

Abstract
Imbalance in neural excitation and inhibition is associated with behavioral dysfunction in individuals with schizophrenia and at risk for this illness. We examined whether targeting increased neural activity with the antiepileptic agent, levetiracetam, would benefit memory performance in a preclinical model of schizophrenia that has been shown to exhibit hyperactivity in the hippocampus. Adult rats exposed to ketamine subchronically during late adolescence showed impaired hippocampal-dependent memory performance. Treatment with levetiracetam dose-dependently improved memory performance of the ketamine-exposed rats. In contrast, the antipsychotic medication risperidone was not effective in this assessment. Levetiracetam remained effective when administered concurrently with risperidone, supporting potential viability of adjunctive therapy with levetiracetam to treat cognitive deficits in schizophrenia patients under concurrent antipsychotic therapy. In addition to its pro-cognitive effect, levetiracetam was also effective in attenuating amphetamine-induced augmentation of locomotor activity, compatible with the need for therapeutic treatment of positive symptoms in schizophrenia.
Keywords: amphetamine, overactivity, radial arm maze, risperidone, SV2A
1. Introduction
Cognitive deficits strongly predict long-term functional disability in schizophrenia, but existing standard-of-care antipsychotic medications lack efficacy for improving cognition and functional outcomes in patients (Corigliano et al., 2014; Eastvold et al., 2007; Green, 1996; Green et al., 2004; Jahshan et al., 2010; Nuechterlein et al., 2014). Disturbance in the balance of neural excitation and inhibition (E/I) is recognized as possibly contributing to both psychotic and cognitive dysfunctions in schizophrenia (Foss-Feig et al., 2017; Krystal et al., 2017). For example, recent evidence from human neuroimaging studies points to heightened neural activity localized to the medial temporal lobe as a potential driver of pathophysiology in schizophrenia, exerting significant adverse effects on cognitive function (Medoff et al., 2001; Sanderson et al., 2012; Schobel et al., 2009; 2013; Tregellas et al., 2014; Zierhut et al., 2010). Specifically, the level of neural hyperactivity correlates with worse cognitive performance in patients (Tregellas et al., 2014), and the hyperactivity in a prodromal phase of illness predicts clinical progression to overt psychosis within two years (Schobel et al., 2009).
Preclinical animal models of schizophrenia have recapitulated key neurobehavioral features of the disease, including E/I imbalance in the neural circuits important for cognitive function. For example, higher metabolic basal activity and neuronal firing rates have been observed in adult animals exposed prenatally to the antimitotic compound methylazoxymethanol acetate or to the NMDA receptor antagonist ketamine during adolescence (Gill et al., 2011; Lodge and Grace, 2007; Schobel et al., 2013). Importantly, those same pharmacological induction protocols that produce changes in neural activity lead to cognitive dysfunction detected by behavioral assessments (Koh et al., 2016; Moore et al., 2006). In vitro slice recordings from such treated animals have also provided evidence for neuronal hyperactivity of principal neurons in the hippocampus that are partially normalized by diazepam administration (Sanderson et al., 2012). Thus, both preclinical and clinical data suggest that increased neural activity is a condition contributing to dysfunction in this illness.
We set out to investigate whether targeting neural overactivity with levetiracetam, an atypical antiepileptic agent that binds with high affinity to the synaptic vesicle 2A (SV2A) protein to regulate synaptic exocytosis and neurotransmitter release, would be effective at improving cognition in an animal model of schizophrenia that has been shown to exhibit heightened neural activity. Levetiracetam has already been evaluated in an animal model of impaired sensory gating, and was found to improve auditory gating in mice with schizophrenia-like gating deficits (Smucny et al., 2015). Low dose treatment with levetiracetam has also been found to improve memory in aging and Alzheimer’s disease that are associated with increased neural activity in the medial temporal lobe (Devi and Ohno, 2013; Hall et al., 2015; Koh et al., 2010; Sanchez et al., 2012; Suberbielle et al., 2013). Taken together, the beneficial effects of levetiracetam on normalizing neural overactivity suggests that reduction in neural overactivity by levetiracetam may have potential efficacy on cognition in schizophrenia.
In the present studies, we assessed the ability of levetiracetam to alleviate memory impairment in a ketamine animal model of schizophrenia that recapitulates neural hyperactivity and memory problems akin to those seen in schizophrenia patients (Koh et al., 2016; Olney et al., 1999; Neill et al., 2010; Schobel et al., 2013). We tested levetiracetam, alongside and in combination with the antipsychotic medication, risperidone, to compare their efficacy to improve cognition under the same testing conditions. We further examined whether levetiracetam alleviates the augmented response to a dopamine agonist amphetamine, a commonly used behavioral assay to assess a dopaminergic perturbation that is central to the illness. Levetiracetam, but not risperidone, was found to improve memory performance dose-dependently in a hippocampal-dependent memory task, and levetiracetam remained effective when administered concurrently with antipsychotic drug treatment. In addition, levetiracetam attenuated the response to amphetamine in the ketamine model, also suggesting a potential link between hippocampal overactivity and dopamine system dysfunction as proposed by other investigators (Grace, 2012; Lodge and Grace, 2007).
2. Materials and methods
2.1 Subjects
Male Long-Evans rats were obtained at approximately 5 weeks old from Charles River Laboratories (Raleigh, NC), and housed individually at 25°C and maintained on a 12-hr light/dark cycle. Food (Purina autoclave laboratory rodent diet) and water were provided ad libitum unless otherwise noted. All procedures in the current investigations were approved by the Institutional Animal Care and Committee in accordance with the National Institutes of Health directive.
2.2 Ketamine exposure
Ketamine (VedCo; 100 mg/ml concentration) was diluted in saline to 30 mg/ml, and injected at a volume of 1 ml/kg of body weight (Enomoto and Floresco, 2009). Rats were injected intraperitoneally twice daily (morning and late afternoon) with saline or ketamine (30 mg/kg) for two weeks starting at 7-weeks of age. Following ketamine exposure, the rats were left undisturbed for at least five days for drug washout before behavioral training.
2.3 Drug treatments
Levetiracetam (synthesized by Tecoland Corporation, Irvine, CA) and risperidone (Sigma, Saint Louis, MO) were tested for their effect on cognition. Levetiracetam was diluted in saline and dosed at 1, 5, and 10 mg/kg, and risperidone was diluted in a vehicle consisting of 0.25% Tween-80 in saline and dosed at 0.1, 0.17, and 0.3 mg/kg. The drugs were administered in a volume of 1ml/kg intraperitoneally 30–40 min prior to test sessions. Levetiracetam doses were chosen based on their efficacy in targeting neural overactivity in rodents (Haberman et al., 2017; Koh et al., 2010), and risperidone doses were chosen based on antipsychotic clinical relevance (50% D2 receptor occupancy and efficacy in animal models of antipsychotic activity; Wadenberg et al., 2001).
2.4 Overview of experiments
A set of ketamine-exposed rats (n = 23) was first tested on a radial arm maze task to assess memory impairment compare to control rats (n = 14). About half of these ketamine rats were then tested with risperidone (n = 11) and the remaining with risperidone-levetiracetam combination (n = 12) on the maze. The entire assessment on the radial arm maze took about 2 months to complete. A different set of ketamine-exposed rats (n = 14) was tested on the radial arm maze under levetiracetam treatment. A subset of these rats (n = 12) was then used to assess the efficacy of levetiracetam in the amphetamine-induced locomotor activity study together with a set of untreated controls (n = 7). The radial arm maze and amphetamine studies took approximately 2.5 months to complete.
2.5 Radial arm maze
A hippocampal-dependent radial arm maze task was used to assess the effect of drug treatment as described in detail elsewhere (Chappell et al., 1998; Koh et al., 2010). The protocol allowed repeated within-subject assessment at different drug doses and in combinations. Pre-training consisted of habituation, standard win-shift training, and win-shift training with delays interposed between information and memory test phases on the eight-arm maze. Drug treatments began a day after the completion of pretraining. Three arms were blocked at the beginning of each test trial (information phase). The identity and configuration of the blocked arms were varied across trials. Food-deprived rats were allowed to retrieve food reward (Kellogg’s Froot Loops cereal) from the five unblocked arms. The rat was then removed from the maze for a retention interval, during which time the barriers on the blocked arms were removed allowing access to all eight arms. Rats were then placed back onto the center platform and allowed to retrieve the remaining food rewards (memory phase). An error consisted of returning to an arm (all four paws on the arm) from which food had already been obtained. The number of errors made in the retention phase was used to assess memory performance. We used a 3-hr retention interval between information and memory test phases in all our drug studies based on our background data showing a reliable difference in memory performance between ketamine-exposed and saline control rats at that retention delay. Rats were tested with a series of drug doses in ascending/descending order; each dose, including vehicle alone, was thus tested twice.
2.6 Amphetamine-induced locomotor activity
Rats were challenged with amphetamine to examine dopamine-mediated hyperlocomotor activity and to determine whether levetiracetam treatment would alleviate the increased response to amphetamine that is characteristic of the ketamine model. Using a within-subject design, each rat was treated with either levetiracetam or saline on different test sessions; the order of drug treatment was counterbalanced such that half of the rats received levetiracetam on the first test session and saline on the second one, and vice versa. The test sessions were separated by at least one day of drug washout. During the test, each rat was injected intraperitoneally with levetiracetam (10 mg/kg) or saline and placed in an open field chamber (42 cm × 42 cm × 30.5 cm) in which locomotion was tracked with the VersaMax animal activity monitoring system (AccuScan Instruments, Columbus, OH). After 30 min of baseline activity, the rat was taken out of the chamber and injected intraperitoneally with a small dose of amphetamine (0.5 mg/kg in a volume of 1 ml/kg; Sigma, Saint Louis, MO). The rat was then returned to the chamber for another 60 min of activity monitoring. Total distance travelled and movement time were the dependent measures.
3. Results
3.1 Ketamine exposure impaired memory performance
We first examined whether rats exposed to ketamine subchronically during late adolescence/early adulthood showed memory impairment as has been reported elsewhere (e.g., Enomoto and Floresco, 2009; Olney et al., 1999; Neill et al., 2010). We used a well-established radial arm maze task to assess cognitive performance that is known to be dependent on the hippocampal memory system (e.g., Olton et al., 1982; Schlesiger et al., 2013), and also allowed for within-subject assessment of different drug doses on memory performance (Koh et al., 2013). Figure 1 shows that under drug-free condition, ketamine-exposed rats had significantly more memory errors than saline control rats, t(35) = 2.22, p = 0.033. Thus, past exposure to ketamine during adolescence induced memory impairment consistent with that found by others using the ketamine animal model of schizophrenia.
Figure 1.
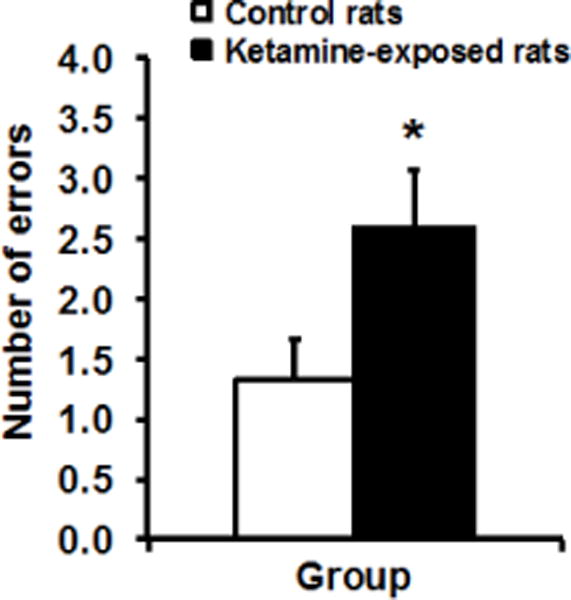
Adult rats exposed to ketamine subchronically during late adolescence (n = 23) had more memory errors than saline control rats (n = 14) in a radial arm maze task, t(35) = 2.22, p = 0.033 (independent samples t-test).
3.2 Levetiracetam, but not risperidone, rescued memory impairment
We next examined the efficacy of risperidone and levetiracetam to improve the memory performance of the ketamine-exposed rats in two parallel studies using the radial arm maze task. Figure 2A shows the performance of ketamine-exposed rats under risperidone treatment at 0 (vehicle), 0.1, 0.17, and 0.3 mg/kg. None of the doses tested were effective at lowering the memory errors relative to vehicle treatment. A repeated measures ANOVA confirmed no effect of risperidone treatment, F(3, 30) = 0.40, p = 0.758, or a dose response function, F(1, 10) = 0.55, p = 0.477.
Figure 2.
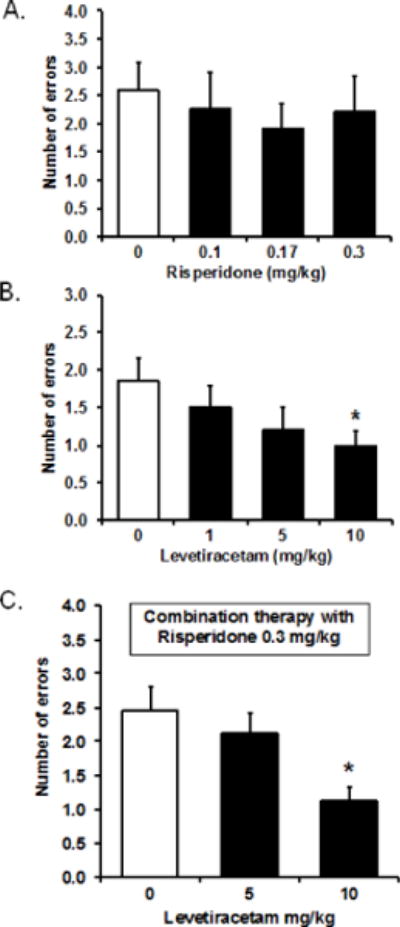
(A) Risperidone treatment at the doses tested had no effect on memory performance in the ketamine-exposed rats (n = 11) in the radial arm maze task. A repeated measures ANOVA showed no effect of treatment, F(3, 30) = 0.40, p = 0.758, or a dose response function, F(1, 10) = 0.55, p =0.477. (B) Levetiracetam treatment dose-dependently improved memory performance in ketamine-exposed rats (n = 14). A repeated measures ANOVA showed a marginally significant within-subject effect, F(3, 39) = 2.49, p = 0.075, with a significant within-subject contrast of the linear dose response function, F(1, 13) = 6.73, p = 0.022. The 10-mg/kg dose significantly reduced memory errors compared to vehicle treatment (0 mg/kg), t(13) = 2.26, p = 0.042 (paired sample t-test). (C) Levetiracetam treatment in the presence of risperidone preserved the efficacy of levetiracetam to dose-dependently lower the number of memory errors, F(2, 22) = 5.97, p = 0.008 for within-subject effect, and F(1, 11) = 13.28, p = 0.004 for linear dose response effect. Ketamine-exposed rats treated with levetiracetam at 10 mg/kg in combination with risperidone at 0.3 mg/kg (n = 12) had significantly fewer memory errors compared to when they were treated with risperidone at 0.3 mg/kg alone (levetiracetam 0 mg/kg), t(11) = 3.65, p = 0.004 (paired sample t-test).
With an independent set of ketamine-exposed rats, we tested levetiracetam at 0 (vehicle), 1, 5, and 10 mg/kg. Figure 2B shows the treatment with levetiracetam improved memory performance in a linear dose-dependent manner. A repeated measures ANOVA indicated a marginally significant within-subject effect, F(3, 39) = 2.49, p = 0.075, with a significant within-subject contrast of the linear dose response function, F(1, 13) = 6.73, p = 0.022. Specifically, within the range of doses tested, levetiracetam improved performance as the doses increased, with significantly reduced memory errors at the 10 mg/kg dose in the ketamine-exposed rats compared to their performance under vehicle saline, t(13) = 2.26, p = 0.042. The same treatment with levetiracetam in saline control rats had no effect on performance (data not shown; see Koh et al., 2010).
To examine whether the pro-cognitive effect of levetiracetam would persist under concurrent antipsychotic drug treatment, we tested levetiracetam with risperidone in a combination therapy in a different set of ketamine-exposed rats. Risperidone was dosed at a constant 0.3 mg/kg and levetiracetam was dosed at either 0 (vehicle), 5, or 10 mg/kg. Figure 2C shows concurrent risperidone treatment did not interfere with the therapeutic action of levetiracetam to improve memory performance. A repeated measures ANOVA indicated a significant within-subject effects, F(2, 22) = 5.97, p = 0.008, with a significant within-subject contrasts of the linear dose response effect, F(1, 11) = 13.28, p = 0.004. Treatment with levetiracetam at 10 mg/kg concomitant with risperidone at 0.3 mg/kg significantly reduced memory errors of the ketamine-exposed rats compared to when the rats were treated with risperidone at 0.3 mg/kg with no levetiracetam, t(11) = 3.65, p = 0.004.
3.3 Ketamine exposure increased amphetamine-induced locomotor activity
Rats with a history of ketamine exposure exhibit dysregulation of dopaminergic function that is thought to mimic the pathophysiology of schizophrenia (e.g., Olney et al., 1999). Here, we challenged our ketamine-exposed and control rats with amphetamine to assess amphetamine-induced augmentation of locomotor activity in this model. Figure 3 shows the activity level of the rats as measured by distance travelled during the first 30 min of baseline assessment followed by 60 min of post-amphetamine evaluation. A repeated measures ANOVA shows an overall significant interaction of group by time, F(8, 136) = 2.99, p = 0.004, a significant effect of time, F(8, 136) = 21.43, p = 0.001, and a marginally significant effect of group, F(1, 17) = 3.42, p = 0.082. Further evaluation of the interaction effect showed that the ketamine-exposed and control groups did not differ at baseline, with levels of activity that decreased over time due to habituation. Amphetamine injection noticeably increased the activity of all rats from the baseline level, with the ketamine-exposed rats showing a greater increase than the control rats. A repeated measures ANOVA of distance travelled post-amphetamine showed a significant effect of group, F(1, 17) = 4.69, p = 0.045, and a significant effect of time, F(5, 85) = 12.36, p = 0.001, but no interaction, F(5, 85) = 1.38, p = 0.239, as the influence of amphetamine peaked and then subsided over time in both groups at roughly similar rates. We measured and analyzed movement time of the rats as a second dependent measure, and found the same pattern of results as that with distance travelled (data not shown).
Figure 3.
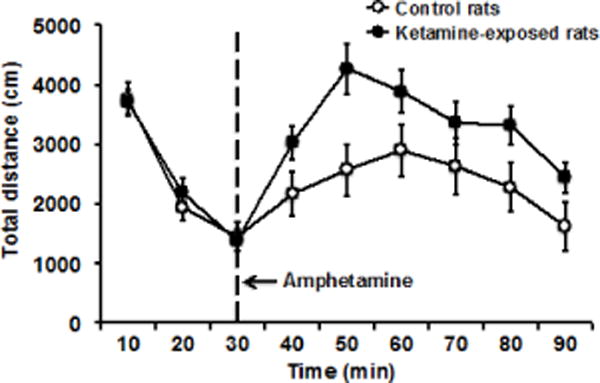
To assess amphetamine-induced augmentation of locomotor activity, ketamine-exposed (n = 12) and control (n = 7) rats were first placed in activity chambers for 30 min of baseline assessment, they were then taken out for amphetamine injection, and returned to the activity chambers for another 60 min of post-amphetamine evaluation. While the two groups did not differ at baseline activity, the ketamine-exposed rats showed greater increased in locomotor activity in response to the amphetamine challenge compared to control rats, F(1, 17) = 4.69, p = 0.045 for group effect (see text for detailed analysis). The vertical (y) axis showed total distance travelled as the dependent measure, and the horizontal (x) axis showed time in 10 min bin.
3.4 Levetiracetam decreased amphetamine-induced locomotor activity
In the context of targeting hippocampal activity for cognitive impairment, we also examined the possible effects of levetiracetam treatment on reduction of positive symptoms of schizophrenia, exemplified by amphetamine augmentation of locomotor activity. Targeting hyperactivity in the hippocampus has previously been shown to have beneficial effects on the augmented response to amphetamine (Gill et al., 2011). Here, we compared the performance of the rats under levetiracetam to saline treatment in response to the same amphetamine challenge. The ketamine-exposed rats were treated with saline 30 min before amphetamine administration in the results described above, and those data served here as a within-subject comparison for treatment with levetiracetam prior to amphetamine challenge in a different test session (see Methods for details including counterbalancing of treatment order).
For distance travelled (Figure 4A), the results showed an overall significant interaction of treatment by time, F(8, 88) = 3.56, p = 0.001, a significant effect of time, F(8, 88) = 19.84, p = 0.001, but no main effect of treatment, F(1, 11) = 2.16, p = 0.169. Further analysis of the interaction showed that activity during baseline did not differ under both treatments, and amphetamine injection markedly boosted locomotor activity of the ketamine-exposed rats under saline treatment as noted before, but the response to amphetamine challenge under levetiracetam treatment was somewhat attenuated (see Figure 4A). A two-way repeated measures ANOVA of distance travelled showed a trend for the treatment by time interaction, F(5, 55) = 2.04, p = 0.087, a significant effect of time, F(5, 55) = 16.89, p = 0.001, but the effect of treatment was not statistically significant, F(1, 11) = 2.99, p = 0.112.
Figure 4.
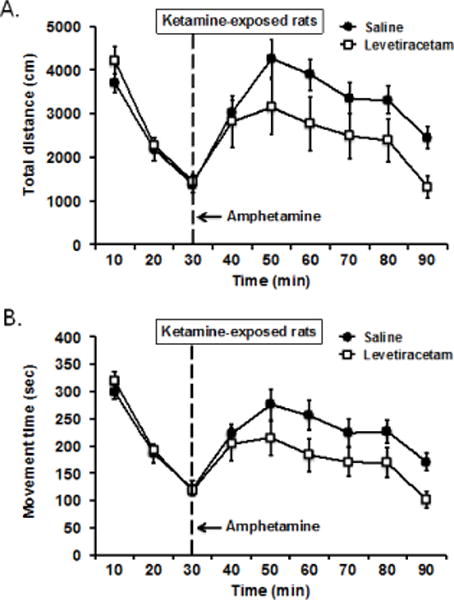
To assess the effect of levetiracetam on amphetamine-induced augmentation of locomotor activity, ketamine-exposed rats (n = 12) were given levetiracetam (10 mg/kg) or vehicle treatment on different test days prior to amphetamine challenge. (A) Compared to vehicle saline treatment, levetiracetam appeared to reduce the increased in distance travelled in response to amphetamine, but difference was not statistically significant, F(1, 11) = 2.99, p = 0.112 for treatment effect. (B) A second dependent measure using movement time showed that levetiracetam significantly lowered the amphetamine-induced increased in locomotor activity relative to vehicle saline, F(1, 11) = 5.01, p = 0.047 for treatment effect (see text for a more complete analysis).
For movement time (Figure 4B), the same analysis showed an overall significant treatment by time interaction, F(8, 88) = 3.34, p = 0.002, a significant effect of time, F(8, 88) = 23.93, p = 0.001, and a marginally significant effect of treatment, F(1, 11) = 3.86, p = 0.075. Although no difference in baseline activity was evident between the two treatment conditions for movement time, levetiracetam treatment after amphetamine injection significantly reduced the response to amphetamine on this measure compared to saline treatment. A two-way repeated measures ANOVA showed a significant effect of treatment, F(1, 11) = 5.01, p = 0.047, a main effect of time, F(5, 55) = 17.34, p = 0.001, but no interaction between the two factors as the activity patterns under both treatment conditions rose and fell over time in response to amphetamine were nearly identical, F(5, 55) = 1.69, p = 0.152. Together, these data indicate that levetiracetam treatment appears to have a modest effect in reducing amphetamine-stimulated locomotor activity in ketamine-exposed rats.
In regard to whether levetiracetam would modulate the antipsychotic effects of risperidone, we have found that risperidone by itself (0.1 and 0.3 mg/kg) significantly reduced baseline locomotor activity in ketamine-treated rats in open field test (data not shown), which prohibits direct assessment of any interaction between risperidone and levetiracetam using the amphetamine challenge paradigm.
4. Discussion
Adult rats exposed to ketamine subchronically during late adolescence showed impaired hippocampal-dependent memory performance. Administration of subchronic ketamine to rodents induces a condition of increased neural activity that is localized to the hippocampus (Schobel et al., 2013). Low doses of the atypical antiepileptic agent levetiracetam aimed at reducing increased neural activity were effective at alleviating cognitive impairment. The antipsychotic medication risperidone in contrast was not effective in this same assessment. Importantly, the benefit provided by levetiracetam was preserved under concurrent risperidone treatment in the ketamine-exposed rats, a finding that supports the potential viability of adjunctive therapy with levetiracetam to treat cognitive deficits in schizophrenia patients under concurrent antipsychotic therapy.
Increased hippocampal activity has been linked to the pathophysiology of schizophrenia in patients (Malaspina et al., 1999; Medoff et al., 2001; Molina et al., 2005; Schobel et al., 2013; Talati et al., 2014; Zierhut et al., 2010). In high-risk patients for developing schizophrenia, for example, increased metabolism was localized to the CA1 of the hippocampus (Talati et al., 2014; Schobel et al., 2013), and this condition predicted hippocampal atrophy as patients progressed to psychosis (Schobel et al., 2013). Such increased hippocampal activity was also correlated with more severe positive and negative symptoms of schizophrenia (Schobel et al., 2009; Zierhut et al., 2010), as well as with worse performance on a broad spectrum of cognitive functional assessments (Tregellas et al., 2014). Evidence for a decrease in hippocampal GABAergic inhibitory interneurons and other biomarkers in patients with schizophrenia further supports a condition of excitatory-inhibitory imbalance in the hippocampus (Konradi et al., 2011; Li et al., 2015; Torrey et al., 2005; Zhang and Reynolds, 2002). These clinical data have led to the suggestion that targeting hippocampal hyperactivity might alleviate cognitive impairment (Gill and Grace, 2014; Heckers and Konradi, 2014; Tamminga et al., 2010).
Our finding that levetiracetam is effective at improving memory performance provides the first evidence that such a treatment strategy targeting hippocampal overactivity may be beneficial for higher-level cognitive function in the context of schizophrenia. In addition to its pro-cognitive effect, levetiracetam was also modestly effective in attenuating amphetamine-induced locomotor activity, providing an additional therapeutic avenue for treating positive symptoms of schizophrenia, perhaps lowering the dose of antipsychotic medications for efficacy. That finding is consistent with other preclinical research. For example, treatment with a GABAA α5 receptor positive allosteric modulator was found to reduce evoked excitatory responses of hippocampal neurons in a neurodevelopmental animal model of schizophrenia (Gill et al., 2011). In that model, the GABAA α5 receptor positive allosteric modulator also attenuated amphetamine-induced augmentation of locomotor activity, consistent with the results we found with levetiracetam on amphetamine challenge in ketamine-exposed rats. As noted before, treatment with levetiracetam has also been found to improve auditory gating in a mouse model of sensory gating deficits in schizophrenia (Smucny et al., 2015).
Levetiracetam, as an atypical antiepileptic agent, has a pharmacological profile that differs from other antiepileptic medications. It does not appear to interact significantly with GABAergic or glutaminergic receptors, benzodiazepine binding sites, or voltage-gated sodium channels that serve as conventional targets of antiepileptic agents (Margineanu and Klitgaard, 2003; Noyer et al., 1995; Zona et al., 2001). Instead, levetiracetam appears to mediate its effects through high affinity for synaptic vesicle glycoprotein 2A (SV2A), which is important for activity-dependent, calcium-induced transmitter release especially during high activation (Bajjalieh et al., 1992; Chang and Südhof, 2009; Crowder et al., 1999; Custer et al., 2006; Lynch et al., 2004). Treatment with levetiracetam has shown a beneficial effect in a number of in vivo and in vitro models, including the ability to decrease excitatory transmission in the CA1 subregion of the hippocampus (Yang et al., 2007; Yang and Rothman, 2009). In the present study, the dose of levetiracetam found to improve memory performance in ketamine-exposed rats is consistent with the low dose efficacy of levetiracetam observed in age-related memory impairment and Alzheimer’s disease preclinical models in rodents in which neural overactivity occurs (Devi and Ohno, 2013; Koh et al., 2010; Sanchez et al., 2012; Spiegel et al., 2013; Suberbielle et al., 2013). In those conditions, the low dose therapeutic window for improving cognition with levetiracetam is well below those used for the treatment of epilepsy (Lyseng-Williamson, 2011), and has successfully translated clinically in patients with amnestic mild cognitive impairment, a prodromal condition in Alzheimer’s disease (Bakker et al., 2012; 2015). It is also notable that the therapeutic efficacy is observed in a dose range that does not alter behavioral or neurophysiological responses in normal young adults or wild type mice lacking Alzheimer’s disease pathophysiology. Our in-house data have similarly shown no effect of levetiracetam treatment on amphetamine-induced locomotor activity in control rats, consistent with the lack of behavioral effect with low-dose levetiracetam treatment in normal animals (Koh et al., 2010).
Existing antipsychotics have limited efficacy in improving cognition and functional outcomes in schizophrenia. The current findings suggest that the use of levetiracetam to target elevated neural activity may confer a dual advantage of ameliorating cognitive impairment and attenuating positive symptoms in a preclinical model of schizophrenia. Success in translating levetiracetam treatment for the clinical indication of amnestic mild cognitive impairment has been encouraging (Bakker et al., 2012; 2015). The current evidence may warrant clinical investigation to normalize E/I imbalance for cognitive therapy in schizophrenia. Disruption in E/I balance has been documented in schizophrenia affecting both hippocampal and prefrontal circuitry. Recent evidence has implicated deficient inhibitory control involving parvalbumin interneurons in the cortex (Chung et al., 2016; Krystal et al., 2017), which has also been proposed as a basis for impairment in models of Alzheimer’s disease pathology (Palop and Mucke, 2016). Thus, the efficacy of levetiracetam in such models (Sanchez et al., 2012) as well as for the treatment of heightened cortical excitability in age-related cognitive impairment (Haberman et al., 2017) would suggest that such treatment would also benefit additional cortical-dependent behavioral functions in schizophrenia.
Acknowledgments
This research was supported by the Silvo O. Conte Center, P50MH094268. YS was a Woodrow Wilson Fellow at Johns Hopkins University.
Role of funding agencies
None of the funding agencies had a role in collection, management, analysis or interpretation of the data or in preparation, review or approval of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
MG is the founder of AgeneBio Incorporated, a biotechnology company that is dedicated to discovery and development of therapies to treat cognitive impairment in aging. She has a financial interest in the company. Her conflict of interest is managed by Johns Hopkins University. MTK and MG are inventors on Johns Hopkins University intellectual property licensed to AgeneBio. MTK and YS have received no financial support or compensation from any individual or corporate entity for research or professional services, and have no financial holdings that could be perceived as constituting a potential conflict of interest. SRL is a consultant to AgeneBio and serves as the Vice President of Research. SRL is a former employee of Pfizer and retains Pfizer stock, and has no additional financial holdings that could be perceived as constituting a potential conflict of interest.
Contributors
MTK, SRL, and MG developed the study design, MTK and YS performed the experiments, and all authors contributed to and approved the final manuscript.
References
- Bajjalieh SM, Peterson K, Shinghal R, Scheller RH. SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science. 1992;257(5074):1271–1273. doi: 10.1126/science.1519064. [DOI] [PubMed] [Google Scholar]
- Bakker A, Albert MS, Krauss G, Speck CL, Gallagher M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin. 2015;7:688–698. doi: 10.1016/j.nicl.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74(3):467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WP, Südhof TC. SV2 renders primed synaptic vesicles competent for Ca2+ - induced exocytosis. J Neurosci. 2009;29(4):883–897. doi: 10.1523/JNEUROSCI.4521-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell J, McMahan R, Chiba A, Gallagher M. A re-examination of the role of basal forebrain cholinergic neurons in spatial working memory. Neuropharmacology. 1998;37(4–5):481–487. doi: 10.1016/s0028-3908(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Chung DW, Fish KN, Lewis DA. Pathological basis for deficient excitatory drive to cortical parvalbumin interneurons in schizophrenia. Am J Psychiatry. 2016;173(11):1131–1139. doi: 10.1176/appi.ajp.2016.16010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corigliano V, De Carolis A, Trovini G, Dehning J, Di Pietro S, Curto M, Donato N, De Pisa E, Girardi P, Comparelli A. Neurocognition in schizophrenia: from prodrome to multi-episode illness. Psychiatry Res. 2014;220(1–2):129–134. doi: 10.1016/j.psychres.2014.07.067. [DOI] [PubMed] [Google Scholar]
- Crowder KM, Gunther JM, Jones TA, Hale BD, Zhang HZ, Peterson MR, Scheller RH, Chavkin C, Bajjalieh SM. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A) Proc Natl Acad Sci USA. 1999;96(26):15268–15273. doi: 10.1073/pnas.96.26.15268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custer KL, Austin NS, Sullivan JM, Bajjalieh SM. Synaptic vesicle protein 2 enhances release probability at quiescent synapses. J Neurosci. 2006;26(4):1303–1313. doi: 10.1523/JNEUROSCI.2699-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M. Effects of levetiracetam, an antiepileptic drug, on memory impairments associated with aging and Alzheimer’s disease in mice. Neurobiol Learn Mem. 2013;102:7–11. doi: 10.1016/j.nlm.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Eastvold AD, Heaton RK, Cadenhead KS. Neurocognitive deficits in the (putative) prodrome and first episode of psychosis. Schizophr Res. 2007;93(1–3):266–277. doi: 10.1016/j.schres.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto T, Floresco SB. Disruptions in spatial working memory, but not short-term memory, induced by repeated ketamine exposure. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(4):668–675. doi: 10.1016/j.pnpbp.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Foss-Feig JH, Adkinson BD, Ji JL, Yang G, Srihari VH, McPartland JC, Krystal JH, Murray JD, Anticevic A. Searching for cross-diagnostic convergence: neural mechanisms governing excitation and inhibition balance in schizophrenia and autism spectrum disorders. Biol Psychiatry. 2017;81(10):848–861. doi: 10.1016/j.biopsych.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill KM, Grace AA. The role of α5 GABAA receptor agonists in the treatment of cognitive deficits in schizophrenia. Curr Pharm Des. 2014;20(31):5069–5076. doi: 10.2174/1381612819666131216114612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill KM, Lodge DJ, Cook JM, Aras S, Grace AA. A novel α5GABA(A)R-positive allosteric modulator reverses hyperactivation of the dopamine system in the MAM model of schizophrenia. Neuropsychopharmacology. 2011;36(9):1903–1911. doi: 10.1038/npp.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA. Dopamine system dysregulation by the hippocampus: implications for the pathophysiology and treatment of schizophrenia. Neuropharmacology. 2012;62(3):1342–1348. doi: 10.1016/j.neuropharm.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153(3):321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- Green MF, Kern RS, Heaton RK. Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr Res. 2004;72(1):41–51. doi: 10.1016/j.schres.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Haberman RP, Koh MT, Gallagher M. Heightened cortical excitability in aged rodents with memory impairment. Neurobiol Aging. 2017;54:144–151. doi: 10.1016/j.neurobiolaging.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AM, Throesch BT, Buckingham SC, Markwardt SJ, Peng Y, Wang Q, Hoffman DA, Roberson ED. Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J Neurosci. 2015;35(15):6221–6230. doi: 10.1523/JNEUROSCI.2552-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Konradi C. GABAergic mechanisms of hippocampal hyperactivity in schizophrenia. Schizophr Res. 2015;167(1–3):4–11. doi: 10.1016/j.schres.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahshan C, Heaton RK, Golshan S, Cadenhead KS. Course of neurocognitive deficits in the prodrome and first episode of schizophrenia. Neuropsychology. 2010;24(1):109–120. doi: 10.1037/a0016791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MT, Haberman RP, Foti S, McCown TJ, Gallagher M. Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive impairment. Neuropsychopharmacology. 2010;35(4):1016–1025. doi: 10.1038/npp.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MT, Rosenzweig-Lipson S, Gallagher M. Selective GABA(A) α5 positive allosteric modulators improve cognitive function in aged rats with memory impairment. Neuropharmacology. 2013;64:145–152. doi: 10.1016/j.neuropharm.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MT, Shao Y, Sherwood A, Smith DR. Impaired hippocampal-dependent memory and reduced parvalbumin-positive interneurons in a ketamine mouse model of schizophrenia. Schizophr Res. 2016;171(1–3):187–194. doi: 10.1016/j.schres.2016.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konradi C, Yang CK, Zimmerman EI, Lohmann KM, Gresch P, Pantazopoulos H, Berretta S, Heckers S. Hippocampal interneurons are abnormal in schizophrenia. Schizophr Res. 2011;131(1–3):165–173. doi: 10.1016/j.schres.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Anticevic A, Yang GJ, Dragoi G, Driesen NR, Wang XJ, Murray JD. Impaired tuning of neural ensembles and the pathophysiology of schizophrenia: a translational and computational neuroscience perspective. Biol Psychiatry. 2017;81(10):874–885. doi: 10.1016/j.biopsych.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Ghose S, Gleason K, Begovic A, Perez J, Bartko J, Russo S, Wagner AD, Selemon L, Tamminga CA. Synaptic proteins in the hippocampus indicative of increased neuronal activity in CA3 in schizophrenia. Am J Psychiatry. 2015;172(4):373–382. doi: 10.1176/appi.ajp.2014.14010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27(42):11424–11430. doi: 10.1523/JNEUROSCI.2847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, Fuks B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci USA. 2004;101(26):9861–9866. doi: 10.1073/pnas.0308208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyseng-Williamson KA. Levetiracetam: a review of its use in epilepsy. Drugs. 2011;71(4):489–514. doi: 10.2165/11204490-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Malaspina D, Storer S, Furman V, Esser P, Printz D, Berman A, Lignelli A, Gorman J, Van Heertum R. SPECT study of visual fixation in schizophrenia and comparison subjects. Biol Psychiatry. 1999;46(1):89–93. doi: 10.1016/s0006-3223(98)00306-0. [DOI] [PubMed] [Google Scholar]
- Margineanu DG, Klitgaard H. Levetiracetam has no significant gamma-aminobutyric acid-related effect on paired-pulse interaction in the dentate gyrus of rats. Eur J Pharmacol. 2003;466(3):255–261. doi: 10.1016/s0014-2999(03)01563-2. [DOI] [PubMed] [Google Scholar]
- Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus. 2001;11(5):543–550. doi: 10.1002/hipo.1070. [DOI] [PubMed] [Google Scholar]
- Molina V, Sanz J, Sarramea F, Benito C, Palomo T. Prefrontal atrophy in first episodes of schizophrenia associated with limbic metabolic hyperactivity. J Psychiatr Res. 2005;39(2):117–127. doi: 10.1016/j.jpsychires.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60(3):253–264. doi: 10.1016/j.biopsych.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill JC, Barnes S, Cook S, Grayson B, Idris NF, McLean SL, Snigdha S, Rajagopal L, Harte MK. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther. 2010;128(3):419–432. doi: 10.1016/j.pharmthera.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Noyer M, Gillard M, Matagne A, Hénichart JP, Wülfert E. The novel antiepileptic drug levetiracetam (ucb L059) appears to act via a specific binding site in CNS membranes. Eur J Pharmacol. 1995;286(2):137–146. doi: 10.1016/0014-2999(95)00436-o. [DOI] [PubMed] [Google Scholar]
- Nuechterlein KH, Ventura J, Subotnik KL, Bartzokis G. The early longitudinal course of cognitive deficits in schizophrenia. J Clin Psychiatry. 2014;75(Suppl 2):25–29. doi: 10.4088/JCP.13065.su1.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33(6):523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Olton DS, Walker JA, Wolf WA. A disconnection analysis of hippocampal function. Brain Res. 1982;233(2):241–253. doi: 10.1016/0006-8993(82)91200-8. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci. 2016;17(12):777–792. doi: 10.1038/nrn.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci USA. 2012;109(42):E2895–2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TM, Cotel MC, O’Neill MJ, Tricklebank MD, Collingridge GL, Sher E. Alterations in hippocampal excitability, synaptic transmission and synaptic plasticity in a neurodevelopmental model of schizophrenia. Neuropharmacology. 2012;62(3):1349–1358. doi: 10.1016/j.neuropharm.2011.08.005. [DOI] [PubMed] [Google Scholar]
- Schlesiger MI, Cressey JC, Boublil B, Koenig J, Melvin NR, Leutgeb JK, Leutgeb S. Hippocampal activation during the recall of remote spatial memories in radial maze tasks. Neurobiol Learn Mem. 2013;106:324–333. doi: 10.1016/j.nlm.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, Inbar BP, Corcoran CM, Lieberman JA, Moore H, Small SA. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78(1):81–93. doi: 10.1016/j.neuron.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, Small SA. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66(9):938–946. doi: 10.1001/archgenpsychiatry.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smucny J, Stevens KE, Tregellas JR. The antiepileptic drug levetiracetam improves auditory gating in DBA/2 mice. NPJ Schizophr. 2015;1:e15002. doi: 10.1038/npjschz.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel AM, Koh MT, Vogt NM, Rapp PR, Gallagher M. Hilar interneuron vulnerability distinguishes aged rats with memory impairment. J Comp Neurol. 2013;521(15):3508–3523. doi: 10.1002/cne.23367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat Neurosci. 2013;16(5):613–621. doi: 10.1038/nn.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talati P, Rane S, Kose S, Blackford JU, Gore J, Donahue MJ, Heckers S. Increased hippocampal CA1 cerebral blood volume in schizophrenia. Neuroimage Clin. 2014;5:359–364. doi: 10.1016/j.nicl.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry. 2010;167(10):1178–1193. doi: 10.1176/appi.ajp.2010.09081187. [DOI] [PubMed] [Google Scholar]
- Torrey EF, Barci BM, Webster MJ, Bartko JJ, Meador-Woodruff JH, Knable MB. Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol Psychiatry. 2005;57(3):252–260. doi: 10.1016/j.biopsych.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Tregellas JR, Smucny J, Harris JG, Olincy A, Maharajh K, Kronberg E, Eichman LC, Lyons E, Freedman R. Intrinsic hippocampal activity as a biomarker for cognition and symptoms in schizophrenia. Am J Psychiatry. 2014;171(5):549–556. doi: 10.1176/appi.ajp.2013.13070981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadenberg ML, Soliman A, VanderSpek SC, Kapur S. Dopamine D(2) receptor occupancy is a common mechanism underlying animal models of antipsychotics and their clinical effects. Neuropsychopharmacology. 2001;25(5):633–641. doi: 10.1016/S0893-133X(01)00261-5. [DOI] [PubMed] [Google Scholar]
- Yang XF, Rothman SM. Levetiracetam has a time- and stimulation-dependent effect on synaptic transmission. Seizure. 2009;18(9):615–619. doi: 10.1016/j.seizure.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Yang XF, Weisenfeld A, Rothman SM. Prolonged exposure to levetiracetam reveals a presynaptic effect on neurotransmission. Epilepsia. 2007;48(10):1861–1869. doi: 10.1111/j.1528-1167.2006.01132.x. [DOI] [PubMed] [Google Scholar]
- Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res. 2002;55(1–2):1–10. doi: 10.1016/s0920-9964(01)00188-8. [DOI] [PubMed] [Google Scholar]
- Zierhut K, Bogerts B, Schott B, Fenker D, Walter M, Albrecht D, Steiner J, Schütze H, Northoff G, Düzel E, Schiltz K. The role of hippocampus dysfunction in deficient memory encoding and positive symptoms in schizophrenia. Psychiatry Res. 2010;183(3):187–194. doi: 10.1016/j.pscychresns.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Zona C, Niespodziany I, Marchetti C, Klitgaard H, Bernardi G, Margineanu DG. Levetiracetam does not modulate neuronal voltage-gated Na+ and T-type Ca2+ currents. Seizure. 2001;10(4):279–286. doi: 10.1053/seiz.2000.0504. [DOI] [PubMed] [Google Scholar]