

Abstract
Since their invention in 1991, peptide nucleic acids (PNAs) have been used in a myriad of chemical and biological assays. More recently, peptide nucleic acids have also been demonstrated to hold great potential as therapeutic agents because of their physiological stability, affinity for target nucleic acids, and versatility. While recent modifications in their design have further improved their potency, their preclinical development has reached new heights due to their combination with recent advancements in drug delivery. This review focuses on recent advances in PNA therapeutic applications, in which chemical modifications are made to improve PNA function and nanoparticles are used to enhance PNA delivery.
Keywords: Peptide Nucleic Acids, nanoparticles, gene editing, gene therapy, antigene, antisense therapy, anti-miR therapy, anti-miRs, oncomiRs, anti-sense
Introduction
Nucleic acids play a critical role in governing various disease states. The expression or overexpression of certain genes can worsen prognosis, as with BCR-ABL in myeloid leukemia (CML) [1] and c-Myc in various cancers [2]. Upregulation of other genetic elements, such as microRNAs (miRNAs) are also seen in several diseases, most notably cancer [3]. Single-base pair mutations in genomic DNA are also the hallmark of several genetic disorders, including cystic fibrosis [4] and sickle cell disease [5]. While small molecule drugs targeting these genetic elements have had some success [6,7], nucleotide-based drugs can take advantage of complementary base pairing, reducing the potential for side effects [8].
To selectively target specific genetic elements, synthetic chemists have developed several oligonucleotide analogs capable of binding to genomic DNA (gDNA), messenger RNA (mRNA), and microRNA (miRNA), and many other non-coding genetic elements [9]. While several of these analogs maintain the specificity of complementary base pairing, many benefit from several structural modifications intended to improve their physiological stability or enhance their binding affinity. For example, 2’fluorine RNA, 2’ methoxide RNA, and O-methyl methoxy RNA are all RNA analogs which benefit from a 2’ ribose modification, making them resistant to endonuclease degradation [9-11]. Similar analogs benefit from direct modification or replacement of the phosphodiester backbone of DNA and RNA. Locked Nucleic Acids (LNAs), for example, contain a “bridge” that connects the 2’ and 4’ carbons on the ribose moiety, which facilitates backbone pre-organization and enhances hybridization with oligonucleotides [12]. Phosphorodiamidate morpholinos (PMOs), on the other hand, contain DNA bases attached to methylenemorpholine rings linked by phosphorodiamidate groups, completely replacing the phosphodiester backbone and rendering resistance to nuclease-mediated degradation [13]. A third analogue, peptide nucleic acids (PNAs), is characterized by a unique peptide backbone, which similarly imparts nuclease resistance [14] (Figure 1). Of these analogs, PNAs have been shown to be especially versatile and have been explored for their utility in various applications including laboratory techniques such as PCR, purification of nucleic acids, southern and northern blotting, and FISH [15-18], as well as numerous potential therapeutic applications. In this review, we focus on PNAs and their therapeutic potential in the realm of genetic regulation.
Figure 1.

Canonical structures of PNA paired with DNA. Adapted from [136].
Principles
To appreciate the recent advancements in using PNAs, it is helpful to understand the fundamental structure and properties of PNAs, as well as the challenges faced in therapeutic applications of these molecules.
Structure and Properties of PNAs
PNAs were first developed in 1991 by Peter E. Nielsen and his colleagues at the University of Copenhagen when they sought to develop a DNA analog with a polyamide backbone that was homomorphous to DNA with respect to the number of backbone bonds and distance between the backbone and nucleobases [19] (Figure 1). The goal was to design a ligand that could recognize double-stranded DNA (dsDNA) via Hoogsteen base pairing in the major groove, thereby avoiding the need for strand invasion. Additionally, they sought to develop a molecule that would be synthetically easy to produce [14]. Using a computer algorithm in combination with advancements in Merrifield solid-phase synthesis, Nielsen et al. arrived at the structure that came to be known as “peptide-nucleic acid” (PNA).
PNAs are composed of N-(2-aminoethyl)-glycine units linked by peptide bonds [8,10,14]. The nucleobases—adenine, guanine, cytosine, and thymine (A,G,C,T)—are attached to this backbone via a methylene carbonyl linkage rather than the traditional sugar moiety such as deoxyribose (in DNA) or ribose (in RNA) (Figure 1). This pseudopeptide backbone, in lieu of the phosphodiester backbone of DNA or RNA, renders PNA molecules charge neutral (compared to negatively charged DNA and RNA molecules), allowing for stronger hybridization with complementary nucleic acids [20,21]. While the PNA affinity for DNA is higher than that of DNA for DNA, PNA hybridization is less tolerant of base pair mismatches [16]. The unnatural, polyamide backbone of PNAs renders them resistant to enzymatic degradation [15,22,23].
As with other synthetic analogs, PNAs bind to complementary DNA or RNA in accordance with both Watson-Crick and Hoogsteen base pairing rules [22] (Figure 2). Under Watson-Crick base pairing, adenine (A) forms two hydrogen bonds with thymine (T), while cytosine (C) forms three hydrogen bonds with guanine (G). Under Hoogsteen base pairing, PNAs can bind duplex DNA along its major groove, accommodating the formation of base triplets. This motif allows thymine to bind to an AT doublet and cytosine to bind to a GC doublet. Through another mechanism, known as reverse Hoogsteen base pairing, guanine is capable of binding to a GC doublet, while adenine is able to bind to an AT doublet [24].
Figure 2.

Binding motifs for triplex-forming oligonucleotides. Triplex-forming oligonucleotides (TFOs) bind in both the parallel and antiparallel direction. In the pyrimidine motif (left), TFOs bind the polypurine DNA strand in the parallel direction. In the purine motif (right), TFOs bind the polypurine DNA strand in the antiparallel direction. Corresponding canonical Watson-Crick and Hoogsteen base pair triplets are shown on below each binding motif. Adapted from [137].
PNAs bind to DNA according to different motifs, which depend on PNA design (Figure 3). A simple PNA/DNA triplex is the result of Hoogsteen base pairing along the major groove of dsDNA, typically observed with cytosine-rich PNAs. Invasion of duplex DNA, on the other hand, can occur when using homopurine PNAs (ones whose sequence are composed entirely of adenine and guanine nucleotides). In binding to one strand of dsDNA by Watson-Crick base-pairing, duplex invasion further results in a displaced D-loop of the complementary strand [25]. Duplex invasion can also occur by triplex formation, where a PNA molecule binds to a strand of dsDNA by both Watson-Crick and Hoogsteen base-pairing, as is observed for homopyridine PNAs (ones whose sequence are composed entirely of cytosine and thymine nucleotides). This 2PNA:1 DNA triplex also results in a locally displaced D-loop [26]. “Tail-clamp” PNAs (tcPNAs)—PNAs joined by a chemical linker—make use of this biding motif, linking homopyridine sequences while extending the Watson-Crick portion to invade mixed homopurine/homopyridine DNA [27]. Lastly, pseudocomplementary PNAs (pcPNAs), which are complementary to each other, but unable to form stable duplexes, can invade dsDNA using Watson-Crick base pairing, resulting in a double-duplex invasion [26].
Figure 3.

Canonical binding motifs of PNAs with double-stranded DNA. Adapted from [25].
Given their stability, charge neutrality, and ease of design, PNAs can be used for a variety of applications. Their versatility and ability to bind to target DNA or RNA by both Watson-Crick and Hoogsteen base pairing rules further enhances their therapeutic potential. However, before considering how PNAs may be used, it is important to consider their limitations.
Limitations
While several therapeutic applications for PNAs exist, as demonstrated by numerous in vitro and in vivo studies (see below), several fundamental challenges still must be considered before these molecules become broadly applicable in a clinical setting.
Solubility
Unlike DNA or RNA, which exist primarily in elongated conformations due to their negatively charged phosphate backbones, the original class of PNAs that were developed were found to have a tendency toward folding into complex globular structures when in solution, presumably due to both the intra- and intermolecular affinity between their hydrophobic nucleobases [28]. Although PNAs in this globular structure have been shown to be effective in various applications, their hydrophobicity and tendency to aggregate confers several limitations. For one, their hydrophobicity, which promotes PNA aggregation, also seems to promote their relatively nonspecific adherence to both other macromolecules and larger surfaces. This adherence often creates technical challenges in the handling of PNAs in complex applications, such as microarrays, or even in simple laboratory manipulations such as transferring between tubes [29].
To combat these tendencies, researchers have attempted to modify PNAs through several means: the addition of amino acid residues at their termini [30], conjugation to charged DNA molecules [31], and conjugation to high molecular weight polymers such as polyethylene glycol (PEG) [32]. While these modifications have overcome solubility issues, installation of hydrophilic (R)-diethylene glycol “miniPEG” chains at the γ-backbone position, was found to improve PNA solubility, and to enhance PNA hybridization and sequence selectivity via induction of helical preorganization (to be further discussed later in the review) [29,33-35].
Intracellular Delivery
While PNAs hold tremendous therapeutic potential, they have been primarily studied using either cell-free systems or with artificial techniques for penetrating the cell membrane, such as electroporation, nucleofection, and microinjection [36]. Though useful for studying PNAs in an experimental setting, these techniques are not readily translatable to the clinic, and thus the challenge of achieving sufficient intracellular delivery of PNAs must still be solved. Without crossing the cellular membrane, PNAs are unable to reach their targeted sequence and thus can neither act on microRNAs or mRNAs in the cytosol nor hybridize with DNA targets in the nucleus. Thus, the ability of PNAs to cross the cell membrane either on their own or by using clinically-applicable techniques has been of great interest to researchers.
PNAs, though charge neutral, are not easily taken up by cells. Using liposomes as a model for cellular membranes, researchers showed that two different PNAs, approximately 10 base pairs in length, had efflux half-times of 5.5 and 11 days, comparable to a molecule of DNA with an efflux half-time of 7 days. With such lengthy transport times, it is unlikely that PNAs would be able to traverse the cellular membrane alone to act on target DNA or RNA [37].
To overcome these limitations in delivery, researchers have explored cell-penetrating peptides (CPPs) to enhance the cellular uptake of PNAs [38-40]. CPPs are short, cationic peptides that vary in length, typically ranging from nine to 30 amino acids [41]. Several CPPs have been investigated for the delivery of PNAs, most notably penetratin, a 16-residue peptide derived from the Drosophila Antennapedia gene [41]. Using PNAs targeted against human telomerase, Corey and his group demonstrated in vitro that PNA-penetratin conjugates could efficiently hybridize with target DNA sequences, showing that the attached peptide did not interfere with binding [42]. Nielsen and his group later demonstrated that penetratin conjugation enhanced PNA uptake in vitro, but that cellular uptake of the conjugates was variable and dependent on cell type, temperature, and concentration [43].
Our lab verified that conjugation of PNA to penetratin significantly enhances PNA cellular uptake in vitro, when compared to delivery by electroporation. Importantly, we found that intraperitoneal administration of PNA-penetratin conjugates resulted in enhanced biodistribution and targeted gene modification compared to PNA alone in several somatic tissues and in several compartments of the hematopoietic system [44] (the therapeutic implications of this finding are discussed below). Other studies have also shown promising in vitro and in vivo delivery using PNAs modified with lysine residues [45] as well as with glycosylated side chains [46].
While CPP-mediated PNA delivery remains a promising approach for in vitro and in vivo applications, the need for excessively high and repeated dosing to achieve therapeutic effects (typically 10 to 50 mg/kg administered several times) renders it impractical for potential translation to the clinic. Considering their large size relative to their PNA cargo, as well as their relatively inefficient conjugation, the need for extensive CPP synthesis presents an additional barrier to their broader use [47]. Limited endosomal escape of CPP-PNA conjugates following their cellular uptake via endocytosis—which often leads to a requirement of co-administration with chloroquine or Ca2+ to facilitate release into the cytosol—also limits the therapeutic efficiency of CPP-PNAs [48]. In considering the application of PNAs for gene editing (discussed later), the need for co-delivery with a donor DNA would require additional conjugation and purification steps to create separate CPP-DNA constructs, further complicating their use.
Physiological Stability and Clearance
As the applications of PNAs have progressed from simple in vitro work to the more complex in vivo systems, researchers have sought to better understand their in vivo stability and physiological clearance.
PNAs have significant stability in human serum and eukaryotic cellular extracts. One study incubated both a homothymidine PNA and an unmodified oligonucleotide in fresh guinea pig serum and demonstrated that the unmodified oligonucleotides had a half-life of only a few minutes, whereas the PNA was still intact after two days [49]. In a separate study, PNAs incubated with mouse tumor cell extracts showed no significant degradation after two hours [23]. This stability is a consequence of the PNAs unnatural backbone, which makes them resistant to enzymatic degradation [23,31]. Unlike their DNA counterparts which depurinate in the presence of strong acids, PNAs are also stable in acidic environments such as tumor microenvironments, making them ideal candidates for genetically-targeted therapies in vivo [36].
Despite their enhanced stability, like other oligonucleotides, PNAs are rapidly cleared from systemic circulation after administration. Following intravenous injection in rats, the circulation half-life of a 12-mer PNA was found to be approximately 3 minutes [50]. Although detectable amounts were observed in the spleen, liver, heart, brain, and kidney at 24 hours, the dose recovered ranged from 0.042 percent to 2.47 percent, with over 90 percent detected in urine [50]. Other studies produced similar results, demonstrating that unmodified PNAs are cleared from the bloodstream within 10 to 30 minutes after intravenous or intraperitoneal administration [51]. With such poor cellular permeability and rapid clearance, it should be no surprise that progress in the therapeutic application of these molecules has been slow.
The attempts to improve cellular uptake by the conjugation of CPPs to PNAs have also demonstrated that the addition of CPPs to PNAs can reduce the physiological clearance rates of these molecules [44]. This result is most likely due to the combined effect of CPPs slowing the inherent physiological clearance of PNAs as well as promoting the cellular internalization of PNAs—making them less available for physiological clearance. However, the previously described limitations on this approach make it an unlikely strategy for increasing physiological retention of PNAs in the clinical setting. Thus, alternative methods—such as delivery via nanocarriers, which is discussed later in this review—must be explored and optimized before PNAs can be robustly used in the in vivo setting.
Applications
While the general challenges outlined in the previous section apply to many potential applications of PNAs, it is important to consider that each specific application will inherently hold an additional set of new challenges. While there are numerous uses of PNAs, here we will focus on their therapeutic applications in the realm of genetic regulation, specifically serving as antigene, pro-gene, antisense, anti-miR, and gene-editing molecules.
Antigene
With their propensity to bind dsDNA [52] and enhanced stability over traditional oligonucleotides, PNAs make the ideal antigene agent (agPNA)—a molecule capable of inhibiting transcription and/or replication of a targeted gene [49]. In cell-free experiments, PNAs targeted to the template strand of dsDNA halted transcriptional elongation when using both prokaryotic and eukaryotic RNA polymerases [53,54]. Other cell-free experiments have shown that PNAs targeted to regions of dsDNA flanked by restrictions enzyme sites blocked restriction enzyme cleavage, demonstrating that PNAs can be used to block DNA recognizing proteins. These experiments highlight the specificity of the PNA sequence to the target site by showing reduced blocking if the PNA had a single mismatch, and no blocking if the PNA had two mismatches [55]. The potential of this antigene activity of PNAs has since been confirmed in many cell culture experiments using PNAs: as examples, PNAs have been shown to inhibit interactions between NFκ-B and the IL2-Rα NFκ-B binding site [56] and PNAs designed to bind to the second exon of the c-Myc gene that were shown to downregulate c-Myc expression in a cell-line model of Burkitt’s lymphoma [57]. More recently, PNAs targeting the transcription start sites of the human progesterone receptors B (hPR-B) and A (hPR-A) were found to inhibit transcription, resulting in distinct morphological changes in breast cancer cells. Notably, phenotypic change was confirmed by use of siRNA, highlighting the potential for combinatorial antigene/antisense therapeutic approaches [58]. Targeting of transcription sites for antigene application may hold additional promise, as subsequent studies have demonstrated that transcriptional activity catalyzes PNA strand invasion [59].
Beyond the suppression of endogenous gene expression, the use of PNAs as inhibitors of viral DNA replication has also been tested. In one such study, researchers were able to suppress HIV-1 replication by using PNAs to target poly(purine) tracts and a 4 base (TTTT) 5’ flanking sequence found in HIV-1 proviral DNA. These PNA molecules suppressed viral replication, and this suppression persisted for 14 days after the PNAs were removed from the culture medium. This finding was in sharp contrast to inhibition by U75875—a previously used gold standard inhibitor of HIV-1 protease—which showed no suppression of HIV replication once it was removed from the medium [60].
Pro-gene
Although many studies have focused on the use of PNAs as suppressors of gene transcription, some studies have shown that PNAs can be used to activate genes when they are targeted to promoter regions. Mollegaard et al., for example, demonstrated that when homopyrimidine PNAs form triplexes with dsDNA, the resulting D-loop can serve as a site for RNA polymerase recognition and thus a local transcription initiation site [61].
Using a similar approach, our lab explored reactivation of fetal hemoglobin as an ameliorative therapy for hemoglobinopathies (i.e., sickle cell and β-thalassemia). Using PNAs designed to bind to a mixed homopurine/homopyridine region of the γ-globin gene, we induced expression of a reporter gene construct both in vitro and in vivo. Importantly, we were able to use this same strategy to achieve γ-globin gene expression in K562 human erythroleukemia cells [62], demonstrating that PNAs 16 to 18mer in length were ideal for inducing gene transcription [63].
More recently, others have used PNAs conjugated to CPPs on one end (such as TAT) and transcription activation domains on the other to reactivate human fetal γ-globin in primary human CD34+ cells, providing further proof that PNAs may indeed play a role in not just the suppression of genes, but in inducing expression of therapeutically significant ones as well [64].
Antisense
Genetic expression can be regulated using antisense agents, such as antisense oligonucleotides, which target mRNA. While most antisense molecules mediate their antisense activity through RNAse H degradation of the mRNA/oligonucleotide hybrid, studies have shown that PNAs targeting mRNA mediate their effects through steric interference [65,66]. Rather than degrading the target mRNA, mRNA bound to PNA is unable to reach cellular translational machinery, thereby inhibiting protein expression.
Although theoretically useful for targeting mRNA, experiments have shown that PNAs are most effective when targeting the start codon in a cell-free assay. PNAs targeted to the AUG initiation region of mRNA were found to be potent inhibitors of mRNA translation, with little to no effect when targeting the coding regions [66]. Similarly PNAs targeting the start codon of the promyelocytic leukemia/retinoic acid receptor-a (PML/RAR) fusion gene achieved an 80 percent inhibition of translation, while 40 times more antisense oligonucleotide control was required to achieve similar levels of inhibition [67].
While most studies have demonstrated that PNAs exhibit significant antisense activity, it is also clear that their potency is dependent on target location and the lifetime of the PNA/mRNA duplex, as mRNA targeted by PNA is not degraded by RNAse H [68]. In comparing PNAs to C-5 propynyl pyrimidine phosphorthioate oligonucleotides (propyne-S-ON) designed to target the 5’ untranslated region of T Ag mRNA, propyne-S-ON exhibited superior antisense effects at lower doses and at longer time points. When targeted to intronic as well as exonic portions of the same mRNA, however, PNAs performed comparably well [68].
Taken together, the results above highlight the role PNAs may play in antisense applications. While further work regarding their precise mechanism of action and how mRNA target regions may influence their antisense activity, their robust stability and strong binding affinity makes these molecules suitable candidates for antisense applications.
Anti-miR
MicroRNAs (miRs) are short (~22 nt long), evolutionarily conserved, single-stranded non-coding RNA molecules that bind to target mRNA, preventing translation by one of two specific mechanisms [69]. miR is first cleaved from a primary miR molecule, which is then incorporated into the RNA-induced silencing complex (RISC). The mature miR then functions as a guide strand, targeting the RISC complex to the appropriate mRNA sequence. Based on the level of complementarity between the miR and mRNA, the mRNA will be subsequently degraded or translationally suppressed [70]. Though much remains to be learned about miRs and their function, it is clear that they play an important regulatory role in many biological processes critical to embryological development and cellular differentiation [3,70]. Dysregulation of miR expression has also been implicated in several disease states, including cardiovascular disease, hepatitis, and cancer [3,69,71]. Given the broad scope and diversity of their roles, miRs have become therapeutic targets of particular interest in a wide range of fields.
Among the efforts to explore methods to target miRs has been the development of anti-miR PNAs. Much like mRNA targeting, anti-miR PNAs appear to work by steric inhibition, in which a PNA molecule binds to its target miR, preventing it from interacting with the RISC complex [72]. The first report of targeting miR using PNAs was by Fabani and Gai, who synthesized PNA molecules against miR-122 in human hepatocellular carcinoma cells as well as primary rat hepatocytes [38]. Similarly, others have shown that PNA-peptide conjugates are able to inhibit miR-16 as well as miR-21, which are aberrantly expressed in certain cancers [73,74]. Interestingly, when compared to LNAs and 2’-OMe oligonucleotides, PNAs were shown to be more potent inhibitors of the targeted miRNA activity [75]. Recently, a PNA-based strategy was developed to target miR-210, which is upregulated under hypoxic conditions [76]. Using both unmodified PNAs and PNAs conjugated to various cell-penetrating peptides, Fabbri et al. found that they were able to decrease miR-210 expression and modulate mRNA expression of miR-210 target genes. Recently, our group has shown that miniPEG-γ modified PNAs are more potent anti-miRs than regular, unmodified PNAs against miR-210 in vivo [77]. We have also now developed a second PNA-based strategy—using peptides that target the PNA to tumors—against miR-155 in an oncomiR-addicted mouse model of lymphoma [78].
Although the literature is still limited, a few other studies have also demonstrated the potential of PNAs as anti-miR therapeutics [38,79-83]. While potency does not appear to be as challenging of an issue in targeting miRNA as it is in targeting mRNA, achieving sufficient intracellular delivery of unmodified PNAs has proven to be problematic [15,38,75]. To overcome these limitations, researchers have attached PNA to lysine residues [38], oligoarginine [79], or cell-penetrating peptides [75] to facilitate intracellular delivery. As discussed earlier, while these delivery methods are effective in vitro, they are difficult to translate to the in vivo setting, particularly when they are administered systemically due to rapid physiological clearance and nonspecific accumulation of PNAs.
Gene Editing
Triplex-forming PNAs are capable of introducing permanent genetic modifications into genomic DNA. When a homopyridine PNA interacts with a homopurine region of DNA, it may bind according to both Watson-Crick and Hoogsteen base pairing rules. To accomplish this process efficiently, homopyrimidine PNAs are joined by a chemical linker, forming a bis-PNA. In this design, one strand of the bis-PNA is designed to bind by a Hoogsteen motif, incorporating a pseudoisocytosine (J) rather than a cytosine (C) to prevent any self-complementarity. Through this modification, one strand is able to bind in the anti-parallel direction by Watson-Crick base pairing, while the other strand is available to bind by Hoogsteen base pairing in the parallel orientation relative to the DNA strand. The resulting triplex leads to DNA helical distortion, which has the capacity to activate DNA repair mechanisms and can result in the recombination of “donor” DNA into the native DNA at a site close to the PNA binding site [84] (Figure 4).
Figure 4.
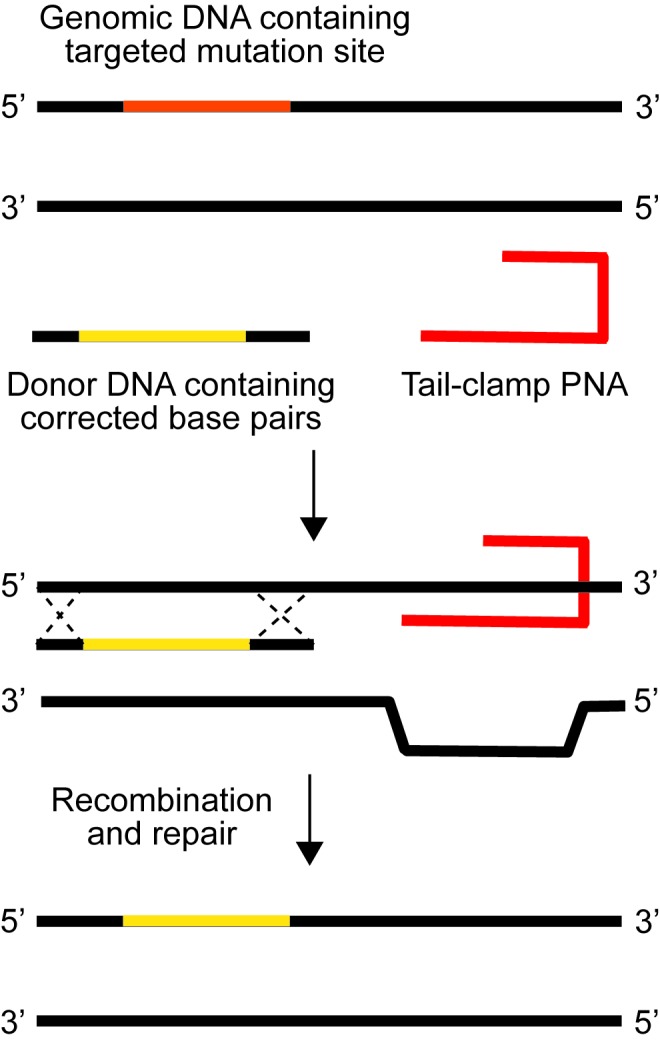
Gene editing by triplex-forming oligonucleotides. PNAs stimulate recombination of short (60 bp) DNA fragments into genomic DNA. Binding of the PNA subsequently produces a structural change within the dsDNA that activates cellular repair mechanisms, which are initiated by nucleotide excision repair. Adapted from [102].
Initial studies from our lab using a supFG1 reporter gene in mouse cells demonstrated that bis-PNAs can induce site-specific mutagenesis at or adjacent to the PNA recognition site [85]. Subsequently, using bis-PNAs conjugated to a short donor DNA fragment, our lab demonstrated that PNA-DNA hybrids were capable of directing site-specific recombination [86]. In this strategy, the PNA molecule served two functions: first, to form a triplex with targeted DNA and second, to position the donor DNA fragment at the appropriate location for recombination. Using a mutated pSupFG1 plasmid, Rogers et al. demonstrated that in human cell-free extracts, they could mediate site-specific sequence changes within the reporter gene at levels higher than those achieved by other triplex-forming oligonucleotide (TFO)-DNA counterparts. They also showed that the induction of gene editing by bis-PNAs depends on recognition of the PNA/DNA/PNA structure by the nucleotide excision repair factor, XPA, to initiate repair of the altered helix “lesion” and thereby catalyze recombination with the donor DNA. Interestingly, we discovered that when the bis-PNA and the donor DNA were mixed, rather than covalently coupled together, the level of recombination was relatively higher [86].
In addition to correcting disease-causing mutations, our lab has also begun to use the gene-editing properties of PNAs to introduce stop codons to prevent HIV-1 infection. Patients with a naturally occurring 32 bp deletion in the CCR5 gene (CCR5-delta32) produce truncated CCR5 proteins, preventing R5 HIV-1 tropic infection. Using this genotype—and the resulting phenotype—as motivation, Schleifman et al. delivered PNAs targeted to the CCR5 gene with a donor DNA fragment designed to introduce a stop codon in human CD34+ hematopoietic stem cells. Using this strategy, Schleifman et al. were able to achieve gene editing (effectively measured as knockout) in 2.46 percent of treated cells. Following subsequent engraftment of these edited cells into mice, it was shown that the gene modification persisted in vivo for longer than four months post-engraftment and led to decreased viral load [87].
While PNAs have proven effective as gene editing agents, important challenges remain with their design, particularly in understanding where to target them with respect to a mutation site of interest. Previous studies from our lab have shown that targeted genome modification can be achieved using bisPNAs designed to bind 100 to 200 bps away from a mutation site of interest [88]. Studies using miniPEG PNAs found that these PNAs, when targeted closest to the mutation site, were more effective at inducing targeted gene correction than those that were targeted further away from the mutation site, but still demonstrated the ability of PNAs to induce gene editing over a distance [89]. These results suggest that repair of PNAs likely involves induction of strand breaks that are subsequently processed by end resection to expose long single-stranded regions to which the donor DNAs can anneal. Work is underway to further elucidate the mechanism by which PNAs confer gene editing as well as how to best design them.
While these initial results for the application of PNAs for gene editing are promising, it is also clear that much work still remains to be done. Until we can gain a clearer understanding of the underlying mechanisms by which PNAs induce gene editing, and the important factors that influence these mechanisms, it seems that for now, optimization of applications in gene editing will require an iterative approach to the design of PNAs to determine sequences and sequence locations that yield the highest efficiency of targeted gene editing.
Opportunities for Modifications
While challenges in PNA delivery, clearance, and efficacious application have slowed progress toward the clinical translation of PNA applications to therapeutic problems, they have also created opportunities for innovation. Efforts to overcome these challenges include direct modification of PNAs themselves, as well as the use of PNA delivery vehicles such as nanocarriers.
PNA Modifications
One approach to overcoming some of the challenges described above, as well as to improving the efficacy of the application of PNAs, has been to directly modify PNA molecules while still attempting to preserve their function. With each modification, its effect on the PNA’s physical, chemical, and physiological properties, as well as on the PNA’s biological function and mechanism of function must be considered.
As discussed above, one of the challenges of working with PNAs is their limited solubility and poor cellular uptake. To overcome these challenges, PNAs have been modified directly with lysine residues [90] and guanidinium functional groups [91], in addition to several other backbone modifications, reviewed elsewhere [92]. PNAs modified with lysine residues have demonstrated superior hybridization, in addition to significantly improved water solubility [90]. The addition of guanidinium functional groups has enhanced their solubility, and significantly enhanced their cellular uptake, while improving the PNAs discrimination for base pair mismatches in target sequences [91].
PNAs have also been modified with several non-standard nucleobases. In particular, pseudoisocytosine, which mimics an N3-protonated cytosine, has been used for pH-independent recognition of DNA-guanine during Hoogsteen binding of triplex forming bis-PNAs [16]. Diaminopurine, in lieu of adenine, has also been explored, as it increases the melting temperature (Tm) of a PNA-DNA duplex by 4°C for each additional substitution [16]. Several conformationally constrained PNAs have also been investigated to improve invasion kinetics and reduce the ambiguity in the direction of binding with DNA (antiparallel vs. parallel) [93-96]. Using PNAs modified with D-Lysine, for instance, Sforza et al. demonstrated that pre-helical organization of PNAs could enhance specificity and direction of binding in the antiparallel direction [94].
Among the many PNA modifications explored thus far, none have enhanced the therapeutic potential of PNAs as drastically as has the addition of diethylene glycol—“miniPEG”—chains at the PNA’s gamma position (Figure 5). Initially, the lab of Dr. Danith Ly of Carnegie Mellon demonstrated that simple L-serine side-chain incorporation into the γ-position of the PNA backbone was capable of preorganizing PNAs into a right-handed helix, increasing its affinity and specificity for DNA and RNA [35]. By extending the length of these γ-PNA oligomers, the group went on to show that these molecules were capable of invading the most commonly found DNA double helical form—B-DNA—thus removing the need for DNA intercalators (acrinide) as had been previously required [97,98]. While placement of a methyl group at the gamma position had previously proven useful in preorganizing PNAs into a right-handed helical structure, they were still poorly soluble and tended to aggregate. To overcome this limitation, the group decided to incorporate miniPEG into the γ-position, which was found to maintain pre-helical organization while improving PNA solubility [29]. Importantly, by comparing unmodified PNAs to ones with miniPEG or methyl groups at the gamma position, Bahal et al. demonstrated that these gamma-miniPEG-PNAs (γMP-PNAs) were capable of invading mixed sequence double helical B-DNA through Watson-Crick base pairing [34].
Figure 5.

Canonical structures of DNA, PNA, and gamma modified miniPEG PNA monomer units.
Although these initial results using γMP-PNAs were promising, these molecules were designed to bind solely through Watson-Crick base pairing rules, resulting in a duplex binding motif (Figure 3). While capable of inducing gene editing, we had previously shown that tail clamp PNAs (tcPNA), which bind by a combined triplex/duplex motif, were much more efficient at invading dsDNA and did so with greater specificity [99]. In an effort to extend the work using γMP-PNAs, Bahal et al. synthesized γMP-tcPNAs and found that in comparison to regular tcPNAs, the corresponding γMP-tcPNA induced nearly 2-fold higher genetic correction in cells treated ex vivo. In CD117+ cells, correction was even higher (~3-fold) following ex vivo treatment [89,100].
These γMP-tcPNAs were then investigated in the in vivo setting, by loading them into degradable polymer nanoparticles and injecting them intravenously into mice with human β-thalassemia. The systemic administration of γMP-tcPNAs was found to confer significantly improved RBC morphology, increased hemoglobin levels into the wild-type range, reduced reticulocyte counts, and a significant reduction in splenomegaly and restoration of normal splenic architecture. Deep sequencing of total bone marrow cells further demonstrated a target gene correction of ~4 percent [100].
While these modifications have certainly enhanced the biophysical properties and applicability of PNAs, issues surrounding their delivery in vitro and in vivo first had to be overcome before their therapeutic applications could be studied.
PNA Delivery Systems
One of the most fundamental challenges in moving PNAs into the clinic—the safe and efficient delivery of PNAs to target sites—was eloquently stated by Peter E. Nielsen, the inventor of PNAs, in 2003. He said: “widespread use of PNA for inhibition of translation still awaits better transfection methodology. We believe enhanced and controlled endosomal release of PNAs taken up by endocytosis to be the most likely solution” [101]. While the above-described results with advancements in PNAs such as the γtcMP-PNAs were promising, these results could not have been achieved without the parallel development of PNA delivery systems that were used to complete these experiments.
In an effort to address the challenge articulated by Nielsen, several delivery systems for PNAs have been explored including cationic liposomes, avidin-labeled protein NPs, zeolite-L-nanocrystals, mesoporous silica NPs, porous silicon NPs, and cationic shell-cross-linked knedels-like NPs (reviewed elsewhere [102]). However, each of these materials and delivery systems is limited by their translatability to humans, as none of these materials has been extensively explored in the clinical setting. Therefore, we have focused on the use of polymeric NPs as PNA delivery systems [103-112]. Importantly, the base materials for these nanoparticles is comprised of poly(lactic-co-glycolic acid) (PLGA), a biocompatible and degradable polymer. Currently, several PLGA-based products have been approved by the FDA for the treatment of numerous disorders including acromegaly (Signifor LAR), schizophrenia (Risperdal Consta), and prostate cancer (Lupron, Trelstar, and Elligard) [113]. Several studies have shown that PLGA NPs are readily tolerated at extremely high doses, both in vitro and in vivo [114,115]. Numerous studies, including several from our lab, have also shown that PLGA NPs can be used to deliver siRNA, plasmid DNA, as well as several other small molecule drugs [103-112].
Using a double emulsion solvent evaporation technique, we are able to encapsulate both PNA and donor DNA (Figure 6). This advantage—the ability to co-deliver molecules—presents a unique opportunity for gene editing by PNAs, which requires the co-delivery of a donor DNA. In addition to delivering the donor DNA, PLGA NPs have been shown to enhance DNA stability, providing protection against enzymatic degradation [116].
Figure 6.

Formulation of PNA/DNA Nanoparticles. PNA and donor DNA are formulated into poly(lactic-co-glycolic acid) nanoparticles using a double emulsion solvent evaporation technique. Once formulated, nanoparticles are collected by centrifugation and dried by lyophilization. These dry nanoparticle powders can be stored and resuspended for later use.
By co-encapsulating PNA and donor DNA into PLGA NPs, we were able to demonstrate, for the first time, that PLGA NPs could be used for the delivery of PNA and DNA to mediate recombination in human CD34+ cells, which resulted in site-specific modification of the β-globin locus [117]. Importantly, there was no change in cell viability after NP treatment, whereas nucleofection resulted in a 60-fold reduction in cell viability. While this study was limited to cultured cells, it provided the first piece of evidence that PLGA NPs could be used for delivery of PNA and donor DNA for gene editing [117].
The advancements in γMP-PNAs, which had previously demonstrated enhanced hybridization, solubility, and sequence-unrestricted binding to double-stranded DNA [34], still presented a challenge to effective intracellular uptake and in vivo delivery. Using a transgenic mouse model carrying a β-globin/eGFP reporter gene, Bahal et al., sought to investigate whether single-stranded γMP-PNAs could be used in vivo to target a splice site in this reporter gene. It was only by using a PLGA NP delivery system that Bahal et al. showed for the first time that γMP-PNAs could be delivered and induce gene modification both ex vivo and in vivo [89].
Since these initial studies, we have similarly shown that our approach using PNA and DNA delivered by PLGA NPs could be also be used to insert an in-frame stop codon, such as for the disruption of the CCR5 gene required for HIV-1 entry into T cells [118]. Human peripheral blood mononuclear cells (PBMCs) treated with these NPs were engrafted into immunodeficient NOD-scid IL-2rγnull mice. After challenging these mice with an R5-tropic strain of HIV-1, mice with treated PBMCs displayed significantly higher levels of CD4+ T cells and reduced plasma viral RNA loads compared with control mice [99].
While we have primarily focused on a relatively simple PLGA NP system for the delivery of PNAs, there have been more recent advancements in polymeric NPs and there remains a huge potential for the exploitation of the NPs functions and capabilities to further enhance their application to the delivery of PNAs. More recent studies in our lab have shown that other FDA-approved materials such as poly(lactic acid) (PLA) can be used to make radiosensitizing nanoparticles and to enhance the local delivery of chemotherapeutics [108,119]. The use of these other polymers has been shown to offer certain advantages such as the capacity to alter cargo release patterns, and/or confer preferential uptake by particular cell types [120]. These results suggest that the use of other FDA-approved materials such as PLA can also potentially be applicable to further optimization of polymeric NP-based PNA delivery systems.
Downstream Directions
While the initial work with PNAs highlighted significant challenges inherent to these molecules such as their limited solubility and hybridization kinetics, PNA modifications—most notably the addition of miniPEG chains to the gamma position of PNAs (γMP-PNAs)—have enabled us to overcome many of the technical challenges previously faced with regular PNAs. The increased hybridization and kinetics of binding with γMP-PNAs make these molecules even more potent, further expanding their therapeutic potential. While PNAs will undoubtedly continue to benefit from chemical modifications, future efforts will likely focus on expanding their roles to other applications, such as antimicrobials [121], or as regulators of previously untargeted genetic elements like long non-coding RNAs (lncRNA) [122]. With recent advancements in delivery technologies, a whole host of previously modified PNAs can now be explored in a therapeutic context such as pcPNAs, PNAs with nucleobase substitutions such as G-clamp in place of cytosine [25,33] and diaminopurine in place of adenine to improve hybridization [16], in addition to other backbone modifications [123].
As compared to other gene modification techniques such as the CRISPR/Cas-9 system, the in vivo delivery of PNA and PNA/DNA has been readily successful, taking advantage of delivery systems initially developed for other oligonucleotides. While CPP-mediated delivery proved invaluable to the initial in vitro and in vivo delivery of these molecules, complicated conjugation strategies, non-specific uptake, and high dosing requirements limited their applicability. Using PLGA NPs, however, we have significantly simplified their delivery, providing a method for delivery of PNA alone [77,124] or PNA co-encapsulated with DNA [125]. Still, to achieve true clinical potential, we must continue to refine our delivery methods and enhance the localization of PNAs to target cells. In recent work, we have begun to investigate the use of new polymer blends for enhanced delivery of PNA/DNA into the lungs of mice [126,127]. As compared to PLGA, we have shown that blends of PLGA and poly(β-amino esters)—a biodegradable cationic polymer with tertiary amines in its backbone—are better able to deliver tcPNA and DNA to the lungs of mice in β-globin/eGFP reporter mice [128] and in mice with cystic fibrosis [127]. In both cases, editing in the lung was enhanced by the addition of CPPs to the surface of the NPs, combining the benefits of CPP-mediated delivery with the advantages of polymeric NPs.
In addition to NP-mediated delivery, we have also begun exploring another delivery strategy using pH low insertion peptides (pHLIP), a peptide which preferentially accumulates in acidic microenvironments [129]. Under normal pH, pHLIP has an affinity for the cell membrane. As the pH is reduced, residues on pHLIP become protonated, enhancing its affinity for the cell membrane and resulting in formation of a transmembrane α-helix [130]. As the peptide flips across the cell-membrane, its N-terminus remains in the extracellular space, while its C-terminus is inserted into the cytosol [130,131]. Using this peptide, we showed that PNAs can be selectively delivered to a tumor’s acidic microenvironment. In this strategy, a PNA directed against miR-155 was delivered to a mouse model of miR-155 addicted lymphoma. To ensure that the anti-miR PNA could bind to its target oncomiR, anti-miR PNA was conjugated to the c-terminus of pHLIP by a disulfide bond which, though stable in the acidic microenvironment, would be reduced once inside the cell. With this strategy, pHLIP-anti miR-155 treatment silenced miR-155 and significantly delayed tumor growth in vivo [78].
The field of genome modification has seen a recent explosion in approaches including zinc finger nucleases, TALENS, and CRISPR. Although these competing technologies, particularly CRISPR, have attracted significant attention, their therapeutic applicability is limited by challenges with delivery [132,133] and concerns over off-target effects [134]. Clinical use of CRISPR will require systemic administration of Cas9 protein, mRNA, or DNA, which present additional challenges in delivery [134]. Although lipid NPs have been recently used to effectively deliver Cas9 mRNA, adeno-associated viruses encoding the guide strand and a donor DNA were also required for gene editing, further complicating delivery [135]. The use of viral vectors also raises safety concerns over oncogenicity, as well as immunogenicity. Although CRISPR has been tremendously effective in vitro and ex vivo, it has not had as much success in vivo. In fact, in a recent study using CRISPR to target liver disease in vivo, there was only editing in ~6 percent of hepatocytes, comparable to our efficiencies using PNAs [135].
While the work reviewed here demonstrates the promise of PNA for the future of gene editing, much work remains to be done before these technologies can be safely translated to human therapeutic applications. We believe that—of the several potential gene editing technologies currently being explored—PNAs offer a safe and clinically translatable approach, with perhaps the greatest potential to affect therapeutic outcomes after simple systemic administration. However, we still have a long way to go before we can turn this potential into a reality. As hinted at by our collaborative work joining nucleic acid chemistry with expertise in nanoparticle delivery systems, successful clinical translation of this technology will most likely continue to rely on the work of interdisciplinary teams willing to collaborate and take risks.
Acknowledgments
This work was supported by the NIGMS Medical Scientist Training Program T32GM07205 (to E.Q.), National Institute of Health grants R01HL125892 and R01AI112443 (to W.M.S. and P.M.G), and institutional training grant 5T32GM007223-43 (to E.Q.).
Glossary
- PNAs
Peptide Nucleic Acids
- tcPNA
Tail-clamp PNAs
- pcPNAs
Pseudocomplementary PNAs
- γMP-PNAs
Gamma MiniPEG Peptide Nucleic Acids
- CPPs
Cell-penetrating peptide
- PLGA
Poly(lactic-co-glycolic acid)
- NPs
Nanoparticles
- PEG
Polyethylene glycol
References
- Gadzicki D, von Neuhoff N, Steinemann D, Just M, Busche G, Kreipe H, et al. BCR-ABL gene amplification and overexpression in a patient with chronic myeloid leukemia treated with imatinib. Cancer Genet Cytogenet. 2005;159(2):164–7. [DOI] [PubMed] [Google Scholar]
- Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203–22. [DOI] [PubMed] [Google Scholar]
- Brown SD, White R, Tobin P. Keep them breathing: cystic fibrosis pathophysiology, diagnosis, and treatment. JAAPA. 2017;30(5):23–7. [DOI] [PubMed] [Google Scholar]
- Hoban MD, Orkin SH, Bauer DE. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood. 2016;127(7):839–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesfeld JM, Neely L, Trauger JW, Baird EE, Dervan PB. Regulation of gene expression by small molecules. Nature. 1997;387(6629):202–5. [DOI] [PubMed] [Google Scholar]
- Gallego J, Varani G. Targeting RNA with small-molecule drugs: therapeutic promise and chemical challenges. Acc Chem Res. 2001;34(10):836–43. [DOI] [PubMed] [Google Scholar]
- Nielsen PE, Appella DH. Peptide Nucleic Acids Methods and Protocols. Methods Mol Biol. 2014;1050:V-V. [Google Scholar]
- Joyce GF. Evolution. Toward an alternative biology. Science. 2012;336(6079):307–8. [DOI] [PubMed] [Google Scholar]
- Chaput JC, Yu H, Zhang S. The emerging world of synthetic genetics. Chem Biol. 2012;19(11):1360–71. [DOI] [PubMed] [Google Scholar]
- Taskova M, Mantsiou A, Astakhova K. Synthetic Nucleic Acid Analogues in Gene Therapy: An Update for Peptide-Oligonucleotide Conjugates. ChemBioChem. 2017 [DOI] [PubMed] [Google Scholar]
- Koshkin AA, Singh SK, Nielsen P, Rajwanshi VK, Kumar R, Meldgaard M, et al. LNA (Locked Nucleic Acids): synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. 1998;54(14):3607–30. [Google Scholar]
- Summerton J, Weller D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic A. 1997;7(3):187–95. [DOI] [PubMed] [Google Scholar]
- Nielsen PE, Egholm M, Buchardt O. Peptide nucleic acid (PNA). A DNA mimic with a peptide backbone. Bioconjug Chem. 1994;5(1):3–7. [DOI] [PubMed] [Google Scholar]
- Pellestor F, Paulasova P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur J Hum Genet. 2004;12(9):694–700. [DOI] [PubMed] [Google Scholar]
- Nielsen PE, Egholm M. An introduction to peptide nucleic acid. Curr Issues Mol Biol. 1999;1(1-2):89–104. [PubMed] [Google Scholar]
- Briones C, Moreno M. Applications of peptide nucleic acids (PNAs) and locked nucleic acids (LNAs) in biosensor development. Anal Bioanal Chem. 2012;402(10):3071–89. [DOI] [PubMed] [Google Scholar]
- Karkare S, Bhatnagar D. Promising nucleic acid analogs and mimics: characteristic features and applications of PNA, LNA, and morpholino. Appl Microbiol Biotechnol. 2006;71(5):575–86. [DOI] [PubMed] [Google Scholar]
- Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254(5037):1497–500. [DOI] [PubMed] [Google Scholar]
- Nielsen PE. DNA analogues with nonphosphodiester backbones. Annu Rev Biophys Biomol Struct. 1995;24:167–83. [DOI] [PubMed] [Google Scholar]
- Ricciardi AS, McNeer NA, Anandalingam KK, Saltzman WM, Glazer PM. Targeted genome modification via triple helix formation. Methods Mol Biol. 2014;1176:89–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, et al. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature. 1993;365(6446):566–8. [DOI] [PubMed] [Google Scholar]
- Demidov VV, Potaman VN, Frank-Kamenetskii MD, Egholm M, Buchard O, Sonnichsen SH, et al. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem Pharmacol. 1994;48(6):1310–3. [DOI] [PubMed] [Google Scholar]
- Bahal R, Gupta A, Glazer PM. Precise Genome Modification Using Triplex Forming Oligonucleotides and Peptide Nucleic Acids. Adv Exp Med Biol. 2016;895:93–110. [Google Scholar]
- Nielsen PE. Targeted gene repair facilitated by peptide nucleic acids (PNA). ChemBioChem. 2010;11(15):2073–6. [DOI] [PubMed] [Google Scholar]
- Egholm M, Christensen L, Dueholm KL, Buchardt O, Coull J, Nielsen PE. Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA. Nucleic Acids Res. 1995;23(2):217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentin T, Larsen HJ, Nielsen PE. Combined triplex/duplex invasion of double-stranded DNA by “tail-clamp” peptide nucleic acid. Biochemistry-Us. 2003;42(47):13987–95. [DOI] [PubMed] [Google Scholar]
- Braasch DA, Corey DR. Synthesis, analysis, purification, and intracellular delivery of peptide nucleic acids. Methods. 2001;23(2):97–107. [DOI] [PubMed] [Google Scholar]
- Sahu B, Sacui I, Rapireddy S, Zanotti KJ, Bahal R, Armitage BA, et al. Synthesis and characterization of conformationally preorganized, (R)-diethylene glycol-containing gamma-peptide nucleic acids with superior hybridization properties and water solubility. J Org Chem. 2011;76(14):5614–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanardi C, Terzi F, Seeber R, Baldoli C, Licandro E, Maiorana S. Peptide nucleic acids tagged with four lysine residues for amperometric genosensors. Artif DNA PNA XNA. 2012;3(2):80–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey DR. Peptide nucleic acids: cellular delivery and recognition of DNA and RNA targets. Lett Pept Sci. 2003;10(3-4):347–52. [Google Scholar]
- Bonora GM, Drioli S, Ballico M, Faccini A, Corradini R, Cogoi S, et al. PNA conjugated to high-molecular weight poly(ethylene glycol): synthesis and properties. Nucleosides Nucleotides Nucleic Acids. 2007;26(6-7):661–4. [DOI] [PubMed] [Google Scholar]
- Rapireddy S, Bahal R, Ly DH. Strand invasion of mixed-sequence, double-helical B-DNA by gamma-peptide nucleic acids containing G-clamp nucleobases under physiological conditions. Biochemistry-Us. 2011;50(19):3913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahal R, Sahu B, Rapireddy S, Lee CM, Ly DH. Sequence-unrestricted, Watson-Crick recognition of double helical B-DNA by (R)-miniPEG-gammaPNAs. ChemBioChem. 2012;13(1):56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragulescu-Andrasi A, Rapireddy S, Frezza BM, Gayathri C, Gil RR, Ly DH. A simple gamma-backbone modification preorganizes peptide nucleic acid into a helical structure. J Am Chem Soc. 2006;128(31):10258–67. [DOI] [PubMed] [Google Scholar]
- Ray A, Norden B. Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for the future. FASEB J. 2000;14(9):1041–60. [DOI] [PubMed] [Google Scholar]
- Wittung P, Kajanus J, Edwards K, Haaima G, Nielsen PE, Norden B, et al. Phospholipid membrane permeability of peptide nucleic acid. FEBS Lett. 1995;375(3):27–9. [PubMed] [Google Scholar]
- Fabani MM, Gait MJ. miR-122 targeting with LNA/2 '-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA. 2008;14(2):336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrian-Herrada G, Desarmenien MG, Orcel H, Boissin-Agasse L, Mery J, Brugidou J, et al. A peptide nucleic acid (PNA) is more rapidly internalized in cultured neurons when coupled to a retro-inverso delivery peptide. The antisense activity depresses the target mRNA and protein in magnocellular oxytocin neurons. Nucleic Acids Res. 1998;26(21):4910–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooga M, Soomets U, Hallbrink M, Valkna A, Saar K, Rezaei K, et al. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat Biotechnol. 1998;16(9):857–61. [DOI] [PubMed] [Google Scholar]
- Shiraishi T, Nielsen PE. Cellular delivery of peptide nucleic acids (PNAs). Methods Mol Biol. 2014;1050:193–205. [DOI] [PubMed] [Google Scholar]
- Simmons CG, Pitts AE, Mayfield LD, Shay JW, Corey DR. Synthesis and membrane permeability of PNA-peptide conjugates. Bioorg Med Chem Lett. 1997;7(23):3001–6. [Google Scholar]
- Koppelhus U, Awasthi SK, Zachar V, Holst HU, Ebbesen P, Nielsen PE. Cell-dependent differential cellular uptake of PNA, peptides, and PNA-peptide conjugates. Antisense Nucleic A. 2002;12(2):51–63. [DOI] [PubMed] [Google Scholar]
- Rogers FA, Lin SS, Hegan DC, Krause DS, Glazer PM. Targeted Gene Modification of Hematopoietic Progenitor Cells in Mice Following Systemic Administration of a PNA-peptide Conjugate. Mol Ther. 2012;20(1):109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazani P, Gemignani F, Kang SH, Maier MA, Manoharan M, Persmark M, et al. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat Biotechnol. 2002;20(12):1228–33. [DOI] [PubMed] [Google Scholar]
- Hamzavi R, Dolle F, Tavitian B, Dahl O, Nielsen PE. Modulation of the pharmacokinetic properties of PNA: preparation of galactosyl, mannosyl, fucosyl, N-acetylgalactosaminyl, and N-acetylglucosaminyl derivatives of aminoethylglycine peptide nucleic acid monomers and their incorporation into PNA oligomers. Bioconjug Chem. 2003;14(5):941–54. [DOI] [PubMed] [Google Scholar]
- Nielsen PE. Addressing the challenges of cellular delivery and bioavailability of peptide nucleic acids (PNA). Q Rev Biophys. 2005;38(4):345–50. [DOI] [PubMed] [Google Scholar]
- Shiraishi T, Pankratova S, Nielsen PE. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic tat and oligoarginine peptides. Chem Biol. 2005;12(8):923–9. [DOI] [PubMed] [Google Scholar]
- Uhlmann E, Peyman A, Breipohl G, Will DW. PNA: synthetic polyamide nucleic acids with unusual binding properties. Angew Chem Int Ed. 1998;37(20):2797–823. [DOI] [PubMed] [Google Scholar]
- McMahon BM, Mays D, Lipsky J, Stewart JA, Fauq A, Richelson E. Pharmacokinetics and tissue distribution of a peptide nucleic acid after intravenous administration. Antisense Nucleic Acid Drug Dev. 2002;12(2):65–70. [DOI] [PubMed] [Google Scholar]
- Nielsen PE. Addressing the challenges of cellular delivery and bioavailability of peptide nucleic acids (PNA). Q Rev Biophys. 2005;38(4):345–50. [DOI] [PubMed] [Google Scholar]
- Iyer M, Norton JC, Corey DR. Accelerated Hybridization of Oligonucleotides to Duplex DNA. J Biol Chem. 1995;270(24):14712–7. [DOI] [PubMed] [Google Scholar]
- Nielsen PE, Egholm M, Buchardt O. Sequence-Specific Transcription Arrest by Peptide Nucleic-Acid Bound to the DNA-Template Strand. Gene. 1994;149(1):139–45. [DOI] [PubMed] [Google Scholar]
- Hanvey JC, Peffer NJ, Bisi JE, Thomson SA, Cadilla R, Josey JA, et al. Antisense and Antigene Properties of Peptide Nucleic-Acids. J Cell Biochem. 1993:212. [DOI] [PubMed] [Google Scholar]
- Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence Specific-Inhibition of DNA Restriction Enzyme Cleavage by Pna. Nucleic Acids Res. 1993;21(2):197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers TA, Griffith MC, Ramasamy K, Risen LM, Freier SM. Inhibition of Nf-Kappa-B Specific Transcriptional Activation by Pna Strand Invasion. Nucleic Acids Res. 1995;23(15):3003–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutrona G, Carpaneto EM, Ulivi M, Roncella S, Landt O, Ferrarini M, et al. Effects in live cells of a c-myc anti-gene PNA linked to a nuclear localization signal. Nat Biotechnol. 2000;18(3):300–3. [DOI] [PubMed] [Google Scholar]
- Janowski BA, Kaihatsu K, Huffman KE, Schwartz JC, Ram R, Hardy D, et al. Inhibiting transcription of chromosomal DNA with antigene peptide nucleic acids. Nat Chem Biol. 2005;1(4):210–5. [DOI] [PubMed] [Google Scholar]
- Larsen HJ, Nielsen PE. Transcription-mediated binding of peptide nucleic acid (PNA) to double-stranded DNA: sequence-specific suicide transcription. Nucleic Acids Res. 1996;24(3):458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesce CD, Bolacchi F, Bongiovanni B, Cisotta F, Capozzi M, Diviacco S, et al. Anti-gene peptide nucleic acid targeted to proviral HIV-1 DNA inhibits in vitro HIV-1 replication. Antiviral Res. 2005;66(1):13–22. [DOI] [PubMed] [Google Scholar]
- Mollegaard NE, Buchardt O, Egholm M, Nielsen PE. Peptide Nucleic-Acid DNA Strand Displacement Loops as Artificial Transcription Promoters. Proc Natl Acad Sci USA. 1994;91(9):3892–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Xu XX, Pace P, Dean DA, Glazer PM, Chan P, et al. Peptide nucleic acid (PNA) binding-mediated induction of human gamma-globin gene expression. Nucleic Acids Res. 1999;27(13):2806–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Xu XX. Peptide nucleic acid (PNA) binding-mediated gene regulation. Cell Res. 2004;14(2):111–6. [DOI] [PubMed] [Google Scholar]
- Chen J, Peterson KR, Iancu-Rubin C, Bieker JJ. Design of embedded chimeric peptide nucleic acids that efficiently enter and accurately reactivate gene expression in vivo. Proc Natl Acad Sci USA. 2010;107(39):16846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean DA. Peptide nucleic acids: versatile tools for gene therapy strategies. Adv Drug Deliv Rev. 2000;44(2-3):81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen H, Nielsen PE. Antisense properties of duplex- and triplex-forming PNAs. Nucleic Acids Res. 1996;24(3):494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambacorti Passerini C, Mologni L, Bertazzoli C, LeCoutre P, Marchesi E, Grignani F, et al. In vitro transcription and translation inhibition by anti-promyelocytic leukemia (PML)/retinoic acid receptor alpha and anti-PML peptide nucleic acid. Blood. 1996;88(4):1411–7. [PubMed] [Google Scholar]
- Bonham MA, Brown S, Boyd AL, Brown PH, Bruckenstein DA, Hanvey JC, et al. An assessment of the antisense properties of RNase H-competent and steric-blocking oligomers. Nucleic Acids Res. 1995;23(7):1197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane LA, Murphy PR. MicroRNA: Biogenesis, Function and Role in Cancer. Curr Genomics. 2010;11(7):537–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. [DOI] [PubMed] [Google Scholar]
- Cheng CJ, Saltzman WM, Slack FJ. Canonical and Non-Canonical Barriers Facing AntimiR Cancer Therapeutics. Curr Med Chem. 2013;20(29):3582–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchelli R, Corradini R, Manicardi A, Sforza S, Tedeschi T, Fabbri E, et al. Gene Modulation by Peptide Nucleic Acids (PNAs) Targeting microRNAs (miRs). In: You Y, editor. Targets in Gene Therapy. Rijeka: InTech; 2011. p. Ch. 02. [Google Scholar]
- Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65(14):6029–33. [DOI] [PubMed] [Google Scholar]
- Kitadate A, Ikeda S, Teshima K, Ito M, Takahashi N, Miyagaki T, et al. Microrna-16 Mediates the Regulation of a Senescence-Apoptosis Switch in Cutaneous T-Cell and Other Non-Hodgkin Lymphomas. Blood. 2015;126(23). [DOI] [PubMed] [Google Scholar]
- Oh SY, Ju Y, Park H. A highly effective and long-lasting inhibition of miRNAs with PNA-based antisense oligonucleotides. Mol Cells. 2009;28(4):341. [DOI] [PubMed] [Google Scholar]
- Fabbri E, Manicardi A, Tedeschi T, Sforza S, Bianchi N, Brognara E, et al. Modulation of the biological activity of microRNA-210 with peptide nucleic acids (PNAs). ChemMedChem. 2011;6(12):2192–202. [DOI] [PubMed] [Google Scholar]
- Gupta A, Quijano E, Liu Y, Bahal R, Scanlon SE, Song E, et al. Anti-tumor Activity of miniPEG-γ-Modified PNAs to Inhibit MicroRNA-210 for Cancer Therapy. Mol Ther Nucleic Acids. 2017;9 Supplement C:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518(7537):107–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brognara E, Fabbri E, Bazzoli E, Montagner G, Ghimenton C, Eccher A, et al. Uptake by human glioma cell lines and biological effects of a peptide-nucleic acids targeting miR-221. J Neurooncol. 2014;118(1):19–28. [DOI] [PubMed] [Google Scholar]
- Brognara E, Fabbri E, Aimi F, Manicardi A, Bianchi N, Finotti A, et al. Peptide nucleic acids targeting miR-221 modulate p27Kip1 expression in breast cancer MDA-MB-231 cells. Int J Oncol. 2012;41(6):2119–27. [DOI] [PubMed] [Google Scholar]
- Fabani MM, Abreu-Goodger C, Williams D, Lyons PA, Torres AG, Smith KG, et al. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010;38(13):4466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambari R, Brognara E, Spandidos DA, Fabbri E. Targeting oncomiRNAs and mimicking tumor suppressor miRNAs: nuew trends in the development of miRNA therapeutic strategies in oncology [Review] Int J Oncol. 2016;49(1):5–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambari R, Fabbri E, Borgatti M, Lampronti I, Finotti A, Brognara E, et al. Targeting microRNAs involved in human diseases: a novel approach for modification of gene expression and drug development. Biochem Pharmacol. 2011;82(10):1416–29. [DOI] [PubMed] [Google Scholar]
- Kalish JM, Glazer PM. Targeted genome modification via triple helix formation. Ann N Y Acad Sci. 2005;1058:151–61. [DOI] [PubMed] [Google Scholar]
- Faruqi AF, Egholm M, Glazer PM. Peptide nucleic acid-targeted mutagenesis of a chromosomal gene in mouse cells. Proc Natl Acad Sci USA. 1998;95(4):1398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers FA, Vasquez KM, Egholm M, Glazer PM. Site-directed recombination via bifunctional PNA-DNA conjugates. Proc Natl Acad Sci USA. 2002;99(26):16695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleifman EB, Bindra R, Leif J, del Campo J, Rogers FA, Uchil P, et al. Targeted disruption of the CCR5 gene in human hematopoietic stem cells stimulated by peptide nucleic acids. Chem Biol. 2011;18(9):1189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JY, Kuan JY, Lonkar PS, Krause DS, Seidman MM, Peterson KR, et al. Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids. Proc Natl Acad Sci USA. 2008;105(36):13514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahal R, Quijano E, McNeer NA, Liu Y, Bhunia DC, Lopez-Giraldez F, et al. Single-stranded gammaPNAs for in vivo site-specific genome editing via Watson-Crick recognition. Curr Gene Ther. 2014;14(5):331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaima G, Lohse A, Buchardt O, Nielsen PE. Peptide nucleic acids (PNAs) containing thymine monomers derived from chiral amino acids: hybridization and solubility properties of D-lysine PNA. Angew Chem Int Ed Engl. 1996;35(17):1939–42. [Google Scholar]
- Zhou P, Wang M, Du L, Fisher GW, Waggoner A, Ly DH. Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA). J Am Chem Soc. 2003;125(23):6878–9. [DOI] [PubMed] [Google Scholar]
- Moccia M, Adamo MF, Saviano M. Insights on chiral, backbone modified peptide nucleic acids: properties and biological activity. Artif DNA PNA XNA. 2014;5(3):e1107176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagriffoule P, Wittung P, Eriksson M, Jensen KK, Norden B, Buchardt O, et al. Peptide nucleic acids with a conformationally constrained chiral cyclohexyl-derived backbone. Chemistry. 1997;3(6):912–9. [Google Scholar]
- Sforza S, Corradini R, Ghirardi S, Dossena A, Marchelli R. DNA binding of a D-lysine-based chiral PNA: direction control and mismatch recognition. Eur J Org Chem. 2000;(16):2905–13. [Google Scholar]
- D’Costa M, Kumar VA, Ganesh KN. Aminoethylprolyl peptide nucleic acids (aepPNA): Chiral PNA analogues that form highly stable DNA : aepPNA(2) triplexes. Org Lett. 1999;1(10):1513–6. [DOI] [PubMed] [Google Scholar]
- Kumar VA, Ganesh KN. Conformationally constrained PNA analogues: structural evolution toward DNA/RNA binding selectivity. Acc Chem Res. 2005;38(5):404–12. [DOI] [PubMed] [Google Scholar]
- He GF, Rapireddy S, Bahal R, Sahu B, Ly DH. Strand invasion of extended, mixed-sequence B-DNA by gPNAs (g-peptide nucleic acid. Abstr Pap Am Chem S. 2010;240. [Google Scholar]
- Rapireddy S, He G, Roy S, Armitage BA, Ly DH. Strand invasion of mixed-sequence B-DNA by acridine-linked, gamma-peptide nucleic acid (gamma-PNA). J Am Chem Soc. 2007;129(50):15596–600. [DOI] [PubMed] [Google Scholar]
- Schleifman EB, McNeer NA, Jackson A, Yamtich J, Brehm MA, Shultz LD, et al. Site-specific Genome Editing in PBMCs With PLGA Nanoparticle-delivered PNAs Confers HIV-1 Resistance in Humanized Mice. Mol Ther Nucleic Acids. 2013;2:e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahal R, Ali McNeer N, Quijano E, Liu Y, Sulkowski P, Turchick A, et al. In vivo correction of anaemia in beta-thalassemic mice by gammaPNA-mediated gene editing with nanoparticle delivery. Nat Commun. 2016;7:13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppelhus U, Nielsen PE. Cellular delivery of peptide nucleic acid (PNA). Adv Drug Deliv Rev. 2003;55(2):267–80. [DOI] [PubMed] [Google Scholar]
- Gupta A, Bahal R, Gupta M, Glazer PM, Saltzman WM. Nanotechnology for delivery of peptide nucleic acids (PNAs). J Control Release. 2016;240:302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devalliere J, Chang WG, Andrejecsk JW, Abrahimi P, Cheng CJ, Jane-wit D, et al. Sustained delivery of proangiogenic microRNA-132 by nanoparticle transfection improves endothelial cell transplantation. FASEB J. 2014;28(2):908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilov K, Seo YE, Tietjen GT, Cui J, Cheng CJ, Saltzman WM. Enhancing potency of siRNA targeting fusion genes by optimization outside of target sequence. Proc Natl Acad Sci USA. 2015;112(48):E6597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LB, Sawyer AJ, Saucier-Sawyer J, Saltzman WM, Kyriakides TR. Nanoparticle delivery of miR-223 to attenuate macrophage fusion. Biomaterials. 2016;89:127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JK, Utsumi T, Seo YE, Deng Y, Satoh A, Saltzman WM, et al. Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomedicine (Lond). 2016;12(5):1365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JM, Seo YE, Saltzman WM. Cell penetrating peptide-modified poly(lactic-co-glycolic acid) nanoparticles with enhanced cell internalization. Acta Biomater. 2016;30:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AR, Corso CD, Chen EM, Song E, Bongiorni P, Chen Z, et al. Local DNA Repair Inhibition for Sustained Radiosensitization of High-Grade Gliomas. Mol Cancer Ther. 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodrow KA, Cu Y, Booth CJ, Saucier-Sawyer JK, Wood MJ, Saltzman WM. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater. 2009;8(6):526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CJ, Saltzman WM. Enhanced siRNA delivery into cells by exploiting the synergy between targeting ligands and cell-penetrating peptides. Biomaterials. 2011;32(26):6194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JM, Weller CE, Booth CJ, Saltzman WM. Polymer nanoparticles encapsulating siRNA for treatment of HSV-2 genital infection. J Control Release. 2012;162(1):102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RJ, Cheng CJ, Quijano E, Weller C, Kristofik N, Duong N, et al. Surface modified poly(beta amino ester)-containing nanoparticles for plasmid DNA delivery. J Control Release. 2012;164(1):41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wen Q, Choi S. FDA's regulatory science program for generic PLA/PLGA-based drug products. Am Pharm Rev. 2016;19(4). [Google Scholar]
- Semete B, Booysen L, Lemmer Y, Kalombo L, Katata L, Verschoor J, et al. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomed-Nanotechnol. 2010;6(5):662–71. [DOI] [PubMed] [Google Scholar]
- Woodrow KA, Cu Y, Booth CJ, Saucier-Sawyer JK, Wood MJ, Saltzman WM. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater. 2009;8(6):526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu XM, Zou WW, Liu CX, Zhang N, Fu CH. Modified nanoprecipitation method to fabricate DNA-loaded PLGA nanoparticles. Drug Dev Ind Pharm. 2009;35(11):1375–83. [DOI] [PubMed] [Google Scholar]
- McNeer NA, Chin JY, Schleifman EB, Fields RJ, Glazer PM, Saltzman WM. Nanoparticles deliver triplex-forming PNAs for site-specific genomic recombination in CD34+ human hematopoietic progenitors. Mol Ther. 2011;19(1):172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeer NA, Schleifman EB, Cuthbert A, Brehm M, Jackson A, Cheng C, et al. Systemic delivery of triplex-forming PNA and donor DNA by nanoparticles mediates site-specific genome editing of human hematopoietic cells in vivo. Gene Ther. 2013;20(6):658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Yang F, Cocco E, Song E, Zhang J, Cui J, et al. Improved i.p. drug delivery with bioadhesive nanoparticles. Proc Natl Acad Sci USA. 2016;113(41):11453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E, Gaudin A, King AR, Seo YE, Suh HW, Deng Y, et al. Surface chemistry governs cellular tropism of nanoparticles in the brain. Nat Commun. 2017;8:15322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AM, Bonke G, Larsen CJ, Yavari N, Nielsen PE, Franzyk H. Antibacterial Peptide Nucleic Acid-Antimicrobial Peptide (PNA-AMP) Conjugates: Antisense Targeting of Fatty Acid Biosynthesis. Bioconjug Chem. 2016;27(4):863–7. [DOI] [PubMed] [Google Scholar]
- Ozes AR, Wang YN, Zong XY, Fang F, Pilrose J, Nephew KP. Therapeutic targeting using tumor specific peptides inhibits long noncoding RNA HOTAIR activity in ovarian and breast cancer. Sci Rep-Uk; 2017. p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka T, Yoshida J, Yamamoto Y, Sumaoka J, Tedeschi T, Corradini R, et al. Chiral introduction of positive charges to PNA for double-duplex invasion to versatile sequences. Nucleic Acids Res. 2008;36(5):1464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahal R, McNeer NA, Ly DH, Saltzman WM, Glazer PM. Nanoparticle for delivery of antisense gammaPNA oligomers targeting CCR5. Artif DNA PNA XNA. 2013;4(2):49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeer NA, Schleifman EB, Glazer PM, Saltzman WM. Polymer delivery systems for site-specific genome editing. J Control Release. 2011;155(2):312–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RJ, Cheng CJ, Quijano E, Weller C, Kristofik N, Duong N, et al. Surface modified poly(beta amino ester)-containing nanoparticles for plasmid DNA delivery. J Control Release. 2012;164(1):41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeer NA, Anandalingam K, Fields RJ, Caputo C, Kopic S, Gupta A, et al. Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nat Commun. 2015:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RJ, Quijano E, McNeer NA, Caputo C, Bahal R, Anandalingam K, et al. Modified poly(lactic-co-glycolic acid) nanoparticles for enhanced cellular uptake and gene editing in the lung. Adv Healthc Mater. 2015;4(3):361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adochite RC, Moshnikova A, Golijanin J, Andreev OA, Katenka NV, Reshetnyak YK. Comparative Study of Tumor Targeting and Biodistribution of pH (Low) Insertion Peptides (pHLIP(A (R)) Peptides) Conjugated with Different Fluorescent Dyes. Mol Imaging Biol. 2016;18(5):686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshetnyak YK, Segala M, Andreev OA, Engelman DM. A monomeric membrane peptide that lives in three worlds: in solution, attached to, and inserted across lipid bilayers. Biophys J. 2007;93(7):2363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreev OA, Engelman DM, Reshetnyak YK. pH-sensitive membrane peptides (pHLIPs) as a novel class of delivery agents. Mol Membr Biol. 2010;27(7):341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross R. CRISPR’s breakthrough problem (vol 95, pg 28, 2017). Chem Eng News. 2017;95(8):4. [Google Scholar]
- Li L, He ZY, Wei XW, Gao GP, Wei YQ. Challenges in CRISPR/CAS9 Delivery: Potential Roles of Nonviral Vectors. Hum Gene Ther. 2015;26(7):452–62. [DOI] [PubMed] [Google Scholar]
- Wang HX, Li M, Lee CM, Chakraborty S, Kim HW, Bao G, et al. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem Rev. 2017 [DOI] [PubMed] [Google Scholar]
- Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34(3):328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler BM, Jansen K, McCormick DJ, Douglas CL, Boules M, Stewart JA, et al. Peptide nucleic acids targeted to the neurotensin receptor and administered i.p. cross the blood-brain barrier and specifically reduce gene expression. Proc Natl Acad Sci USA. 1999;96(12):7053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya DP. New approaches toward recognition of nucleic acid triple helices. Acc Chem Res. 2011;44(2):134–46. [DOI] [PMC free article] [PubMed] [Google Scholar]