

Abstract
NMDA receptors are tetrameric ligand-gated ion channels that are crucial for neurodevelopment and higher order processes such as learning and memory, and have been implicated in numerous neurological disorders. The lack of a structure for the channel open state has greatly hampered the understanding of the normal gating process and mechanisms of disease-associated mutations. Here we report the structural modeling for the open state of an NMDA receptor. Staring from the crystal structure of the closed state, we repacked the pore-lining helices to generate an initial open model. This model was revised to ensure tight packing between subunits and then refined by a molecular dynamics simulation in explicit membrane. We identify Cα-H⋯O hydrogen bonds, between the Cα of a conserved glycine in one transmembrane helix and a carbonyl oxygen of a membrane-parallel helix, at the extracellular side of the transmembrane domain as important for stabilizing the open state. This observation explains why mutations of the glycine are associated with neurological diseases and lead to significant decrease in channel open probability.
Keywords: ionotropic glutamate receptor, channel gating, open state, disease-associated mutations, Cα-H⋯O hydrogen bond
1. Introduction
NMDA and AMPA receptors are two main subtypes of postsynaptic ionotropic glutamate receptors (iGluRs). These receptors form tetrameric ligand-gated ion channels, and the ion channel activity is crucial for neurodevelopment and higher order processes such as learning and memory (Citri and Malenka, 2008). Recently, numerous missense mutations of NMDARs have been associated with an array of neurological disorders (e.g., autism, epilepsy, intellectual disability, and schizophrenia) (Hardingham and Do, 2016; Yuan et al., 2015). Thus, deep understanding of iGluR functional mechanisms is critical to both basic and clinical neuroscience. The central mechanistic question is how agonist binding triggers the opening of the ion channel in the cell membrane. In the past few years, structure determination by both X-ray crystallography and single-particle electron cryomicroscopy has led to the deposition of some 50 entries in the Protein Data Bank (PDB) for NMDA and AMPA receptors missing the intracellular, disordered C-terminal domain (CTD) (Mayer, 2016). However, the ion channel has not been captured in the open conformation, possibly because solubilizing conditions in sample preparations for structure determination did not model well key biophysical properties of the cell membrane.(Zhou and Cross, 2013). The lack of an open state structure has greatly hampered mechanistic understanding of iGluRs (Amin et al., 2017; Chen et al., 2017; Ogden et al., 2017; Zhou and Wollmuth, 2017).
NMDA receptors form obligatory heteromers consisting typically of two GluN1 and two GluN2A-D subunits, whereas AMPA receptors form both homo- and heterotetramers of GluA1-4 subunits. iGluR subunits have a modular architecture with four distinct structural domains (Fig. 1): extracellular amino-terminal (ATD) and ligand-binding (LBD) domains; a transmembrane domain (TMD) forming the ion channel upon tetramer assembly, with the M3 helix lining the channel pore and harboring the activation gate near the helix C-terminus; and an intracellular, disordered C-terminal domain (CTD) (Karakas and Furukawa, 2014; Lee et al., 2014; Sobolevsky et al., 2009). The LBD is shaped like a clam shell (Armstrong et al., 1998). Binding of agonists (glutamate for GluN2 and GluA subunits and glycine for GluN1) to the cleft results in closure of the two lobes (Armstrong and Gouaux, 2000). Structural studies have defined the basic outline of the activation mechanism: closure of the LBD lower (or D2) lobe upon the upper (or D1) lobe produces, via the M3-D2 linker, an outward pull of the M3 helix, and hence the opening of the ion channel.
Fig. 1.
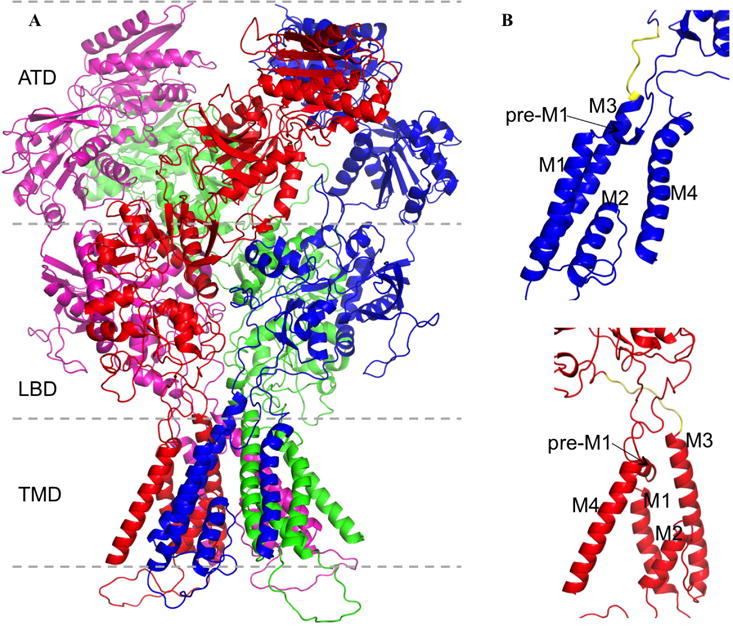
Modular architecture of NMDA receptors. (A) Subunit and domain organizations of a GluN1/GluN2B receptor, with residues missing in PDB 4TLM modeled (Amin et al., 2017). The two GluN1 subunits are colored in blue and purple; the two GluN2B subunits are in red and green. (B) Close-up views of the transmembrane regions of a GluN1 (top) and a GluN2B (bottom) subunit. The transmembrane helices M1, M3, and M4, the membrane-parallel helix pre-M1, and the reentrant helix M2 are labeled. The M3-D2 linker is highlighted in yellow.
Computational studies have filled in important details on the activation mechanism (Zhou, 2017). In targeted molecular dynamics simulations where the LBDs were forced to close, the displacements of the M3-D2 linkers and other elements leading to channel pore opening were visualized (Dai and Zhou, 2013; Dong and Zhou, 2011). The displacements were validated by residue-specific information from electrophysiological (including substituted cysteine accessibility method, or SCAM) data (Chang and Kuo, 2008; Sobolevsky et al., 2002a; Sobolevsky et al., 2007; Sobolevsky et al., 2002b; Sobolevsky et al., 2003; Sobolevsky et al., 2004; Talukder et al., 2010). Importantly, the extents of the outward movement of the M3-D2 linkers were different in the four subunits, greater in the B/D subunits (occupied by GluN2 for NMDA receptors) than in the A/C subunits (occupied by GluN1). This difference was attributed to the near orthogonal orientations of the M3-D2 linkers, parallel and perpendicular, respectively, to the membrane plane in the B/D and A/C subunits (Fig. 1). The unevenly splayed M3-D2 linkers resulted in symmetry breaking of the TMD layer from four-fold to two-fold upon channel opening. A further critical test was made through introducing glycine insertions in the M3-D2 linkers (Kazi et al., 2014). Molecular dynamics simulations showed that, with lengthening by glycine insertions, the linkers became less effective in pulling the M3 helices, leading to weaker stabilization of the channel open state. In full agreement, single-channel recordings showed that the channel open probability decreased progressively as the linkers were lengthened by glycine insertions. Still, the targeted molecular dynamics simulations did not produce a fully open channel.
An alternative approach is de novo repacking of the pore-lining helices (Heymann et al., 2013), developed initially to correct for excessive intersubunit crevices in a crystal structure of the trimeric, ATP-gated P2X4 receptor (Hattori and Gouaux, 2012). In this approach, the pore-lining helix of one subunit was positioned and oriented relative to the symmetry axis by sampling values for the radial distance (r), tilt angle (θ), azimuthal angle (ϕ), and self-rotation angle (ρ) over specified ranges. The other subunits were generated by replicating the first subunit while observing rotational symmetry. Plausible models were screened for by avoiding both clashes and excessive crevices between the subunits. The latter requirements were found to eliminate most combinations in the rθϕ space, so the number of plausible models for the TMD would be dramatically reduced (Dai and Zhou, 2014). Moreover, increase in pore radius follows distinct paths in the rθϕ space, such that the directions of changes in rθϕ required for channel opening can be predicted.
Here we report the application of the helix repacking approach to generate a structural model for the open state of an NMDA receptor. Initial assessment of the model finds some features that can explain and others that seem incompatible with functional observations. Our modeling hopefully will form the basis for formulating mechanistic hypotheses and spur the integration of computational and functional studies for advancing iGluR physiology.
2. Modeling and simulation methods
We repacked the TMD of the closed state structure for a GluN1/GluN2B receptor in PDB entry 4TLM to produce plausible models for the open channel and selected an initial open model based on correlation with SCAM data. Unfortunately this model did not have the M4 helix of one subunit packed against the M1 or M3 helix of a neighboring subunit, a packing pattern that has been found to be essential for proper NMDA receptor gating (Amin et al., 2017). We thus built a revised open model by running a targeted molecular dynamics simulation, starting from the crystal structure for the closed state but forcing the M3 helices to move toward their conformation in the initial open model. As we hoped, the revised model maintained tight M4-M1-M3 packing. We finally refined the revised open model in a 900-ns molecular dynamics simulation in explicit membrane and solvent.
2.1 Generation of initial open model by M3 repacking
Missing residues in 4TLM were modeled (Amin et al., 2017). The repacking procedure was as described in our previous study (Heymann et al., 2013), but with an important difference. Instead of choosing the position and orientation for the M3 helix of one subunit and replicating to create the M3 helices of the other three subunits to produce an M3 bundle with 4-fold rotational symmetry, we independently chose the positions and orientations of the M3 helices of one GluN1 subunit and one GluN2B subunit. The resulting GluN1-GluN2B dimer was then replicated to produce a 2-fold symmetric tetramer.
The TMD was comprised of 4TLM residues 543–660 (containing M1, M2, and M3) and 798–834 (containing M4) of GluN1 and 538–658 and 802–838 of GluN2B. Of these, residues 624–654 of GluN1 and 622–652 for GluN2B were taken as constituting the M3 helixes. In 4TLM, the radial distance r, tilt angle θ, and azimuthal angle ϕ, respectively, had values of 10.7 Å, 31.2°, and −46.4 for GluN1 and 9.7 Å, 36.8°, and − 50.0° for GluN2B. Starting from these values, increases in each of the three parameters are expected to increase the pore radius (Dai and Zhou, 2014). We thus sampled the rθϕ space by stepping r in the range of 10–16 Å with 1 Å increments, θ in the range of 31°–45° with 3° increments, and ϕ in the range of −50°–0° with increments of 3° for both GluN1 and GluN2B. Theoretically this sampling would produce (7 × 5 × 17)2 = 354,025 models. The helix rotational angle was kept at that in 4TLM so the same face of each M3 helix as in 4TLM was oriented toward the symmetry axis.
For each subunit, the M3 helix was used to place the whole transmembrane region. We then used the following criteria to screen for plausible open models for the TMD: (1) no clash (i.e., heavy atom distance < 3 Å) between any two subunits; (2) some contact between neighboring subunits, i.e., at least one pair of backbone heavy atoms at < 7 Å distance; (3) increase in pore radius by > 1 Å from the value in 4TLM; (4) greater outward displacement for the GluN2B M3 helices than for GluN1 M3 helices. A total of 21,388 models passed the screening.
Further selection of models was based on correlation with SCAM data. As in our previous studies (Dai and Zhou, 2013; Dong and Zhou, 2011), we compared the differences in accessible surface area between a putative open model and 4TLM for individual residues with the agonist-induced changes in modification rate for substituted cysteines (Chang and Kuo, 2008; Sobolevsky et al., 2002a; Sobolevsky et al., 2002b). The latter SCAM data for the following positions were used: pre-M1 residues F552 and L560 and M3 C-terminus proximal residues V642, T646, N648, L649, A651, F652, L655 for GluN1, and M2 tip residue N612 and M3 C-terminus proximal residues V637, L640, T644, N646, M651 for GluN2B. The 10 models showing the highest correlations with the SCAM data were attained for visual inspection and further modification.
2.2 Generation of a revised open model by targeted molecular dynamics simulation
Visual inspection showed that the attained models did not position M4 helices for packing against neighboring subunits. In this procedure, the transmembrane region of each subunit was treated as rigid. We reasoned that the correct open model should have the M3 helices similar in conformation to those in an attained model but have transmembrane regions deformed to preserve M4 packing. We decided to obtain such a revised open model by a targeted molecular dynamics simulation, with one attained model showing the second highest correlation with the SCAM data and a robust pore chosen as the target. Since we also needed the LBD-TMD linkers for the subsequent refinement stage (see next subsection), we chose to run the targeted molecular dynamics simulation for a full-length receptor (actually with CTD missing).
We built the full-length target model using Modeller (Eswar et al., 2007). The template for the TMD was the chosen initial open model; the template for the ATD and LBD was PDB entry 5FXG, which is the structure for the extracellular domains of an activated GluN1/GluN2B receptor (Tajima et al., 2016). The targeted molecular dynamics simulation in vacuum was run using NAMD 2.11 (Phillips et al., 2005), for 1 ns with a time step of 1 fs at 310K. Starting from 4TLM (with missing residues modeled), the Cα atoms of the ATD and LBD (residues 23–538 and 669–791 for GluN1 and residues 26–533 and 667–795 for GluN2B) as well as the M3 helices (residue 625–652 for GluN1 and residue 623–651 for GluN2B) were steered toward the target model by a root-mean-square-deviation (RMSD)-based restraint. The force constants were 5 kcal mol−1 Å−2 for the ATD and LBD and 1 kcal mol−1 Å−2 for M3. Additional restraints were used to maintain the secondary structure of the TMD. Specifically, the backbone dihedral angles of helical residues were restrained to ideal values (−60°, −45°) with a force constant of 10 kcal mol−1 deg−2 and the backbone hydrogen bonds were restrained (O–N and O-HN distances at 3.0 Å and 2.06 Å, respectively) with a force constant of 2 kcal mol−1 Å−2. Lastly to open the pore around the M2 loop (residues 614–619 for GluN1 and residues 612–617 for GluN2B), all the M2 loop atoms were pushed outside a cylinder in the center of the pore with radius of 5 Å, by applying a force in the radial direction and with a magnitude of 5 − r kcal mol−1 Å−1, for 10 ps.
2.3 Refinement of open TMD by molecular dynamics simulation in explicit membrane
For further refinement, a truncated TMD (residues 539–668 and 792–834 for GluN1 and residue 534–666 and 796–839 for GluN2B) from the above full-length open model was embedded in a bilayer of 202 POPC lipids and solvated by 16,440 water molecules and 0.15 M NaCl. The system was prepared using CHARMM-GUI (Jo et al., 2008). A molecular dynamics simulation was carried out with Amber14 on GPUs (Salomon-Ferrer et al., 2013). In each subunit, the four residues on each of the three LBD-TMD linkers most proximal to the LBD were restrained to mimic the stabilization of the open channel by the agonist-bound LBD.
The default protocol from CHARMM-GUI was used for the initial steps of the simulation: energy minimization followed by equilibration steps 1 through 6 at a time step of 1 fs, with the restraints listed in Table 1. Three more equilibration steps were added, followed finally by the production run. The time step was increased to 2 fs and all restraints had a force constant of 5 kcal mol−1 Å−2 unless otherwise indicated. Step 7 for 20 ns had all the protein Cα restrained; step 8 for 100 ns had only Cα restraints on the LBD-proximal linker residues and M3 residues; and step 9 had the restraints on the M3 residues gradually removed, with the force constant set to 5.0, 2.5, 1.0, 0.5, 0.25, 0.1, and 0.0 kcal mol−1 Å−2 in blocks of 2 ns each. The final production run was 900 ns.
Table 1.
Force constants for restraints in the initial steps of refinement by molecular dynamics simulation.
| Protein backbone heavy atoms (kcal mol−1 Å−2 | P atoms of POPC (kcal mol−1 Å−2) | Dihedral angles of POPC (kcal mol−1 deg−2) | Number of steps or time (ps) | |
|---|---|---|---|---|
| Minimization | 10.0 | 2.5 | 250.0 | 5,000 steps |
| Step 1 | 10.0 | 2.5 | 250.0 | 25 |
| Step 2 | 10.0 | 2.5 | 100.0 | 25 |
| Step 3 | 10.0 | 1.0 | 50.0 | 250 |
| Step 4 | 5.0 | 0.5 | 50.0 | 250 |
| Step 5 | 5.0 | 0.1 | 25.0 | 100 |
| Step 6 | 5.0 | 0.0 | 0 | 100 |
We also carried out a simulation of the TMD truncate in the closed state. The initial steps (up to step 6) were exactly the same as described above. The subsequent production run was 74.8 ns.
3. Results and discussion
3.1 Channel pore remains open in the refinement simulation
In the refinement simulation in explicit membrane, the pore-lining M3 helix bundle continued to adjust. After 300 ns, the M3 Cα RMSD from the initial open model from repacking stabilized to around 2.5 Å (Fig. 2A). Throughout the simulation, the channel pore remained filled with water molecules and sealed from the side, as illustrated by a snapshot at 544.9 ns (Fig. 2B). Water molecules diffused across the pore. Ions from the extracellular side also diffused across the activation gate to the bottom of central vestibule, but passage through the selectivity filter formed by the M2-M3 loops was not observed in the simulation. Compared to the close state, the channel pore in the open model is widened throughout the entire length, largely by outward splaying of the GluN2B M3 helices (Fig. 3).
Fig. 2.
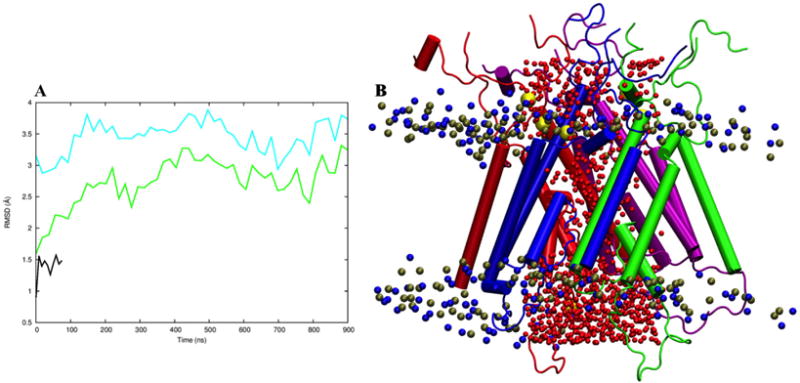
Refinement simulation of the open TMD in explicit membrane. (A) The Cα RMSD of the M3 bundle as a function of simulation time, referenced to either the closed structure 4TLM (cyan curve) or to our initial open model from helix repacking (green curve). Also shown is the Cα RMSD of the M3 bundle in the simulation of the closed TMD (black curve). (B) A snapshot at 544.9 ns of the simulation of the open TMD. The two GluN1 subunits are colored in blue and purple; the two GluN2B subunits are in red and green. Oxygen atoms of water molecules in the pore, P and N atoms in POPC headgroups, and sodium ions are shown as spheres colored in red, ochre, blue, and yellow, respectively.
Fig. 3.
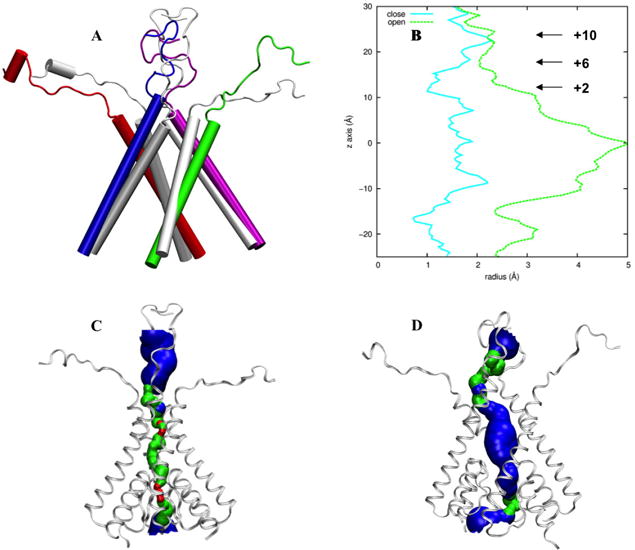
Comparison of the TMD closed structure 4TLM and the refined open model. (A) The M3 helices and the M3-D2 linkers. The closed structure is shown in gray whereas the open model in color. (B) Pore radius along the channel axis. The horizontal arrows indicate the positions of three residues forming the activation gate. The numbers are those defined according to the conserved sequence SYTANLAAF, with the residue “S” at number 0. (C) “HOLE” image (Smart et al., 1996) of the closed structure. (D) HOLE image of the open model.
3.2 GluN2B but not GluN1 M3 helices maintain splayed pose
During the refinement simulation, the GluN2B M3 helices maintained the splayed pose in the initial open model, but the GluN1 M3 helices retracted inward. The inward retraction of GluN1 M3 led to a decrease of the pore radius at the activation gate from the initial open model, but the activation gate opened wide enough to allow for passage of ions in the refinement simulation (see preceding paragraph). The different behaviors of the GluN1 and GluN2B M3 helices in the simulation can be attributed to the near orthogonal orientations of the M3-D2 linkers, horizontal in GluN2B while vertical in GluN1 (Figs. 1B and 3A). Keeping GluN1 M3 helices more splayed will be an important issue for future model improvements. In particular, stronger constraints of LBD residues most proximal to the TMD will allow for better mimicking of the stabilization of the open channel by the agonist-bound LBD.
In Fig. 4 we compare the changes in residue accessible surface area from the closed structure 4TLM to our open model and SCAM data for 15 positions in the pre-M1 and M3 helices of GluN1 and the M2 tip and M3 helices of GluN2. Our initial open model shows good correlation, with R2 = 0.64. In the open model refined by molecular dynamics simulation, the correlation R2 is reduced to 0.29, mainly due to inward retraction of the GluN1 M3 helices. Several GluN1 residues, including F552, V642, L649, and L655, are outliers in the correlation plot. Future experimental studies will allow for more stringent tests of the open model, e.g., by designing cysteine substitutions that are predicted to form metal coordination or disulfide cross-linking (Heymann et al., 2013; Kazi et al., 2013).
Fig. 4.
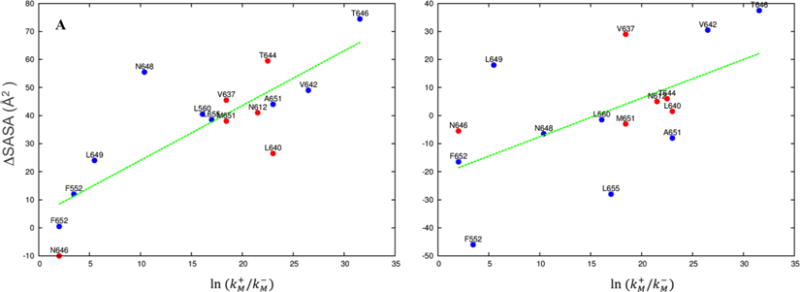
Comparison of ΔSASA, the differences in solvent accessible surface area between a putative open model and 4TLM for individual residues, with , where and denote the modification rates for substituted cysteines in the activated and resting states, respectively (Chang and Kuo, 2008; Sobolevsky et al., 2002a; Sobolevsky et al., 2002b). (A) Correlation plot for the initial open model, with the energy minimized 4TLM as reference. R2 = 0.64. Blue and red symbols are for GluN1 and GluN2B residues, respectively. (B) Correlation plot for the refined open model, with the last snapshot of the molecular simulation of 4TLM (Fig. 2A) as reference. R2 = 0.29.
3.3 Intrasubunit pre-M1 and M4 tight contacts are important for channel activation
The external positions of the TMD are rich in missense mutations associated with a variety of neurological disorders (Yuan et al., 2015). Electrophysiological studies have shown significant impairment in channel gating properties by mutations in these positions (Chen et al., 2017; Ogden et al., 2017). Moreover, the external half of the M4 helix, partly via intrasubunit interactions with the membrane-parallel pre-M1 helix, contributes significantly to channel activation (Amin et al., 2017). We therefore closely inspected the pre-M1 and M4 intrasubunit interactions in both the closed structure 4TLM and in our open model.
We found that, in the closed structure, pre-M1 tightly packs against M4, with a conserved Gly residue (position 813 in GluN1 and position 817 in GluN2B) potentially forming a Cα-H⋯O hydrogen bond, which is known to stabilize transmembrane helix association (Senes et al., 2001). The hydrogen bond acceptor is a carbonyl oxygen, of Asp550 in GluN1 and Ser545 in GluN2B. Mutations of this conserved Gly residue are associated with diseases (Yuan et al., 2015) and resulted in significant decreases in channel open probability (Amin et al., 2017). We thus reasoned that this Gly residue mediates tight interactions between pre-M1 and M4 in the open state to maintain the latter’s stability.
Consistent with this expectation, during the refinement simulation of the open model, GluN2B Gly817 consistently formed Cα-H⋯O hydrogen bonds, with the most prevalent acceptor being Pro544 of the same subunit (19.1% of all snapshots) followed by Ser545 (5.7% of all snapshots) (Fig. 5). GluN1 Gly813 likewise formed Cα-H⋯O hydrogen bonds.
Fig. 5.
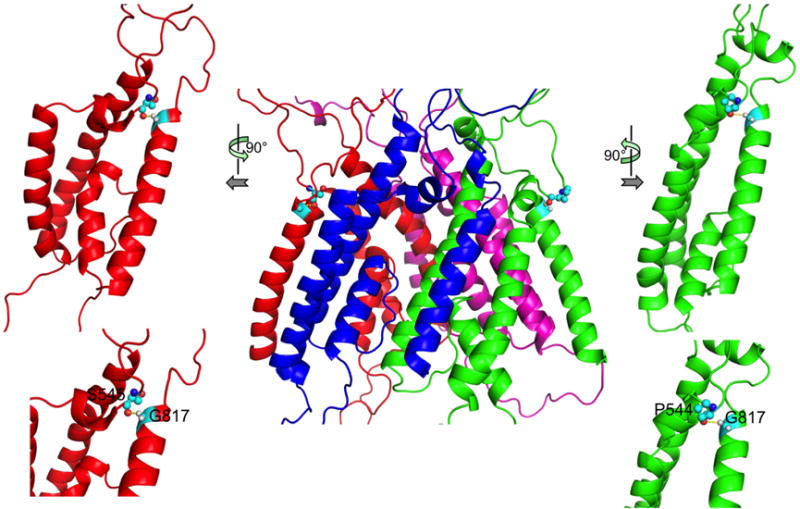
Hydrogen bonds between M4 Gly817 Cα and the carbonyl oxygen of pre-M1 Pro544 and Ser545 in GluN2B. The front view is shown in the middle; side views (top) and close-up views (bottom) are shown on the two sides.
Our modeling of the open state can thus be the basis for formulating hypotheses regarding the gating process and mechanisms of disease-associated mutations. To help address how the ATD and LBD communicate with the TMD, we merged the refined open model with PDB entry 5FXG, which putatively represent the extracellular domains in the activated state (Tajima et al., 2016) (Fig. 6). The full-length model will allow us to start probing questions like why X-ray crystallography and single-particle electron cryomicroscopy have failed to capture the ion channel in the open conformation and how disease-associated mutations affect functional properties. These future efforts will require iteration between computational and electrophysiological studies.
Fig. 6.
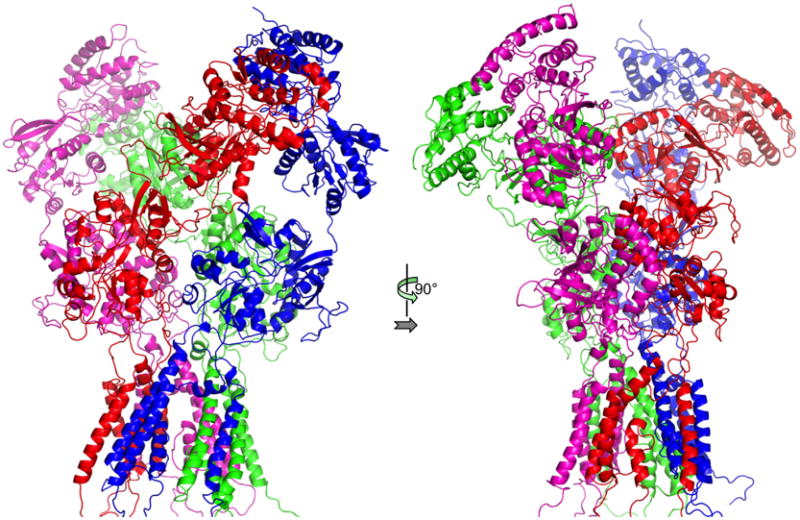
Full-length open model, by merging the refined open model for the TMD and the structure in 5FXG for the ATD and LBD.
Acknowledgments
This work was supported by National Institutes of Health Grant GM118091. It gives us great pleasure in dedicating this work to our inspiring colleague Don Caspar for his 90th birthday.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amin J, Salussolia CL, Chan K, Regan MC, Dai J, Zhou HX, Furukawa H, Bowen ME, Wollmuth LP. Divergent roles of a peripheral transmembrane segment in AMPA and NMDA receptors. J Gen Physiol. 2017 doi: 10.1085/jgp.201711762. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong N, Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Sun Y, Chen GQ, Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- Chang HR, Kuo CC. The activation gate and gating mechanism of the NMDA receptor. J Neurosci. 2008;28:1546–1556. doi: 10.1523/JNEUROSCI.3485-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Tankovic A, Burger PB, Kusumoto H, Traynelis SF, Yuan H. Functional evaluation of a de novo GRIN2A mutation identified in a patient with profound global developmental delay and refractory epilepsy. Mol Pharmacol. 2017;91:317–330. doi: 10.1124/mol.116.106781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- Dai J, Zhou HX. An NMDA receptor gating mechanism developed from MD simulations reveals molecular details underlying subunit-specific contributions. Biophys J. 2013;104:2170–2181. doi: 10.1016/j.bpj.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Zhou HX. General rules for the arrangements and gating motions of pore-lining helices in homomeric ion channels. Nat Commun. 2014;5:4641. doi: 10.1038/ncomms5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Zhou HX. Atomistic mechanism for the activation and desensitization of an AMPA-subtype glutamate receptor. Nat Commun. 2011;2:354. doi: 10.1038/ncomms1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Current protocols in protein science Chapter 2. 2007 doi: 10.1002/0471140864.ps0209s50. Unit 2.9. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Do KQ. Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat Rev Neurosci. 2016;17:125–134. doi: 10.1038/nrn.2015.19. [DOI] [PubMed] [Google Scholar]
- Hattori M, Gouaux E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature. 2012;485:207–212. doi: 10.1038/nature11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann G, Dai J, Li M, Silberberg SD, Zhou HX, Swartz KJ. Inter- and intrasubunit interactions between transmembrane helices in the open state of P2X receptor channels. Proc Natl Acad Sci U S A. 2013;110:E4045–4054. doi: 10.1073/pnas.1311071110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi R, Dai J, Sweeney C, Zhou HX, Wollmuth LP. Mechanical coupling maintains the fidelity of NMDA receptor-mediated currents. Nat Neurosci. 2014;17:914–922. doi: 10.1038/nn.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi R, Gan Q, Talukder I, Markowitz M, Salussolia CL, Wollmuth LP. Asynchronous movements prior to pore opening in NMDA receptors. J Neurosci. 2013;33:12052–12066. doi: 10.1523/JNEUROSCI.5780-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lu W, Michel JC, Goehring A, Du J, Song X, Gouaux E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML. Structural biology of glutamate receptor ion channel complexes. Curr Opin Struct Biol. 2016;41:119–127. doi: 10.1016/j.sbi.2016.07.002. [DOI] [PubMed] [Google Scholar]
- Ogden KK, Chen W, Swanger SA, McDaniel MJ, Fan LZ, Hu C, Tankovic A, Kusumoto H, Kosobucki GJ, Schulien AJ, Su Z, Pecha J, Bhattacharya S, Petrovski S, Cohen AE, Aizenman E, Traynelis SF, Yuan H. Molecular mechanism of disease-associated mutations in the pre-M1 helix of NMDA receptors and potential rescue pharmacology. PLoS Genet. 2017;13:e1006536. doi: 10.1371/journal.pgen.1006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- Senes A, Ubarretxena-Belandia I, Engelman DM. The Calpha —H…O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc Natl Acad Sci U S A. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MS. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph. 1996;14:354–360. 376. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, Beck C, Wollmuth LP. Molecular rearrangements of the extracellular vestibule in NMDAR channels during gating. Neuron. 2002a;33:75–85. doi: 10.1016/s0896-6273(01)00560-8. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, Prodromou ML, Yelshansky MV, Wollmuth LP. Subunit-specific contribution of pore-forming domains to NMDA receptor channel structure and gating. J Gen Physiol. 2007;129:509–525. doi: 10.1085/jgp.200609718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rooney L, Wollmuth LP. Staggering of subunits in NMDAR channels. Biophys J. 2002b;83:3304–3314. doi: 10.1016/S0006-3495(02)75331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Yelshansky MV, Wollmuth LP. Different gating mechanisms in glutamate receptor and K+ channels. J Neurosci. 2003;23:7559–7568. doi: 10.1523/JNEUROSCI.23-20-07559.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Yelshansky MV, Wollmuth LP. The outer pore of the glutamate receptor channel has 2-fold rotational symmetry. Neuron. 2004;41:367–378. doi: 10.1016/s0896-6273(04)00008-x. [DOI] [PubMed] [Google Scholar]
- Tajima N, Karakas E, Grant T, Simorowski N, Diaz-Avalos R, Grigorieff N, Furukawa H. Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature. 2016;534:63–68. doi: 10.1038/nature17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukder I, Borker P, Wollmuth LP. Specific sites within the ligand-binding domain and ion channel linkers modulate NMDA receptor gating. J Neurosci. 2010;30:11792–11804. doi: 10.1523/JNEUROSCI.5382-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF. Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol Pharmacol. 2015;88:203–217. doi: 10.1124/mol.115.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HX. Gating motions and stationary gating properties of ionotropic glutamate receptors: computation meets electrophysiology. Acc Chem Res. 2017;50:814–822. doi: 10.1021/acs.accounts.6b00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HX, Cross TA. Influences of membrane mimetic environments on membrane protein structures. Annu Rev Biophys. 2013;42:361–392. doi: 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HX, Wollmuth LP. Advancing NMDA receptor physiology by integrating multiple approaches. Trends Neurosci. 2017;40:129–137. doi: 10.1016/j.tins.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]