

Abstract
Toll-like receptors (TLRs) play important roles in initiation of innate immune responses and promotion of pathological forms of inflammation. Recent technological advances have enabled the visualization of transcription factor binding and histone modifications in response to TLR signaling at genome-wide levels. Findings emerging from these studies are beginning to provide a picture of how signal-dependent transcription factors regulate the inflammatory response in a cell-specific manner by controlling the recruitment of nucleosome remodeling factors and histone modifying enzymes. Of particular interest, new small molecule inhibitors have been developed that influence inflammatory responses by altering the reading or erasure of histone modifications required for inflammatory gene activation. These findings suggest new approaches for treatment of inflammatory diseases.
Introduction
Precise control of inflammation is essential for effective immunity and the maintenance of normal tissue homeostasis. Inadequate inflammatory responses confer risk of overwhelming infection, while excessive or inappropriate responses contribute to a diverse spectrum of cancers and chronic inflammatory diseases. Members of the Toll-like-receptor (TLR) family play important roles as initiators of inflammation by responding to structurally conserved lipid, carbohydrate, peptide and nucleic-acid molecules that are components of microbial pathogens[1,2]. There have been 10 and 12 functional Toll-like receptors identified in human and mouse, respectively, that are characterized as type 1 transmembrane proteins. The ectodomain contains leucine-rich repeats, which allow for recognition of microbial pathogens. The intracellular domains couple to Myd88 and/or TRIF adapter proteins required for downstream signaling pathways. TLRs can also function as receptors for endogenous ligands that are danger signals of tissue injury and damage [3]. Consistent with these findings, genetic studies have documented important roles of TLRs in a number of inflammation-related disease models, including atherosclerosis and type 2 diabetes [4–7]. In addition, there is substantial evidence that TLRs can play both stimulatory and inhibitory roles in tumor biology [8,9]. Understanding the molecular mechanisms that underlie positive and negative regulation of TLR-dependent gene expression is therefore likely to facilitate the development of novel therapeutic strategies for diseases that are influenced by TLR signaling and other pro-inflammatory mediators.
The emergence of massively parallel DNA sequencing technologies has recently enabled the development of a number of unbiased genome-wide approaches for interrogation of transcriptional mechanisms controlling signal-dependent gene regulation, including chromatin immunoprecipitation coupled to deep sequencing (ChIP-Seq) and global RNA sequencing (RNA-Seq)[10]. ChIP-Sequencing approaches not only enable the definition of the binding sites for transcription factors at a genome-wide level, they also enable interrogation of the large number of histone modifications that are ‘written’ and ‘erased’ by a diverse array of histone modifying enzymes and are ‘read’ by a similarly large number of proteins that play essential roles in chromatin-dependent processes that include transcription, DNA replication and DNA repair [11–13]. In general, the recruitment of histone modifying enzymes required for transcriptional activation or repression is mediated by sequence-specific transcription factors that interact with DNA recognition motifs in promoters and/or enhancers [14]. We refer to the role of histone modifications in the regulation of gene expression as ‘epigenetic’ control.
Application of ChiP-Seq and RNA-Seq methods to TLR4 signaling in macrophages has resulted in a number of insights into the molecular mechanisms that enable rapid, high-magnitude transitions in rates of gene expression. The conclusions emerging from these studies are likely to be relevant to the understanding of signal-dependent gene activation in diverse cell types. In this review we will focus on recent advances in defining the epigenetic features that distinguish promoters from enhancers and evaluate their impact on regulating inflammatory gene expression in macrophages.
TLR-dependent gene expression
TLRs represent a family of conserved proteins that serve to recognize ‘danger’ and ‘stranger’ signals (Figure 1). Stranger signals are exemplified by the lipopolysaccharide (LPS) component of gram-negative bacteria, which serves as a pathogen-associated molecular pattern that is recognized with high affinity by TLR4[15]. Danger signals are exemplified by oxidized phospholipids that are generated in the context of injury and chronic disease are ligands for TLR4[15]. Upon ligation, TLRs couple to Myd88 and/or TRIF-dependent signal transduction pathways that activate latent transcription factors including NFkB, AP-1 and interferon regulatory factors (IRFs)[16,17]. Upon activation, these factors bind to regulatory elements in promoters and/or enhancers of target genes where they function to recruit various co-activators required for gene activation. The biological consequence of TLR signaling is the up-regulation of a large cohort of genes that include interferons alpha and beta (IFNα/β), Nitric Oxide Synthase 2 (NOS2A), and Tumor Necrosis Factor (TNF) which play critical roles in initiating innate immune responses to bacterial and viral infection.
Figure 1. General scheme for TLR-dependent regulation of gene expression.
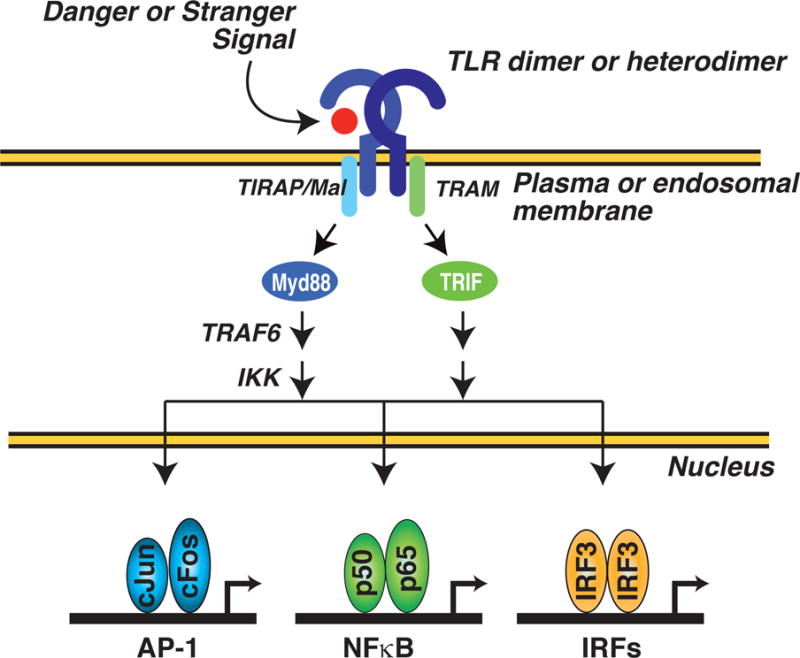
TLR dimers or heterodimers are activated by ‘danger’ (e.g., products of tissue injury) or ‘stranger’ (e.g., components of bacteria or viruses) signals. The liganded receptors couple to Myd88 and/or TRIF-dependent signal transduction pathways that function to activate latent transcription factors such as NFkB, AP-1 and interferon regulatory factors (IRFs). Upon activation, these factors bind to regulatory elements in target genes and positively regulate gene expression.
Epigenomic features of TLR-dependent promoters
Cellular DNA is organized in the nucleus through interaction with nucleosome complexes consisting of a dimer of tetramers for histone H3, H4, H2A and H2B[18,19]. These interactions enable the marked compaction of DNA required for packaging into the nucleus, but also impose a barrier to transcription. The transition of the chromatin template from transcriptionally silent to active requires a combination of nucleosome remodeling and histone modification. Nucleosome remodeling is accomplished through the recruitment of ATP-dependent nucleosome remodeling complexes by sequence-specific transcription factors[20]. Similarly, sequence specific transcription factors are required for the recruitment of histone modifying enzymes that function to erase repressive marks and write activating marks[14]. Over 60 residues on histone tails are known to be post-translationally modified by one of at least seven identified covalent chromatin modifications which include acetylation, methylation, phosphorylation, ubiquitinylation, sumoylation, ADP ribosylation, deimination, and proline isomerization[21].
A full description of the many post-translational modifications that occur on histone tails is beyond the scope of this review, but acetylation and methylation of lysine residues located on the tails of histone H3 and H4 are among the key modifications that have been linked to regulation of gene expression (Figure 2).
Figure 2. Epigenomic features of TLR-responsive promoters and enhancers under basal and activated conditions.
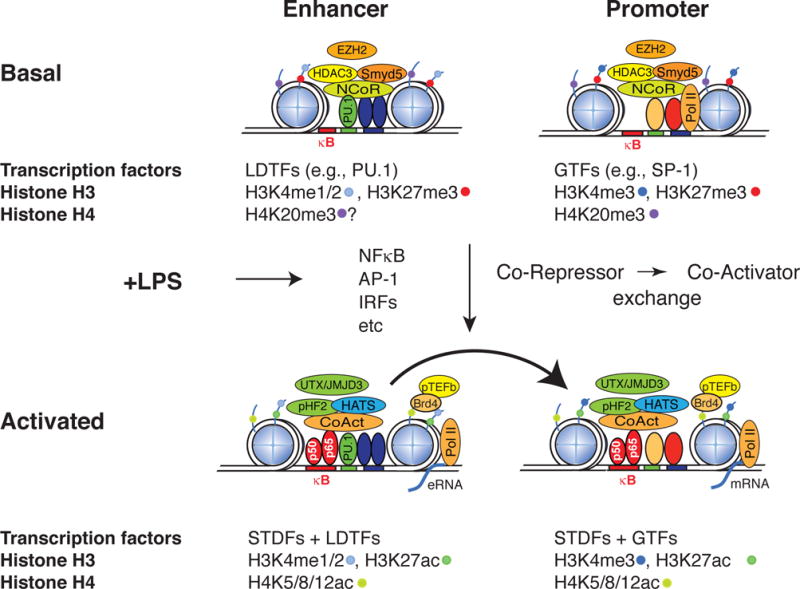
A prototypic enhancer is illustrated at left and a prototypic promoter at right. Blue circles represent histone octomers. Lines emerging from the circles represent histone tails. LDTF; lineage determining transcription factor, GTF; general transcription factor, STDF; signal-dependent transcription factor. H3K4me1/2; histone H3 mono or di-methylated at lysine 4. H3K4me3; histone H3 trimethylated at lysine 4. H3K27me3; Histone H3 tri-methylated at lysine 27. H4K20me3; histone H4 trimethylated at lysine 20. H3K27ac; histone H3 acetylated at lysine 27.
The chromatin states of LPS-inducible promoters provide the ability for immediate activation of inflammatory gene expression during a bacterial insult, while also strictly imposing regulatory checkpoints to prevent spurious, unregulated gene activation. The nucleosome structure surrounding promoters is highly dependent on the GC content, as low GC content regions form stable nucleosomes not observed in high GC promoters[22,23]. These stable assembled nucleosomes require chromatin remodeling as a step in gene activation and as a result are typically activated with slower kinetics than high GC containing promoters. Therefore genes activated by TLR signaling can be divided into primary response genes (PRGs) that are rapidly induced by signal-dependent activation of latent transcription factors, such as NF-κB, and secondary response genes (SRGs) that require additional transcription factors and chromatin remodelers that are induced/activated as a consequence of the primary response[24]. ChIP-Seq studies have revealed that the promoters for both PRGs and SRGs exhibit H3K4me3 under basal conditions by the Mll family of histone methyltransferases and relatively little change in this mark is observed upon TLR4 activation[25,26]. These findings are consistent with H3K4me3 being established by signal-independent transcription factors that initiate promoter selection and result in low but detectable levels of transcription under basal conditions.
Inflammatory cytokines are repressed in the basal state by the NCoR and SMRT co-repressor complexes, which consist of NCoR or SMRT, Tbl1 and/or TblR1 (exchange factors), GPS2 and Histone deacetylase 3 (HDAC3)[27–29]. The presence of HDAC3 as a core component of both the NCoR and SMRT complexes functions to keep H3K9/14 acetylation (H3K9/14ac, a mark of transcriptional activation) levels low under resting conditions and increased levels following signal-dependent NCoR/SMRT turnover and transcriptional activation. Surprisingly, the activation of hundreds of inflammatory genes is greatly impaired in HDAC3 −/− macrophages and this effect has been thought to contribute to the loss of interferon β gene activation[30]. These observations suggest that HDAC3 deacetylates substrates other than H3K9/14ac and/or the NCoR complex exerts repressive effects through mechanisms independent of HDAC3.
The RNA polymerase II complex is recruited to the promoters of PRG genes under resting conditions, where it is maintained in an inactive, poised state [22,23,26]. Upon TLR4 activation, the histone acetyltransferases GCN5 and PCAF are recruited to pro-inflammatory gene promoters to acetylate H4K5/K8/K12 which serves as a molecular beacon for BRD4, a component of the P-TEFb complex[22,31]. Once recruited, cyclin-dependent kinase 9 (CDK9), a component of the P-TEFb complex, phosphorylates the carboxy terminal domain of the large subunit of RNA Pol II, causing the release of the negative elongation factor (NELF) and promoting the transition into productive elongation[32]. Therefore acetylation of H4K5/K8/K12 serves an essential role in controlling the elongation of inflammatory gene transcripts (Figure 2) and recently has been the focus of a new class of pharmacological inhibitors. Small molecules called I-BETs that mimic acetylated histone H4 have recently been shown to be capable of blocking the recruitment of BRD4 and the P-TEFb complex to TLR4-regulated promoters, thereby stalling the elongation of RNA polymerase II[33]. These inhibitors effectively suppress a subset of LPS-inducible genes and protect against lipopolysaccharide-induced endotoxic shock[33].
Genome-wide localization studies for several activation and repressive epigenetic marks have revealed that extensive methylation and demethylation events are components of the activation program at promoters and distal regulatory elements[11,34]. Recent studies have demonstrated that pro-inflammatory gene programs are under the control of the histone methyltransferase Smyd5, which establishes the repressive mark H4K20me3[35] (Figure 2). Smyd5 is recruited to target promoters through its interaction with NCoR, and represses the basal expression of inflammatory genes. Interestingly, knockdown of Smyd5 results in hyper-response to LPS further supporting its role as a transcriptional repressor. The H4K20me3 epigenetic mark is removed upon TLR4 activation through the signal-dependent delivery of the histone demethylase Phf2 by the NFκB component p65 to inflammatory promoters. Whether similar events occur at enhancers remains to be established.
In addition, recent studies have found that inflammatory gene promoters become methylated by EZH2 [36,37] on H3K27me3 under basal conditions, but the mechanisms by which EZH2 is recruited to these promoters and the extent to which EZH2 modulates inflammatory gene expression in vivo remain unknown. The histone demethylase Jmjd3, responsible for removing H3K27me3, is required for activation of a subset of TLR4-dependent genes. Small molecules targeting the family of H3K27me3 demethylases Jmjd3 and UTX reduces lipopolysaccharide-induced pro-inflammatory cytokine production in macrophages[38]. Surprisingly, genome-wide studies for Jmjd3 failed to establish a clear link between demethylation of H3K27me3 and Jmjd3 recruitment to inflammatory gene promoters suggesting the potential for additional substrates for Jmjd3 or important roles of UTX[36].
Cell-specific and epigenomic features of TLR-regulated enhancers
Physiologic programs of regulated gene expression require collaboration between gene promoters and enhancers. A striking finding to emerge from ChIP-Seq studies of a variety of signal dependent transcription factors is the observation the great majority of binding sites for such factors are in enhancers rather than in promoters[39]. This observation has important functional consequences in that enhancers are selected in a cell-specific manner to a much greater extent than promoters[40–44]. Therefore, binding of signal-dependent transcription factors to cell-specific enhancers provides an important mechanism for enabling a broadly expressed transcription factor, such as NFκB, to exert cell-specific effects on gene expression. Recent studies in macrophages indicate that a relatively small set of macrophage lineage determining transcription factors (LDTFs), that include PU.1 and members of the C/EBP and AP1 families of transcription factors, function in a collaborative manner to establish a large fraction of the enhancer-like elements in macrophages (Figure 2)[42,45]. These factors are capable of opening up closed regions of chromatin and enabling the coordinate or subsequent binding of signal-dependent factors such as nuclear receptors and the p65 component of NFκB[42,45]. Although many signal dependent genes harbor binding sites for the corresponding signal dependent transcription factor within their promoters, this is often not the case, and signal-dependent activation of these promoters therefore requires communication with enhancer elements that are under direct control of such factors.
In contrast to promoters, which exhibit relatively high levels of H3K4me3 as compared to H3K4me1 or H3K4me2, enhancers are characterized as having relatively low H3K4me3, and high H3K4me1 (Figure 2)[26,40,46–48]. Interestingly enhancers have also been shown to recruit RNA pol II, which results in active enhancer transcription[26,49–52]. Enhancer RNAs (eRNAs) are generally short transcripts (<1000 bp), lack polyadenylation, and appear to be unstable. This observation has raised the key question of whether such transcripts represent ‘noise’ due to spurious transcriptional activation at regions of open chromatin, are a consequence of an important role of enhancer transcription itself, or actually play direct roles in enhancer function. Recently, eRNAs have been shown to modulate the levels of adjacent mRNAs and may thus represent a novel layer of transcriptional control of inflammatory responses[53].
Although there is less knowledge of how epigenomic processes control enhancer function in comparison to promoters, enhancers have been proposed to exist in at least three functional states defined by the levels of H3K4me1 and H3K27ac levels in response to cell stimulation. Enhancers can be poised for activation and have high basal H3K4me1, with levels either remaining the same or elevated upon stimulation. These enhancers are broadly active across many cell types. Repressed enhancers represent the second class and are characterized by high basal levels of both H3K4me1 and H3K27ac levels which decrease upon cell stimulation (Figure 2). Finally, latent enhancers have low basal levels of H3K4me1 and H3K27ac levels which increase in response to signal-dependent macrophage activation. These latent enhancers are established by the macrophage lineage determining transcription factors which allow the macrophage to selectively activate a subset of eRNAs and mRNAs in response to specific cellular signals. This epigenetic signature remains at enhancers after stimulation has ceased and permits faster and more robust gene activation upon subsequent stimulation[54].
Conclusions
Genome-wide analysis of transcription factor binding and histone modifications in response to TLR activation have provided striking insights into molecular events underlying the transcriptional program that drives the innate immune response. These findings are likely to be applicable to other signal dependent programs of gene expression and to the understanding of pathogenic patterns of gene expression that are associated with chronic inflammatory diseases. The recognition that gene activation requires specific histone tail modifications that are carried out by diverse families of histone modifying enzymes suggests that pharmacologic manipulation of such modifications might provide new therapeutic strategies. Substantial efforts have previously been directed at developing inhibitors of histone deacetylases. More recent efforts indicate promise in approaches that act through inhibiting histone demethylases (e.g., Jmjd3/UTX inhibitors) and mimicking histone tails (e.g. the I-BETs). These efforts remain in their infancy, and substantial work will be required to define the identities of what are likely to be a large number of histone modifying enzymes that control the expression of inflammatory genes in macrophages and other relevant cell types. The development of new small molecule inhibitors of specific histone modifying enzymes will be important chemical tools for exploration of these questions and determining the extent to which specific effects on gene expression can be achieved in vivo. Since histone modifying enzymes control the expression of inflammatory gene expression whose aberrant regulation contributes to several human diseases, such chemical tools may become the basis for new therapeutic approaches to combat inflammatory driven illnesses.
Highlights.
TLR activation of latent transcription factors drives innate immune responses
Massively parallel sequencing technologies permit global analysis of mechanisms
Signal-dependent gene activation requires alterations in histone modifications
Some histone modification-dependent mechanisms can be inhibited by drugs
Epigenomic mechanisms may provide new targets for anti-inflammatory therapies
References
- 1.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol Rev. 2009;227:248–263. doi: 10.1111/j.1600-065X.2008.00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caricilli AM, Nascimento PH, Pauli JR, Tsukumo DM, Velloso LA, Carvalheira JB, Saad MJ. Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J Endocrinol. 2008;199:399–406. doi: 10.1677/JOE-08-0354. [DOI] [PubMed] [Google Scholar]
- 5.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and-independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25:1213–1219. doi: 10.1161/01.ATV.0000159891.73193.31. [DOI] [PubMed] [Google Scholar]
- 7.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen R, Alvero AB, Silasi DA, Steffensen KD, Mor G. Cancers take their Toll-the function and regulation of Toll-like receptors in cancer cells. Oncogene. 2008;27:225–233. doi: 10.1038/sj.onc.1210907. [DOI] [PubMed] [Google Scholar]
- 9.Huang B, Zhao J, Unkeless JC, Feng ZH, Xiong H. TLR signaling by tumor and immune cells: a double-edged sword. Oncogene. 2008;27:218–224. doi: 10.1038/sj.onc.1210904. [DOI] [PubMed] [Google Scholar]
- 10.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;11:476–486. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 15.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 16.Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 2006;6:44–55. doi: 10.1038/nri1748. [DOI] [PubMed] [Google Scholar]
- 17.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 18.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 19.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 20.Glatt S, Alfieri C, Muller CW. Recognizing and remodeling the nucleosome. Curr Opin Struct Biol. 2011;21:335–341. doi: 10.1016/j.sbi.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 22.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, Smale ST. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 2009;138:114–128. doi: 10.1016/j.cell.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clin Immunol. 2009;130:7–15. doi: 10.1016/j.clim.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raetz CR, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC, Jr, Ribeiro AA, Murphy RC, Ulevitch RJ, Fearns C, et al. Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J Lipid Res. 2006;47:1097–1111. doi: 10.1194/jlr.M600027-JLR200. [DOI] [PubMed] [Google Scholar]
- 26••.Escoubet-Lozach L, Benner C, Kaikkonen MU, Lozach J, Heinz S, Spann NJ, Crotti A, Stender J, Ghisletti S, Reichart D, et al. Mechanisms establishing TLR4-responsive activation states of inflammatory response genes. PLoS Genet. 2011;7:e1002401. doi: 10.1371/journal.pgen.1002401. The authors utilize genome-wide sequencing methods to provide a comprehensive analysis of the histone modifications and mechanisms driving inflammatory gene expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guenther MG, Lane WS, Fischle W, Verdin E, Lazar MA, Shiekhattar R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000;14:1048–1057. [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 2003;22:1336–1346. doi: 10.1093/emboj/cdg120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, Mietton F, Matteoli G, Hiebert S, Natoli G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A. 2012;109:E2865–2874. doi: 10.1073/pnas.1121131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 32.Sims RJ, 3rd, Belotserkovskaya R, Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes Dev. 2004;18:2437–2468. doi: 10.1101/gad.1235904. [DOI] [PubMed] [Google Scholar]
- 33••.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. The authors identify a new class of small molecules that blocks the recruitment of a class of proteins required for the activation of inflammatory gene expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35•.Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, Boutros M, Perrimon N, Rosenfeld MG, Glass CK. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol Cell. 2012;48:28–38. doi: 10.1016/j.molcel.2012.07.020. The authors identity and characterize a novel repression checkpoint on inflammatory gene promoters involving the methylation and demethylation of H4K20me3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, Bucci G, Caganova M, Notarbartolo S, Casola S, et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 2009;28:3341–3352. doi: 10.1038/emboj.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 38•.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. Using structure-function analysis, the authors identify a small molecular inhibitor for the Jmjd3 family of histone demethylases that reduces the TLR4-dependennt gene activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trompouki E, Bowman TV, Lawton LN, Fan ZP, Wu DC, DiBiase A, Martin CS, Cech JN, Sessa AK, Leblanc JL, et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell. 2011;147:577–589. doi: 10.1016/j.cell.2011.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mercer EM, Lin YC, Benner C, Jhunjhunwala S, Dutkowski J, Flores M, Sigvardsson M, Ideker T, Glass CK, Murre C. Multilineage priming of enhancer repertoires precedes commitment to the B and myeloid cell lineages in hematopoietic progenitors. Immunity. 2011;35:413–425. doi: 10.1016/j.immuni.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mullen AC, Orlando DA, Newman JJ, Loven J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, Young RA. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell. 2011;147:565–576. doi: 10.1016/j.cell.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei CL, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 46.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 47.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 48.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–634. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Leveille N, Kalluri R, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell. 2013;49:524–535. doi: 10.1016/j.molcel.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 54•.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. These studies define the distinct repertoire of newly selected enhancers in macrophages in response to activation of several toll-like receptors. [DOI] [PubMed] [Google Scholar]