

Abstract
Background
Complement is a central part of both the innate and adaptive immune response and its activation has traditionally been considered part of the immunosurveillance response against cancer. Its pro-inflammatory role and its contribution to the development of many illnesses associated with inflammatory states implicate complement in carcinogenesis.
Methods
We evaluated the role of three protein inhibitors of complement—cobra venom factor, humanized cobra venom factor, and recombinant staphylococcus aureus superantigen-like protein 7—in the setting of a transplantable murine colon cancer model. Outcomes were evaluated by monitoring tumor growth, and flow cytometry, ELISPOT, and quantitative real-time PCR were used to determine the impact of complement inhibition on the host immune response.
Results
Complement inhibitors were effective at depleting complement component C3 in tumor bearing mice and this was temporally correlated with a decreased rate of tumor growth during the establishment of tumors. Treatment with cobra venom factor resulted in increased CD8+ T cells as a percentage of tumor-infiltrating cells as well as a reduced immunosuppressive environment evidenced by decreased myeloid derived suppressor cells in splenocytes of treated mice. Complement inhibition resulted in increased expression of the chemoattractive cytokines CCL5, CXCL10, and CXCL11.
Discussion
Complement depletion represents a promising mode of immunotherapy in cancer by its ability to impair tumor growth by increasing the host’s effective immune response to tumor and diminishing the immunosuppressive effect created by the tumor microenvironment and ultimately could be utilized as a component of combination immunotherapy.
Complement, comprising more than 30 proteins and fragments, is part of the innate and adaptive immune system.1,2 All three pathways of complement activation (antibody mediated, mannose binding lectin mediated, and alternative) converge on C3 and C5, anaphylatoxins with various immune responses.1
Complement activation has traditionally been considered part of the immunosurveillance response against cancer as a result of its ability to tag, clear, and lyse altered cells as well as mediate antibody responses through complement-dependent cytotoxicity.3 Recent studies, however, have linked complement activation to a suppressive response.
Complement is pro-inflammatory and plays a role in chronic inflammation, which has been associated with carcinogenesis.4,5 More specific mechanisms, including increasing tumorigenic growth factors and cytokines, preventing apoptosis, enhancing angiogenesis, and promoting immunosuppression contribute to complement’s role in tumorigenesis.4 Cancer cells themselves can promote the conversion of C5 to C5a to enhance invasiveness.6 Further, the interaction between tumor and complement may protect tumors from complement mediated lysis through expression of complement-regulatory proteins via continuous, low-level complement activation resulting in sublytic levels of the membrane attack complex protecting cells from lysis.7–11
Complement deposition and/or activation has been demonstrated in many cancers. Its components have been shown to activate cellular responses involved in tumor growth.12–17 Differentiation of regulatory T cells is correlated with C5a concentration within the tumor and C5a released as a result of complement activation on tumor cells is connected to the recruitment and activation of myeloid-derived suppressor cells (MDSCs) into tumors.5,18
Inhibition of complement can be achieved via antibody or protein inhibitors. The protein inhibitors cobra venom factor (CVF) and humanized cobra venom factor (hCVF) inhibit complement by continuous activation and ultimately depletion. Recombinant Staphylococcus aureus superantigen-like protein 7 (SSL7) directly inhibits complement. On the basis of complement’s potential for promoting tumor growth, we investigated transient complement depletion to target complement signaling pathways, decrease pathologic inflammation, and improve host response to tumor. To ensure observed effects were due to complement inhibition and were not specific to the protein, we examined the effects of the three aforementioned protein inhibitors in a transplantable murine colon cancer model.
MATERIALS AND METHODS
Cell Lines
MC38 (originally induced with oral dimethylhydrazine in C57/BL6 mice) and MC38-luc (transfected with plasmid carrying the luciferase gene, obtained from Dr. Stephen Thorne) murine colon cancer cells were grown in Dulbecco modified Eagle medium supplemented with 10 % fetal bovine serum, L-glutamine, and penicillin/streptomycin (Invitrogen) at 37 °C under 5 % CO2.
Viable Cell Assay
Using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega), MTS tetrazolium compound was added to culture wells. After 4 h, absorbance at 490 nm with a 96-well plate reader was measured with quantity of colored formazan product directly proportional to the number of living cells.
Mice
Five- to 6-week-old female C57/BL6 mice were obtained from Taconic Corporation. Animal studies were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh Cancer Institute (protocol 1110916).
Complement Protein Inhibitors
Purified CVF (Naja naja kaouthia) was obtained from Quidel Corporation. hCVF was obtained from Dr. Carl Wilhelm Vogel (Cancer Research Center, Honolulu, HI). Both agents deplete complement by continuous activation. Recombinant SSL7, a direct C5 inhibitor, was obtained from John Fraser, University of Auckland, New Zealand.
Animal Models
MC38 subcutaneous tumors were established by injection of 5.0 × 105 cells in the right flank of mice.
ELISA
ELISA was performed to detect C3 (R&D) in pericardial blood of tumor-bearing mice.
Flow Cytometry
Three-color immunostaining of cell surface and intracellular markers was performed utilizing the Accuri C6 flow cytometer. Isolated splenocytes were stained for CD11b-PE, Gr1-APC, and F480-FITC to evaluate MDSCs; CD3-PE, CD8-FITC, and Granzyme-B-PECy7 (intracellular) to evaluate CD8 cells; and CD4-APC, CD25-FITC, and FoxP3-PE (intracellular) to evaluate Treg cells (eBio-science). Before intracellular staining, cells were fixed and permeabilized using the FoxP3 Fix/Perm Buffer Set solution (eBioscience). Tumors from mice were morselized into a single cell suspension to evaluate tumor-infiltrating lymphocytes and probed using APC-conjugated anti-mouse antibody directed against CD4 with isotype control (mouse IgG) and PE-conjugated anti-mouse mAb directed against CD8 with isotype control (mouse IgG1) (eBioscience).
Gamma Interferon ELISPOT
Splenocytes (5 × 105) from tumor-bearing animals were plated on interferon (IFN) γ Ab (Mabtech) precoated Millipore membrane multiwell plates and restimulated with 2000 Gy irradiated 2 × 104 MC38 cells for 48 h. IFN-γ production was detected with secondary IFN-γ antibody and spots developed using Vectastain ABC kit (Vector Laboratories). Spots were analyzed using the ImmunoSpot Analyzer (Cellular Technology).
Quantitative Real-Time PCR (qRT-PCR)
RNA was obtained from tumors and spleens according to manufacturer’s protocol (Invitrogen, Life Sciences), followed by cDNA synthesis with the qScript cDNA supermix (Quanta Biosciences). qRT-PCR was performed utilizing Lifetech primers.
Statistical Analysis
Statistical analyses were performed by Student’s t test (expressed as mean ± SD). A p value of 0.05 was considered significant.
RESULTS
Complement Inhibitors Have No Direct Cytotoxic Effect In Vitro
Before utilizing complement inhibitors in vivo, we evaluated whether CVF, hCVF, and SSL7 had direct cytotoxic effects on tumor cells (Supplementary Fig. 1). Doses of 40 mg/mL of CVF, 1 mg/mL of hCVF, and 75 μg/mL SSL7 (clinically relevant concentrations) were used to treat MC38-luc cells, which were examined by fluorescent microscopy and viable cell assay after 48 h of incubation. There was no difference in morphology or viability between control and treated cells.
Complement Depletion Decreases the Rate of Tumor Growth During the Establishment of Subcutaneous Tumors
CVF depletes C3 by continuous activation. After daily administration of CVF, treated mice had significantly lower levels (37 %) of C3 compared to controls (p = 0.049), with an effect until day 12. At later time points (day 20), there remained a trend toward reduced C3 in CVF-treated mice (35 % less).
A subcutaneous colon cancer model was established via injection of 1e5 MC38 cells in the right flank of C57/BL6 mice. Two days later, mice were treated with CVF (500 μg/kg per mouse i.p. qod), hCVF (1 mg/kg hCVF i.p. qd), or phosphate-buffered saline (PBS; 100 μL per mouse i.p.). Mice treated with CVF (Fig. 1a) had significantly smaller tumors compared to controls on posttumor implantation days 6 (average tumor sizes 44 vs. 78 mm3), 12 (164 vs. 345 mm3), and 18 (389 vs. 652 mm3). At later time points, this difference was lost. There was no survival advantage. Findings were similar in mice treated with hCVF (Fig. 1b).
FIG. 1.
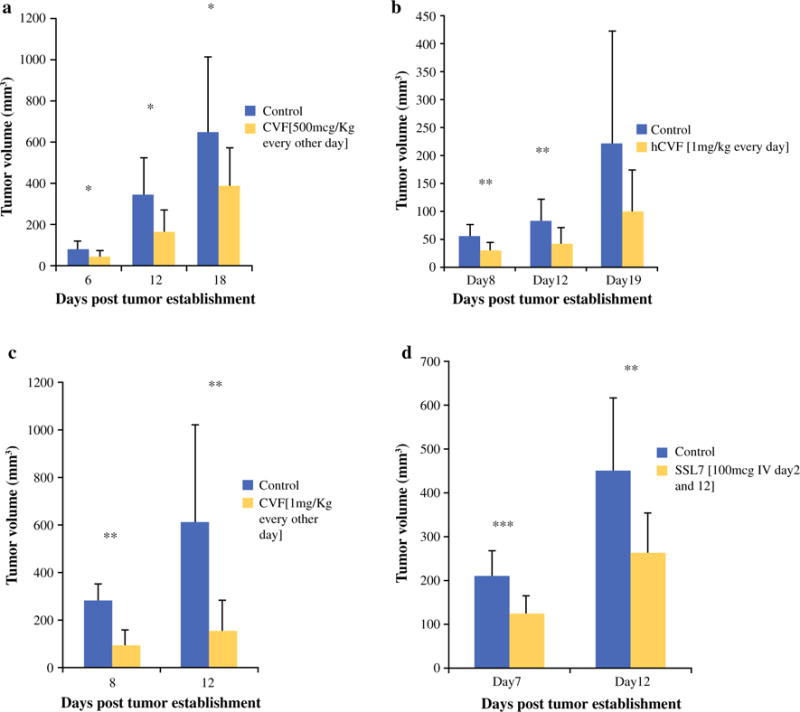
Complement inhibition decreases tumor volume. a Tumor volumes of mice treated with PBS or CVF (500 μg/kg per mouse) measured at days 6 (78 vs. 44 mm3; p = 0.049), 12 (343 vs. 164 mm3; p = 0.019), and 16 (652 vs. 390 mm3; p = 0.03) after tumor establishment. Mice treated with CVF had significantly smaller tumors than those treated with PBS at all 3 time points. b Tumor volumes of mice treated with PBS or hCVF (1 mg/kg per mouse) measured at days 8 (54 vs. 29 mm3; p = 0.0015), 12 (82 vs. 42 mm3; p = 0.002), and 19 (222 vs. 99 mm3; p = 0.06) after tumor establishment. Mice treated with hCVF had significantly smaller tumors at days 8 and 12 after tumor establishment, but after this time point, significance was lost. c Tumor volumes of mice treated with PBS or CVF at a higher dose (1 mg/kg per mouse) measured at days 8 (282 vs. 90 mm3; p = 0.009) and 12 (613 vs. 152 mm3; p = 0.004) after tumor establishment. Mice treated with this dose of CVF had smaller tumors at days 8 and 12, but this difference was lost at later time points. d Tumor volumes of mice treated with PBS or SSL7 (100 μg IV day 2 and 12 after tumor establishment) measured at days 7 and 12 after tumor establishment. Mice treated with SSL7 had smaller tumors at days 7 (207 vs. 121 mm3; p = 0.001) and 12 (447 vs. 263 mm3; p = 0.005), but this difference was lost at later time points. *p < 0.05, **p < 0.01, ***p ≤ 0.001
This experiment was repeated with a higher dose of CVF (1 mg/kg per mouse) (Fig. 1c), and treated mice had smaller tumors than control mice at days 8 (91 vs. 282 mm3) and 12 (152 vs. 613 mm3). The difference was lost at later time points, confirming that C3 depletion associated with CVF administration noted on day 12 after tumor implantation correlated with decreased tumor growth in CVF-treated animals.
SSL7 (100 μg SSL7 per mouse i.v. days 2 and 15 after tumor implantation) also reduced tumor progression compared to control mice, with significant differences in tumor volume noted at days 8 (121 vs. 207 mm3; p = 0.001) and 12 (263 vs. 447 mm3; p = 0.005) (Fig. 1d).
Complement Depletion with CVF Reduces Tumor-Associated Immunosuppression but Alters Effector T Cells in Peripheral Lymphoid Organs
To evaluate the immune responses of tumor-bearing mice treated with CVF (i.p. 500 mcg/kg per mouse every second day), flow cytometry was performed on splenocytes isolated from mice 12 days after tumor implantation. Splenocytes of both CVF-treated and control mice demonstrated similar percentages of CD4+ and CD8+ T cells.
Surprisingly, there was an increased population of granzyme B+CD8+ T cells from splenocytes of control mice (48.6 vs. 37.4 %; p = 0.001) (Supplementary Fig. 2a). ELISPOT evaluation of IFN-γ production after 48 h of incubation with irradiated MC38 cells revealed no difference in the number of spots per 500,000 splenocytes in control or CVF-treated mice at day 7 (Supplementary Fig. 2b), but by day 12, control mice demonstrated greater tumor-specific immune responses from splenocytes (7.7 vs. 4.4; p = 0.037) (Supplementary Fig. 2c).
There was a significantly higher percentage of MDSCs (CD11b+Gr1+ cells) in the spleens of control mice compared to CVF-treated mice (p = 0.045) (Fig. 2a). No difference in Treg cells was noted (CD25+FoxP3+ of CD4+ cells). The increased predominance of MDSCs in control mice compared to CVF-treated mice despite increased granzyme B+CD8+T cells and increased IFN-γ response of control mice suggest a reduction in immunosuppressive cells may play a role in complement’s inhibition of tumor growth.
FIG. 2.
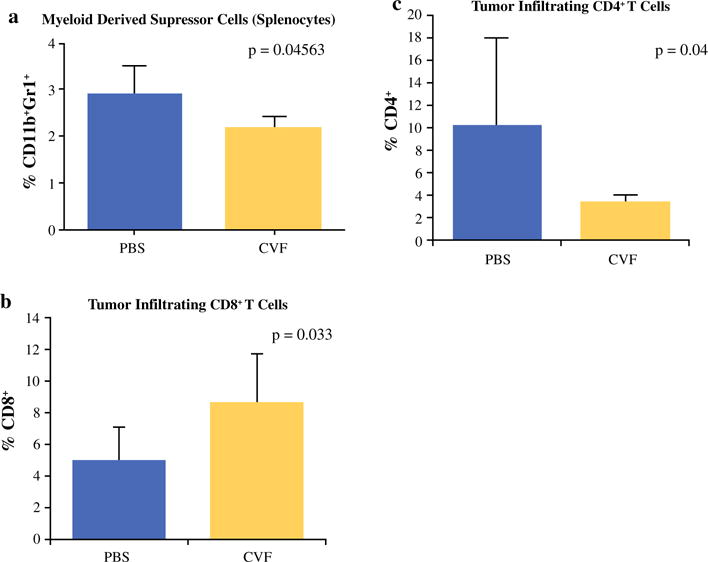
Complement depletion with CVF reduces immunosuppressive MDSCs and results in increased tumor infiltration of effector T cells via increased production of chemoattractive mediators. a FACS analysis shows increased percentage of CD11b+Gr1+ cells in splenocytes of PBS-treated mice vs. CVF-treated mice (p = 0.04563). b FACS analysis of tumor-infiltrating lymphocytes shows a higher percentage of CD8+ T cells in CVF-treated mice compared to control mice (p = 0.033) and c decreased percentage of CD4+ T cells infiltrating tumors in CVF-treated mice (p = 0.04). *p < 0.05, **p < 0.01, ***p ≤ 0.001
Complement Depletion Results in Increased Chemoattraction and Infiltration of CD8+ Cells in the Tumor Microenvironment
The tumor microenvironment was examined focusing on CD4+ and CD8+ T cell infiltration. Flow cytometry of tumor infiltrated cells revealed a higher percentage of CD8+ cells in the tumors of CVF-treated mice (i.p. 500 μg/kg per mouse every second day) on day 12 after tumor implantation (p = 0.033) (Fig. 2b). There were significantly lower CD4+ cells in the tumors of CVF-treated mice (p = 0.04) (Fig. 2c). These differences were not apparent at day 20. Similar differences with nearly a twofold percentage increase in CD8+ cells (p = 0.004) (Fig. 3a) and a trend of less CD4+ cells were observed in the hCVF-treated mice compared to control mice (p = 0.06) (Fig. 3b).
FIG. 3.
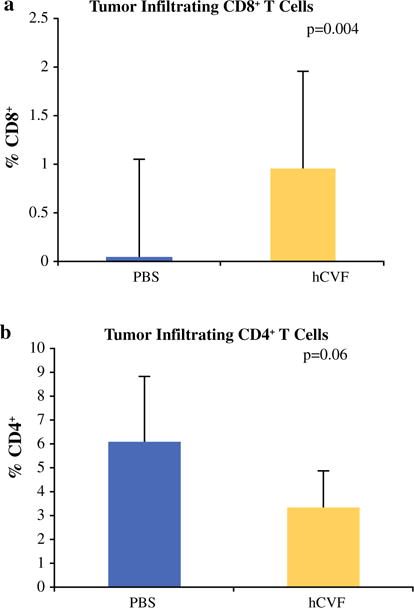
Complement inhibition with SSL7 results in increased tumor infiltration of effector T cells via increased production of chemoattractive mediators. a Using complement inhibitor, SS7, similar effects on tumor infiltrating lymphocytes were found with increased percentage of CD8+ cells in SSL7-treated mice (p = 0.004). b Trend was exhibited toward decreased infiltration of CD4+ cells in SSL7-treated mice compared to controls (p = 0.06)
The chemokines CCL5, CXCL10, and CXCL11 demonstrated increased expression in the tumors of CVF-treated (Fig. 4) mice at 12 days after tumor implantation when evaluated by qRT-PCR. These findings were similar in SSL7-treated mice (Fig. 5) compared to controls.
FIG. 4.
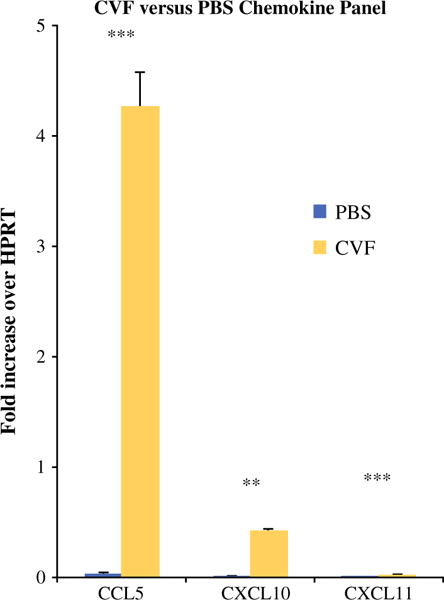
Complement depletion with CVF results in increased production of chemoattractive chemokines in tumor microenvironment. qRT-PCR performed on tumor tissues of mice treated with PBS or CVF showed significantly higher expression [fold increase over HPRT (hypoxanthine guanine phosphoribosyl transferase) (murine housekeeping gene)] of chemokines CCL5 (0.03-fold vs. 4.3-fold; p = 0.001), CXCL10 (0.004 vs. 0.42; p = 0.002), and CXCL11 (0.001 vs. 0.023; p = 0.001) in CVF-treated mice 12 days after tumor establishment. *p < 0.05, **p < 0.01, ***p ≤ 0.001
FIG. 5.
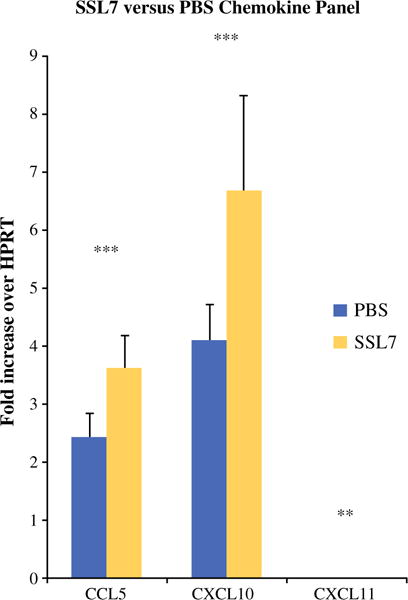
Complement inhibition with SSL7 results in increased production of chemoattractive chemokines in tumor microenvironment. qRT-PCR performed on tumor tissues of mice treated with PBS or CVF showed significantly higher expression [fold increase over HPRT (hypoxanthine guanine phosphoribosyl transferase) (murine housekeeping gene)] of chemokines: CCL5 (2.42 vs. 3.61; p = 0.001), CXCL10 (4.1 vs. 6.7; p = 0.001), and CXCL11 (0.038 vs. 0.06; p = 0.008). *p < 0.05, **p < 0.01, ***p ≤ 0.001
Although the spleens of control mice demonstrated more granzyme B+ effector T cells and ELISPOT analysis revealed a stronger IFN-γ response to MC38 cells, there was a reduction in tumor size noted in the mice treated with complement inhibitors. This may be explained not only by reduced MDSCs in the mice treated by complement inhibition but also by the increased infiltration of effector T cells into the tumor microenvironment, which may be promoted by increased expression of the chemoattractive mediators CCL5, CXCL10, and CXCL11. Further studies are needed to fully elucidate the mechanism of differential CD8+ T cell recruitment in complement-inhibited mice.
DISCUSSION
The tumor-promoting potential of complement has been recently demonstrated in complement-deficient mice.12 Here we describe a novel approach to inhibit the complement system that results in impaired tumor growth via increased tumor chemoattraction and infiltration of CD8+ T cells and decreased MDSCs.
Using two methods of complement depletion (CVF, hCVF) and one method of inhibition (SSL7), we demonstrated decreased tumor growth in a transplantable murine model of colon cancer. Suppressed tumor growth correlated with a nadir in C3 levels, and tumor progression was restored when complement component C3 was replenished. Our findings support recent studies evaluating the role of complement in cancer. Using a murine model of cervical cancer and mice deficient in various complement components, Markiewski et al. confirmed complement’s role in cancer, showing that C3 is deposited in tumors and its deficiency resulted in decreased tumor growth.12 Moreover, a peptide antagonist of the C5a receptor enhanced CD8+ T cell antitumor responses and was as effective as the chemotherapeutic paclitaxel (Taxol) in retarding tumor growth.12
Tumor-induced immunosuppression mediated by cells such as MDSCs and Treg cells contributes to the failure of many immunotherapies in clinical trials.19 It has been shown that activation of the complement system by transformed cells promotes tumor growth, whereby complement protects tumors from the host immune response by inducing MDSC differentiation.20 Reduced tumor growth was associated with decreased percentage of MDSCs, but not Treg cells, in the spleens of tumor-bearing CVF-treated mice compared to control tumor-bearing mice. Splenocytes of control mice did have greater granzyme B+ T cells and a stronger IFN-γ response to MC38 cells; however, mice treated with CVF and hCVF demonstrated enhanced tumor CD8+ T cell infiltration as well as enhanced expression of chemoattractive cytokines CCL5, CXCL10, and CXCL11 in the tumor microenvironment. Accordingly, in the study by Markiewski and Lambris, mice treated with C5aR antagonist had fewer MDSCs and greater effector T cells compared to their complement-replenished counterparts.12 This has been corroborated by a study using a murine lung cancer model, where blockade of C5a resulted in slower tumor growth and reduced MDSC; the study concluded that C5a promotes a favorable tumor microenvironment via enhanced MDSC recruitment.15
Additionally, Hsieh et al. have demonstrated that C3 is essential for the differentiation of MDSCs and that complement cleavage factors divert the differentiation from dendritic cells into potent immunosuppressive MDSCs.20 We and others have linked inflammation and MDSCs by demonstrating that key pro-inflammatory mediators, including interleukin (IL) 1β and IL-6, pros-taglandin E2 (PGE2), and S100A8/A9 proteins regulate the accumulation and suppressive activity of MDSCs, which in turn promote tumor growth by preventing the activation of CD4+ and CD8+ T lymphocytes, inhibiting natural killer cell cytotoxicity, stimulating tumorigenic cytokine production, and increasing angiogenesis.21–24 Deficiency or inhibition of the pro-inflammatory components of the complement system was associated with reduced tumor-induced accumulation of MDSCs as well as increased expression of chemokines CCL5, CXCL10, and CXCL11, which may contribute to the observed increased infiltration of CD8+ T cells into tumors of complement-inhibited mice despite increased peripheral CD8+ T cell response in control mice. On the basis of differential tumor growth, we assume the decreased peripheral MDSC population observed in complement inhibited mice is reflective of the MDSC composition in the tumor microenvironment of these mice; however, this is an important finding to confirm in future studies.
These data demonstrate that complement depletion represents a promising mode of cancer therapy. Although we have shown that complement depletion correlates with impaired tumor growth, we did not demonstrate a survival benefit. Additionally, the effect of complement inhibition on tumor growth was transient, likely the result of immunogenicity of the various inhibitors and ultimate clearance by the host immune system.25 In humans, extensive use of eculizumab (monoclonal C5Ab) for paroxysmal nocturnal hematuria has demonstrated prolonged (more than 12 weeks), continuous, and complete complement inhibition without any immune reaction against the antibody.26 Although we could not use this product in our murine studies, the sequential use of different protein inhibitors may allow for a more prolonged effect. It is also possible that the immune microenvironment adapts to complement inhibition and overcomes its effects through other mechanisms of immune suppression. In that case, complement inhibition is likely best used as combination immunotherapy. Ideally, complement inhibition could be combined with an agent that required transient immunosuppression for effectiveness such as an oncolytic virus which requires time to infect and replicate to be effective.
On the bases of the knowledge that complement can lead to direct viral destruction, even in the absence of antiviral antibodies, and our previously published in vitro work demonstrating enhanced vaccinia virus infectivity when paired in vitro with an inhibitor of the C5 component of complement, we believe that pairing complement inhibition with the tumor-selective oncolytic vaccinia virus could be highly successful for enhancing viral infectivity and replication in vivo.27 A recently published study demonstrated that in immune hosts, antibody-mediated vaccinia virus neutralization is dependent on the complement system, and in both small and large immunized animals, the combination of complement inhibition with vaccinia virus treatment results in increased delivery to tumor and virus stability in the blood.28 Ultimately, further evaluation of complement inhibition in combination with other immunotherapies such as viral oncolytic therapy will determine the ideal role for complement inhibition in the clinical setting.
Supplementary Material
Acknowledgments
Supported in part by U.S. National Institutes of Health (NIH) Grant R01CA155925 (to DLB) and by David C. Koch Regional Therapy Cancer Center. DM and SDC are supported by training grant T32CA113263 from the National Cancer Institute. This project used University of Pittsburgh Cancer Institute shared resources that are supported in part by NIH grant award P30CA047904. Special thanks to Dr. Stephen Thorne’s group at the University of Pittsburgh for providing MC38-luc cells, Dr. John Fraser’s group in Auckland, New Zealand, for their collaboration and for providing SSL7 protein for our study, and Dr. Carl Wilhelm Vogel for collaboration and providing hCVF for our study.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1245/s10434-015-4778-7) contains supplementary material, which is available to authorized users.
DISCLOSURE
The authors declare no conflict of interest.
References
- 1.Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011;343:227–35. doi: 10.1007/s00441-010-1034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–42. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dechant M, Weisner W, Berger S, et al. Complement-dependent tumor cell lysis triggered by combinations of epidermal growth factor receptor antibodies. Cancer Res. 2008;68:4998–5003. doi: 10.1158/0008-5472.CAN-07-6226. [DOI] [PubMed] [Google Scholar]
- 4.Rutkowski MJ, Sughrue ME, Kane AJ, et al. Cancer and the complement cascade. Mol Cancer Res. 2010;8:1453–65. doi: 10.1158/1541-7786.MCR-10-0225. [DOI] [PubMed] [Google Scholar]
- 5.Markiewski MM, Lambris JD. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol. 2009;30:286–92. doi: 10.1016/j.it.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nitta H, Murakami Y, Wada Y, et al. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol Rep. 2014;32:1715–9. doi: 10.3892/or.2014.3341. [DOI] [PubMed] [Google Scholar]
- 7.Yu J, Caragine T, Chen S, Morgan BP, Frey AB, Tomlinson S. Protection of human breast cancer cells from complement-mediated lysis by expression of heterologous CD59. Clin Exp Immunol. 1999;115:13–8. doi: 10.1046/j.1365-2249.1999.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) Mol Immunol. 2003;40:109–23. doi: 10.1016/s0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 9.Kempshall E, Thebault S, Morgan BP, Harris CL, Gallimore A. Complement-induced protectin: an explanation for the limitations of cell-based tumour immunotherapies. Immunol Cell Biol. 2012;90:869–71. doi: 10.1038/icb.2012.30. [DOI] [PubMed] [Google Scholar]
- 10.Reiter Y, Ciobotariu A, Fishelson Z. Sublytic complement attack protects tumor-cells from lytic doses of antibody and complement. Eur J Immunol. 1992;22:1207–13. doi: 10.1002/eji.1830220515. [DOI] [PubMed] [Google Scholar]
- 11.Reiter Y, Ciobotariu A, Jones J, Morgan BP, Fishelson Z. Complement membrane attack complex, perforin, and bacterial exotoxins induce K562 cells calcium-dependent cross-protection from lysis. J Immunol. 1995;155:2203–10. [PubMed] [Google Scholar]
- 12.Markiewski MM, DeAngelis RA, Benencia F, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9:1225–35. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Y, Hu XB. C5a stimulates the proliferation of breast cancer cells via Akt-dependent RGC-32 gene activation. Oncol Rep. 2014;32:2817–23. doi: 10.3892/or.2014.3489. [DOI] [PubMed] [Google Scholar]
- 14.Niculescu F, Rus HG, Retegan M, Vlaicu R. Persistent complement activation on tumor cells in breast cancer. Am J Pathol. 1992;140:1039–43. [PMC free article] [PubMed] [Google Scholar]
- 15.Corrales L, Ajona D, Rafail S, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189:4674–83. doi: 10.4049/jimmunol.1201654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lucas SD, Karlsson-Parra A, Nilsson B, et al. Tumor-specific deposition of immunoglobulin G and complement in papillary thyroid carcinoma. Hum Pathol. 1996;27:1329–35. doi: 10.1016/s0046-8177(96)90346-9. [DOI] [PubMed] [Google Scholar]
- 17.Ytting H, Jensenius JC, Christensen IJ, et al. Increased activity of the mannan-binding lectin complement activation pathway in patients with colorectal cancer. Scand J Gastroenterol. 2004;39:674–9. doi: 10.1080/00365520410005603. [DOI] [PubMed] [Google Scholar]
- 18.Gunn L, Ding C, Liu M, et al. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. 2012;189:2985–94. doi: 10.4049/jimmunol.1200846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kempshall E, Thebault S, Morgan BP, et al. Complement-induced protection: an explanation for the limitations of cell-based tumour immunotherapies. Immunol Cell Biol. 2012;90:869–71. doi: 10.1038/icb.2012.30. [DOI] [PubMed] [Google Scholar]
- 20.Hsieh CC, Chou HS, Yang HR, et al. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood. 2013;121:1760–8. doi: 10.1182/blood-2012-06-440214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song X, Krelin Y, Dvorkin T, et al. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol. 2005;175:8200–8. doi: 10.4049/jimmunol.175.12.8200. [DOI] [PubMed] [Google Scholar]
- 22.Bunt SK, Sinha P, Clements VK, et al. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176:284–90. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 23.Obermajer N, Muthuswamy R, Lesnock J, et al. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011;118:5498–505. doi: 10.1182/blood-2011-07-365825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Obermajer N, Muthuswamy R, Odunsi K, et al. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011;71:7463–70. doi: 10.1158/0008-5472.CAN-11-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vogel CW, Fritzinger DC. Cobra venom factor: structure, function, and humanization for therapeutic complement depletion. Toxicon. 2010;56:1198–222. doi: 10.1016/j.toxicon.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 26.Hillmen P, Hall C, Marsh JC, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350:552–9. doi: 10.1056/NEJMoa031688. [DOI] [PubMed] [Google Scholar]
- 27.Magge D, Guo ZS, O’Malley ME, et al. Inhibitors of C5 complement enhance vaccinia virus oncolysis. Cancer Gene Ther. 2013;20:342–50. doi: 10.1038/cgt.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evgin L, Acuna SA, Tanese de Souza C, et al. Complement inhibition prevents oncolytic vaccinia virus neutralization in immune humans and cynomolgus macaques. Mol Ther. 2015;23:1066–76. doi: 10.1038/mt.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.