

ABSTRACT
The difficulty involved in quantifying biogeochemically significant microbes in marine sediments limits our ability to assess interspecific interactions, population turnover times, and niches of uncultured taxa. We incubated surface sediments from Cape Lookout Bight, North Carolina, USA, anoxically at 21°C for 122 days. Sulfate decreased until day 68, after which methane increased, with hydrogen concentrations consistent with the predicted values of an electron donor exerting thermodynamic control. We measured turnover times using two relative quantification methods, quantitative PCR (qPCR) and the product of 16S gene read abundance and total cell abundance (FRAxC, which stands for “fraction of read abundance times cells”), to estimate the population turnover rates of uncultured clades. Most 16S rRNA reads were from deeply branching uncultured groups, and ∼98% of 16S rRNA genes did not abruptly shift in relative abundance when sulfate reduction gave way to methanogenesis. Uncultured Methanomicrobiales and Methanosarcinales increased at the onset of methanogenesis with population turnover times estimated from qPCR at 9.7 ± 3.9 and 12.6 ± 4.1 days, respectively. These were consistent with FRAxC turnover times of 9.4 ± 5.8 and 9.2 ± 3.5 days, respectively. Uncultured Syntrophaceae, which are possibly fermentative syntrophs of methanogens, and uncultured Kazan-3A-21 archaea also increased at the onset of methanogenesis, with FRAxC turnover times of 14.7 ± 6.9 and 10.6 ± 3.6 days. Kazan-3A-21 may therefore either perform methanogenesis or form a fermentative syntrophy with methanogens. Three genera of sulfate-reducing bacteria, Desulfovibrio, Desulfobacter, and Desulfobacterium, increased in the first 19 days before declining rapidly during sulfate reduction. We conclude that population turnover times on the order of days can be measured robustly in organic-rich marine sediment, and the transition from sulfate-reducing to methanogenic conditions stimulates growth only in a few clades directly involved in methanogenesis, rather than in the whole microbial community.
IMPORTANCE Many microbes cannot be isolated in pure culture to determine their preferential growth conditions and predict their response to changing environmental conditions. We created a microcosm of marine sediments that allowed us to simulate a diagenetic profile using a temporal analog for depth. This allowed for the observation of the microbial community population dynamics caused by the natural shift from sulfate reduction to methanogenesis. Our research provides evidence for the population dynamics of uncultured microbes as well as the application of a novel method of turnover rate analysis for individual taxa within a mixed incubation, FRAxC, which stands for “fraction of read abundance times cells,” which was verified by quantitative PCR. This allows for the calculation of population turnover times for microbes in a natural setting and the identification of uncultured clades involved in geochemical processes.
KEYWORDS: microbial ecology
INTRODUCTION
Marine sediment microbial communities are phylogenetically diverse and abundant, totaling ∼1029 cells globally (1–4). However, little is known about the abundance dynamics of individual clades in situ or about the metabolisms of the abundant deeply branching clades of uncultured bacteria and archaea. Many marine sediments shift from sulfate reduction to methane production as they are buried. The identification of microorganisms involved in these processes and their growth rates are known only from pure cultures (5, 6). However, pure cultures usually experience plentiful, well-defined substrates and no competition or mutualism. In contrast, natural marine sediments contain a diverse and poorly characterized array of organic matter substrates and a diverse ecosystem of uncultured bacteria and archaea, so in situ population dynamics and the identities of microbes involved in a particular metabolism may differ from those of cultures (5, 7–10).
Inferring the rates of metabolism or changes in abundance from down-core concentration profiles requires the application of sediment age models as well as accounting for molecular diffusion/advection and depositional changes (11–15). To obviate using sediment depth as a proxy for time, we incubated sediments from Cape Lookout Bight, North Carolina, to measure metabolic processes and population changes in real time. This shallow methane seep experiences rapid organic matter deposition and high microbial activity, so oxygen and nitrate are depleted within several millimeters of the sediment-water interface and sulfate can be depleted on a reasonable laboratory time scale (16–19).
To measure changes in population sizes of individual clades over time, we multiplied relative 16S rRNA gene amplicon abundances by total cell counts (FRAxC, which stands for “fraction of read abundance times cells”) and made quantitative PCR (qPCR) measurements. These data are not absolutely quantitative, since the outcomes are biased by DNA extraction and primer-based amplification (20, 21). They are, however, relatively quantitative within a single clade over time, which allows for the calculation of turnover times. This is because the difficult-to-measure parameter accounting for extraction and amplification bias is mathematically removed in turnover rate calculations, assuming that the parameter does not change over the course of the experiment (see Materials and Methods). We hypothesized that taxa closely related to cultured sulfate reducers would decrease after sulfate depletion and taxa closely related to cultured methanogens would increase, both of them at rates that are different from those in pure culture. We further hypothesized that many currently uncultured clades would shift populations at this inflection point as well, since even if they are not directly involved in methane or sulfur cycling, they may be indirectly dependent on anaerobic respiration to remove their reduced compounds.
RESULTS
Concentrations of sulfate and pH were similar in each of the three microcosm incubations and were plotted as the means of the results for three incubations for each time point (Fig. 1). Hydrogen and methane concentrations were more variable between incubations, so individual data points were plotted separately. Sulfate decreased at a rate of 0.20 ± 0.01 mM/day for the first 68 days, increased at 0.04 ± 0.00 mM/day during days 68 to 94, and was steady from day 94 to day 122 (Fig. 1A). There was no identifiable difference in the rate of sulfate decrease due to the added methane in incubation 3, suggesting that a leak, not sulfate-dependent methane oxidation, was responsible for the methane disappearance. The slight increase in sulfate concentrations after day 68 could have been the result of sulfide reoxidation with iron (22) or sulfide reoxidation during sample handling, which may inadvertently introduce atmospheric oxygen (13). The average pH of the three incubations decreased steadily, beginning at 8.23 ± 0.09 on day 0 and 7.36 ± 0.06 on day 107, before increasing to 7.56 ± 0.03 on day 122 (Fig. 1B).
FIG 1.
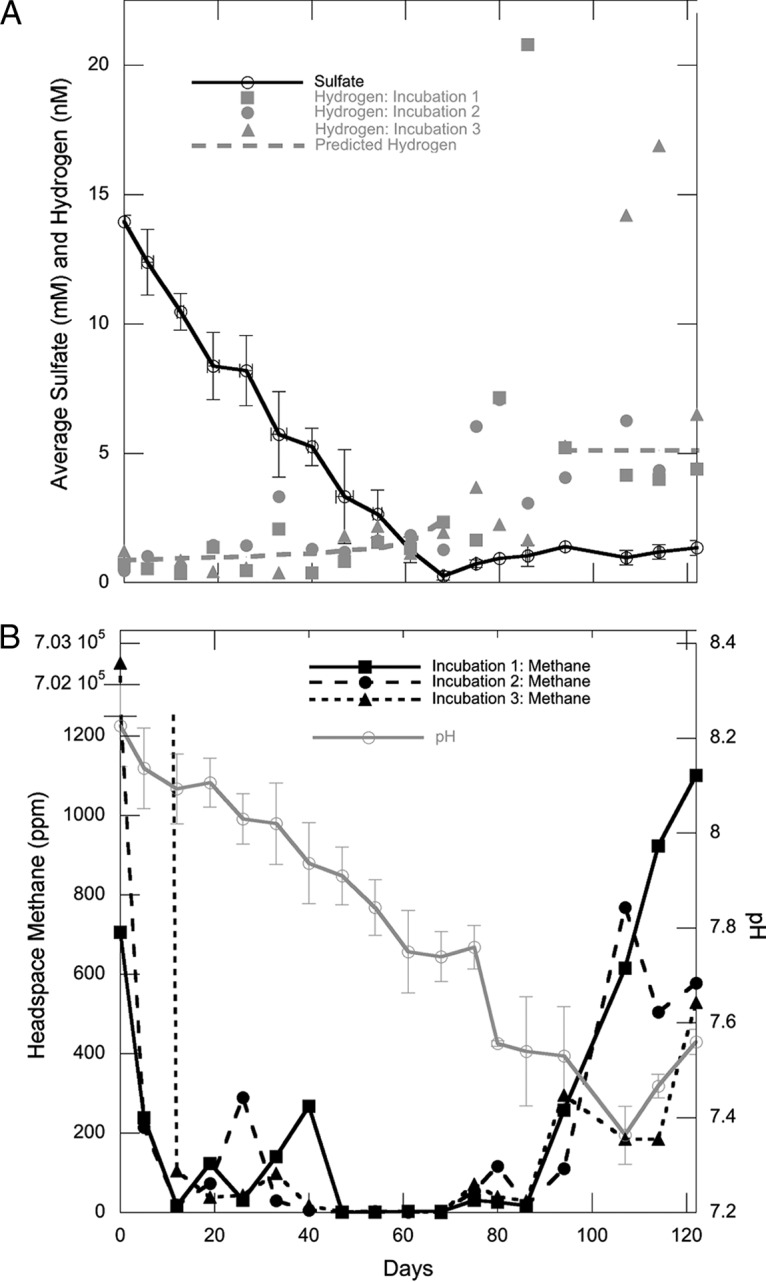
(A) Porewater concentrations of sulfate (black) and hydrogen (gray). Dashed lines represent hydrogen calculated for sulfate reducers operating in thermodynamic equilibrium (0 to 68 days) and for methanogens (96 to 122 days). The hydrogen values from days 68 to 96 appear to be out of equilibrium with methanogenesis since methanogen populations have not grown yet. (B) Headspace methane concentrations (black) and pH (gray). The x axis label applies to both panels. Sulfate and pH are shown as the means of the values from all three incubations with error bars of 1 standard deviation. The initial methane concentrations in incubation 3 are discussed in Results.
Hydrogen concentrations were extremely low during days 0 to 68 (1.21 ± 0.70 nM, n = 33), during sulfate reduction (Fig. 1A). These values are consistent with those predicted for sulfate reducers operating at their minimum energy (1.22 ± 0.45 nM) (18). Incubation 3 had a hydrogen value of 54 nM on day 40 that is not plotted because experimental error cannot be ruled out for such an extreme single outlier. After sulfate depletion on day 68, hydrogen increased in all three incubations to 4 to 21 nM. These values are as high as, or higher than, the value measured for methanogen maintenance energy of 5.11 nM (18).
In order to disturb the sediment as little as possible during mixing, methane was not subjected to even careful purging from the incubations during the setup. Therefore, on day zero, headspaces of incubations 1 and 2 contained methane (1,276.1 and 703.7 ppm), and incubation 3 contained a high quantity of methane (700,490.8 ppm) since it was added manually. Methane rapidly decreased to similar concentrations (78.3 ± 42.2 ppm) in all three incubations by day 19 (Fig. 1B); an intermediate data point of 11,044 ppm on day 5 for incubation 3 was not plotted because it would have required a second y axis break. Methane varied between time points and incubations from days 19 to 47, likely resulting from the remaining in situ methane coming gradually out of porewater solution during this time. Evidence for this is that heavy agitation for 1 min is usually required to release all methane from 3 ml sediment during routine measurements (13). No such agitation was applied to our incubations, in order to decrease sediment disturbance. From days 47 to 68, methane was extremely low in all three incubations (2.14 ± 0.4 ppm), suggesting that the residual methane left over from the in situ concentrations had outgassed by day 47. Methane increased slightly in all three incubations after day 68 but was not continuously produced until day 86 through the end of the experiment (18.7 ± 25.3 ppm/day). The switch in pH trend from decreasing to increasing is potentially influenced by the switch from CO2-producing sulfate reduction and fermentation to CO2-consuming methanogenesis and fermentation, thereby pushing the carbonate balance away from carbonic acid. The autoclaved control had high sulfate (12.60 mM), low hydrogen (0.64 nM), low methane (2.70 ppm), and neutral pH (7.55) after 260 days, suggesting that the decrease in sulfate and the increase in hydrogen and methane observed in our incubations were due to biological activity.
Quantitative PCR and direct cell counts of the total microbial community.
For all three incubations, total cell abundance determined by direct cell counts remained on the order of 109 cells/g sediment throughout the experiment (see Fig. S1A in the supplemental material) but decreased in the first 33 days, remained steady from days 33 through 80, and then decreased on day 86, although it is impossible to determine whether this single time point is significant. After this, cell counts increased until 122 days but varied over time and between incubations. Archaeal 16S rRNA gene copies/g sediment increased in all three incubations after 80 days and decreased again by 120 days (Fig. S1B). Bacterial 16S rRNA gene copies/g sediment remained steady throughout the experiment, with one high value on day 94 in incubation 2 (Fig. S1C).
High-abundance 16S rRNA gene amplicon sequences.
DNA sequencing of 16S rRNA sequences suggests that about 75% were uncultured at the genus level. Throughout the 122 days, bacteria represented ∼90% of the amplicons (Fig. 2). The 12 most-abundant bacterial genera in all three incubations accounted for ∼40% of sequences and demonstrated no observable response to sulfate depletion that was consistent between the three replicates (Fig. 3). Of these, the FRAxC of Spirochaeta and the unclassified JTB255 group within Xanthamonodales increased gradually throughout the incubation. The Spirochaeta, including members such as Spirochaeta sp. and Haliea sp., as well as several others, are capable of fermenting a diverse range of hydrocarbon, proteinaceous, and carbohydrate substrates (23–25). The uncultured JTB255 organisms are putative sulfur oxidizers and have recently been implicated in “dark carbon fixation” in marine sediments (26, 27). The rest of the 12 most-abundant bacterial genera were Anaerolineaceae in the phylum Chloroflexi, which did not change in FRAxC during the incubation and are believed to be obligate anaerobes associated with the degradation of oil-related compounds (28–30), as well as uncultured members of the Desulfobacteraceae, which decreased gradually by FRAxC throughout the incubation, and are likely sulfate reducers.
FIG 2.

FRAxC for phylum level classification as determined by the Silva reference database for archaea (A) and the 10 most-abundant bacteria (B); the remaining taxa were pooled into “Other Bacteria,” all shown as the means of results for the three incubations at each time point. The x axis label applies to both panels.
FIG 3.

FRAxC for the 12 most-abundant genera, shown as the means of results for the three incubations at each time point. Some taxa were scaled in order to be plotted concurrently.
The most-abundant archaeal genera were from the uncultured phylum Woesearchaeota. and two uncultured groups within the Thermoplasmatales order in the Euryarchaeota, CCA47 and marine benthic group D (MBG-D), none of which changed in FRAxC after sulfate depletion. Woesearchaeota have been hypothesized to be symbionts due to apparent genome reductions (31), MBG-D has genomic features suggesting protein fermentation (32), and CCA47 has no hypothesized functions but is associated with anoxic environments (33). The coverage estimates using Good's coverage estimator were 87% or above for all samples.
Methanogenic communities.
The following eight genera exhibited strong positive trends in relative 16S rRNA gene amplicon abundance from days 68 to 122 in all three incubations: three uncultured genera from Methanomicrobiales, two uncultured genera from Methanosarcinales, Methanosaeta (within the Methanosarcinales), an uncultured genus from Syntrophaceae, and an uncultured genus from the archaeal group Kazan-3A-21, which contains organisms that are related to the class WSA2 but uncultured at the order level (Fig. 4A and B) (34). On day 80, sequences for organisms from the Methanosarcinales and Methanomicrobiales decreased in all three incubations, which may be responsible for the drop in methane observed the following week, but it is difficult to conclude this from a single data point. qPCR measurements of the 16S rRNA genes of Methanomicrobiales and Methanosarcinales were low during days 0 to 80, with the exception of an isolated data point in incubation 1, and increased at day 86 for all three incubations (Fig. 4C and D). Quantitative PCR measurements for anaerobic methanotrophic archaea group 1 (ANME-1) and group 2 ANME-2 were too close to the quantification limit to be significant. Three potentially methanogenic groups did not increase noticeably after day 68 and received very few total reads: Methanococcoides sp., Methanolobus sp., and Methanosarcina sp.
FIG 4.

FRAxC of methanogenic orders Methanomicrobiales (black) and Methanosarcinales (gray) (A) and of unclassified Syntrophaceae (red) and uncultured Kazan-3A-21 group (blue) members (B). Copies of 16S rRNA genes per gram dry weight measured by qPCR for Methanomicrobiales (C) and Methanosarcinales (D) are shown as the means of replicate measurements for each incubation. Error bars represent the range of each sample. The x axis label “Days” applies to all panels. Using a single-factor analysis of variance (ANOVA) (P < 0.05), biological qPCR replicates were statistically significant for Methanosarcinales and not for Methanomicrobiales, and FRAxC values were statistically significant for Methanosarcinales and Kazan-3A-21, but not for Methanomicrobiales and uncultured Syntrophacaea. The vertical line at day 68 in each panel denotes the time of sulfate depletion.
Turnover times for Methanomicrobiales, Methanosarcinales, Kazan-3A-21, and Syntrophaceae calculated from days 80 through 122 ranged from 9.2 to 12.6 days and were not significantly different from each other (Table 1). Within the Methanosarcinales, Methanosaeta species were abundant enough to have a separate turnover time measurement, which was 7.4 ± 4.5 days.
TABLE 1.
Turnover times
| Quantification method | Putative metabolism | Taxon | Turnover time (days) |
Period of exponential growth (days) | |
|---|---|---|---|---|---|
| Avg (n = 3) | SD | ||||
| qPCR | Methanogenesis | Methanomicrobiales | 9.7 | 3.9 | 80–122 |
| Methanogenesis | Methanosarcinales | 12.6 | 4.1 | 80–122 | |
| 16S rRNA amplicon | Methanogenesis | Methanomicrobiales | 9.4 | 5.8 | 80–122 |
| Methanogenesis | Methanosarcinales | 9.2 | 3.5 | 80–122 | |
| Methanogenesis | Methanosaeta sp. | 7.4 | 4.5 | 80–122 | |
| Unknown | Kazan-3A-21 | 10.6 | 3.6 | 80–122 | |
| Fermentation | Unclassified Syntrophaceae | 14.7 | 6.9 | 80–122 | |
| Sulfate reduction | Desulfobacterium sp. | 24.5 | 31.4 | 0–19 | |
| Sulfate reduction | Desulfovibrio sp. | 35.7 | 38.2 | 0–19 | |
Groups related to cultured sulfate-reducing bacteria.
Twenty-five genera from orders with cultured members capable of sulfate reduction were present throughout the 122-day incubation, but none changed after sulfate depletion on day 68. The general trend among these sequences was either a slow decline over the course of 122 days (Desulfobacula sp., Desulfosarcina sp., Desulfatirhabdium sp., and Sva0081 sediment group) (Fig. 5) or no clear trend over time. The only increase in these groups that was consistent for all three incubations occurred in the first 19 days for Desulfovibrio sp., Desulfobacter sp., and Desulfobacterium sp., with turnover times of 4.5 to 79.7 days (Table 1; Fig. 5), after which they declined. Putative sulfate reducers accounted for 7 to 12% of the total sequences initially, and this fraction decreased to 5 to 10% by day 47 and remained there for the duration of the 122-day incubation. Orders containing sulfide-oxidizing bacteria, such as Campylobacterales (Sulfurimonas sp.) and Thiotrichales (Thiomicrospira sp.), either were low in abundance or not detected or demonstrated no significant increases in abundance over time. Obligate iron- and manganese-reducing bacteria were not abundant in sequence libraries and did not increase during the 122-day incubation. We were unable to quantify sulfate reduction functional genes using qPCR in order to support the trends in sulfate-reducing bacteria observed with 16S rRNA gene amplicon abundance.
FIG 5.
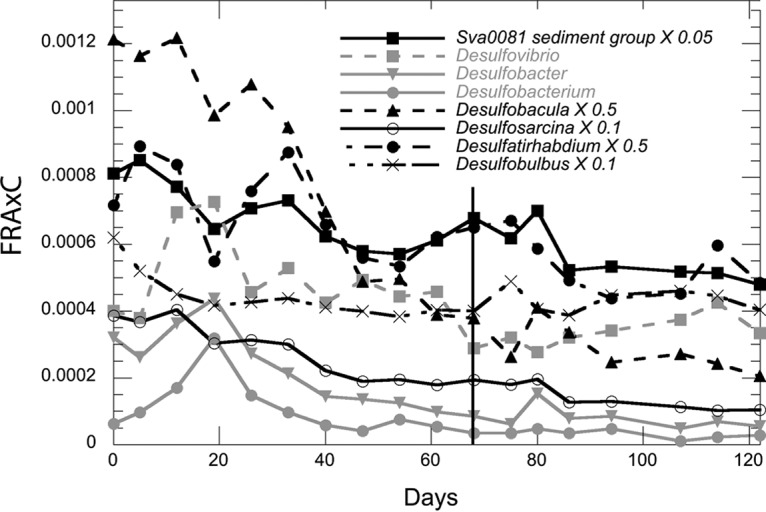
FRAxC for putative sulfate-reducing bacteria that decline steadily with time as determined by a net loss in relative abundance. Desulfobacula, Desulfosarcina, the Sva0081 sediment group, Desulfatirhabdium, and an unclassified member of Desulfarculaceae were scaled in order to be plotted concurrently. The vertical line at day 68 notes the point of sulfate depletion. Data points are the means of results for three incubations. For clarity, error bars are not shown, but they support the lack of trends over time as shown by the means.
DISCUSSION
In all three microcosm incubations, methane, sulfate, and hydrogen dynamics agreed with previous studies showing that sulfate reduction prevents methanogenesis (either hydrogenotrophic or acetoclastic) through the thermodynamic control of hydrogen (18, 35). Sulfate reducers, although phylogenetically diverse, did not increase in relative abundance consistently throughout the phase of sulfate reduction. This supports the prediction from geochemistry that sulfate reducers held hydrogen at a level meeting their maintenance energy requirements, which are too low to allow appreciable growth. Our results suggest that the initial community of sulfate reducers was already at the carrying capacity for sulfate reducers in a maintenance state before the experiment started. As sulfate concentrations decreased, fewer cells could be supported by sulfate reduction, and the relative abundance of total sulfate reducers trended downward (Fig. 5). Although total sulfate reducers decreased over the course of sulfate reduction, Desulfovibrio sp., Desulfobacter sp., and Desulfobacterium sp. increased over the first 19 days. This suggests that these three groups briefly outcompeted other sulfate reducers when they were first placed into the incubation chamber. The long turnover times for these clades (ranging from 5 to 80 days) of sulfate reducers may suggest that several factors, such as predation and competition, may attenuate their growth rates in a natural setting (discussed below). Interestingly, the rapid decreases seen for these three sulfate-reducing bacteria are among the only significantly negative trends that we see in these data sets, suggesting that factors such as viral lysis may be involved in their growth dynamics (36).
Sulfate depletion had little immediate effect on the abundance of any sulfate reducers. Many sulfate reducers can also ferment short-chain fatty acids or aromatics, and this metabolic shift may explain the lack of response by sulfate-reducing bacteria (37). Nonfermentative sulfate reducers exist in the genera that we found, and yet no rapid decline of FRAxC was observed from microbial taxa associated with sulfate reduction. These molecular techniques cannot determine the viable state of an organism, so the post-sulfate-reduction sulfate reducers might be metabolically inactive (38, 39). Lastly, the growth advantage in stationary-phase (GASP) response, in which subspecies dynamics underlie abundances that are stable or declining at the population level, could help explain some of the apparent stability among sulfate-reducing bacteria (40).
Relatives of cultured methanogens grew at the onset of methanogenesis, with increases in FRAxC and qPCR values for Methanomicrobiales and Methanosarcinales (Fig. 4). Together with Kazan-3A-21 (discussed below), these methanogenic clades were the only archaea that increased during methanogenesis. In addition to the increases in these clades, there were corresponding increases in the total archaea and total cell counts after day 80 (Fig. S1). 16S rRNA gene sequences and isolates from Methanomicrobiales and Methanosarcinales are commonly found in marine sediments (41, 42). All cultured members of the Methanomicrobiales and most Methanosarcinales use H2-CO2 or H2-formate as the substrates, and many Methanosarcinales often use acetate and other methylated compounds (43). One of the most commonly recovered Methanosarcinales in our incubations was Methanosaeta sp., which is an obligate acetoclastic methanogen (44). The simultaneous inhibition of methanogenic clades during sulfate reduction supports predictions from geochemistry that both hydrogenotrophic and acetoclastic methanogens were thermodynamically prevented from meeting their maintenance energies in extremely low hydrogen concentrations, and the variable amount of methane in the headspace during the first 40 days was the result of outgassing, not biological production. Even if acetate concentrations were high enough to allow methanogenesis, loss of hydrogen during intracellular metabolic shuttling would decrease the energy available from acetoclastic methanogenesis (45). Immediately after net sulfate reduction ceased, hydrogen increased to large amounts that varied between replicates and between time points. This is consistent with a persistent production of hydrogen by fermenters that was no longer being efficiently oxidized by sulfate reducers. At this point, when hydrogen rose above the thermodynamic requirements of methanogens, sustained methane production commenced and acetoclastic and hydrogenotrophic methanogens grew simultaneously, with little evidence for competition between them.
One taxon of uncultured archaea (Kazan-3A-21) and a bacterial taxon (an unclassified genus of Syntrophaceae) increased in FRAxC in concert with methanogens (Fig. 4). In previous studies, 16S rRNA gene sequences from Kazan-3A-21 have been found in diverse methane-rich environments, although no potential functions have been proposed (46, 47). Kazan 3A-21 organisms are phylogenetically related to the Thermoplasmata, some members of which can use methylamines as a methanogenic substrate (47, 48). Additionally, another uncultured group, WSA2, phylogenetically related to the Thermoplasmata, contains the genetic potential for methane production from methylated compounds (34). Although it is possible that Kazan-3A-21 bacteria are fermenters that feed hydrogen and/or acetate to methanogens, their phylogenetic similarity to methane-producing organisms suggests that they may be methanogens themselves. All cultivated members of the family Syntrophaceae are syntrophic with H2-utilizing methanogens or sulfate reducers (49). Natural sediments and anaerobic digesters become enriched with Syntrophaceae during methanogenesis from hydrocarbon degradation (50, 51). We hypothesize that uncultured members of Syntrophaceae are potentially responsible for the syntrophic degradation of natural organic matter with methanogens in this study, even though the composition of the organic matter is diverse.
Turnover times for Methanomicrobiales and Methanosarcinales measured from 16S rRNA gene amplicon data were very similar to those measured for the same clades with qPCR (Table 1), even though the two methods are subject to different extraction, amplification, and cell count biases (20, 21). The fact that the rates measured with these two methods match well suggests that, even though neither of these methods is absolutely quantitative, relative changes in total cell abundance can be measured from FRAxC. This method is therefore useful for measuring changes in cell abundance of any clade observed in a 16S rRNA gene library, without the need to make specific qPCR primers.
Populations of Methanomicrobiales and Methanosarcinales increased extremely slowly (turnover time [td] = 9 to 13 days) relative to pure culture analogs, most of which double in less than 0.5 day at 21°C (42). Even some of the slowest-growing oceanic bacteria and archaea, Prochlorococcus sp. and Nitrosopumilus sp., have turnover times in pure culture of only 1 to 4 days and 0.5 to 1 day, respectively (52, 53). Only Methanosaeta pelagica, which was isolated from tidal flat sediments, has a turnover time of 12 days at 21°C (54). At least some portion of the differences between pure culture and in situ growth rates may be explained by the fact that our techniques target a population that may not be clonal; however, our data suggest that as has been seen in gut environments (55), even cultured clades have lower turnover rates in nature, in this case, marine sediment, than they do in culture.
A precedent for slower growth in natural oceanic environments than in pure culture comes from Leucothrix mucor with turnover times of 11 h versus 1.5 h, respectively (56). This slower growth in natural settings may result from limitations of electron donors or other nutrients. Decreasing hydrogen partial pressure in pure cultures of Methanocaldococcus jannaschii from 178 kPa to 0.650 kPa slowed turnover times from 0.8 h to 3.0 h (57). Pure cultures of hydrogenotrophic methanogens are generally grown in 80% hydrogen, but our methanogenic sediments had 0.05 to 0.25% hydrogen, so this substrate limitation may at least partially explain the deviation from pure culture growth rates. Other contributing factors could be competition from other microbes or predation from bacteriovores or viruses, which are prevalent in marine sediment environments (58). Bacteriovores such as Bdellovibrio sp. and Peredibacter sp. (59–62) were present in the 16S rRNA gene amplicon libraries.
Known methanogenic and sulfate-reducing clades accounted for about 15% of total assigned sequences, and only the clades discussed above increased during methanogenesis. There is no reason to suspect primer bias against methanogenic and sulfate-reducing groups, since our primers matched them well. So, it is likely that the majority of microbial taxa in our incubation were not directly involved in sulfate reduction or methanogenesis. These other taxa were diverse, representing the major groups of uncultured bacteria and archaea commonly found in anoxic marine sediments. Some evidence that these uncultured groups participate in slow fermentation, which could function independently of large redox shifts, is present in the genomic composition of common marine sediment taxa (10, 32, 63–65). The lack of community shift of any other clade besides those mentioned above was unexpected because, even if these communities are largely dependent on fermentation of organic matter, they should be energetically influenced by a shift from sulfate as the terminal acceptor for their reducing equivalents to CO2. This shift increased the concentration of the fermentatively derived hydrogen (Fig. 1), which would certainly decrease the energy available to fermenters that produce it. The lack of response from the abundant uncultured clades to this shift in hydrogen has multiple possible explanations. The first is that none of these clades participate in fermentation. This seems unlikely, given that the genomes of many of the uncultured clades predict at least the capability of a fermentative lifestyle (32, 66, 67). A second possibility is that these clades are fermenters but are not energetically limited and are unaffected by an increase in hydrogen. This seems unlikely, since we would expect the populations to grow if they were not energetically limited. A third possibility is that these clades are in long-term persistent states lacking some required substrate or their DNA is otherwise protected from degradation, which could explain the lack of response to large changes in available energy, positive or negative (68). This is consistent with recent suggestions that long-term persistence is a general feature of subsurface microbes (69).
Our results show that populations of Methanosarcinales and Methanomicrobiales increased simultaneously during methane production along with Syntrophaceae and an uncultured clade called Kazan-3A-21, which we suggest was involved in methanogenesis. These microbes quickly responded after a thermodynamic barrier was removed but grew much more slowly than in pure culture. The results of this study also indicate that a majority of observed microbes, including most clades of sulfate reducers, do not change abruptly in FRAxC in response to the major redox shift from sulfate reduction to methanogenesis. This suggests that future studies should focus on other metabolisms, not closely linked to sulfate reduction or methanogenesis, for this uncultured majority. A limitation of FRAxC analysis is the need for the observation of an exponential growth of a microbial taxon, which is often rare in energy-limited natural environments.
MATERIALS AND METHODS
Sample collection and incubation setup.
Samples were collected by scuba divers at Cape Lookout Bight, North Carolina (34.6205°N, 76.5500°W), on 2 October 2013, in 10-m water depth with an estimated bottom water temperature of ∼20°C. Thirty 20-cm-long and 7.62-cm-diameter polyvinyl chloride (PVC) push cores were collected, capped, refrigerated, and then returned to the lab on water ice in Tennessee within 48 h. The first three centimeters of sediment from each core were placed in a 2-liter Erlenmeyer flask through a funnel. Approximately 10 core tubes were needed to fill 1.5 liters of sediment. No liquid or solid amendments were made to the natural sediments. Cell counts and concentrations of hydrogen, sulfate, and methane were measured for the negative control on day 260. All measurements described below were performed for 18 weekly time points for all three incubations (0 to 122 days). A 100-ml volume of sediment was autoclaved and incubated alongside the experiments under anoxic conditions as a negative control.
Each of the three flasks was fitted with a custom butyl rubber stopper with a hole drilled through the center to accommodate a wide-bore (6-mm) glass and Teflon stopcock for the removal of sediment samples (Fig. 6). Two 18-gauge needles with stainless steel stopcocks were inserted into the stopper as well for gas sampling. Using the Luer-Lok (BD) fitting on the needles, ultrahigh-purity nitrogen gas (99.999%) that had been scrubbed of oxygen using heated copper fillings was flowed through the bottles using the second needle for the outflow to make the headspace anoxic. Incubation 3 was then flushed for a period of 15 min with ≥99.0% methane gas (Sigma-Aldrich, St. Louis, MO). Then, the flasks were inverted so that sediment covered the gas ports, stopper, and stopcock and were placed in a ring stand at constant room temperature (21.4°C) in the dark. The porosity of the sediment added to the incubations was found to be 0.833 and was determined by allowing preweighed sediment samples to dry over the course of several weeks at 40°C.
FIG 6.

(A) Diagram of a 2-liter incubation bottle containing anoxic sediments from Cape Lookout Bight, North Carolina. Sampling ports consist of a glass stopcock for sediment removal and two luer lock stainless steel needles for gasses, both through the black butyl stopper. (B) Picture of the same setup. The bottles were stored in the dark and inverted on a ring stand in an attempt to prevent any gas leakage.
Sampling and geochemical measurements.
The incubations were turned over once every 7 days just before sampling. Prior to gas sampling, 2 ml of anoxic N2 gas (99.999%) was used to blow the needle clear of sediment. Separate hydrogen and methane gas samples were collected in glass gastight Hamilton syringes using the steel needle ports in the custom stopper. Approximately 32 ml of sediment was removed through the glass and Teflon stopcock using a sterile 60-ml plastic catheter tip syringe. From this, two 15-ml conical centrifuge tubes were filled and capped, one used for porewater analysis and the other frozen at −80°C for later molecular analysis. One milliliter of sediment was placed in a 2-ml screw-cap tube with 500 μl of 3% paraformaldehyde, where it sat overnight before being washed twice with phosphate-buffered saline (PBS), centrifuged at 3,000 relative centrifugal force (rcf) between steps, and placed into a 1:1 PBS-ethanol solution. After sampling, 30 ml of oxygen- and hydrogen-scrubbed N2 was injected into the bottle to replace the lost volume. The 15-ml tube destined for porewater analysis was centrifuged at 5,000 × g for 5 min. A syringe was used to remove the supernatant, which was filtered using a 0.2-μm syringe filter into 100 μl of 10% HCl to a final volume of 1 ml that was used to determine sulfate concentrations by ion chromatography (Dionex, Sunnyvale, CA), with the remaining porewater used for determining the pH.
To measure hydrogen concentrations, 500 μl of headspace gas was injected into a Peak Performer 1 reducing compound photometer (Peak Laboratories, Mountain View, CA). Premixed hydrogen ppm lab bottles (Airgas, Radnor, PA) were used as standards. Hydrogen was assumed to be equilibrated between headspace and porewater, since the equilibration time for Cape Lookout Bight sediments is <2 days (18). Therefore, gas phase partial pressures were converted to aqueous hydrogen concentrations using the solubility coefficient of hydrogen corrected for salinity of 35 ppt and temperature of 21.4°C (70). Methane was determined by injecting 500 μl of gas from the headspace into an evacuated glass bottle to be later analyzed on a gas chromatograph with a flame ionization detector (Agilent, Santa Clara, CA). Methane concentrations were not assumed to be equilibrated with the aqueous phase; therefore, concentrations are presented as headspace partial pressures.
Hydrogen concentrations for sulfate reducers and methanogens operating at their energetic minima were calculated from the relationships in Hoehler et al. (18). Here these calculations have been used to determine the theoretical minima in the microcosm incubations. For sulfate reduction, the following formula was used:
| (1) |
No temperature correction was necessary, since equation 1 was determined at the same temperature as our experiments. For methanogenesis, the predicted hydrogen concentrations were calculated from the following temperature relationship formula, which was reported in Hoehler et al. (18) and applied here:
| (2) |
where T is temperature in degrees Celsius.
Cell quantification.
Total cell numbers were determined by direct epifluorescence microscopy with SYBRGold DNA stain (Invitrogen, Carlsbad, CA) by a single individual. Sediments were sonicated using a Microson ultrasonic cell disruptor (Misonix Inc., Farmingdale, NY) at 20% power for 40 s to disaggregate cells from sediments and diluted 40-fold into PBS prior to filtration onto a 0.2-μm black 25-mm polycarbonate filter (Fisher Scientific, Waltham, MA) and mounted onto a slide for manual counting of 30 fields on a Zeiss epifluorescence microscope.
Quantitative PCR.
DNA was extracted from frozen sediment samples using the Fast DNA kit for soil (MP Bio, Santa Ana, CA). Autoclaved sediment and water blanks were used as negative controls and had qPCR values below the detection level. Quantitative PCR was used on a Bio-Rad IQ5 machine to determine 16S rRNA gene copy numbers of several taxa with Quantifast SYBRGreen kit (Qiagen) for 35 cycles with a denaturation temperature of 95°C and combined annealing/extension of 60°C using 1 μl template for a total reaction volume of 25 μl. DNA standards with an R2 of >0.98 in a range of 109 to 102 were prepared either from existing stocks of TOPO plasmids containing an insert of the target 16S gene (13) or from TOPO plasmids (Invitrogen, Carlsbad, CA) containing PCR-amplified 16S rRNA gene products of closely related relatives in the clades Methanomicrobiales and Methanosarcinales synthesized by Invitrogen (accession numbers AB236118 [1,324 bp] and AB679168 [1,407 bp], respectively). Plasmid standards, nonlinearized, were quantified in the same manner as the extracted DNA from the incubations, which used Hoechst dye in a fluorometer (Hoefer, Holliston, MA). In addition to general Archaea and Bacteria primers, 16S rRNA-specific primers for Methanomicrobiales and Methanosarcinales (Table 2) were selected to have good coverage of the taxa (71). Each qPCR amplification was individually checked with a melt curve, and any amplification exhibiting primer dimers was not used. The amplification efficiency factor was variable between sample runs and ranged from 51% to 124%.
TABLE 2.
Quantitative PCR primers used for taxonomic quantification of 16S rRNA gene
| Primer name | Sequence | Target | Reference | Amplification efficiency (%) |
|---|---|---|---|---|
| A915f | GTGCTCCCCCGCCAATTCCT | Archaea | 76 | |
| A1059r | GCCATGCACCWCCTCT | Archaea | 77 | 59 |
| B340f | TCCTACGGGAGGCAGCAGT | Bacteria | 78 | |
| B515r | CGTATTACCGCGGCTGCTGGCAC | Bacteria | 78 | 73 |
| ANME-2-240f | CTATCAGGTTGTAGTGGG | ANME-2 | 79 | |
| ANME-2-538r | CGGCTACCACTCGGGCCGC | ANME-2 | 79 | 95 |
| ANME-1-830r | TCGCAGTAATGCCAACAC | ANME-1 | 79 | |
| ANME-1-628f | GCTTTCAGGGAATACTGC | ANME-1 | 79 | 101 |
| A1100f | TGGGTCTCGCTCGTTG | Methanomicrobiales | 71 | |
| MG1200r | CGGATAATTCGGGGCATGCTG | Methanomicrobiales | 71 | 124 |
| MSMX860f | AGGGAAGCCGTGAAGCGCC | Methanosarcinales | 71 | |
| A1100r | TGGGTCTCGCTCGTTG | Methanosarcinales | 71 | 51 |
16S rRNA gene amplicon sequencing.
The V4 region of each DNA extraction was amplified using primers 806r and 515f (72), as a universal primer pair for Bacteria and Archaea. Library preparations via the Nextera kit and sequencing using an Illumina MiSeq were performed at the Center for Environmental Biotechnology at the University of Tennessee in Knoxville (73). The mothur MiSeq Standard Operating Procedure was used with default settings to make contigs of bidirectional sequences, clustered concurrently into operational taxonomic units (OTUs) at 97% similarity, and classify them with the Silva reference set 126 (74, 75). A number of sequences amounting to 7.6% of total sequences were removed as chimeric, and then approximately 5% of the remaining sequences were removed for not being classified as Archaea or Bacteria. Analyses were considered only for taxa with more than 20 reads when summed from the 19 time points from each of the three incubations; 927 taxonomically classified genus level clades of bacteria and archaea met these criteria. Total reads ranged from 20,922 to 329,380 for the 57 libraries with an average of 144,957. Estimates of community coverage were determined for a rarefied set of 90,848 OTUs using the mothur software v.1.37.0 (75). Coverage estimates were similar for all samples, at 84% or above using Good's coverage estimator. Relative read abundances of 16S rRNA sequences were calculated by dividing their reads by the sum of bacterial and archaeal reads. Negative samples containing DNA extracted from autoclaved sediment control yielded no amplification.
Population turnover time calculations.
Described below is the derivation for the formula used to calculate population turnover times. The density of cells, C, of taxon i at time t is given by the following:
| (3) |
where Ctot,t is the density of total cells at time t, enumerated by epifluorescence cell counts using a general DNA stain, z is a factor that cannot be absolutely measured that encompasses all biases related to cell lysis, extraction, and amplification, and FRA is the fraction read abundance of 16S rRNA genes, calculated as the number of reads of taxon i at time t divided by the total reads for the sample at time t. We assume that zi is constant through time because the factors that affect zi (taxon being examined, sediment matrix, and procedures) are constant. The population rate constant for taxon i, ki, is proportional to the log change in the cell density of taxon i between time zero and time t,
| (4) |
which, combined with equation 3, is
| (5) |
where zi has cancelled out, since it is identical in the numerator and the denominator, allowing a calculation of population turnover time td:
| (6) |
which is ln(2) divided by the slope of the natural log of FRAi,tCtot,t (simplified as FRAxC) or qPCR values versus time during the period of exponential increase. It is important to note that this FRAxC method depends on the assumption of constant z for microbe i over the course of the experiment, which would not be the case when comparing two samples from different locations or after the addition of a substance between time points that affects DNA extraction efficiency.
Availability of data.
16S rRNA gene sequences can be found at the NCBI GenBank Sequence Read Archive with project accession number SRP097059. Geochemistry and qPCR data can be found at www.bco-dmo.org with project number 649807.
Supplementary Material
ACKNOWLEDGMENTS
We thank Pennylloyd and John Baldridge and Andrew Steen for assistance in retrieving the sediment samples, Frank Löffler for the use of ion and flame-ionized detector chromatographs, the Center for Dark Energy Biosphere Investigations for the computational resources, and Frank Löffler and Terry Hazen for use of their Illumina Miseq.
This work was funded by the NSF Center for Dark Energy Biosphere Investigations (OCE-0939564) (publication no. 400), NSF (OCE-1431598), and NASA Exobiology (NNX16AL59G).
We declare that we have no conflicts of interest.
Footnotes
This article is C-DEBI publication no. 400.
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01443-17.
REFERENCES
- 1.Parkes RJ, Webster G, Cragg BA, Weightman AJ, Newberry CJ, Ferdelman TG, Kallmeyer J, Jørgensen BB, Aiello IW, Fry JC. 2005. Deep sub-seafloor prokaryotes stimulated at interfaces over geological time. Nature 436:390–394. doi: 10.1038/nature03796. [DOI] [PubMed] [Google Scholar]
- 2.Kallmeyer J, Pockalny R, Adhikari RR, Smith DC, D'Hondt S. 2012. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc Natl Acad Sci U S A 109:16213–16216. doi: 10.1073/pnas.1203849109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huber JA, Welch DM, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML. 2007. Microbial population structures in the deep marine biosphere. Science 318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- 4.Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2011. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc Natl Acad Sci U S A 103:12115–12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Köpke B, Wilms R, Engelen B. 2005. Microbial diversity in coastal subsurface sediments: a cultivation approach using various electron acceptors and substrate gradients. Appl Environ Microbiol 71:7819–7830. doi: 10.1128/AEM.71.12.7819-7830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kendall MM, Wardlaw GD, Tang CF, Bonin AS, Liu Y, Valentine DL. 2007. Diversity of archaea in marine sediments from Skan Bay, Alaska, including cultivated methanogens, and description of Methanogenium boonei sp. nov. Appl Environ Microbiol 73:407–414. doi: 10.1128/AEM.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rappé MS, Giovannoni SJ. 2003. The uncultured microbial majority. Annu Rev Microbiol 57:369–394. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- 8.Süß J, Engelen B, Cypionka H, Sass H. 2004. Quantitative analysis of bacterial communities from Mediterranean sapropels based on cultivation-dependent methods. FEMS Microbiol Ecol 51:109–121. doi: 10.1016/j.femsec.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Freitag TE, Klenke T, Krumbein WE, Gerdes G, Prosser JI. 2003. Effect of anoxia and high concentrations on heterotrophic microbial communities in reduced surface sediments (black spots) in sandy intertidal flats of the German Wadden Sea. FEMS Microbiol Ecol 44:291–301. doi: 10.1016/S0168-6496(03)00076-X. [DOI] [PubMed] [Google Scholar]
- 10.Kubo K, Lloyd KG, Biddle FJ, Amann R, Teske A, Knittel K. 2012. Archaea of the Miscellaneous Crenarchaeotal Group are abundant, diverse and widespread in marine sediments. ISME J 6:1949–1965. doi: 10.1038/ismej.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berner RA. 1980. Early diagenesis: a theoretical approach. No. 1. Princeton University Press, Princeton, NJ. [Google Scholar]
- 12.Reeburgh WS. 1980. Anaerobic methane oxidation: rate depth distributions in Skan Bay sediments. Earth Planet Sci Lett 47:345–352. doi: 10.1016/0012-821X(80)90021-7. [DOI] [Google Scholar]
- 13.Lloyd KG, Alperin MJ, Teske A. 2011. Environmental evidence for net methane production and oxidation in putative ANaerobic MEthanotrophic (ANME) archaea. Environ Microbiol 13:2548–2564. doi: 10.1111/j.1462-2920.2011.02526.x. [DOI] [PubMed] [Google Scholar]
- 14.Ravenschlag K, Sahm K, Knoblauch C, Jørgensen B, Amann R, Jørgensen BOB. 2000. Community structure, cellular rRNA content, and activity of sulfate-reducing bacteria in marine arctic sediments community structure, cellular rRNA content, and activity of sulfate-reducing bacteria in marine arctic sediments. Appl Environ Microbiol 66:3592–3602. doi: 10.1128/AEM.66.8.3592-3602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leloup J, Fossing H, Kohls K, Holmkvist L, Borowski C, Jørgensen BB. 2009. Sulfate-reducing bacteria in marine sediment (Aarhus Bay, Denmark): abundance and diversity related to geochemical zonation. Environ Microbiol 11:1278–1291. doi: 10.1111/j.1462-2920.2008.01855.x. [DOI] [PubMed] [Google Scholar]
- 16.Martens C, Albert DB, Alperin MJ. 1998. Biogeochemical processes controlling methane in gassy coastal sediments. Part 1. A model coupling organic matter flux to gas production, oxidation and transport. Cont Shelf Res 18:1741–1770. [Google Scholar]
- 17.Hoehler TM, Alperin MJ, Albert DB, Martens CS. 1994. Field and laboratory studies of methane oxidation in an anoxic marine sediment: evidence for a methanogen-sulfate reducer consortium. Global Biogeochem Cycles 8:451–463. doi: 10.1029/94GB01800. [DOI] [Google Scholar]
- 18.Hoehler TM, Alperin MJ, Albert DB, Martens CS. 1998. Thermodynamic control on hydrogen concentrations in anoxic sediments. Geochim Cosmochim Acta 62:1745–1756. doi: 10.1016/S0016-7037(98)00106-9. [DOI] [Google Scholar]
- 19.Alperin MJ, Blair NE, Albert DB, Hoehler TM, Martens CS. 1992. Factors that control the stable carbon isotopic composition of methane produced in an anoxic marine sediment. Global Biogeochem Cycles 6:271–291. doi: 10.1029/92GB01650. [DOI] [Google Scholar]
- 20.Suzuki MT, Giovannoni SJ. 1996. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62:2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mumy KL, Findlay RH. 2004. Convenient determination of DNA extraction efficiency using an external DNA recovery standard and quantitative-competitive PCR. J Microbiol Methods 57:259–268. doi: 10.1016/j.mimet.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Holmkvist L, Ferdelman TG, Jørgensen BB. 2011. A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochim Cosmochim Acta 75:3581–3599. doi: 10.1016/j.gca.2011.03.033. [DOI] [Google Scholar]
- 23.Aksenova HYU, Rainey FA, Janssen PH, Zavarzin GA, Morgan HW. 1992. Spirochaeta thermophila sp. nov., an obligately anaerobic, polysaccharolytic, extremely thermophilic bacterium. Int J Syst Evol Microbiol 42:175–177. [Google Scholar]
- 24.Romanenko LA, Tanaka N, Frolova GM, Mikhailov VV. 2010. Marinicella litoralis gen. nov., sp. nov., a gammaproteobacterium isolated from coastal seawater. Int J Syst Evol Microbiol 60:1613–1619. doi: 10.1099/ijs.0.016147-0. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, Nakamura T, Fuse H. 2012. Isolation of two novel marine ethylene-assimilating bacteria, haliea species ETY-M and ETY-NAG, containing particulate methane monooxygenase-like genes. Microbes Environ 27:54–60. doi: 10.1264/jsme2.ME11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dyksma S, Bischof K, Fuchs BM, Hoffmann K, Meier D, Meyerdierks A, Pjevac P, Probandt D, Richter M, Stepanauskas R, Mußmann M. 2016. Ubiquitous Gammaproteobacteria dominate dark carbon fixation in coastal sediments. ISME J 10:1939–1953. doi: 10.1038/ismej.2015.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowman JP, McCammon SA, Dann AL. 2005. Biogeographic and quantitative analyses of abundant uncultivated gamma-proteobacterial clades from marine sediment. Microb Ecol 49:451–460. doi: 10.1007/s00248-004-0070-2. [DOI] [PubMed] [Google Scholar]
- 28.Sutton NB, Maphosa F, Morillo JA, Al-Soud WA, Langenhoff AAM, Grotenhuis T, Rijnaarts HHM, Smidt H. 2013. Impact of long-term diesel contamination on soil microbial community structure. Appl Environ Microbiol 79:619–630. doi: 10.1128/AEM.02747-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sekiguchi Y, Yamada T, Hanada S, Ohashi A, Harada H, Kamagata Y. 2003. Anaerolinea thermophila gen. nov., sp. nov. and Caldilinea aerophila gen. nov., sp. nov., novel filamentous thermophiles that represent a previously uncultured lineage of the domain bacteria at the subphylum level. Int J Syst Evol Microbiol 53:1843–1851. doi: 10.1099/ijs.0.02699-0. [DOI] [PubMed] [Google Scholar]
- 30.Yamada T, Sekiguchi Y, Hanada S, Imachi H, Ohashi A, Harada H, Kamagata Y. 2006. Anaerolinea thermolimosa sp. nov., Levilinea saccharolytica gen. nov, sp nov and Leptolinea tardivitalis gen nov, sp nov, novel filamentous anaerobes, and description of the new classes Anaerolineae classis nov and Caldilineae classis nov in the bacterial phylum Chloroflexi. Int J Syst Evol Microbiol 56:1331–1340. doi: 10.1099/ijs.0.64169-0. [DOI] [PubMed] [Google Scholar]
- 31.Castelle CJ, Wrighton KC, Thomas BC, Hug LA, Brown CT, Wilkins MJ, Frischkorn KR, Tringe SG, Singh A, Markillie LM, Taylor RC, Williams KH, Banfield JF. 2015. Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol 25:690–701. doi: 10.1016/j.cub.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 32.Lloyd KG, Schreiber L, Petersen DG, Kjeldsen KU, Lever MA, Steen AD, Stepanauskas R, Richter M, Kleindienst S, Lenk S, Schramm A, Jørgensen BB. 2013. Predominant archaea in marine sediments degrade detrital proteins. Nature 496:215–218. doi: 10.1038/nature12033. [DOI] [PubMed] [Google Scholar]
- 33.Ferrer M, Guazzaroni ME, Richter M, García-Salamanca A, Yarza P, Suárez-Suárez A, Solano J, Alcaide M, van Dillewijn P, Molina-Henares MA, López-Cortés N, Al-Ramahi Y, Guerrero C, Acosta A, de Eugenio LI, Martínez V, Marques S, Rojo F, Santero E, Genilloud O, Pérez-Pérez J, Rosselló-Móra R, Ramos JL. 2011. Taxonomic and functional metagenomic profiling of the microbial community in the anoxic sediment of a sub-saline shallow lake (Laguna de Carrizo, Central Spain). Microb Ecol 62:824–837. doi: 10.1007/s00248-011-9903-y. [DOI] [PubMed] [Google Scholar]
- 34.Nobu MK, Narihiro T, Kuroda K, Ran M, Liu W-T. 2016. Chasing the elusive Euryarchaeota class WSA2: genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J 10:2478–2487. doi: 10.1038/ismej.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoehler TM, Albert DB, Alperin MJ, Martens CS. 1999. Acetogenesis from CO2 in an anoxic marine sediment. Limnol Oceanogr 44:662–667. doi: 10.4319/lo.1999.44.3.0662. [DOI] [Google Scholar]
- 36.Thingstad TF. 2000. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol Oceanogr 45:1320–1328. doi: 10.4319/lo.2000.45.6.1320. [DOI] [Google Scholar]
- 37.Zhou J, He Q, Hemme CL, Mukhopadhyay A, Hillesland K, Zhou A, He Z, Van Nostrand JD, Hazen TC, Stahl DA, Wall JD, Arkin AP. 2011. How sulphate-reducing microorganisms cope with stress: lessons from systems biology. Nat Rev Microbiol 9:452–466. doi: 10.1038/nrmicro2575. [DOI] [PubMed] [Google Scholar]
- 38.Cangelosi GA, Meschke JS. 2014. Dead or alive: molecular assessment of microbial viability. Appl Environ Microbiol 80:5884–5891. doi: 10.1128/AEM.01763-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lomstein BA, Langerhuus AT, D'Hondt S, Jørgensen BB, Spivack AJ. 2012. Endospore abundance, microbial growth and necromass turnover in deep sub-seafloor sediment. Nature 484:101–104. doi: 10.1038/nature10905. [DOI] [PubMed] [Google Scholar]
- 40.Finkel SE. 2006. Long-term survival during stationary phase: evolution and the GASP phenotype. Nat Rev Microbiol 4:113–120. doi: 10.1038/nrmicro1340. [DOI] [PubMed] [Google Scholar]
- 41.Nunoura T, Oida H, Miyazaki J, Miyashita A, Imachi H, Takai K. 2008. Quantification of mcrA by fluorescent PCR in methanogenic and methanotrophic microbial communities. FEMS Microbiol Ecol 64:240–247. doi: 10.1111/j.1574-6941.2008.00451.x. [DOI] [PubMed] [Google Scholar]
- 42.Jaboski S, Rodowicz P, Lukaszewicz M. 2015. Methanogenic archaea database containing physiological and biochemical characteristics. Int J Syst Evol Microbiol 65:1360–1368. doi: 10.1099/ijs.0.000065. [DOI] [PubMed] [Google Scholar]
- 43.Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R. 2008. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6:579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 44.Smith KS, Ingram-Smith C. 2007. Methanosaeta, the forgotten methanogen? Trends Microbiol 15:150–155. doi: 10.1016/j.tim.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 45.Finke N, Hoehler TM, Jørgensen BB. 2007. Hydrogen “leakage” during methanogenesis from methanol and methylamine: implications for anaerobic carbon degradation pathways in aquatic sediments. Environ Microbiol 9:1060–1071. doi: 10.1111/j.1462-2920.2007.01248.x. [DOI] [PubMed] [Google Scholar]
- 46.Heijs SK, Haese RR, Van Der Wielen PWJJ, Forney LJ, Van Elsas JD. 2007. Use of 16S rRNA gene based clone libraries to assess microbial communities potentially involved in anaerobic methane oxidation in a Mediterranean cold seep. Microb Ecol 53:384–398. doi: 10.1007/s00248-006-9172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(Database issue):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Offre P, Spang A, Schleper C. 2013. Archaea in biogeochemical cycles. Annu Rev Microbiol 67:437–457. doi: 10.1146/annurev-micro-092412-155614. [DOI] [PubMed] [Google Scholar]
- 49.Kuever J. 2014. The family Syntrophaceae, p 281–288. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The prokaryotes. Springer, Berlin, Germany. [Google Scholar]
- 50.Gray ND, Sherry A, Grant RJ, Rowan AK, Hubert CRJ, Callbeck CM, Aitken CM, Jones DM, Adams JJ, Larter SR, Head IM. 2011. The quantitative significance of Syntrophaceae and syntrophic partnerships in methanogenic degradation of crude oil alkanes. Environ Microbiol 13:2957–2975. doi: 10.1111/j.1462-2920.2011.02570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng L, Ding C, Li Q, He Q, Dai L, Zhang H. 2013. DNA-SIP reveals that Syntrophaceae play an important role in methanogenic hexadecane degradation. PLoS One 8(7):e66784. doi: 10.1371/journal.pone.0066784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Könneke M, Bernhard AE, de la Torre JR, Walker CB, Waterbury JB, Stahl DA. 2005. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437:543–546. doi: 10.1038/nature03911. [DOI] [PubMed] [Google Scholar]
- 53.Tolonen AC, Aach J, Lindell D, Johnson ZI, Rector T, Steen R, Church GM, Chisholm SW. 2006. Global gene expression of Prochlorococcus ecotypes in response to changes in nitrogen availability. Mol Syst Biol 2:53. doi: 10.1038/msb4100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mori K, Iino T, Suzuki KI, Yamaguchi K, Kamagata Y. 2012. Aceticlastic and NaCl-requiring methanogen “Methanosaeta pelagica” sp. nov., isolated from marine tidal flat sediment. Appl Environ Microbiol 78:3416–3423. doi: 10.1128/AEM.07484-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marino S, Baxter NT, Huffnagle GB, Petrosino JF, Schloss PD. 2014. Mathematical modeling of primary succession of murine intestinal microbiota. Proc Natl Acad Sci U S A 111:439–444. doi: 10.1073/pnas.1311322111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brock TD. 1967. Bacterial growth rate in the sea: direct analysis by thymidine autoradiography. Science 155:81–83. doi: 10.1126/science.155.3758.81. [DOI] [PubMed] [Google Scholar]
- 57.Mukhopadhyay B, Johnson EF, Wolfe RS. 2000. A novel pH2 control on the expression of flagella in the hyperthermophilic strictly hydrogenotrophic methanarchaeaon Methanococcus jannaschii. Proc Natl Acad Sci U S A 97:11522–11527. doi: 10.1073/pnas.97.21.11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Danovaro R, Dell'Anno A, Trucco A, Serresi M, Vanucci S. 2001. Determination of virus abundance in marine sediments. Appl Environ Microbiol 67:1384–1387. doi: 10.1128/AEM.67.3.1384-1387.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rendulic S, Jagtap P, Rosinus A, Eppinger M, Baar C, Lanz C, Keller H, Lambert C, Evans KJ, Goesmann A, Meyer F, Sockett RE, Schuster SC. 2004. A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science 303:689–692. doi: 10.1126/science.1093027. [DOI] [PubMed] [Google Scholar]
- 60.Davidov Y, Jurkevitch E. 2004. Diversity and evolution of Bdellovibrio-and-like organisms (BALOs), reclassification of Bacteriovorax starrii as Peredibacter starrii gen. nov., comb. nov., and description of the Bacteriovorax-Peredibacter clade as Bacteriovoracaceae fam. nov. Int J Syst Evol Microbiol 54:1439–1452. doi: 10.1099/ijs.0.02978-0. [DOI] [PubMed] [Google Scholar]
- 61.Suttle CA. 2005. Viruses in the sea. Nature 437:356–361. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- 62.Breitbart M, Rohwer F. 2005. Here a virus, there a virus, everywhere the same virus? Trends Microbiol 13:278–284. doi: 10.1016/j.tim.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 63.Seitz KW, Lazar CS, Hinrichs K-U, Teske AP, Baker BJ. 2016. Genomic reconstruction of a novel, deeply branched sediment archaeal phylum with pathways for acetogenesis and sulfur reduction. ISME J 10:1696–1705. doi: 10.1038/ismej.2015.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lazar CS, Baker BJ, Seitz KW, Hyde AS, Dick GJ, Hinrichs K-U, Teske AP. 2016. Genomic evidence for distinct carbon substrate preferences and ecological niches of Bathyarchaeota in estuarine sediments. Environ Microbiol 18:1200–1211. doi: 10.1111/1462-2920.13142. [DOI] [PubMed] [Google Scholar]
- 65.Baker BJ, Lazar CS, Teske AP, Dick GJ. 2015. Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3:14. doi: 10.1186/s40168-015-0077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He X, McLean JS, Edlund A, Yooseph S, Hall AP, Liu S-Y, Dorrestein PC, Esquenazi E, Hunter RC, Cheng G, Nelson KE, Lux R, Shi W. 2015. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc Natl Acad Sci U S A 112:244–249. doi: 10.1073/pnas.1419038112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rinke C, Lee J, Nath N, Goudeau D, Thompson B, Poulton N, Dmitrieff E, Malmstrom R, Stepanauskas R, Woyke T. 2014. Obtaining genomes from uncultivated environmental microorganisms using FACS-based single-cell genomics. Nat Protoc 9:1038–1048. doi: 10.1038/nprot.2014.067. [DOI] [PubMed] [Google Scholar]
- 68.Lennon JT, Jones SE. 2011. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol 9:119–130. doi: 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
- 69.Starnawski P, Batallion T, Ettema TJG, Jochum LM, Schreiber L, Chen X, Lever MA, Polz MF, Jørgensen BB, Schramm A, Kjeldsen KU. 2017. Microbial community assembly and evolution in subseafloor sediment. Proc Natl Acad Sci U S A 114:2940–2945. doi: 10.1073/pnas.1614190114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crozier TE, Yamamoto S. 1974. Solubility of hydrogen in water, seawater, and NaCl solutions. J Chem 19:242–244. [Google Scholar]
- 71.Narihiro T, Sekiguchi Y. 2011. Oligonucleotide primers, probes and molecular methods for the environmental monitoring of methanogenic archaea. Microb Biotechnol 4:585–602. doi: 10.1111/j.1751-7915.2010.00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.DeLong EF. 1992. Archaea in coastal marine environments. Proc Natl Acad Sci U S A 89:5685–5689. doi: 10.1073/pnas.89.12.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu Y, Lee C, Hwang S. 2005. Analysis of community structures in anaerobic processes using a quantitative real-time PCR method. Water Sci Technol 52:85–91. [PubMed] [Google Scholar]
- 78.Nadkarni M, Martin FE, Jacques N, Hunter N. 2002. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148:257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 79.Boetius A, Ravenschlag K, Schubert CJ, Rickert D, Widdel F, Gieseke A, Amann R, Jørgensen BB, Witte U, Pfannkuche O. 2000. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407:623–626. doi: 10.1038/35036572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.