

ABSTRACT
Cattle are the primary carrier of Escherichia coli O157:H7, a foodborne human pathogen, and those shedding >104 CFU/gram of feces of E. coli O157:H7 are defined as supershedders (SS). This study investigated the rectoanal junction (RAJ) mucosa-associated microbiota and its relationship with host gene expression in SS and in cattle from which E. coli O157:H7 was not detected (nonshedders [NS]), aiming to elucidate the mechanisms involved in supershedding. In total, 14 phyla, 66 families, and 101 genera of RAJ mucosa-associated bacteria were identified and Firmicutes (61.5 ± 7.5%), Bacteroidetes (27.9 ± 6.4%), and Proteobacteria (5.5 ± 2.1%) were the predominant phyla. Differential abundance analysis of operational taxonomic units (OTUs) identified 2 OTUs unique to SS which were members of Bacteroides and Clostridium and 7 OTUs unique to NS which were members of Coprococcus, Prevotella, Clostridium, and Paludibacter. Differential abundance analysis of predicted microbial functions (using PICRUSt [phylogenetic investigation of communities by reconstruction of unobserved states]) revealed that 3 pathways had higher abundance (log2 fold change, 0.10 to 0.23) whereas 12 pathways had lower abundance (log2 fold change, −0.36 to −0.20) in SS. In addition, we identified significant correlations between expression of 19 differentially expressed genes and the relative abundance of predicted microbial functions, including nucleic acid polymerization and carbohydrate and amino acid metabolism. Our findings suggest that differences in RAJ microbiota at both the compositional and functional levels may be associated with E. coli O157:H7 supershedding and that certain microbial groups and microbial functions may influence RAJ physiology of SS by affecting host gene expression.
IMPORTANCE Cattle with fecal E. coli O157:H7 at >104 CFU per gram of feces have been defined as the supershedders, and they are responsible for the most of the E. coli O157:H7 spread into farm environment. Currently, no method is available for beef producers to eliminate shedding of E. coli O157:H7 in cattle, and the lack of information about the mechanisms of supershedding greatly impedes the development of effective methods. This study investigated the role of the rectoanal junction (RAJ) mucosa-associated microbiome in E. coli O157:H7 shedding, and our results indicated that the compositions and functions of RAJ microbiota differed between supershedders and nonshedders. The identified relationship between the differentially abundant microbes and 19 previously identified differentially expressed genes suggests the role of host-microbial interactions involved in E. coli O157:H7 supershedding. Our findings provide a fundamental understanding of the supershedding phenomenon which is essential for the development of strategies, such as the use of directly fed microbials, to reduce E. coli O157:H7 shedding in cattle.
KEYWORDS: cattle, E. coli O157:H7, rectoanal junction, microbiome, gene expression, E. coli O157
INTRODUCTION
Gut microbiota has been suggested to regulate the host immune function; nutrient metabolism; energy harvest; and liver, muscle, and even brain function (1), with a metagenome more than a hundred times greater in size than the host genome (2). It has been suggested that gut commensal microbiota can inhibit pathogenic microbes through both direct effects, such as releasing antimicrobials and competitive exclusion, and indirect effects, such as activation of host immune protection (3). Thus, investigating the composition and function of gut microbiota can provide understanding of the interaction between the host and gut commensal/pathogenic microorganisms.
Commonly, the foodborne pathogen Escherichia coli O157:H7 is known to inhabit the intestinal tract of cattle without causing symptoms of illness. However, it has been suggested that it can occasionally cause pathogenesis in cattle evidenced by the formation of attaching and effacing lesions at the rectoanal junction (RAJ), the primary colonization site of E. coli O157:H7 of calves, and by damage to intestinal epithelium of yearling cattle (4, 5). Cattle shedding >104 CFU of E. coli O157:H7 per gram of feces have been defined as supershedders (SS) (6). These cattle usually account for less than 10% of the animals in a feedlot but are the source of most of E. coli O157:H7 shed into the farm environment. Furthermore, the E. coli O157:H7 strains that are most commonly isolated from SS have also been reported to be associated with human infection (6) and to cause severe human diseases, including hemolytic uremic syndrome and even death. Therefore, there is an urgency to better understand supershedding phenomena to develop control strategies against this pathogen on farm.
Studies on SS cattle have been conducted for more than a decade, but the mechanisms of supershedding remain unclear, especially from the perspective of gut microbiota and its interaction with host. A previous study reported that the levels of diversity of fecal microbiota differed between SS and cattle with negative fecal E. coli O157:H7 results (NS), suggesting a potential linkage between gut microbiota and E. coli O157:H7 shedding (7). In addition, it has been speculated that the mucosa-associated microbiota plays a role in host-microbe interaction in its direct interaction with the host and that it differs in composition from luminal microbiota (8). We speculate that the RAJ mucosa-associated microbiota plays a role in regulating shedding of E. coli O157:H7, as this bacterium cohabitates the host's gut with other commensal species. Our previous study (9) revealed that host gene expression profiles differed between SS and NS and that the differentially expressed (DE) genes were associated with immune function, but whether such a difference is caused by host or microbial factors is unknown. In this study, we hypothesized that the RAJ mucosa-associated microbiota of SS is different from that of NS and that such difference could affect host gene expression at the RAJ, leading to a more favorable environment for growth of E. coli O157:H7 in SS. Taxonomic profiling was performed to characterize the mucosa-associated microbiota by 16S rRNA gene amplicon sequencing, and the functions of the microbiome were also predicted using PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states) based on the identified bacterial taxa. Correlation analysis was conducted to identify potential relationships between bacterial groups/predicted functions and the differentially expressed RAJ genes that are involved in supershedding.
RESULTS
Taxonomic assessment of the RAJ mucosa-associated microbiota of beef steers.
From joined paired-end MiSeq sequencing reads, 94,512 reads passed quality filtering, with a length of 478 ± 14 bases (Table 1). In total, 1,278 operational taxonomic units (OTUs) were identified from all nine RAJ samples, and the number of OTUs identified in each sample ranged from 223 (SS 294) to 523 (SS 310) (Table 1; see also Tables S1 and S2 in the supplemental material). From all the samples, >99% reads were classified into 14 phyla, with Firmicutes (61.5 ± 7.5%), Bacteroidetes (27.9 ± 6.4%), Proteobacteria (5.5 ± 2.1%), and Spirochaetes (2.9 ± 3.3%) being the predominant phyla (relative abundance, >1%). Only four phyla, including Firmicutes, Bacteroidetes, Proteobacteria, and Tenericutes, were identified from RAJ of all the steers (see Table S1 in the supplemental material). At the family level, 66 families were detected from RAJ of all the steers, with 56 classified and 10 unclassified. Ruminococcaceae (28.9 ± 6.8%) and Lachnospiraceae (9.9 ± 4.4%) from the Firmicutes phylum and Bacteroidaceae (8.2 ± 3.4%) from the Bacteroidetes phylum were the most abundant families, and 13 families were identified in all the steers (Table S2 in the supplemental material). At the genus level, 101 genera, including 44 unclassified and 57 classified, were detected in all animals, with genera 5-7N15 (6.9 ± 2.4%, belonging to family of Bacteroidaceae), Prevotella (4.7 ± 4.1%), and Ruminococcus (3.8 ± 6.1%) being the most abundant classified genera (Table S2). Seven genera, including 5-7N15, Prevotella, Ruminococcus, CF231 (belonging to family of Paraprevotellaceae), Dorea, Clostridium, and Oscillospira, were detected in all steers. The most abundantly detected genera (36 genera) belonged to Firmicutes. These included 27 classified and 9 unclassified genera, and Ruminococcus, Dorea, and Clostridium were the most abundant (relative abundance ranged from 1% to 7%) (Table S2 in the supplemental material). Twenty-three detected genera, including 11 classified and 12 unclassified, belonged to Bacteroidetes. Of these, 5-7N15, Prevotella, and CF231 were the most abundant (relative abundance ranged from 2% to 6%) (Table S2). Twenty-three genera, including 11 classified and 12 unclassified genera, belonged to Proteobacteria, and Pseudomonas, Succinivibrio, and Sutterella were the most abundant (average abundance ranged from 0.5% to 0.9%) (Table S2). At the species level, only 1.4 ± 1.6% reads could be classified, and 11 species were detected. The most abundant species was Edaphobacter modestum (0.5% ± 1%), and none of classified species were shared by all the animals.
TABLE 1.
Sequencing results and alpha diversity indices for all steers
| Animal_id | No. of readsa | No. of OTUs | Good's coverage | Chao1 value | Shannon index value |
|---|---|---|---|---|---|
| NS108_NS | 10,199 | 407 | >99% | 435.5 | 7.6 |
| NS152_NS | 7,795 | 239 | >99% | 260.0 | 7.2 |
| NS165_NS | 10,175 | 275 | >99% | 285.5 | 7.3 |
| NS242_NS | 10,515 | 296 | >99% | 310.0 | 7.0 |
| SS274_SS | 9,919 | 372 | >99% | 391.3 | 7.6 |
| SS287_SS | 12,468 | 409 | >99% | 431.7 | 7.1 |
| SS294_SS | 10,308 | 223 | >99% | 255.7 | 6.8 |
| SS299_SS | 11,339 | 225 | >99% | 241.3 | 6.8 |
| SS310_SS | 11,794 | 523 | >99% | 539.2 | 7.7 |
Number of reads that passed quality filtering.
Comparison of rectal mucosa-associated microbiotas between supershedders and nonshedders.
The Good's coverage was >99% for all samples, indicating adequate sequencing depth that represents the RAJ-attached bacterial community. Neither of the alpha-diversity indices Chao1 (321.1 ± 71.4 for NS and 368.8 ± 130.6 for SS) and Shannon (7.2 ± 0.2 for NS and 7.2 ± 0.5 for SS) showed statistical significance (t test; P value, >0.05) in comparisons between SS and NS, indicating no statistically significant difference in microbial diversity at the RAJ. Steer SS 310 had the highest Chao1 value (539.2), while SS 299 had the lowest Chao1 value (241.3), indicating that mucosa-associated bacterial populations exhibited great individual variation within SS animals. According to beta-diversity analysis results, the SS animals formed two clusters. One cluster included SS 310, SS 274, and SS 287, and the other cluster included SS 299 and SS 294 (Fig. 1A).
FIG 1.

(A) Principal-coordinate analysis (PCOA) based on UniFrac distance of OTUs identified from rectoanal junction tissue samples. NS, nonshedders; SS, supershedders. (B) Phyla detected in nonshedders and supershedders. Detected phyla, relative abundance of >0.1% in at least one nonshedder or supershedder. (C) Genera detected in nonshedders and supershedders. Detected genera, relative abundance of >0.1% in at least one nonshedder or supershedder. NS, nonshedders; SS, supershedders.
Although the alpha indices were similar between NS and SS, there were differences in the composition of the RAJ-associated microbiota. At the phylum level, 9 phyla were detected in both NS and SS, while 4 phyla, including Elusimicrobia, Fusobacteria, Lentisphaerae, and OD1, were detected only in NS steers (Fig. 1B). At the genus level, 11 genera were specific to NS and 10 were specific to SS (Table 2), with 36 of them shared between NS and SS (Fig. 1C). At the OTU level, differential abundance analysis identified significant differences in 9 OTUs (P value, <0.1), including 7 unique to NS and 2 unique to SS (Fig. 2). However, only OTU108 and OTU260 were classified (Coprococcus and Prevotella, respectively), and the rest of OTUs could not be classified to the genus level. The phylogenic analysis was then performed, and the results showed that OTU56 was closely related to Bacteroides; OTU121 and OTU66 were closely related to Clostridium; OTU45 and OTU180 were closely related to Paludibacter; and OTU228 and OTU336 were clustered together and shared common ancestors with microbes within Proteobacteria (Fig. 3).
TABLE 2.
Nonshedder- and supershedder-specific genera and the microbial families and phyla to which these genera belonged
| Phylum | Family | Genus |
|---|---|---|
| NS-specific microbes | ||
| Actinobacteria | Intrasporangiaceae | Janibacter |
| Bacteroidetes | Paraprevotellaceae | Paraprevotella |
| Bacteroidetes | Sphingobacteriaceae | Pedobacter |
| Firmicutes | Lachnospiraceae | Epulopiscium |
| Firmicutes | Veillonellaceae | Mitsuokella |
| Firmicutes | Ruminococcaceae | Faecalibacterium |
| Fusobacteria | Fusobacteriaceae | u114 |
| Proteobacteria | Bradyrhizobiaceae | Bradyrhizobium |
| Proteobacteria | Alcaligenaceae | Pelistega |
| Proteobacteria | Bdellovibrionaceae | Bdellovibrio |
| Proteobacteria | Shewanellaceae | Shewanella |
| SS-specific microbes | ||
| Actinobacteria | Corynebacteriaceae | Corynebacterium |
| Actinobacteria | Gordoniaceae | Gordonia |
| Actinobacteria | Propionibacteriaceae | Luteococcus |
| Firmicutes | Lactobacillaceae | Lactobacillus |
| Firmicutes | Erysipelotrichaceae | Coprobacillus |
| Firmicutes | Staphylococcaceae | Jeotgalicoccus |
| Firmicutes | Veillonellaceae | Acidaminococcus |
| Firmicutes | Mogibacteriaceae | Anaerovorax |
| Proteobacteria | Moraxellaceae | Acinetobacter |
| Proteobacteria | Rhodobacterales | Rhodobacter |
FIG 2.

OTUs differentially abundant between nonshedders and supershedders. The blue bars indicate that the OTUs were more highly abundant in supershedders, and red bars indicate that the OTUs were less abundant in supershedders.
FIG 3.

Phylogenetic tree built based on sequences of OTUs assigned to known bacterial genera.
Functional prediction of RAJ mucosa-associated microbiota using PICRUSt.
Due to the challenges to obtaining functional aspects for mucosa-associated microbiota using metagenomics, KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways were predicted by the use of PICRUSt and the 16S rRNA gene data set. In total, 23 KEGG categories (abundance, >0.1%) (Table S3 in the supplemental material) were identified, with the 10 most abundant categories shown in Fig. 4A. Among these categories, six, including metabolism of amino acids, carbohydrate, energy, lipid, cofactors and vitamins, and nucleotides, were related to metabolism; three, including replication and repair as well as translation and transcription, were related to genetic information processing; and one was related to membrane transport. Comparing SS and NS, two pathways, including signal transduction (log2 fold change, −0.15) and cell motility (log2 fold change, −0.25), had lower abundance (P value, <0.1) in SS, whereas one pathway (replication and repair [log2 fold change, 0.07]) had higher abundance in SS.
FIG 4.
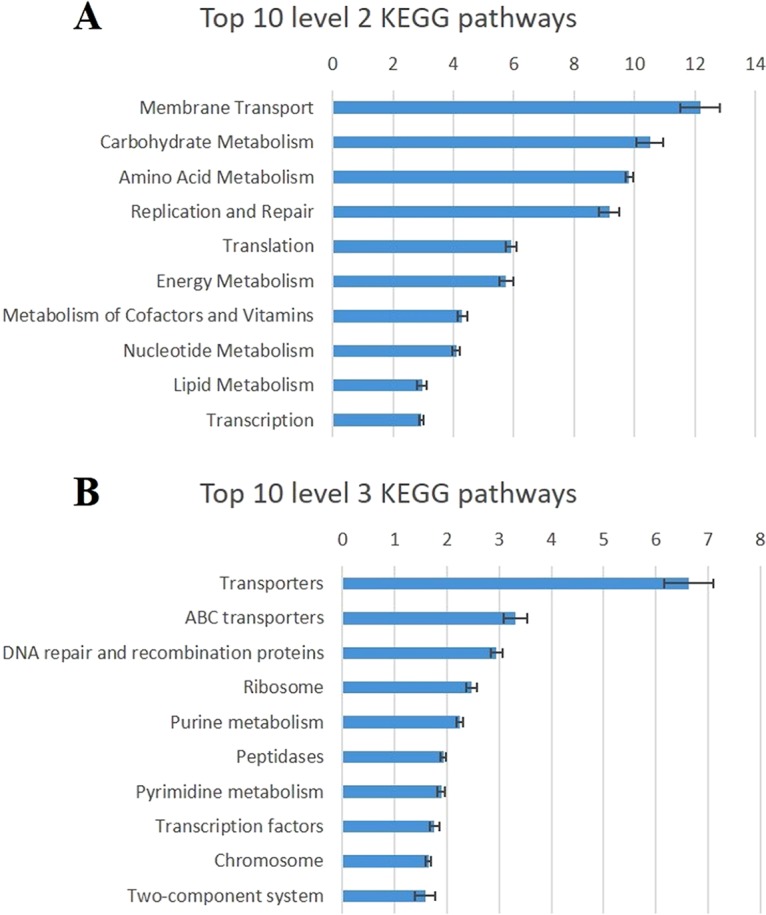
(A) The top 10 predicted metagenomic functions at level 2 of the KEGG pathway. (B) The top 10 predicted metagenomic functions at level 3 of the KEGG pathway. The blue bars stand for the percentage of relative abundance of each predicted function.
In total, 120 individual pathway maps were predicted to be associated with the RAJ microbiota (abundance, >0.1%) (Table S4), and the 10 most abundant pathways included 3 pathways associated with environmental information processes, including ABC transporters, transporters, and two-component systems; 4 pathways associated with genetic information processes, including chromosome, DNA repair, and recombination proteins, transcription factors, and ribosome; and 3 pathways associated with metabolism, including peptidases, purine metabolism, and pyrimidine metabolism (Fig. 4B). In further comparisons of the predicted pathways between SS and NS, 12 of the pathways had lower abundance (P < 0.1) and three of them had higher abundance (P < 0.1) in SS (Table 3). Three low-abundance pathways in SS were related to cell motilities, including flagellar assembly (log2 fold change, −0.36), bacterial motility proteins (log2 fold change, −0.33), and bacterial chemotaxis (log2 fold change, −0.32). Nutrient metabolism was also predicted to differ between the RAJ microbiotas of SS and NS. For example, pyruvate metabolism pathways (log2 fold change, 0.10) had a higher abundance in SS, while pathways associated with glycan biosynthesis and metabolism (log2 fold change −0.29 to −0.28), tryptophan metabolism (log2 fold change, 0.17), and lipid metabolism (log2 fold change, −0.27 to −0.21) showed lower abundance in SS (Table 3). The abundance of cellular signaling, such as the function of two-component systems, was predicted to be lower (log2 fold change, −0.20) in SS, and the biosynthesis of ansamycins (log2 fold change, 0.21) exhibited a higher abundance in SS (Table 3).
TABLE 3.
KEGG pathways that showed differential abundance between the rectal microbiota of SS and NS
| Level 2 | Level 3 | Abundancea | Log FCb | P value |
|---|---|---|---|---|
| Amino acid metabolism | Tryptophan metabolism | 0.17 | −0.32 | 0.09 |
| Carbohydrate metabolism | Pyruvate metabolism | 1.07 | 0.10 | 0.09 |
| Cell motility | Bacterial chemotaxis | 0.62 | −0.32 | 0.05 |
| Bacterial motility proteins | 1.27 | −0.33 | 0.04 | |
| Flagellar assembly | 0.55 | −0.36 | 0.06 | |
| Glycan biosynthesis and metabolism | Lipopolysaccharide biosynthesis | 0.21 | −0.28 | 0.07 |
| Lipopolysaccharide biosynthesis proteins | 0.30 | −0.29 | 0.02 | |
| Lipid metabolism | Biosynthesis of unsaturated fatty acids | 0.14 | −0.27 | 0.07 |
| Sphingolipid metabolism | 0.21 | −0.21 | 0.06 | |
| Membrane transport | Secretion system | 1.35 | −0.14 | 0.07 |
| Metabolism of other amino acids | Glutathione metabolism | 0.18 | −0.25 | 0.04 |
| Metabolism of terpenoids and polyketides | Biosynthesis of ansamycins | 0.12 | 0.21 | 0.07 |
| Signal transduction | Two-component system | 1.58 | −0.20 | 0.02 |
| Xenobiotics biodegradation and metabolism | Aminobenzoate degradation | 0.13 | −0.26 | 0.06 |
| Naphthalene degradation | 0.16 | 0.23 | 0.07 |
Data represent average abundances among all the steers.
Log FC, log2 fold change.
Differing relationships between host gene expression and the RAJ mucosa-associated microbiome in SS.
To further understand the association between altered RAJ mucosa-associated microbiota and host functions, the expression of 58 genes identified as DE between SS and NS (obtained from our previous study [9]) was subjected to analysis of correlations with the relative abundance of microbial taxa and predicted functions (described above). The relative abundances of the members of the Succinivibrionaceae family were negatively correlated with the presence of four DE genes, including those encoding lysyl oxidase-like 1 (LOXL1) (rho value, −0.80; P value, 0.009), matrix metallopeptidase 16 (MMP16) (rho value, −0.88; P value, 0.001), matrix remodeling associated 8 (MXRA8) (rho value, −0.92, P value, <0.001), and heat shock protein family B (small) member 6 (HSPB6) (rho value, −0.88; P value, 0.002) (Fig. 5A). The expression of three S100 protein genes showed correlations with the members of several bacterial families, including positive correlations between the gene encoding S100 calcium binding protein A8 (S100A8) and Pseudomonadaceae (rho value, 0.80; P value, 0.009), the gene encoding S100 calcium binding protein A9 (S100A9) and Paraprevotellaceae (rho value, 0.87; P value, 0.002), and the gene encoding S100 calcium binding protein A12 (S100A12) and Lachnospiraceae (rho value, 0.85; P value, 0.004) and a negative correlation between the gene encoding S100A9 and Ruminococcaceae (rho value, −0.85; P value, 0.003) (Fig. 5A). For the relationship between bacterial genera and the expression of DE genes, three S100 proteins showed correlations with six genera, including S100A8 with Pseudomonas (rho value, 0.82; P value, 0.007) and Clostridium (rho value, 0.92; P value, <0.001); S100A9 with Parabacteroides (rho value, 0.80; P value, 0.009) and Clostridium (rho value, 0.83; P value, 0.005); and S100A12 with YRC22 (rho value, 0.82; P value, 0.007), Dorea (rho value, 0.80; P value, 0.009), and Blautia (rho value, 0.80; P value, 0.009) (Fig. 5B).
FIG 5.

(A) Results of correlation analysis of differentially expressed genes identified in rectal-anal junction samples and microbial abundance at the family level. (B) Results of correlation analysis of differentially expressed genes identified in rectal-anal junction samples and microbial abundance at the genus level. The color bars indicate Spearman correlation coefficient rho values; only correlations with rho values of >0.8 (blue) and <−0.8 (red) with P values of <0.01 are indicated (blue and red data). Data for genes/microbial families that did not show a significant correlation are not shown.
Moreover, the expression of DE genes was correlated with the abundance of predicted microbial functions. In total, the expression of 19 DE genes showed a correlation with at least one bacterial function. Both the expression of S100A8 and that of S100A9 were negatively correlated with the relative abundances of five microbial functions, including those corresponding to chromosome, DNA replication proteins, homologous recombination, mismatch repair, and RNA polymerase (rho values below −0.8; P values, <0.01) (Fig. 6A). The expression of ficolin 2 (FCN2) was negatively correlated with the relative abundances of metabolic pathways, including those corresponding to amino acid-related enzymes, one carbon pool by folate, terpenoid backbone biosynthesis, and thiamine metabolism (rho values below −0.8; P value < 0.01), and was positively correlated with glyoxylate and dicarboxylate metabolism and base excision repair (rho values, >0.8; P value, <0.01) (Fig. 6B). The expression of secreted frizzled related protein 2 (SFRP2) was positively correlated with the relative abundances of microbial amino acid metabolism pathways (i.e., valine, leucine and isoleucine, tryptophan, and lysine metabolism; rho values, >0.8; P values, <0.01) (Fig. 6C). The expression of cytokine genes encoding C-C motif chemokine ligand 19 (CCL19) and C-X-C motif chemokine ligand 13 (CXCL13) was positively correlated with microbial glycosyltransferases (rho values, >0.8; P values, <0.01), the enzymes involved in glycan biosynthesis (Fig. 6D). The relative abundance of butanoate metabolism was positively correlated with the expression of GTPase, IMAP family member 5 (GIMAP5), CD69 molecule (CD69), and thymocyte selection associated (THEMIS) proteins (rho value, below −0.8; P values, <0.01) (Fig. 6D).
FIG 6.

Correlation networks for differentially expressed genes identified in rectal-anal junction samples and predicted microbial functions at KEGG pathway hierarchical level 3. The green circles represent genes differentially expressed in the RAJ, and the red diamonds represent the predicted microbial pathways. The blue lines indicate positive correlation, and red lines indicate negative correlation; only correlations with rho values of >0.8 and P values of <0.01 are shown. Data for genes/functions that did not show a significant correlation are not shown.
DISCUSSION
This study characterized the RAJ mucosa-associated bacterial community of NS and SS yearling beef steers and its relationship with host gene expression, with the aim of identifying whether the differences in the composition of gut microbiota might be associated with E. coli O157:H7 shedding. The findings indicated that members of Firmicutes, Bacteroidetes, and Proteobacteria were the core microbes of RAJ mucosa-associated microbiota, which is consistent with previous studies on the rectum content of dairy cattle (10) and on fecal microbiota of beef and dairy cattle (11, 12). Xu et al. (7) investigated the rectal content microbiota of the same beef steers, showing that Firmicutes, Bacteroidetes, and Proteobacteria were the most predominant phyla in feces, similarly to the RAJ mucosa-associated microbiota identified in this study. Nevertheless, the relative abundances of Firmicutes, Bacteroidetes, and Proteobacteria were 53.9%, 35.6%, and 1.7% in fecal samples (7), respectively, while they were 61.5%, 27.9%, and 5.5% in mucosa, respectively, suggesting the presence of higher numbers of Firmicutes and Proteobacteria and lower numbers of Bacteroidetes associated with the mucosa of RAJ than with the fecal microbiota. In addition, studies on humans and mice also indicated that these microbial groups represent the predominant phyla in the gut, suggesting that they play an important role in the intestine of mammals (13). The current study on beef steers and that of Mao et al. on dairy cows (10) showed consistent findings, with predominance of Bacteroidetes and Firmicutes genera in the mucosa of the rectum. For example, the Prevotella and unclassified genera of Prevotellaceae and Rikenellaceae were the predominant genera belonging to Bacteroidetes, although the relative abundance of Prevotella was >5% in this study but was <2% in the study by Mao et al. (10). Also, in that study, the unclassified genera of Ruminococcaceae and Lachnospiraceae were the most abundant Firmicutes, which was in agreement with our findings. However, for Proteobacteria, Mao et al. (10) reported that Campylobacter, Desulfobulbus, and Acinetobacter were the predominant Proteobacteria genera, while Pseudomonas, Succinivibrio, and Sutterella were more predominant in this study, suggesting that beef and dairy cattle may have similar levels of gut microbial richness but that the abundance of each microbial taxon may differ. The higher abundance of Prevotella in rectal contents of beef cattle compared with dairy cattle was also reported by Durso et al. (14), further supporting this conclusion.
For the RAJ mucosa-associated microbiota, large animal-to-animal variations were observed from both taxonomic and beta-diversity analyses. Even for the predominant taxa shared by all the animals, the relative abundances differed from individual to individual. Such individual variation was also previously reported in cattle fecal microbiota by Durso et al. (14), who suggested that individual animal variation cannot be attributed simply to breed, gender, diet, age, or environmental factors. As several of these variables, including diet, environment, and gender, were controlled in this study, we suggest that the host animal may be the main driver for the individualization of the RAJ mucosa-associated microbiota.
Comparing NS and SS, the diversity indices did not differ in a statistically significant manner, suggesting that the levels of estimated richness and of evenness of identified species at the RAJ of two groups were comparable. However, the identification of unique microbes associated with NS suggests that the microbial groups that made up the RAJ microbial communities of SS and NS differ, which may play a role in the reduced colonization of E. coli O157:H7 in the NS. The presence of OTU108 (aligned to Coprococcus), which was unique in NS, has been previously documented in the feces of cattle (15). Coprococcus spp. produce butyrate and can lower the pH of the media when cultured in vitro (16). OTU45 and OTU180 were also unique to NS and were closely related to Paludibacter, a genus previously detected in colonic mucosa of cattle (17) and reported to produce propionate (18). The short-chain fatty acids, including butyrate and propionate, have been suggested to decrease the shedding of E. coli O157:H7 in cattle (19). Thus, the presence of Coprococcus-like and Paludibacter-like microbes (OTU108, OTU45, and OTU180) in NS may lead to the formation of a gut environment unfavorable to E. coli O157:H7. Indeed, the production of short-chain fatty acids (SCFA) by gut microbiota was suggested by many studies to be an important factor influencing the shedding of E. coli O157:H7 in cattle (20–22). Further study is needed to evaluate the SCFA in the RAJ content of NS and SS, as well as in vitro tests to confirm the ability of these microbes to inhibit the growth of E. coli O157:H7 and to confirm the role of these NS-unique microbes in E. coli O157:H7 shedding.
The further differential abundance analysis of microbial pathways revealed significant differences between the microbiotas of SS and NS, including lipopolysaccharide (LPS) biosynthesis, biosynthesis of ansamycins, and carbohydrate metabolism. The gene families associated with LPS biosynthesis had lower abundance for the RAJ microbiota of SS, which suggests a potential lower abundance of Gram-negative bacteria attached to the RAJ mucosa of SS. Although lower levels of LPS biosynthesis could be beneficial to the host because LPS is an endotoxin that induces the release of proinflammatory cytokines (23), a lower level of Gram-negative bacteria at the RAJ of SS may not create an environment that competitively excludes E. coli O157:H7. As Zhao et al. (24) reported, 18 fecal bacterial species (screened from 1,200 isolates from gut tissues and fecal samples of nonshedder cattle) capable of inhibiting the growth of E. coli O157:H7 were Gram-negative species and 17 of them were nonpathogenic E. coli strains (24). Therefore, the role of nonpathogenic E. coli and LPS in E. coli O157:H7 super shedding should be further studied in the future. In addition, a higher abundance of microbial gene families associated with biosynthesis of ansamycins was shown in SS. Ansamycins were previously reported to have antimicrobial and antiviral activity toward many Gram-positive bacteria and bacteriophages (25, 26). Some Gram-positive bacteria such as Lactobacillus (27) and Bifidobacterium (28), and bacteriophages such as T1-like bacteriophages (29), have been reported to have an inhibitory effect on the growth and shedding of E. coli O157:H7 in cattle. Thus, a higher potential of production of ansamycins may put beneficial Gram-positive bacteria and bacteriophage at a disadvantage, contributing to the growth of E. coli O157:H7 in SS. It is also interesting that the predicted microbial gene families associated with carbohydrate, lipid, and amino acid metabolism had levels of abundance that differed between SS and NS. This indicates that microbial nutrient metabolism by RAJ mucosal microbiota maybe an important factor associated with shedding of E. coli O157:H7. The products of such metabolisms, such as short-chain/branched-chain fatty acids and biogenic amines, can cause changes in the intestinal environment that may mediate the growth of E. coli O157:H7 (30). These bacterial metabolites, by providing energy to epithelial cells and modulating the development of the mucosal immune system and intestinal barriers, can also influence the physiology of the host (31), eventually influencing the colonization of E. coli O157:H7. It is notable that PICRUSt predicts the functions of the gut microbiota based on 16S rRNA only and that the functional classification database (KEGG Orthology) within this tool is not specific to gut microbiota. Therefore, further studies are required to assess the functional composition of rectal mucosa-attached microbiotas using metagenomics with methods to remove host DNA as well as metabolomics approaches.
The gut microbiota can influence host gut physiologies, such as energy metabolism, mucosal immune system development, gut epithelial cell proliferation, and gene expression (1). Thus, it is possible that certain microbial groups and microbial functions influence the RAJ physiology of SS by affecting host gene expression, leading to supershedding. In our previous study, the DE genes showed expression levels that were significantly different between SS and NS (9). The identified multiple correlations between the expression levels of some DE genes and the relative abundances of microbial taxa and function highlight that host-microbe interactions could play an important role in regulating E. coli O157:H7 shedding. It is worth noting that the genes encoding the members of the S100 family of proteins, including S100A8, S100A9, and S100A12, showed significant correlations with the abundances of several microbial genera, such as Clostridium and Pseudomonas. The members of the S100 protein family are associated with various functions, including protein phosphorylation, cell trafficking, and cell proliferation and differentiation (32). Clostridium has been suggested to be one of the butyrate producers in the human gut microbiota (33), and butyrate-producing Clostridium spp. specifically colonized the mucins based on an in vitro gut model (34). It has been suggested that microbial butyrate enhances the tight junctions of the epithelium, acts as a chemoattractant to neutrophils, and provides energy to the epithelial cells (35). The neutrophils and epithelial cells represent the two major types of host cells that express S100A8 and S100A9 (32). Also, S100 proteins expressed and released by epithelial cells have been suggested to serve as antimicrobials to maintain intestinal mucosal homeostasis (36). Thus, the observed positive correlation between the presence of S100A8/S100A9 and Clostridium suggests that it is possible that there is a higher abundance of butyrate-producing Clostridium at the RAJ of NS (as its level was positively correlated with the presence of S100A8/S100A9, which had higher expression in NS), leading to the development of a stronger intestinal barrier against colonization by pathogens, including E. coli O157:H7 (37). However, the quantification of Clostridium using quantitative PCR (qPCR) measurement did not show a statistically significant difference (P > 0.05) between SS and NS in the levels of RAJ mucosa-associated bacteria (data not shown), which is also in agreement with the sequencing data (compared using the Mann-Whitney U test; P > 0.05). The identification of bacterial taxa that were unique to SS and NS (OTU121 for SS and OTU66 for NS) and that were closely related to Clostridium suggests that it is possible that, although the total numbers of Clostridium were comparable between SS and NS, different Clostridium spp. were harbored by SS and NS, leading to differential abundances of butyric acid production from Clostridium. Further characterization of the Clostridium genus at the RAJ of SS and NS is needed to identify the role of Clostridium in supershedding phenomena.
The abundances of microbial pathways involved in bacterial DNA replication and RNA expression were negatively correlated with expression of S100A8 and S100A9, indicating that bacterial proliferation and gene expression may influence host gene expression. Also, the identified correlation between the abundance of bacterial metabolism of amino acids, vitamin B, and butyrate and expression of host genes suggests that bacterial metabolism plays a regulatory role in host cell gene expression and physiology. The shift of gut microbiota has long been suggested to be associated with host innate (38) and adaptive (39) immune system development. The findings presented here suggest that the differences between SS and NS in the functions of gut microbiota may contribute to the differential levels of expression of genes in the RAJ, especially those associated with microbial functions, such as cell proliferation and nutrient, vitamin, and butyrate metabolism. Our previous speculation was that the host genetic variation could lead to differential levels of expression of host genes in SS (9, 50), and the results of our current study further suggest that the gut microbiota and their functions also represent an important factor leading to differential levels of gene expression between SS and NS.
It is also noticeable that studies on intervention strategies to reduce E. coli O157:H7 shedding using probiotics and dietary modification have had inconsistent results. As reviewed by Sargeant el al. (40), only 16 of 27 trials showed decreased E. coli O157:H7 shedding that occurred as a consequence of probiotic inoculation or dietary changes. On the basis of our current study, we speculate that if interventions target the composition of the gut microbiota and host gene expression, there may be a higher possibility of reducing shedding through modifying the gut environment and/or enhanced host intestinal barriers. Thus, studies using probiotics and dietary modification should be targeted to be more efficient. Nevertheless, our findings highlight the validity of using microbial treatment and dietary modification to control E. coli O157:H7 shedding, with the focus on modifying the gut microbiota and gut environment to be less favorable for the survival of E. coli O157:H7.
Conclusion.
In conclusion, this study characterized the RAJ mucosa-associated microbiome of beef steers with different E. coli O157:H7 shedding phenotypes. Even with the small sample size, we still identified potential microbial members phenotypically associated with the supershedding phenomenon. The results showed that the members of Firmicutes, Bacteroidetes, and Proteobacteria were the predominant phyla of RAJ microbiota and that metabolism, genetic information processing, and cellular signaling were the most abundant microbial functions. Compared between SS and NS, unique microbial taxa were identified for each group, which could possibly lead to different abundances of the gene families that constitute the metagenome. The difference in microbiota composition could influence the growth and colonization of E. coli O157:H7 at the RAJ through two mechanisms. First, certain commensal bacteria, such as the short-chain fatty-acid-producing Coprococcus, Clostridium, and Paludibacter species, may enhance host immune system and intestinal barriers to reduce E. coli O157:H7 growth and colonization. Second, microbial metabolites, such as short-chain fatty acids and antimicrobials, may shape the gut environment of cattle to inhibit/enhance the growth of E. coli O157:H7. Our findings highlight the possibility of altering the gut environment to control E. coli O157:H7 shedding through modifying the gut microbiota and host gene expression. However, future studies focusing on the relationship between the identified potential microbes and the shedding of E. coli O157:H7 in cattle using larger numbers of animals are needed to verify our findings due to the small sample size in this study.
MATERIALS AND METHODS
Animal tissue sample collection and sample preparation for sequencing.
The details of the animal trial have been reported previously (7, 41). The trial followed the Canadian Council of Animal Care Guidelines (Animal Care Committee protocol number 1120) and was reviewed and approved by the Animal Care Committee of Lethbridge Research and Development Centre, Agriculture Agri-food Canada. Animal tissue sample collection and SS identification protocols have also been described in previous publications (9, 41). Briefly, the fecal samples (50 g) were collected from 400 beef steers in an Alberta feedlot. Plate counting was used to enumerate E. coli O157:H7 using MacConkey agar with sorbitol, cefixime, and tellurite (CT-SMAC) (Dalynn Biologicals, Calgary, AB, Canada). The positive culture isolate results were then confirmed with an E. coli O157:H7 Latex Test kit (Oxoid Ltd., Basingstoke, Hampshire, United Kingdom), followed by further verification performed using a multiplex PCR targeting the VT gene, eaeA, and fliC. The primer sequences for multiplex PCR were as follows: for VT1f, CATTGTCTGGTGACAGTAGCT; for VT1r, CCCGTAATTTGCGCACTGAG; for VT2f, CCATGACAACGGACAGCAGTT; for VT2r, CCTGTCAACTGAGCACTTTG; for eaeAf, GTGGCGAATACTGGCGAGACT; for eaeAr, CCCCATTCTTTTTCACCGTCG; for fliCf, GCGCTGTCGAGTTCTATCGAGC; for fliCr, CAACGGTGACTTTATCGCCATTCC. The details of the multiplex procedures were previously described by Gannon et al. (42). Five of the selected SS (among 11 identified SS) were slaughtered within 11 days of being purchased, and the fecal shedding of E. coli O157:H7 was monitored daily until slaughter. All the SS steers were feces positive for E. coli O157:H7 before slaughter. For animals with negative results by plating, 1 g of fecal sample was mixed with 9 ml of tryptic soy broth (TSB) and incubated for 6 h at 37°C, followed by immunomagnetic enrichment performed using anti-E. coli O157:H7 Dynabeads. The 50 μl of bead-bacteria mixture was then plated onto CT-SMAC and incubated at 37°C for 18 to 24 h. Positive colonies (non-sorbitol-fermenting clear colonies) were randomly selected for latex and PCR confirmation as described above. Cattle with negative results from both enumeration (by culture) and immunomagnetic enrichment were classified as nonshedders (NS), and five NS were used as control animals.
The RAJ tissue samples were ground into fine powder prior to DNA extraction. DNA extraction was performed using about 100 mg of tissue and a QIAamp DNA Stool minikit (Qiagen, Germantown, MD, USA). The amplicon of partial bacterial 16S rRNA gene was generated using a pair of primers (27f AGAGTTTGATCMTGGCTCAG and 519r GWATTACCGCGGCKGCTG) targeting the V1–V3 hypervariable region of the 16S rRNA gene (43). The amplicon sequencing (paired end, 2 × 300) was carried out by the Genome Quebec Innovation Centre (Montreal, Quebec, Canada) using a MiSeq platform.
Sequence data processing and microbial community analysis.
The removal of adapter sequences and quality control for raw sequencing reads were performed using fastq-mcf (44). The pairs of reads were joined using QIIME (quantitative insights into microbial ecology) (45), and the joined reads were filtered with the following criteria: minimum length, 400; maximum length of homopolymer, 8. Removal of reverse primers was enabled, and the default settings were used for the rest of the parameters. Chimera detection and removal were performed using usearch61 (46). The method used for picking the operational taxonomic units (OTUs) followed the de novo protocol using usearch61 at 97% identity, followed by taxonomic assignment. To evaluate the adequacy of the sequencing depth for detecting the microbes present in the sequenced samples, the Good's coverage index was calculated using the formula Good's coverage = 1 − S/N, where S is the number of singleton OTUs and N is the total number of OTUs in a sample. The alpha-diversity (within-sample diversity) was evaluated using Chao1 and Shannon indices. The Chao1 index estimates the number of species present in a sample and was calculated with the formula Chao1 index value = Sobs + N12/2N2, where Sobs is the total number of species detected in a sample, N1 is the number of species observed once, and N2 is the number of species observed twice. The Shannon index estimates both the richness and evenness of a microbial community and was calculated with the formula Shannon index value = −ΣPi * ln(Pi), where Pi is the proportion of the ith OTU to the total number of OTUs in a sample. To evaluate beta-diversity (between-samples diversity), a principal-coordinate analysis was performed based on a weighted UniFrac distance. All the index and beta-diversity data were calculated using the script implemented in QIIME. For the taxonomy analysis, only taxa with a relative abundance of >0.1% in at least one sample were defined as detectable. The phylogenetic tree was built using the FastTree algorithm implemented by QIIME.
Microbial function prediction.
PICRUSt was used to predict the functional composition of the metagenome using KEGG (Kyoto Encyclopedia of Genes and Genomes) orthology classification schemes at the second and third KEGG Pathway hierarchy levels (47). KEGG Pathway is a collection of hierarchically classified pathway maps divided into four classification levels, with the higher levels being more specific and the third and fourth levels corresponding to individual pathway maps and KEGG orthology entries, respectively. For PICRUSt analysis, pathways belonging to human diseases, organismal systems, drug development, and plant functions were filtered out, as they do not reflect microbial functions.
Differential abundance analysis and correlation analysis.
All the data were presented as means ± standard deviations unless otherwise indicated. The differential abundance analysis was performed using the edgeR statistical analysis package (48). The edgeR package modeled the read counts as a negative binomial distribution and normalized the read counts according to sequencing depth by calculating the scaling factors computed by internal functions of edgeR. After data fitting to a negative binomial distribution and dispersion estimation were performed, the differential abundance was determined by an exact test implemented in edgeR. The differential abundance analysis was performed at the OTU level for taxonomic data and at the second and third KEGG pathway hierarchical levels for functional comparisons (49). Only OTUs/microbial functions with a relative abundance of >0.1% in >50% of steers of either group (NS or SS) were subjected to differential abundance analysis using edgeR. A P value of less than 0.1 was considered to represent a statistically significant difference between NS and SS. The correlation was performed using Spearman's rank correlation, with a Spearman's correlation coefficient rho value of >0.8 and a P value of <0.01 used as the cutoff values to select significantly correlated pairs.
Accession number(s).
The microbiome sequencing data are described and available under NCBI BioProject accession no. PRJNA379625.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Munns for her contribution to the identification of the supershedder steers and for fecal E. coli O157:H7 quantitation.
We appreciate the financial support provided by Alberta Innovates Bio Solutions (Project FSC-12-017) and a NSERC discovery grant. The funders did not participate in study design, data collection and interpretation, and the decision to submit the work for publication.
O.W. and L.L.G. conceived and designed the experiments. O.W., T.A.M., and B.S. performed the experiments. O.W. and L.L.G. analyzed the data. L.L.G. contributed reagents/materials/analysis tools. O.W., T.A.M., G.P., K.S., B.S., and L.L.G. wrote and edited the paper.
We have declared that we have no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01738-17.
REFERENCES
- 1.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. 2012. Host-gut microbiota metabolic interactions. Science 336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 2.Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, et al. . 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buffie CG, Pamer EG. 2013. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol 13:790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naylor SW, Roe AJ, Nart P, Spears K, Smith DGE, Low JC, Gally DL. 2005. Escherichia coli O157:H7 forms attaching and effacing lesions at the terminal rectum of cattle and colonization requires the LEE4 operon. Microbiology 151:2773–2781. doi: 10.1099/mic.0.28060-0. [DOI] [PubMed] [Google Scholar]
- 5.Baines D, Lee B, McAllister T. 2008. Heterogeneity in enterohemorrhagic Escherichia coli O157:H7 fecal shedding in cattle is related to Escherichia coli O157:H7 colonization of the small and large intestine. Can J Microbiol 54:984–995. doi: 10.1139/W08-090. [DOI] [PubMed] [Google Scholar]
- 6.Chase-Topping M, Gally D, Low C, Matthews L, Woolhouse M. 2008. Super-shedding and the link between human infection and livestock carriage of Escherichia coli O157. Nat Rev Microbiol 6:904–912. doi: 10.1038/nrmicro2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Y, Dugat-Bony E, Zaheer R, Selinger L, Barbieri R, Munns K, McAllister TA, Selinger LB. 2014. Escherichia coli O157:H7 super-shedder and non-shedder feedlot steers harbour distinct fecal bacterial communities. PLoS One 9:e98115. doi: 10.1371/journal.pone.0098115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malmuthuge N, Li M, Chen Y, Fries P, Griebel PJ, Baurhoo B, Zhao X, Guan LL. 2012. Distinct commensal bacteria associated with ingesta and mucosal epithelium in the gastrointestinal tracts of calves and chickens. FEMS Microbiol Ecol 79:337–347. doi: 10.1111/j.1574-6941.2011.01220.x. [DOI] [PubMed] [Google Scholar]
- 9.Wang O, Liang G, McAllister TA, Plastow G, Stanford K, Selinger B, Guan LL. 2016. Comparative transcriptomic analysis of rectal tissue from beef steers revealed reduced host immunity in Escherichia coli O157:H7 super-shedders. PLoS One 11:e0151284. doi: 10.1371/journal.pone.0151284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mao S, Zhang M, Liu J, Zhu W. 2015. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci Rep 5:16116. doi: 10.1038/srep16116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaevska M, Videnska P, Sedlar K, Bartejsova I, Kralova A, Slana I. 2016. Faecal bacterial composition in dairy cows shedding Mycobacterium avium subsp. paratuberculosis in faeces in comparison with nonshedding cows. Can J Microbiol 62:538–541. doi: 10.1139/cjm-2015-0814. [DOI] [PubMed] [Google Scholar]
- 12.Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL, Huse SM, Sogin ML. 2011. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl Environ Microbiol 77:2992–3001. doi: 10.1128/AEM.02988-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. 2005. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durso LM, Harhay GP, Smith TPL, Bono JL, DeSantis TZ, Harhay DM, Andersen GL, Keen JE, Laegreid WW, Clawson ML. 2010. Animal-to-animal variation in fecal microbial diversity among beef cattle. Appl Environ Microbiol 76:4858–4862. doi: 10.1128/AEM.00207-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim M, Wells JE. 2016. A meta-analysis of bacterial diversity in the feces of cattle. Curr Microbiol 72:145–151. doi: 10.1007/s00284-015-0931-6. [DOI] [PubMed] [Google Scholar]
- 16.Holdeman LV, Moore WEC. 1974. New genus, Coprococcus, twelve new species, and emended descriptions of four previously described species of bacteria from human feces. Int J Syst Evol Microbiol 24:260–277. doi: 10.1099/00207713-24-2-260. [DOI] [Google Scholar]
- 17.Reti KL, Thomas MC, Yanke LJ, Selinger LB, Inglis GD. 2013. Effect of antimicrobial growth promoter administration on the intestinal microbiota of beef cattle. Gut Pathog 5:8. doi: 10.1186/1757-4749-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueki A, Akasaka H, Suzuki D, Ueki K. 2006. Paludibacter propionicigenes gen. nov., sp. nov., a novel strictly anaerobic, Gram-negative, propionate-producing bacterium isolated from plant residue in irrigated rice-field soil in Japan. Int J Syst Evol Microbiol 56:39–44. doi: 10.1099/ijs.0.63896-0. [DOI] [PubMed] [Google Scholar]
- 19.Jacob ME, Callaway TR, Nagaraja TG. 2009. Dietary interactions and interventions affecting Escherichia coli O157 colonization and shedding in cattle. Foodborne Pathog Dis 6:785–792. doi: 10.1089/fpd.2009.0306. [DOI] [PubMed] [Google Scholar]
- 20.Bach SJ, Selinger LJ, Stanford K, McAllister TA. 2005. Effect of supplementing corn- or barley-based feedlot diets with canola oil on faecal shedding of Escherichia coli O157:H7 by steers. J Appl Microbiol 98:464–475. doi: 10.1111/j.1365-2672.2004.02465.x. [DOI] [PubMed] [Google Scholar]
- 21.Kudva IT, Hunt CW, Williams CJ, Nance UM, Hovde CJ. 1997. Evaluation of dietary influences on Escherichia coli O157:H7 shedding by sheep. Appl Environ Microbiol 63:3878–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cobbold RN, Desmarchelier PM. 2004. In vitro studies on the colonization of bovine colonic mucosa by Shiga-toxigenic Escherichia coli (STEC). Epidemiol Infect 132:87–94. doi: 10.1017/S0950268803001432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti J-F, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. 2007. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 24.Zhao T, Doyle MP, Harmon BG, Brown CA, Mueller POE, Parks AH. 1998. Reduction of carriage of enterohemorrhagic Escherichia coli O157:H7 in cattle by inoculation with probiotic bacteria. J Clin Microbiol 36:641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price PJ, Suk WA, Skeen PC, Spahn GJ, Chirigos MA. 1977. Geldanamycin inhibition of 3-methylcholanthrene-induced rat embryo cell transformation. Proc Soc Exp Biol Med 155:461–463. doi: 10.3181/00379727-155-39830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wehrli W. 1977. Ansamycins. Chemistry, biosynthesis and biological activity. Top Curr Chem 72:21–49. [DOI] [PubMed] [Google Scholar]
- 27.Peterson RE, Klopfenstein TJ, Erickson GE, Folmer J, Hinkley S, Moxley RA, Smith DR. 2007. Effect of Lactobacillus acidophilus strain NP51 on Escherichia coli O157:H7 fecal shedding and finishing performance in beef feedlot cattle. J Food Prot 70:287–291. doi: 10.4315/0362-028X-70.2.287. [DOI] [PubMed] [Google Scholar]
- 28.Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H. 2011. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469:543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- 29.Niu YD, McAllister TA, Nash JHE, Kropinski AM, Stanford K. 2014. Four Escherichia coli O157:H7 phages: a new bacteriophage genus and taxonomic classification of T1-like phages. PLoS One 9:e100426. doi: 10.1371/journal.pone.0100426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neis EPJG, Dejong CHC, Rensen SS. 2015. The role of microbial amino acid metabolism in host metabolism. Nutrients 7:2930–2946. doi: 10.3390/nu7042930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tremaroli V, Bäckhed F. 2012. Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 32.Donato R. 2001. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol 33:637–668. doi: 10.1016/S1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 33.Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ. 2002. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 217:133–139. doi: 10.1111/j.1574-6968.2002.tb11467.x. [DOI] [PubMed] [Google Scholar]
- 34.Van den Abbeele P, Belzer C, Goossens M, Kleerebezem M, De Vos WM, Thas O, De Weirdt R, Kerckhof F-M, Van de Wiele T. 2013. Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. ISME J 7:949–961. doi: 10.1038/ismej.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A. 2013. Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog 5:23. doi: 10.1186/1757-4749-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cua DJ, Tato CM. 2010. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol 10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 37.Peng L, Li Z-R, Green RS, Holzman IR, Lin J. 2009. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr 139:1619–1625. doi: 10.3945/jn.109.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu H, Mazmanian SK. 2013. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol 14:668–675. doi: 10.1038/ni.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brestoff JR, Artis D. 2013. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol 14:676–684. doi: 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sargeant JM, Amezcua MR, Rajic A, Waddell L. 2007. Pre-harvest interventions to reduce the shedding of E. coli O157 in the faeces of weaned domestic ruminants: a systematic review. Zoonoses Public Health 54:260–277. doi: 10.1111/j.1863-2378.2007.01059.x. [DOI] [PubMed] [Google Scholar]
- 41.Munns KD, Selinger L, Stanford K, Selinger LB, McAllister TA. 2014. Are super-shedder feedlot cattle really super? Foodborne Pathog Dis 11:329–331. doi: 10.1089/fpd.2013.1621. [DOI] [PubMed] [Google Scholar]
- 42.Gannon VP, D'Souza S, Graham T, King RK, Rahn K, Read S. 1997. Use of the flagellar H7 gene as a target in multiplex PCR assays and improved specificity in identification of enterohemorrhagic Escherichia coli strains. J Clin Microbiol 35:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.St-Pierre B, Wright A-DG. 2014. Comparative metagenomic analysis of bacterial populations in three full-scale mesophilic anaerobic manure digesters. Appl Microbiol Biotechnol 98:2709–2717. doi: 10.1007/s00253-013-5220-3. [DOI] [PubMed] [Google Scholar]
- 44.Aronesty E. 2013. Comparison of sequencing utility programs. Open Bioinform J 7:1–8. doi: 10.2174/1875036201307010001. [DOI] [Google Scholar]
- 45.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 47.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang O, McAllister TA, Plastow G, Stanford K, Selinger B, Guan LL. 2017. Host mechanisms involved in cattle Escherichia coli O157 shedding: a fundamental understanding for reducing foodborne pathogen in food animal production. Sci Rep 7:7630. doi: 10.1038/s41598-017-06737-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.