the evidence presented here support the hypothesis that the water network in the ZA channel can be rationally exploited for bromodomain selectivity. Moreover, it is plausible that bromodomain selectivity could be further enhanced by replacing the isopropyl carbamate group in 9–14 with the aniline from 4 and thereby, simultaneously take advantage of selectivity elements at V2 and V4.
Compound 9 demonstrated activity in several cancer cell lines with EC50 values similar to 2, which currently is being evaluated in clinical trials. Furthermore, while maximizing interactions in the ZA channel increases selectivity for BD2 in BRD2-4, they may also provide a route to decrease binding to one or both domains of BRDT, as the residue types are juxtaposed for this BET family member. Taken together, the optimization of BD2-selective compounds has great potential to deliver high quality clinical candidates unburdened by safety concerns associated with pan-BETi.
4. Methods
Synthetic Chemistry
All solvents were purchased from Sigma-Aldrich (anhydrous grade). All reagents and control compounds used were purchased at the highest grade available. All palladium catalyst and ligands were purchased from Strem Chemicals. All melting points were recorded with Buchi MP B-545 instrument and were not corrected. All reactions were performed under an atmosphere of nitrogen and were monitored by ultra-performance liquid chromatography (UPLC) analysis. UPLC analysis was performed on a Waters Acquity instrument and was carried out with a BEH C18 2.1 × 50 mm column using gradient elution with stationary phase: BEH C18, 1.7 mm, solvents: A: 0.1% formic acid in water, B: 0.1% formic acid in acetonitrile, detector types: PDA (210 to 400 nm) and ELS (evaporating light scattering). Chiral purity was ascertained via Waters UPC2 supercritical fluid chromatography (SFC); column: ADH, method: 15–40% methanol: liquid carbon dioxide gradient, time = 11 minutes. High-resolution mass spectrometric (HRMS) analyses were accomplished using a UPLC/Q-ToF MS with stationary phase: BEH C18, 1.7 mm, solvents: A: 0.1% formic acid in water, B: 0.1% formic acid in acetonitrile. All single runs, normal phase flash chromatography or reverse phase purifications employed automated chromatography using a Biotage Isolera One or Four (Biotoage SNAP® Ultra cartridges, solvent system for normal phase: EtOAc in hexane; for reverse phase; 0.1% aqueous formic acid in acetonitrile). Nuclear magnetic resonance (NMR) experiments were conducted using a 400 MHz II instrument (Bruker Avance II). Abbreviations for multiplicities observed in NMR spectra: s; singlet; br s, broad singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet. The purity of all compounds was determined by UPLC (UV and ELSD purity average). QC for control compounds was conducted on the aforementioned UPLC instrument (method: formate). 1 retention time is 1.32 min, [M + H]+ = 456.87.
Chemical Synthesis
Isopropyl (E)-but-2-enoylcarbamate (5)
To a solution of isopropyl carbamate (15.85 g, 154 mmol) in anhydrous THF (150 mL) in a flame-dried flask at −78 °C was added crotonoyl chloride (14.16 mL, 146 mmol), followed by LiHMDS (300 mL, 1.0 M in THF). Reaction was permitted to slowly warm to rt and stir at rt overnight. Reaction was quenched via addition of chilled (~0 °C) saturated aq ammonium chloride (175 mL). Upon warming, solution was extracted with EtOAc (3 × 75 mL). Combined organics were washed with sat NaCl and dried with MgSO4, filtered and concentrated under reduced pressure. Crude product was purified via automated normal phase chromatography (EtOAc: hexanes, 20:80) to provide the desired imide as a white solid (12.3 g, 64% yield, m.pt.: 91 °C). LCMS/UPLC (method: formate) retention time 0.82 min, [M + H]+ = 172.27. 1H NMR (400 MHz, Chloroform-d) δ 7.55 (s, 1H), 7.14 (dq, J = 15.3, 6.9 Hz, 1H), 6.86 (dq, J = 15.2, 1.7 Hz, 1H), 4.99 (p, J = 6.3 Hz, 1H), 1.94 (dd, J = 6.9, 1.7 Hz, 3H), 1.30 (d, J = 6.3 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 166.03, 151.36, 146.34, 122.94, 70.23, 21.78, 18.40.
Isopropyl (S)-(3-((4-bromophenyl)amino)butanoyl)carbamate (6)
To a suspension of imide 5 (5.0 g, 29.2 mmol) and (R)-(+)-BINAP-Pd(OH)2(OTf)2 (1.45 g, 1.36 mmol) in anhydrous toluene (80 mL) at rt was added 4-bromoaniline (3.35 g, 19.47 mmol) slowly over 1 h (alternatively aniline can be dissolved in toluene and added dropwise). Resulting reaction was permitted to stir at rt for 17–72 h. Catalyst can become gummy during the course of the reaction39. Loading catalyst and aniline in portions can help avoid gumminess. Heating the reaction gently (temperature = 45 °C) can help as well without negatively influencing the ee of the reaction. Once reaction was deemed complete, the reaction was concentrated under vacuum. Residual was purified via automated normal phase chromatography (EtOAc: hexanes, 20:80) to provide the desired Michael adduct as an off-white solid (6.12 g, 92% yield, m.pt.: 131 °C). Chiral SFC (method: methanol: liquid CO2) retention time 7.40 min (major enantiomer, 92.2% UV), 10.58 min (minor enantiomer, 7.8% UV); 85% ee. 1H NMR (400 MHz, Chloroform-d) δ 7.42 (s, 1H), 7.25 – 7.21 (m, 2H), 6.54 – 6.46 (m, 2H), 4.97 (hept, J = 6.3 Hz, 1H), 3.99 (q, J = 6.3 Hz, 1H), 3.90 (s, 1H), 3.09 (dd, J = 16.0, 5.7 Hz, 1H), 2.90 (dd, J = 16.0, 6.0 Hz, 1H), 1.29 (m, 9H). 13C NMR (126 MHz, CDCl3) δ 172.71, 151.28, 145.87, 132.02, 115.21, 109.22, 70.56, 45.98, 41.94, 21.76, 20.69.
Isopropyl ((2S,4R)-6-bromo-2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (7)
To a solution of amino imide 6 (6.12 g, 17.83 mmol) in EtOH (90 mL) at −10 °C was added NaBH4 (0.51 g, 13.37 mmol) in one portion, followed by an aqueous solution of MgCl2.6H2O (3.99 g, 19.61 mmol, dissolved in 12 mL water) dropwise such that the internal temperature did not exceed −10 °C. The resulting solution was permitted to stir at 0 °C for 1 h, followed by rt for 1 h. The reaction was quenched via addition of citric acid (6.5 g, 2.5 eq) dissolved in aq 1M HCl (54 mL), followed by treatment of CH2Cl2 (50 mL). After 1 h of heterogeneous stirring, organic solution was collected, followed by aqueous extractions with CH2Cl2 (2 × 30 mL). Combined organics were washed with sat aq NaCl, dried over MgSO4, filtered and concentrated. Crude product was purified via automated normal phase chromatography (EtOAc: hexanes) to provide cyclized amine as a white solid (2.1 g, 91% yield, m.pt.: 167 °C). LCMS/UPLC (method: formate) retention time 1.19 min, [M + H]+ = 327.01. 1H NMR (400 MHz, Chloroform-d) δ 7.26 (d, J = 1.1 Hz, 1H), 7.08 (dd, J = 8.5, 0.9 Hz, 1H), 6.34 (dd, J = 8.5, 0.9 Hz, 1H), 4.98 (m, J = 5.4 Hz, 2H), 4.70 (d, J = 9.5 Hz, 1H), 3.74 (s, 1H), 3.62 – 3.48 (m, 1H), 2.26 (ddd, J = 12.6, 6.0, 2.3 Hz, 1H), 1.43 (q, J = 11.7 Hz, 1H), 1.28 (dd, J = 14.6, 6.3 Hz, 6H), 1.20 (d, J = 6.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 156.26, 143.96, 130.94, 129.44, 123.73, 115.67, 109.04, 68.49, 47.72, 46.74, 37.97, 22.20, 22.14.
Isopropyl ((2S,4R)-1-acetyl-6-bromo-2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (8)
To a solution of amine 7 (5.32 g, 16.26 mmol) in CH2Cl2 (35 mL) and pyridine (3.94 mL) under nitrogen at rt was added dropwise acetyl chloride (1.89 mL, 24.39 mmol). Reaction was permitted to stir at rt for 3–17 h. Reaction was then treated with CH2Cl2 (50 mL) and sat aq. NaHCO3 (50 mL). The phases were separated; aqueous solution was extracted with CH2Cl2 (2 × 20 mL). Combined organics were washed with sat. NaCl, dried over MgSO4, filtered and concentrated. Residual was purified via automated normal phase chromatography (EtOAc:hexanes, 25:75) to provide amide product as a white solid (1.8 g, 95% yield, m.pt.: 163 °C). Chiral SFC (method: methanol: liquid CO2) retention time 5.66 min (major enantiomer, 95.2% UV), 6.06 min (minor enantiomer, 4.8% UV); 90.2% ee. Optical rotation. [α]D = 251.1° (c = 6.1 mg/1 mL EtOH, temperature = 23.1 °C, l = 0.5 dm). Literature: [α]D = 281.1° (c = 0.508 g/100 mL, EtOH temperature = 20 °C, l = 0.1 dm)39. Optical purity (according to optical rotation) = 89.3% ee. LCMS/UPLC (method: formate) retention time 1.11 min, [M + H]+ = 368.88.1H NMR (400 MHz, Methanol-d4) δ 7.49 (dd, J = 8.4, 2.2 Hz, 1H), 7.37 (s, 1H), 7.24 (d, J = 8.4 Hz, 1H), 4.95 (dq, J = 12.5, 6.1 Hz, 1H), 4.81 (d, J = 7.2 Hz, 1H), 4.48 (dd, J = 12.5, 4.1 Hz, 1H), 2.55 (ddd, J = 12.8, 8.7, 4.4 Hz, 1H), 2.15 (s, 3H), 1.39 – 1.21 (m, 7H), 1.13 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, Acetone) δ 169.22, 155.78, 138.21, 133.75, 128.64, 126.59, 124.46, 118.09, 67.08, 46.79, 46.62, 38.39, 20.07, 19.74, 18.76.
General procedure for parallel Suzuki couplings (9–14)
To a microwave reaction vessel flame-dried and purged with nitrogen was added bromide 8 (35 mgs, 0.10 mmol), boronic acid (20–40 mgs, 0.15 mmol), K3PO4 (70 mgs, 0.50 mmol), BrettPhos (5 mgs, 0.01 mmol) and BrettPhos Palladacycle Gen. 3 (10 mgs, 0.01 mmol). The vessel(s) were treated with anhydrous 2-methyl-2-butanol (0.5 mL), sealed under nitrogen and heated to 100 °C for 17 h. Reaction were quenched via addition of EtOAc (2 mL) and water (2 mL). Organic solution was collected and concentrated. Crude products were purified via automated normal phase chromatography (EtOAc: hexane). Reactions can also be purified in an automated reverse phase (acetonitrile: 0.1% formic acid) manner as well (singular or parallel format). The yields of the reported compounds vary from 31–89%. See below for individual reaction yields, % purity and characterization, respectively. For compounds 11 and 14, the pyrrole boronic acids, respectively, were protected with a boc- or tips-group.
Isopropyl ((2S,4R)-1-acetyl-6-(furan-2-yl)-2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (9)
Yield: 31%.1H NMR (400 MHz, Methanol-d4) δ 7.66 (dd, J = 8.2, 1.8 Hz, 1H), 7.62 – 7.57 (m, 2H), 7.31 (d, J = 8.2 Hz, 1H), 6.78 (d, J = 3.3 Hz, 1H), 6.55 (dd, J = 3.4, 1.8 Hz, 1H), 4.97 (p, J = 6.3 Hz, 1H), 4.84 (d, J = 6.7 Hz, 1H), 4.53 (dd, J = 12.5, 4.0 Hz, 1H), 2.57 (ddd, J = 12.7, 8.7, 4.4 Hz, 1H), 2.17 (s, 3H), 1.46 – 1.20 (m, 7H), 1.15 (t, J = 5.7 Hz, 3H).13C NMR (126 MHz, MeOD) δ 170.62, 157.22, 153.26, 142.34, 137.57, 134.71, 129.02, 126.37, 122.09, 117.63, 111.47, 105.09, 68.27, 48.05, 47.88, 39.78, 21.42, 21.01, 20.05. HRMS: calculated: 357.1814 m/z found: 357.1814 m/z.
Isopropyl ((2S,4R)-1-acetyl-2-methyl-6-(thiophen-2-yl)-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (10)
Yield: 83%. 1H NMR (400 MHz, Methanol-d4) δ 7.61 (dd, J = 8.2, 1.7 Hz, 1H), 7.51 (s, 1H), 7.46 – 7.36 (m, 2H), 7.30 (d, J = 8.2 Hz, 1H), 7.12 (dd, J = 5.0, 3.7 Hz, 1H), 4.97 (p, J = 6.2 Hz, 1H), 4.52 (dd, J = 12.5, 4.0 Hz, 1H), 2.56 (ddd, J = 12.8, 8.7, 4.4 Hz, 1H), 2.17 (s, 3H), 1.43 – 1.22 (m, 7H), 1.15 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 170.59, 157.20, 143.32, 137.70, 134.94, 132.56, 127.87, 126.52, 124.86, 124.02, 123.16, 119.78, 68.27, 39.69, 21.46, 21.02, 20.08. HRMS: calculated: 373.1586, found: 373.1580 m/z.
Isopropyl ((2S,4R)-1-acetyl-2-methyl-6-(1H-pyrrol-2-yl)-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (11)
General Suzuki conditions provided N-BOC protected compound, tert-butyl 2-((2S,4R)-1-acetyl-4-((isopropoxycarbonyl)amino)-2-methyl-1,2,3,4-tetrahydroquinolin-6-yl)-1H-pyrrole-1-carboxylate, 1H NMR (400 MHz, Methanol-d4) δ 7.38 (dd, J = 3.3, 1.8 Hz, 1H), 7.34 – 7.25 (m, 2H), 7.21 (s, 1H), 6.26 (t, J = 3.3 Hz, 1H), 6.22 (dd, J = 3.2, 1.8 Hz, 1H), 4.99 – 4.90 (m, 1H), 4.55 (dd, J = 12.6, 4.0 Hz, 1H), 2.56 (ddd, J = 12.7, 8.7, 4.3 Hz, 1H), 2.18 (s, 3H), 1.37 (s, 9H), 1.29 (t, J = 7.2 Hz, 6H), 1.15 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 170.54, 157.03, 149.38, 136.45, 134.71, 134.18, 132.73, 127.56, 125.28, 123.36, 122.45, 114.42, 110.40, 83.66, 68.15, 48.47, 39.95, 26.52, 21.47, 21.02, 20.08. To freshly purified N-BOC pyrrole was added TFA (0.2 mL) and CH2Cl2 (0.2 mL). Reaction was permitted to stir for 1 h, then concentrated and product was purified via automated normal phase chromatography to provide desired product (16 mgs, 48% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 10.82 (s, 1H), 7.63 – 7.43 (m, 2H), 7.24 (d, J = 8.2 Hz, 1H), 6.90 – 6.79 (m, 1H), 6.48 (s, 1H), 6.26 – 6.13 (m, 1H), 4.96 (dt, J = 12.8, 6.4 Hz, 1H), 4.52 (d, J = 8.0 Hz, 1H), 2.64 – 2.41 (m, 1H), 2.16 (s, 3H), 1.34 (dd, J = 19.6, 6.3 Hz, 7H), 1.15 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 171.73, 157.21, 137.37, 132.93, 131.91, 130.96, 126.28, 121.71, 119.00, 117.67, 108.90, 105.41, 68.27, 39.96, 21.38, 21.05, 19.99.
Isopropyl ((2S,4R)-1-acetyl-6-(furan-3-yl)-2-methyl-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (12)
Yield: 37% yield. 1H NMR (400 MHz, Methanol-d4) δ 7.91 (s, 1H), 7.59 (t, J = 1.7 Hz, 1H), 7.53 (dd, J = 8.2, 1.6 Hz, 1H), 7.42 (s, 1H), 7.29 (d, J = 7.9 Hz, 1H), 6.80 (s, 1H), 4.96 (dt, J = 12.5, 6.2 Hz, 1H), 4.52 (d, J = 11.9 Hz, 1H), 2.57 (ddd, J = 12.6, 8.6, 4.2 Hz, 1H), 1.43 – 1.25 (m, 7H), 1.15 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 170.62, 157.22, 153.26, 142.34, 137.57, 134.72, 129.02, 126.37, 122.09, 117.63, 111.47, 105.09, 68.27, 39.78, 21.42, 21.01, 20.05. HRMS: calculated:357.1814 m/z, found: 357.1813 m/z.
Isopropyl ((2S,4R)-1-acetyl-2-methyl-6-(thiophen-3-yl)-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (13)
Yield: 89%. 1H NMR (400 MHz, Methanol-d4) δ 7.70 – 7.58 (m, 2H), 7.51 (dd, J = 5.1, 3.0 Hz, 2H), 7.47 – 7.43 (m, 1H), 7.30 (d, J = 8.1 Hz, 1H), 4.96 (hept, J = 6.3 Hz, 1H), 4.54 (dd, J = 12.4, 3.9 Hz, 1H), 2.56 (ddd, J = 12.7, 8.7, 4.4 Hz, 1H), 2.16 (s, 3H), 1.33 (dd, J = 14.4, 6.3 Hz, 6H), 1.15 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 170.60, 157.20, 141.48, 137.39, 134.59, 134.07, 126.41, 126.25, 125.65, 124.74, 120.28, 68.26, 39.93, 21.46, 21.04, 20.08. HRMS: calculated: 373.1586 m/z, found: 373.1581 m/z
Isopropyl ((2S,4R)-1-acetyl-2-methyl-6-(1H-pyrrol-3-yl)-1,2,3,4-tetrahydroquinolin-4-yl)carbamate (14)
General Suzuki conditions provide N-TIPS protected compounds, Isopropyl ((2S,4R)-1-acetyl-2-methyl-6-(1-(triisopropylsilyl)-1H-pyrrol-3-yl)-1,2,3,4-tetrahydroquinolin-4-yl)carbamate 1H NMR (400 MHz, Methanol-d4) δ 7.50 (dd, J = 8.1, 1.5 Hz, 1H), 7.41 (s, 1H), 7.20 (d, J = 8.1 Hz, 1H), 7.15 (s, 1H), 6.91 – 6.85 (m, 1H), 6.65 – 6.57 (m, 1H), 4.97 (p, J = 6.2 Hz, 1H), 4.49 (dd, J = 12.4, 3.8 Hz, 1H), 2.54 (ddd, J = 12.7, 8.7, 4.3 Hz, 1H), 2.15 (s, 3H), 1.55 (p, J = 7.5 Hz, 3H), 1.34 (dd, J = 16.4, 6.2 Hz, 6H), 1.16 (d, J = 7.5 Hz, 18H). 13C NMR (126 MHz, MeOD) δ 170.64, 157.22, 137.13, 134.63, 132.84, 126.33, 126.14, 125.24, 123.30, 120.44, 118.76, 108.26, 68.08, 39.72, 21.43, 21.25, 21.01, 20.02, 16.87, 11.43. To freshly purified silyl-protected pyrrole was added a solution of TBAF (0.1 mL, 1.0 M in THF). Reaction was permitted to stir for 1 h, then concentrated and product was purified via automated normal phase chromatography to provide desired product (54 % yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 7.48 (dd, J = 8.1, 1.7 Hz, 1H), 7.41 (s, 1H), 7.17 (d, J = 8.1 Hz, 1H), 7.12 (s, 1H), 6.80 (dd, J = 2.7, 2.0 Hz, 1H), 6.49 – 6.41 (m, 1H), 4.96 (dq, J = 12.6, 6.3 Hz, 1H), 4.50 (dd, J = 12.5, 3.9 Hz, 1H), 2.54 (ddd, J = 12.7, 8.7, 4.3 Hz, 1H), 2.15 (s, 2H), 1.31 (ddd, J = 22.9, 16.1, 7.5 Hz, 7H), 1.14 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 170.66, 157.25, 137.04, 135.23, 132.39, 126.07, 123.42, 123.01, 118.62, 114.45, 105.04, 68.17, 39.85, 21.38, 21.01, 19.98. HRMS: calculated: 356.1974 m/z, found: 356.1971 m/z
Commercial compounds
1 (catalog number 11187) was purchased from Cayman Chemical, 2 was sourced from Calbiochem (catalog number 401010), 3 was purchased from Selleck Chemicals (catalog number S7295), and 4 was obtained from MedChemExpress (catalog number HY-13960). Compound purity was confirmed to be ≥95% as defined above for synthetic compounds.
Molecular docking
The Glide v5.545,46 molecular docking module (Schrodinger, 2012) was used to generate unbiased binding modes in the BRD2-BD1 and BRD2-BD2 bromodomains. GW841819X bound to the BRD2-BD1 structure (PDB ID: 2YDW) and 1 bound to BRD2-BD2 (PDB ID: 3ONI) guided construction of the pre-computed grid. Four conserved water molecules present in both crystal structures were also included in the docking studies. The docking protocol started with systematic conformational expansion of the ligand, followed by placement in the receptor site. Minimization of the ligand in the field of the receptor was then carried out using the OPLS-AA force field with the default distance-dependent dielectric. The lowest energy poses were then subjected to a Monte Carlo procedure that sampled nearby torsional minima. Posed were ranked using GlideScore, a modified version of the ChemScore function that includes terms for steric clashes and buried polar groups. The default van der Waal’s scaling was used (1.0 for the receptor and 0.8 for the ligand). Next, the docked poses for THQ analogs 9–14 generated above were aligned to their respective bromodomain using the crystal structures of 4 bound to BRD2-BD1 (PDB ID: 4UYF) and BRD2-BD2 (PDB ID: 4UYG). All crystallographic waters were preserved and the docked ligand was minimized in the context of these bound waters using the ‘Energy Minimize’ module in the MOE application (v2016.08, Chemical Computing Group) and default parameters. Water molecules were kept rigid during minimization, and protein backbone atoms were restrained using the ‘Tether’ option.
Hydrogen-bond network analysis
The Protonate 3D module47 in the MOE application (v2016.08, Chemical Computing Group) was used to assign ionization states and to position hydrogens (rotamers and tautomers). The program optimizes the titration free energy of all titratable groups in the context of an all-atom model of the macromolecular structure (including ligands and solvent). The Generalized Born/Volume Integral48 electrostatics model is used for longer range interactions and solvation effects. After the system composed of protein and ligand is optimized, incidental water molecules are usually oriented one by one. For this study, the ‘precise’ flag was set for water molecules in the ZA channel: this treats the water molecules as part of the main calculation where geometric orientation is optimized in the context of all other system atoms. While substantially increasing computation time, the ‘precise’ flag enables better determination of water-mediated protein-ligand interactions. Only hydrogen bonds exceeding 0.5 kcal/mol as calculated by MOE are reported as bona fide interactions.
Time-resolved fluorescence energy transfer (TR-FRET)
Time-resolved fluorescence energy transfer (TR-FRET) kits for BRD2-BD1 (catalog number 600500) and BRD2-BD2 (catalog number 600510) were was purchased from Cayman Chemical. The buffer components in these kits is proprietary, but we have confirmed the presence of DMSO.
Compound Screening
The TR-FRET conditions were adjusted from the vendor’s instructions to simplify screening by combining all reagents into a single stock (final volume of 26 mL per kit). A Precision Pipetting System (BioTek Instruments, Inc, Winooski, VT) was used to dispense the combined reagents into 384-well solid black plates (20 μL/well). Test compounds dissolved in DMSO were pin-transferred by pintool (V&P Scientific Inc, San Diego, CA). Screening at single final concentration of 35 μM was carried out by transferring 70 nL, while dose-response experiments were conducted on compounds diluted at 10 different concentrations (3 fold series dilutions) in DMSO by transferring 140 nL to assay plates for final concentrations ranging from 70 μM to 3.6 nM. Biological reagents and compounds were incubated at RT for 1h prior to TR-FRET measurements.
With an excitation filter of 337 nm, fluorescence emissions were measured at 620 and 665 nm using a PHERAStar plate reader (BMG Labtech, Durham, NC). The delay time was set to 100 μs and the integration time to 200 μs. The signal ratio was calculated by dividing the emission signal at 665 nm by that at 620 nm. DMSO was used as the negative control (0% inhibition) and 280 μM 1 as the positive control (100% inhibition). A 4-parameter logistic equation was applied to fit the dose-response data to determine IC50 values using in-house software.
Binding by Surface Plasmon Resonance (SPR)
SPR experiments were conducted at 15° C using a SensiQ Pioneer optical biosensor (SensiQ Technologies). Poly-His tagged BRD2 constructs were immobilized on polycarboxylate-coated gold chips preimmobilized with nitrilotriacetic acid (His-Cap chips; SensiQ Technologies) by capture-coupling, a hybrid method of capture and amine coupling chemistry49. The His-Cap chip was preconditioned and charged with Ni2+ in equilibration buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 50 μM EDTA, 0.005% Tween20) at a flow rate of 10 μL/min. The chip was washed with three 60 s injections of regeneration buffer (10 mM HEPES pH 8.3, 150 mM NaCl, 350 mM EDTA, 0.05% Tween20) and one 60 s injection of equilibration buffer and was charged with a 60 s injection of 500 mM NiCl2 in equilibration buffer, which added ~ 55 RU of Ni2+ per channel. The charged chip was primed with binding buffer (20 mM HEPES pH 7.0, 150 mM NaCl, 1 mM TCEP, 0.005% Tween20, 1% DMSO) prior to protein immobilization. Carboxyl groups on the dextran were activated with N-ethyl-N′-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS), and BRD2 constructs were injected until immobilization levels of ~450–530 RU were achieved. One channel on the chip was charged with Ni2+ and activated with EDC/NHS without adding protein to be used as a reference cell. The chip was equilibrated for 1–2 h to allow any remaining active sites to hydrolyze, eliminating the need to apply a blocking agent (e.g. ethanolamine). Compounds SJ830599 and SJ852845 were prepared in binding buffer as a 3-fold dilution series starting at 2 μM and were injected in duplicate at each concentration at a flow rate of 150 μL/min. A series of buffer-only (blank) injections was included throughout the experiment to account for instrumental noise. The data were processed, double-referenced, solvent corrected and analyzed using the software package Qdat50 (version 2.5.3.5, SensiQ Technologies). The kinetic rate constants were determined by globally fitting the data to a 1:1 interaction model. Equilibrium dissociation constants (KD) were calculated as the quotient kd/ka.
Binding by Isothermal Titration Calorimetry (ITC)
Binding of compounds to BRD2 bromodomaims was monitored using an ITC200 (Microcal, Piscataway, NJ). Either BRD2-BD1 (74–194) or BRD2-BD2 (348–455) was dialyzed overnight into a solution containing 50 mM HEPES, pH 7.0 and 150 mM NaCl at 15 °C. Enzyme concentrations were determined after dialysis by absorbance at 280 nm using a molar extinction coefficient (ε280) of 25,565 M−1cm−1 (2.1) or 16,055 M−1cm−1 (2.2). Enzyme was diluted in dialysis buffer (to the concentration listed in the table) and 39.6 μL was loaded into the syringe. Compounds were dissolved in the same dialysis buffer, and loaded in the cell to a volume of 204 μL. Injections were carried out by serial injections of enzyme; first, 1 injection of 1 μL, followed by 19 incremental injections of 2 μL, at 120 second intervals. Data from the first injection was excluded, due to pre-equilibration mixing between the contents of cell and syringe at the syringe tip. Peak areas were integrated, normalized, and then fitted by non-linear regression using the independent sites model in Origin™ (version 2.3.6, Microcal, Piscataway, NJ).
Protein Expression and purification
Proteins were cloned, expressed and purified as previously described9.
Crystallization
BRD2-BD2 construct (Uniprot identifier as BRD2_HUMAN P25440-1 fragment 344–455) was used for crystallographic studies in complex with the inhibitors. Aliquots of the purified proteins were set up for crystallization using a mosquito® crystallization robot (TTP Labtech). Coarse screens were typically setup onto Greiner 3-well plates using three different drop ratios of precipitant to protein per condition (200+100 nL, 150+150 nL and 100+200 nL). All crystallizations were carried out using the sitting drop vapour diffusion method at 277.15 K. BRD2-BD2 in complex with 9 (3 mM final concentration) were obtained by mixing 150 nL of the protein (14 mg/ml) and 150 nL crystallization buffer (20% PEG6000, 0.1 M citrate pH 5).
Data Collection and Structure solution
Complex crystals were cryo-protected using the well solution supplemented with additional 20 % ethylene glycol and was flash frozen in liquid nitrogen. Data was collected at Diamond Light Source I02 and I04 beamlines at wavelengths of 0.9794 and 0.9795 Å, respectively. Indexing and integration was carried out using XDS51 and scaling was performed with AIMLESS52. Initial phases were calculated by molecular replacement with PHASER53 using the apo template BRD2 structure 2DVV.pdb and apo template BRD4 structure 2OSS.pdb. Unique and initial solutions were improved in a total of 50 cycles of automated protein chain tracing starting from existing model and computed using ARP/wARP54. Further manual building with COOT55 and refinement against maximum likelihood target using REFMAC556. Thermal motions were analysed using TLSMD and hydrogen atoms were included in late refinement cycles. GRADE57 was used to generate compound coordinates and cif files. All model validations were carried out using MolProbity58. Models and structure factors have been deposited with PDB accession code: 5EK9.pdb. Additional information regarding data collection and refinement statistics is presented in the Supporting Information.
Cytotoxicity
One human medulloblastoma and four Acute Lymphoblastic Leukemia (ALL) cell lines were evaluated with the bromodomain compounds. The human Group3 medulloblastoma line, HD-MB03, was obtained from Drs Milde, Witt and Deubzer from German Cancer Research Center (DKFZ)42. NALM-16 and 697 cells were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). Loucy and NALM-6 cells were purchased from the American Type Culture Collection (ATCC, Manassas, Virginia).
Leukemic cells were grown in RPMI-1640 media while the other cell lines were cultured in EMEM. Both media were purchased from ATCC and supplemented with 10% fetal bovine serum (GE Healthcare Life Sciences, Hyclone Laboratories, Logan, Utah). Cells were cultured in a humidified 5% CO2, incubator at 37°C according to vendor recommendations. Cells were routinely tested for mycoplasma with the MycoAlert Mycoplasma Detection Kit (Lonza, Walkersville, MD).
Approximately 1000 NALM-16 and 500 Loucy exponentially growing cells were plated per well (150μl) in white polystyrene flat bottom sterile 96-well tissue culture treated plates (Corning, Tewksbury, MA, USA), and incubated overnight at 37 °C in a humidified 5% CO2 incubator. The 697 and NALM-6 cell lines were plated at a concentration of 250 cells per well (150 μl).
HD-MB03 cells were grown as neurospheres in neurobasal medium supplemented with B27, glutamine, streptomycin, penicillin, heparin (1ug/mL, #07980, Stem Cell Technologies), human recombinant basic FGF (25ng/mL, AF-100-18, Peprotech) and EGF (25ng/mL, AF-100-15, Peprotech) on ultra-low attachment dish.
Compound solutions (DMSO) were pin-transferred (V&P Scientific, San Diego, CA, USA) the following day. Cytotoxicity was determined following 5 day incubation using Promega Cell Titer Glo Reagent according to the manufacturer’s recommendations. Luminescence was measured on an Envision plate reader (Perkin-Elmer).
BJ, HEK293, and HEPG2 cells at 400 cells per well (30ml) in white polystyrene flat bottom 384-well sterile tissue culture treated plates (Corning, Tewksbury, MA, USA). Compounds were added as above and cytotoxicity was determined at 3 days after compound addition.
Quantification of MYC protein
3000 human G3 medulloblastoma cells per well were plated into 384-well plates (6007710, Perkin Elmer) in 30 μL of supplemented neurobasal medium using a Wellmate automated dispenser (Thermo Matrix). After 24 hours, 125nl of compound was transferred via pin tool, resulting in a final drug concentration from 2 nM to 40 μM. 72 hours after adding the drug, the plates were fixed with 4% formaldehyde for 20 min. After fixation the cells were then washed 3 times with 75 μl of PBS. The cells were then permeabilized with 0.1% Triton-X 100 for 15 minutes at 25°C and blocked using 1% BSA in PBS for 1 hour at 25°C. The primary antibody against MYC Ab (#9402, Cell Signaling) at a 1:400 dilution was diluted in 1% BSA in PBS. This mixture was added to each well before incubation overnight at 4°C. Each well was then washed 3 times with PBS using a Biotek ELX405 Select plate washer, and incubated for 1 hour at 25°C with a solution containing 1/400 goat α-rabbit-Alexa-488 (4412S, Cell Signaling) and 1 μM Hoechst 33342 to detect nuclear material (H3570, Molecular Probes). Two images were captured of each well at 10x using a GE Healthcare InCell 6000 at 405 nM to detect nuclear staining and 488 nM to detect MYC. All data were analyzed using the multi-target analysis algorithm of the GE InCell Analyzer Workstation software. The number of nuclear objects in each well, as well as all nuclear measurements, was determined through nuclear masks over the Hoechst staining. MYC staining was then measured using top-hat segmentation in the entire cell.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported in part by NIH grant CA-096832 (MFR), a Cancer Center Core grant CA21765, and by the American Lebanese Syrian Associated Charities (ALSAC). SK and CT are grateful for support by the SGC, a registered charity that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute, IMI [115766], Wellcome Trust, Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, and Takeda and the Centre of Excellence Macromolecular complexes (CEF) at Frankfurt University.
Abbreviations Used
- BET
Bromodomain and Extra-Terminal
- BD1
bromodomain 1
- BD2
bromodomain 2
- KAc
acetylated-lysine
- THQ
tetrahydroquinoline
- BETi
Bromodomain inhibitors
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- TR-FRET
time-resolved fluorescence resonance energy transfer
- SPR
surface plasmon resonance
- ITC
isothermal calorimetry
Footnotes
Accession Codes
Structure factors and coordinates of the BRD2-BD2 complex with 9 were deposited with the Protein Data Bank (PDB, www.rcsb.org/pdb/) under accession code 5ek9. Authors will release the atomic coordinates and experimental data upon article publication
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16:258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 2.Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochimica et biophysica acta. 2014;1839:627–643. doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marushige K. Activation of chromatin by acetylation of histone side chains. Proc Natl Acad Sci USA. 1976;73:3937–3941. doi: 10.1073/pnas.73.11.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marmorstein R. Protein modules that manipulate histone tails for chromatin regulation. Nature Reviews Molecular Cell Biology. 2001;2:422–432. doi: 10.1038/35073047. [DOI] [PubMed] [Google Scholar]
- 5.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nature Reviews Drug Discovery. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 6.Wolffe AP, Hayes JJ. Chromatin disruption and modification. Nucleic Acids Research. 1999;27:711–720. doi: 10.1093/nar/27.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Letters. 2002;513:124–128. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez R, Meslamani J, Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochimica et Biophysica Acta, Gene Regulatory Mechanisms. 2014;1839:676–685. doi: 10.1016/j.bbagrm.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French C, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones MH, Numata M, Shimane M. Identification and characterization of BRDT: A testis-specific gene related to the bromodomain genes RING3 and Drosophila fsh. Genomics. 1997;45:529–534. doi: 10.1006/geno.1997.5000. [DOI] [PubMed] [Google Scholar]
- 11.Belkina AC, Blanton WP, Nikolajczyk BS, Denis GV. The double bromodomain protein Brd2 promotes B cell expansion and mitogenesis. Journal of Leukocyte Biology. 2014;95:451–460. doi: 10.1189/jlb.1112588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zang K, Wang J, Dong M, Sun R, Wang Y, Huang Y, Liu X, Li Y, Wang F, Yu M. Brd2 inhibits adipogenesis via the ERK1/2 signaling pathway in 3T3-L1 adipocytes. PLoS One. 2013;8:e78536. doi: 10.1371/journal.pone.0078536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang FN, Liu HS, Blanton WP, Belkina A, Lebrasseur NK, Denis GV. Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochemical Journal. 2009;425:71–83. doi: 10.1042/BJ20090928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. Journal of Immunology. 2013;190:3670–3678. doi: 10.4049/jimmunol.1202838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Umehara T, Nakamura Y, Jang MK, Nakano K, Tanaka A, Ozato K, Padmanabhan B, Yokoyama S. Structural basis for acetylated histone H4 recognition by the human BRD2 bromodomain. Journal of Biological Chemistry. 2010;285:7610–7618. doi: 10.1074/jbc.M109.062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umehara T, Nakamura Y, Wakamori M, Ozato K, Yokoyama S, Padmanabhan B. Structural implications for K5/K12-di-acetylated histone H4 recognition by the second bromodomain of BRD2. FEBS Letters. 2010;584:3901–3908. doi: 10.1016/j.febslet.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang H, Zhang J, Shen W, Wang X, Wu J, Wu J, Shi Y. Solution structure of the second bromodomain of Brd2 and its specific interaction with acetylated histone tails. BMC Structural Biology. 2007;7 doi: 10.1186/1472-6807-7-57. No pp given. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LeRoy G, Rickards B, Flint SJ. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Molecular Cell. 2008;30:51–60. doi: 10.1016/j.molcel.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Walker SR, Nelson EA, Cerulli R, Xiang M, Toniolo PA, Qi J, Stone RM, Wadleigh M, Bradner JE, Frank DA. Targeting STAT5 in hematologic malignancies through inhibition of the bromodomain and extra-terminal (BET) bromodomain protein BRD2. Molecular Cancer Therapeutics. 2014;13:1194–1205. doi: 10.1158/1535-7163.MCT-13-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamonica JM, Deng W, Kadauke S, Campbell AE, Gamsjaeger R, Wang H, Cheng Y, Billin AN, Hardison RC, MacKay JP, Blobel GA. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc Natl Acad Sci USA. 2011;108:E159–E168. doi: 10.1073/pnas.1102140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gamsjaeger R, Webb SR, Lamonica JM, Billin A, Blobel GA, MacKay JP. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Molecular and Cellular Biology. 2011;31:2632–2640. doi: 10.1128/MCB.05413-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci USA. 2003;100:8758–8763. doi: 10.1073/pnas.1433065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, Rusinova E, Zhang G, Wang C, Zhu H, Yao J, Zeng YX, Evers BM, Zhou MM, Zhou BP. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210–225. doi: 10.1016/j.ccr.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J, Cao J, Zhou BP. Twist-BRD4 complex: potential drug target for basal-like breast cancer. Current Pharmaceutical Design. 2015;21:1256–1261. doi: 10.2174/1381612821666141211153853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert Reviews in Molecular Medicine. 2011;13:e29/1. doi: 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Molecular Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z, Yik JHN, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Molecular Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 28.Shang E, Nickerson HD, Wen D, Wang X, Wolgemuth DJ. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development. 2007;134:3507–3515. doi: 10.1242/dev.004481. [DOI] [PubMed] [Google Scholar]
- 29.Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, Knapp S, Bradner JE. Small-molecule inhibition of BRDT for male contraception. Cell. 2012;150:673–684. doi: 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature Reviews Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 31.Atkinson SJ, Soden PE, Angell DC, Bantscheff M, Chung C-w, Giblin KA, Smithers N, Furze RC, Gordon L, Drewes G, Rioja I, Witherington J, Parr NJ, Prinjha RK. The structure based design of dual HDAC/BET inhibitors as novel epigenetic probes. Med Chem Commun. 2014;5:342–351. [Google Scholar]
- 32.Chung C-w, Coste H, White JH, Mirguet O, Wilde J, Gosmini RL, Delves C, Magny SM, Woodward R, Hughes SA, Boursier EV, Flynn H, Bouillot AM, Bamborough P, Brusq JMG, Gellibert FJ, Jones EJ, Riou AM, Homes P, Martin SL, Uings IJ, Toum J, Clement CA, Boullay AB, Grimley RL, Blandel FM, Prinjha RK, Lee K, Kirilovsky J, Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011;54:3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 33.Sachchidanand, Resnick-Silverman L, Yan S, Mutjaba S, Liu W-j, Zeng L, Manfredi JJ, Zhou MM. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chemistry and Biology. 2006;13:81–90. doi: 10.1016/j.chembiol.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 34.Amans D, Atkinson SJ, Harrison LA, Hirst DJ, Law RP, Lindon M, Preston A, Seal JT, Wellaway CR. GlaxoSmithKline Intellectual Property (No.2) Limited; UK: WO2014140076 A1. Application. 2014:479.
- 35.Amans D, Demont EH, Mitchell DJ, Seal JT. GlaxoSmithKline LLC; USA: WO2012143415 A1. Application. 2012:129.
- 36.Amans D, Demont EH, Mitchell DJ, Watson RJ. GlaxoSmithKline LLC; USA: WO2012143413 A1. Application. 2012:70.
- 37.Demont EH, Garton NS, Gosmini RLM, Hayhow TGC, Seal J, Wilson DM, Woodrow MD. GlaxoSmithkline LLC; USA: WO2011054841 A1. Application. 2011:205.
- 38.Demont EH, Gosmini RLM. GlaxoSmithkline LLC; USA: WO2011054848 A1. Application. 2011:199.
- 39.Gosmini R, Nguyen Van L, Toum J, Simon C, Brusq Jean-Marie G, Krysa G, Mirguet O, Riou-Eymard Alizon M, Boursier Eric V, Trottet L, Bamborough P, Clark H, Chung CW, Cutler L, Demont Emmanuel H, Kaur R, Lewis Antonia J, Schilling Mark B, Soden Peter E, Taylor S, Walker Ann L, Walker Matthew D, Prinjha Rab K, Nicodeme E. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J Med Chem. 2014;57:8111–8131. doi: 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- 40.Nobeli I, Price SL, Lommerse JPM, Taylor R. Hydrogen bonding properties of oxygen and nitrogen acceptors in aromatic heterocycles. Journal of Computational Chemistry. 1997;18:2060–2074. [Google Scholar]
- 41.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ., III Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci USA. 2011;108(40):16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milde T, Lodrini M, Savelyeva L, Korshunov A, Kool M, Brueckner LM, Antunes AS, Oehme I, Pekrun A, Pfister SM, Kulozik AE, Witt O, Deubzer HE. HD-MB03 is a novel Group 3 medulloblastoma model demonstrating sensitivity to histone deacetylase inhibitor treatment. J Neurooncol. 2012;110:335–348. doi: 10.1007/s11060-012-0978-1. [DOI] [PubMed] [Google Scholar]
- 43.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, Campbell RM, Carter AJ, Cohen P, Copeland RA, Cravatt B, Dahlin JL, Dhanak D, Edwards AM, Frye SV, Gray N, Grimshaw CE, Hepworth D, Howe T, Huber KVM, Jin J, Knapp S, Kotz JD, Kruger RG, Lowe D, Mader MM, Marsden B, Mueller-Fahrnow A, Muller S, O’Hagan RC, Overington JP, Owen DR, Rosenberg SH, Roth B, Ross R, Schapira M, Schreiber SL, Shoichet B, Sundstrom M, Superti-Furga G, Taunton J, Toledo-Sherman L, Walpole C, Walters MA, Willson TM, Workman P, Young RN, Zuercher WJ. The promise and peril of chemical probes. Nature Chemical Biology. 2015;11:536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geschwindner S, Ulander J, Johansson P. Ligand Binding Thermodynamics in Drug Discovery: Still a Hot Tip? J Med Chem. 2015;58:6321–35. doi: 10.1021/jm501511f. [DOI] [PubMed] [Google Scholar]
- 45.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 46.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 47.Labute P. Protonate3D: Assignment of Ionization States and Hydrogen Coordinates to Macromolecular Structures. Proteins. 2008;75:187–205. doi: 10.1002/prot.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Labute P. The Generalized Born/Volume Integral (GB/VI) Implicit Solvent Model: Estimation of the Free Energy of Hydration Using London Dispersion Instead of Atomic Surface Area. J Comp Chem. 2008;19:1693–1698. doi: 10.1002/jcc.20933. [DOI] [PubMed] [Google Scholar]
- 49.Rich RL, Errey J, Marshall F, Myszka DG. Biacore analysis with stabilized G-protein-coupled receptors. Anal Biochem. 2011;409:267–72. doi: 10.1016/j.ab.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Myszka DG. Improving biosensor analysis. J Mol Recognit. 1999;12:279–84. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 51.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 56.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 57.Smart OS, Womack TO, Sharff A, Flensburg C, Keller P, Paciorek W, Vonrhein C, Bricogne G. Grade v1102. Global Phasing Ltd; Cambridge, United Kingdom: 2011. http://www.globalphasing.com. [Google Scholar]
- 58.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.