

ABSTRACT
Homology searches indicate that Saccharomyces cerevisiae strain BY4741 contains seven redundant genes that encode putative aryl-alcohol dehydrogenases (AAD). Yeast AAD genes are located in subtelomeric regions of different chromosomes, and their functional role(s) remain enigmatic. Here, we show that two of these genes, AAD4 and AAD14, encode functional enzymes that reduce aliphatic and aryl-aldehydes concomitant with the oxidation of cofactor NADPH, and that Aad4p and Aad14p exhibit different substrate preference patterns. Other yeast AAD genes are undergoing pseudogenization. The 5′ sequence of AAD15 has been deleted from the genome. Repair of an AAD3 missense mutation at the catalytically essential Tyr73 residue did not result in a functional enzyme. However, ancestral-state reconstruction by fusing Aad6 with Aad16 and by N-terminal repair of Aad10 restores NADPH-dependent aryl-alcohol dehydrogenase activities. Phylogenetic analysis indicates that AAD genes are narrowly distributed in wood-saprophyte fungi and in yeast that occupy lignocellulosic niches. Because yeast AAD genes exhibit activity on veratraldehyde, cinnamaldehyde, and vanillin, they could serve to detoxify aryl-aldehydes released during lignin degradation. However, none of these compounds induce yeast AAD gene expression, and Aad activities do not relieve aryl-aldehyde growth inhibition. Our data suggest an ancestral role for AAD genes in lignin degradation that is degenerating as a result of yeast's domestication and use in brewing, baking, and other industrial applications.
IMPORTANCE Functional characterization of hypothetical genes remains one of the chief tasks of the postgenomic era. Although the first Saccharomyces cerevisiae genome sequence was published over 20 years ago, 22% of its estimated 6,603 open reading frames (ORFs) remain unverified. One outstanding example of this category of genes is the enigmatic seven-member AAD family. Here, we demonstrate that proteins encoded by two members of this family exhibit aliphatic and aryl-aldehyde reductase activity, and further that such activity can be recovered from pseudogenized AAD genes via ancestral-state reconstruction. The phylogeny of yeast AAD genes suggests that these proteins may have played an important ancestral role in detoxifying aromatic aldehydes in ligninolytic fungi. However, in yeast adapted to niches rich in sugars, AAD genes become subject to mutational erosion. Our findings shed new light on the selective pressures and molecular mechanisms by which genes undergo pseudogenization.
KEYWORDS: aryl-alcohol dehydrogenases, AKR superfamily, subtelomeric, evolution, lignin, pseudogenization
INTRODUCTION
Functional characterization of predicted genes remains one of the chief tasks of the postgenomic era. Two decades after the initial release of the Saccharomyces cerevisiae genome (1), uncertainty remains as to the exact number of genes, the boundaries between them, and their function (2–7). According to the most recent estimates in the Saccharomyces Genome Database (SGD), the yeast genome consists of 6,603 open reading frames (ORFs), of which 4,848 are “verified,” 944 are “uncharacterized,” and 811 are “dubious.” Genes are considered verified once any of their products (transcript or protein) are detected, and indeed, many of these 4,848 genes have only been verified by global transcriptional analyses. To completely characterize the yeast genome and achieve an accurate assessment of gene content and function, high-throughput analyses must be complemented by careful case-by-case experimental analysis of those genes still classified as putative and those proteins still annotated as hypothetical. The yeast aryl-alcohol dehydrogenases (AAD) family is an outstanding example of this class of genes.
Prediction of the 7 putative aryl-alcohol dehydrogenases (Aad) in yeast was based on in silico analyses that showed ≥85% amino acid similarity to an aryl-alcohol dehydrogenase purified from the white rot fungus Phanerochaete chrysosporium (8–10). Aad proteins belong to the aldo-keto-reductase (AKR) superfamily, which has more than 190 members (11). AKR enzymes can reduce a variety of substrates, such as sugar aldehydes, keto-steroids, keto-prostaglandins, retinals, and quinones (11). AKR proteins are usually monomeric proteins of low molecular mass (in the range of 34 kDa) and have an (α/β)8-barrel motif, a conserved catalytic tetrad consisting of four amino acids (Tyr, Asp, Lys, and His), and a conserved cofactor binding domain for NAD(P)H (11). To explore the biological function of this gene family in S. cerevisiae, Delneri et al. constructed single, double, triple, quadruple, quintuple, sextuple, and septuple aad knockouts (12). Surprisingly, these mutants exhibited no pronounced phenotype when tested for their ability to mate, sporulate, or degrade aromatic aldehydes, and neither did they differ with respect to cell composition, in particular, ergosterol content and phospholipid profile (12). Later, the same authors reported that all putative AAD genes contained sequences similar to the oxidation-reactive yap1 transcriptional factor binding sites either upstream of or within their coding sequences; however, transcripts were only detectable for AAD4 and AAD6 under oxidation challenge by diamide, diethyl maleic acid ester, or H2O2 (13). While microarray analyses also suggest that putative Aad proteins may be involved in yeast's response to oxidative damage, heavy-metal stress, and certain fungicides (14–21), a mechanistic understanding of the role(s) they might play in doing so is lacking. To gain this understanding, detailed biochemical analyses are needed.
Previously, we showed that a homologue of yeast AAD, Phanerochaete chrysosporium Aad1p, can reduce aryl-aldehyde derivatives to their corresponding less-toxic alcohol forms (10). This result suggested that yeast Aad could serve to detoxify aromatic inhibitors produced in lignocellulosic ethanol production (22), as well as in the bioremediation of environmental pollutants, such as benzene, toluene, ethylbenzene and xylene (BTEX) derivatives (23). To explore these possibilities and to enlarge our understanding of the function and phylogeny of the yeast Aad gene family, we performed detailed biochemical and molecular genetic analyses. In this study, we show that only two genes of this family (AAD4 and AAD14) encode enzymatic activities on aliphatic and aryl-aldehydes, whereas the other five putative members are being pseudogenized, a finding that informs our speculation about the evolutionary trajectory that created this subtelomeric gene family.
RESULTS
Among the seven-member yeast AAD gene family, only AAD4 and AAD14 encode functional aryl-aldehyde dehydrogenases.
The NADPH-dependent P. chrysosporium Aad1 protein (PcAad1p) has an average of ∼85% residue similarity with all S. cerevisiae Aadp protein (ScAadp protein) members (sequence alignment shown in Fig. S1). We therefore adopted PcAad1p as a positive reference during the expression, purification, and enzymatic assay of the putative yeast ScAad proteins. Highly purified recombinant proteins were obtained following elution from glutathione affinity chromatography resin (Fig. S2). Enzymatic activity of the seven purified yeast Aad recombinant proteins showed that only ScAad14p and ScAad4p were able to reduce a group of candidate aryl-aldehydes with the consumption of NADPH (Fig. 1), validating their predicted enzyme category as aryl-alcohol dehydrogenases (EC 1.1.1.90). Under the same assay conditions, reference PcAad1p was active on a broader spectrum of aryl-aldehyde substrates than either ScAad14p or ScAad4p (Fig. 1). Notably, the reference PcAad1p and the active yeast ScAadp proteins shared substrate affinities for aliphatic and aryl-aldehydes with other previously reported aldo-keto reductase and aldehyde reductases (24–28) (Table 1). Interestingly, each of the two active ScAadp proteins demonstrated a unique pattern of substrate specificity. ScAad4p is more active on aryl-aldehyde-bearing substituents at positions 3, 4, and 5, or even those bearing double and triple substitutions on the aromatic ring, while ScAad4p was inactive on aryl-aldehydes that have position 2 substitutions (Fig. 1C). In contrast, ScAad14p exhibited its highest activity on aryl-aldehydes substituted at position 2 (Fig. 1B). Regarding catalytic efficiency, ScAad4p and ScAad14p have activity in the range of micromoles per minute per milligrams toward their preferred aryl-aldehyde substrates, e.g., 1.76 μmol · min−1 · mg−1 for ScAad4p on hexanal and 2.10 μmol · min−1 · mg−1 for ScAad14p on 2-nitrobenzaldehyde. These values are 3-fold lower than the PcAad1p activity measured on 3,4-dimethoxybenzaldehyde.
FIG 1.

ScAad4p and ScAad14p have aryl-aldehyde reductase activity. Activities were assayed in morpholineethanesulfonic acid (MES) buffer (50 mM [pH 6.1]) containing 0.3 mM NADPH and 0.3 mM substrate. The standard activity of PcAad1p in reducing 3,4-dimethoxybenzaldehyde (being 5.4 μmol · min−1 · mg−1) was set at 100%. Data represent the means ± standard deviations of the results from triplicate experiments.
TABLE 1.
Yeast aldehyde reductases and their kinetic constants toward preferred substrates
| Enzyme (reference) | Cofactor preference | Km (μM) | Aliphatic aldehyde substratea |
Aryl-aldehyde substratea |
||||
|---|---|---|---|---|---|---|---|---|
| Substrate | Km (μM) | kcat (min−1) | Substrate | Km (μM) | kcat (min−1) | |||
| PcAad1 (10) | NADPH | 39 | Hexanal | NA | 247 | 3,4-Dimethoxybenzaldehyde | 12 | 530 |
| Heptaldehyde | NA | 138 | Benzaldehyde | 1,700 | 430 | |||
| Cinnamaldehyde | 3,400 | 670 | ||||||
| Aad4b | NADPH | NA | Hexanal | NA | 172 | 3,4-Dimethoxybenzaldehyde | NA | 142 |
| Heptaldehyde | NA | 64 | 4-Nitrobenzaldehyde | NR | NR | |||
| Aad14b | NADPH | NA | Hexanal | NA | 50 | 4-Nitrobenzaldehyde | NA | 22 |
| Heptaldehyde | NA | 46 | Cinnamaldehyde | NR | NR | |||
| Aad10-35Cb | NADPH | NA | Hexanal | NR | NR | 4-Nitrobenzaldehyde | NR | NR |
| Heptaldehyde | NR | NR | Cinnamaldehyde | NR | NR | |||
| Aad6518Gb | NADPH | NA | Hexanal | NR | NR | 4-Nitrobenzaldehyde | NR | NR |
| Heptaldehyde | NR | NR | Cinnamaldehyde | NR | NR | |||
| Adh6 (25) | NADPH | 29 | Hexanal | 152 | 21,270 | Cinnamaldehyde | 172 | 18,400 |
| Pentanal | 60 | 22,700 | Veratraldehyde | 73 | 15,800 | |||
| Adh7 (26) | NADPH | NA | Pentanal | 49 | 11,915 | Cinnamaldehyde | 43 | 7,913 |
| 3-Methylbutanal | 48 | 9,581 | Veratraldehyde | 58 | 6,000 | |||
| Gre3 (27) | NADPH | 13 | Hexanal | 3,100 | 109 | 4-Nitrobenzaldehyde | 120 | 142 |
| Gcy1 (28) | NADPH | 12 | Butyraldehyde | 5,400 | 81 | 4-Nitrobenzaldehyde | 130 | 71 |
| Benzaldehyde | 5,200 | 58 | ||||||
| Ypr1 (29) | NADPH | 8.7 | Hexanal | 390 | 354 | 4-Nitrobenzaldehyde | 1,070 | 1,776 |
| 2-Methylbutyraldehyde | 1,090 | 524 | 9,10-Phenanthrequinone | 2,600 | 272 | |||
| Yjr096w (28) | NADPH | 370 | Butyraldehyde | 1,800 | 0.5 | 4-Nitrobenzaldehyde | 500 | 88 |
| Benzaldehyde | 4,700 | 4 | ||||||
| Ydl124w (28) | NADPH | 23 | Butyraldehyde | 210,000 | 14 | 4-Nitrobenzaldehyde | 30 | 3.3 |
| Benzaldehyde | 240 | 4 | ||||||
kcat as reported or normalized based on reported Vmax. NA, data not available; NR, not reactive.
Results of this study.
While PcAad1p can use both NADPH (Km, 39 μM) and NADH (Km, 220 μM), neither of the yeast Aad proteins was active with NADH as a reduction cofactor. PcAad1p can also act as an oxidase against several aliphatic and aromatic alcohols (at pH 10.3, with NADP+ as a cofactor). ScAad4p and ScAad14p showed no oxidation activities on any of the following alcohols: pentanol, hexanol, heptanol, 3,4-dimethoxybenzyl alcohol, benzyl alcohol, cinnamyl alcohol, 4-methoxybenzyl alcohol, 4-hydroxybenzyl alcohol, 3,5-dimethoxybenzyl alcohol, 3-hydroxy-4-methoxybenzyl alcohol, 4-hydroxy-3-methoxybenzyl alcohol (vanillyl alcohol), 3,4,5-trimethoxybenzyl alcohol, and 2-phenylethanol (2-tailed t test, P < 0.05; n = 3).
In ancestral-state reconstruction, fusion of Aad6 and Aad16 results in an active Aad enzyme.
Sequence alignment suggested that AAD6 and AAD16 were originally one open reading frame split into two by a nucleotide deletion in the AAD6 coding sequence at positions G517 to C518 (Fig. 2 and S1). The insertion of a guanine nucleotide at this position places AAD6 and AAD16 in frame, which results in a new hypothetical protein (termed ScAad6518Gp) that shares >80% amino acid sequence similarity with other Aad family members. We reconstructed the hypothetical ancestral state (29) of ScAad6518Gp by site-directed mutagenesis and heterologously expressed the translation product as a glutathione S-transferase (GST) tag fusion protein in Escherichia coli, using the pGS-21a vector. The recombinant ScAad6518Gp protein was purified to high homogeneity (Fig. S3) and subjected to enzymatic assay against the same panel of aryl-aldehydes described for PcAad1p (10). Assays of the purified protein yielded weak but detectable enzymatic activity toward several aldehydes, relative to the background. Kinetic (Km and Vmax) measurements on the reconstructed ScAad6518Gp protein revealed affinities for phenylacetaldehyde (Km = 296 ± 30 μM) and 4-methoxybenzaldehyde (Km = 217 ± 26 μM). The maximum rate of reaction (Vmax) of ScAad6518Gp on these substrates was one order of magnitude lower than that of PcAad1p (Table 2 and Fig. S5).
FIG 2.

Single-base-pair substitutions, truncations, and deletions in the AAD gene family of yeast S288C. (A) Amino acid sequence alignments of PcAad1p (reference protein) and the seven putative ScAadp proteins. Blue asterisks (*) denote positions of four strictly conserved essential amino acids in PcAad1p: Asp71, Tyr76, Lys103, and His152. Structural details of the reference protein are provided in Fig. S4. Red asterisks (*) denote inferred point mutations to relative ancestral ScAAD genes. Green color denotes the truncated part of the ancestral gene. Point mutations result in (i) substitution of the conserved Tyr73 in ScAad3p, (ii) truncation of β-sheet 1 in ScAad4p, (iii) truncation of two α-helices and two β-sheets in ScAad10p, and (iv) the split of one ORF into two ORFs (ScAad6p and ScAad16p). The crossed-out ScAad16 indicates that this previously annotated ORF is a truncated part of AAD6 (in chromosome VI), not an independent ORF. The 5′ coding sequence of the ScAad15p was completely deleted from the genome of yeast lab strain S288C (in red). (B) Positions of the corresponding mutations at the nucleic acid and amino acid levels. Minus symbol (−) indicates nucleotide positions upstream of the ORFs as they were annotated in SGD at the time of submission.
TABLE 2.
Recovery of enzyme activity from pseudogenized ScAad10p and ScAad6/16p following ancestral state reconstructiona
| Enzyme |
PcAad1p |
ScAad10-35Cp |
ScAad6518Gp |
|||
|---|---|---|---|---|---|---|
| Vmax | Km | Vmax | Km | Vmax | Km | |
| 4-Methoxybenzaldehyde | 6.63 ± 0.12 | 87.3 ± 116 | 0.08 ± 0.01 | 157 ± 39.8 | 0.55 ± 0.02 | 217 ± 26.1 |
| Hydroxymethylfurfural | 2.40 ± 0.27 | 171 ± 36.7 | 0.37 ± 0.07 | 608 ± 246 | NR | |
| Phenylacetaldehyde | 9.00 ± 0.27 | 527 ± 72.5 | 0.30 ± 0.08 | 17.6 ± 2.20 | 0.49 ± 0.02 | 296 ± 29.6 |
Vmax (in micromoles per minute per milligram) and Km (micromolar) values are shown as the mean ± standard error (SE) of the results from triplicate experiments. NR, not reactive.
Tyr76 is essential for the function of PcAad1p but is missing in ScAad3p.
We identified the catalytic tetrad (Asp71, Tyr76, Lys103, and His152) in the reference enzyme PcAad1p (Fig. S4). However, alignment showed that in ScAad3p, a cysteine73 residue was present at the corresponding catalytic site tyrosine73. This substitution in ScAad3p could have been caused by a single nucleotide mutation from A218 to G218 of its coding sequence (Fig. 2 and S1). We therefore performed two site-directed mutagenesis experiments. In PcAad1p, the functional Tyr76 was mutated into Cys76 (TG227C to TA227C), whereas in ScAad3p, the Cys73 was replaced by Tyr73 (TA218C to TG218C). Following heterologous expression and purification, the activities of the recombinant proteins were assayed on a variety of aryl-aldehyde substrates. As expected, the PcAad1Tyr76Cysp variant was completely inactive on all of these substrates; however, correction of the missense mutation in ScAadCys73Tyrp failed to produce a functional enzyme (data not shown).
In ancestral-state reconstruction, repair of the ScAad10p N-terminal domain results in an enzyme having aryl-aldehyde activity.
We failed to detect any catalytic activity for the putative ScAad10p. However, sequence alignment suggested that a 264-bp sequence upstream of the SGD-annotated ScAAD10 may have once been in frame (detailed alignment is shown in Fig. S1), interrupted by a T−35AG-to-C−35AG substitution, counting from the SGD-annotated start codon. This point mutation (denoted by asterisk in Fig. 2) may have introduced a nonsense mutation in the ancestral ScAAD10. According to our structural modeling of PcAad1p (see Fig. S4), the truncated 88 residues contain the β1, β2, α1, and α2 domains of the classical (α8β8) motif, as well as two essential amino acids (Asp71 and Tyr76) found in all aldo-keto reductases. We therefore investigated whether replacing T by C at position −35 from ATG could resurrect an active Aad10 protein. The reconstructed polypeptide, ScAad10−35Tp, was expressed in E. coli as a GST-tagged protein and purified to near homogeneity (see Fig. S3), and its kinetics were tested on a variety of aryl-aldehydes. The reductase activity of the reconstructed ScAad10−35C protein was observed on hydroxymethylfurfural, phenylacetaldehyde, and 4-methoxybenzaldehyde with NADPH as a cofactor. In all instances, the Vmax values of reconstructed ScAadp were (10 to 80 times) lower than those of PcAad1p, and in 4 of 5 instances, the Km values for these substrates were higher than that of the PcAad1p (Table 2). Interestingly, reconstructed ScAad10p exhibits an even higher affinity for phenylacetaldehyde (Km, 17.6 μM) than does PcAad1p, suggesting that perhaps phenylacetaldehyde was a native substrate for ScAad10p at some point in its evolutionary past.
Yeast AAD gene expression is not induced by aryl-aldehydes, and AAD overexpression does not increase yeast resistance to these compounds.
Aromatic aldehyde derivatives are abundant in nature, being released as saprophytes degrade and digest plant matter (23, 30, 31). These aldehydes also occur in industrial decomposition of lignocellulosic biomass, where they inhibit yeast growth (32–35). We confirmed this inhibitory effect for four aryl-aldehydes: veratraldehyde, hydroxymethylfurfural, vanillin, and trans-cinnamaldehyde, at concentrations ranging from 25 to 50 mM (data not shown). As ScAadp proteins have the capacity to transform these compounds into less-toxic alcoholic derivatives, we investigated whether exposure of cells to aryl-aldehydes could induce AAD gene expression. Contrary to expectation, none of the seven AAD transcript levels increased following 1 to 2 h treatment with aryl-aldehydes, relative to a no-aldehyde control (2-tailed t test, P < 0.05; n = 6).
To test whether overexpression of AAD genes conferred higher resistance of yeast to aryl-aldehyde toxicity, we individually subcloned ScAAD3, ScAAD4, ScAAD14, and PcAAD1 into the multicopy YEplac195 plasmid, placing each under the control of the strong constitutive PGK1 promoter. These constructs were then transformed into wild-type BY4741 as well as into an adh6 knockout, as the expression of Adh6 has been shown by itself to confer aryl-aldehyde resistance (36). None of the transformants exhibited growth improvement on the four aryl-aldehydes, even those bearing AAD1 from P. chrysosporium. Indeed, while activity in BY4741 cell crude extracts expressing PcAad1p was 2-fold higher than that of the blank-plasmid control, activities in cells expressing ScAad3p, ScAad4p, and ScAd14p did not significantly differ (Fig. S7). We did not test whether the overexpression of reconstructed Aad proteins improved growth on aryl-aldehyde, as the in vitro activity of resurrected Aad proteins is even lower than those of ScAad4p and ScAad14p.
Across sequenced S. cerevisiae genomes, AAD genes vary in both copy number and nucleotide sequence.
Our observation that the majority of BY4741 AAD genes were undergoing pseudogenization prompted us to survey their distribution and sequence variation in diverse sequenced strains, including those adapted to the laboratory (BY4741 and Sigma1278b), to wine fermentation (EC1118, AWRI1631, AWRI796, Lalvin QA23, and VL3), to the brewing of beer (Fosters B and Fosters O) and sake (Kyokai no. 7), and to life as an opportunistic human pathogen (YJM789) (Table 3). Using reconstructed full-length AAD genes as query sequences, our analysis uncovered extensive variation in AAD copy number as well as in the number and location of AAD single nucleotide polymorphisms (SNPs). These polymorphisms may be related to the ecology of the species in which they are found. For example, compared to lab strain BY4741, industrial yeasts have one to five AAD homologs missing from their genomes: in Fosters B and Fosters O, which are used for beer production, 5 of 7 AAD genes are missing, while in the wine yeast Lalvin QA23, AAD6, AAD16, and AAD15 are missing. Unlike the missense or nonsense mutations in ScAAD3, ScAAD10, and ScAAD6/16, the 5′ coding sequence of AAD15 has been deleted from the BY4741 genome. AAD15 was entirely absent from the genomes of eight of 11 yeast conspecifics. BY4741 AAD6/AAD16 was not observed in any other genome, although a full-length AAD6 was detected in S. cerevisiae strain T7, which was isolated from oak tree exudate (Fig. S6).
TABLE 3.
AAD ORFs are highly varied among sequenced S. cerevisiae genomes
| Strain | Nucleic acid sequence similarity/amino acid sequence similarity (%)a |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lab |
Pathogen |
Wine |
Beer |
Sake |
|||||||
| BY4741 | Sigma1278b | YJM789 | EC1118 | AWRI1631 | AWRI796 | Lalvin QA23 | VL3 | Fosters B | Fosters O | Kyokai no. 7 | |
| AAD3 | 100/100 | 98.2/82.7 | 96.4/93.7 | 99.8/99.5 | 99.9/99.5 | 99.4/82.1 | 99.7/67.6 | 99.8/85.2 | NR | NR | NR |
| AAD4-136G | 99.9/83.5 | NR | 93.2/93.9 | 94.3/84.6 | 94.4/84.3 | 94.3/84.3 | 94.2/71.3 | 94.2/84.3 | 95.9/84.3 | 94.3/84.3 | 93.4/94.7 |
| AAD6518G | 99.9/- | NR | NR | NR | NR | NR | NR | NR | NR | NR | 99.8/- |
| AAD10-35C | 99.9/76.4 | 87.3/55.2 | 99.9/76.4 | 99.7/76.4 | 99.8/76.1 | 99.6/99.5 | NR | 92.6/72.9 | NR | NR | */* |
| AAD14 | 100/100 | 100/100 | 99.2/99.5 | 97.1/97.6 | NR | 97.1/97.6 | 96.9/52.0 | 96.9/65.3 | 97.3/97.1 | 97.5/97.3 | 100/100 |
| AAD15 | 100/100 | 100/100 | NR | 97.7/95.1 | NR | NR | NR | NR | NR | NR | NR |
Nucleic acid/amino acid sequence similarities are relative to query sequences from lab strain S288C. NR, AAD homologs were not retrieved. *, percentage similarity not calculated when only partial sequences were found at the end of a sequencing contig with missing 3' or 5' coding sequences. (Note: high similarity in nucleic acids can result in low amino acid similarity due to ORF truncation.)
AAD gene phylogeny indicates preferential distribution among wood-saprophyte fungi.
ScAAD14 encodes a full-length active protein whose sequence is highly similar (>80%) to other predicted yeast AAD genes. We therefore conducted a BLASTn search (expected [Exp.] threshold = 10; score, ≥60) for ScAAD14 orthologs in the NCBI database. An unrooted phylogeny (Fig. 3) shows that the most closely related 101 AAD orthologs fall into the Ascomycota and the Basidiomycota, groups that shared a common ancestor 400 million years ago (mya) (37). Species harboring AAD orthologs include lignin-scavenger specialists, such as the white rot fungus Phanerochaete carnosa, the poroid crust fungus Dichomitus squalens, and the mushroom Trametes versicolor, as well as plant pathogens, such as Neofusicoccum parvum (38–41). About one-third of species harboring AAD orthologs are yeast or filamentous fungi found in plant-associated habitats, such as wood debris, leaves, or bark exudate. Two AAD orthologs were retrieved from the millet-isolated fission yeast Schizosaccharomyces pombe, which diverged from the Saccharomyces lineage around 350 mya (Fig. 4) (42). Orthologs exhibiting 90% amino acid sequence similarity to ScAad14p were retrieved from the Mochi tree-isolated (43) pre-whole-genome duplication (WGD) yeast Lachancea waltii, which diverged from the S. cerevisiae progenitor 150 mya (44). Multiple AAD homologs are distributed among the Saccharomyces sensu stricto species S. bayanus, S. kudriavzevii, S. mikatae, and S. paradoxus. Significantly, all AAD-bearing species were isolated from environments rich in decaying plant matter, such as leaves and oak bark (45–49). AAD genes are altogether missing from yeast that have either colonized animal hosts (e.g., Candida glabrata) or have been domesticated for dairy production (Kluyveromyces lactis).
FIG 3.

AAD orthologs are distributed within Basidiomycota and Ascomycota fungi typically associated with plant habitats. Orthologs were identified by BLAST using yeast AAD14 as the query sequence against the NCBI database. Phylogeny of the resulting sequences was constructed using a k-mer (k = 15)-based neighbor-joining method.
FIG 4.
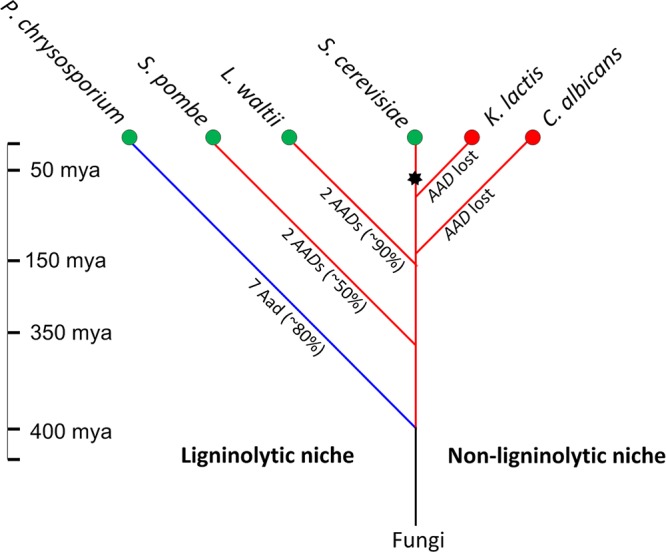
Hypothesized evolution of fungal aryl-aldehyde reductases, enzymes that detoxify lignin by-products. AAD orthologs are observed among wood and leaf litter saprophytes but not among fungi adapted to animal niches. Four hundred million years ago, split of Basidiomycota (blue branch) and the Ascomycota (red branches) (37) coincides with emergence of woody plants in the form of gymnosperms (56–58); 350 mya, divergence of S. pombe and Saccharomyces spp. (42); 150 mya, divergence of L. waltii and S. cerevisiae (44); 50 to 100 mya, adaptation to sugar-rich niches via Adh neofunctionalization (black asterisk) and ethanologenesis (22, 68, 69, 70). Green nodes denote species found in ligninolytic habitats and red nodes denote species found in nonligninolytic habitats. Text along the branches signifies AAD copy numbers and their average nucleotide similarity to ScAAD14.
DISCUSSION
Based on their homology to AAD1 in the white-rot fungus P. chrysosporium, seven AAD genes are predicted to occur in the genome of S. cerevisiae strain BY4741 (8, 12). However, to date, the function(s) of these genes have proven elusive. Here, we show that two members of this family, ScAAD4 and ScAAD14, encode functional enzymes that reduce aryl-aldehydes to their corresponding aryl-alcohols with NADPH as a cofactor. ScAad4p and ScAad14p exhibit overlapping but nonidentical substrate preferences, as often seen when genes arise via duplication from a common ancestor (50, 51). Other members of the BY4741 AAD gene family appear to be undergoing pseudogenization, and the mechanisms by which this is likely occurring offer clues as to how protein function might be restored. In silico analysis of extant AAD coding and protein sequences, coupled with three-dimensional (3D) modeling of enzymatically active PcAad1p suggested that a split ORF had inactivated AAD6/AAD16, that an N-terminal deletion had inactivated AAD10, and that a missense mutation at a key catalytic residue had abolished AAD3-encoded activity. Ancestral-state reconstruction (29) via site-directed mutagenesis enabled us to resurrect aryl-aldehyde dehydrogenase activity for two of these ScAad proteins (ScAad6518Gp and ScAad10−35Cp).
Pseudogenization of AAD genes is also evident in the extensive polymorphism observed among other strains of S. cerevisiae isolated from diverse habitats. Gene copy number and gene sequence are more variable in subtelomeric AAD genes relative to nonsubtelomeric aldehyde reductases, such as ADH genes and AKR genes listed in Table S1. We suggest that pseudogenization of AAD genes is facilitated by their location, as genes located in subtelomeric regions undergo both meiotic and mitotic recombination at elevated rates relative to genes located elsewhere in the genome (52). Indeed, subtelomeric instability has been hypothesized as a mechanism that accelerates adaptation via rapid functional divergence of novel alleles (53). Thus, in yeast, the present distribution of highly polymorphic and variably functional AAD genes reflects their evolutionary history.
S. cerevisiae Aad in vitro activities are about 3-fold lower than those of PcAad1p from the saprophytic white rot fungus Phanerochaete chrysosporium. Also, they are one to two orders of magnitude lower than the broad-specificity NADPH-dependent aldehyde reductases encoded by S. cerevisiae aldehyde dehydrogenase VI (ADH6, ca. 183 μmol · min−1 · mg−1 for cinnamaldehyde) (24) and aldehyde dehydrogenase VII (ADH7, ca. 90 μmol · min−1 · mg−1 for cinnamaldehyde) (25). ScAAD expression is not induced by aryl-aldehydes, and ScAAD overexpression does not confer resistance to toxic aryl-aldehyde derivatives. The data presented in Table 1 suggest that the capacity of ScAadp proteins to detoxify aldehydes has been superseded by promiscuous yeast aldehyde reductases encoded by members of the Akr and Adh families. Unlike yeast AAD genes, yeast ADH genes are transcriptionally upregulated in the presence of such compounds (36), and overexpression of ADH6 and ADH7 increases yeast resistance to veratraldehyde, anisaldehyde, and 5-hydroxymethylfurfural (24, 36). Yeast aldehyde reductases catalyze diverse reactions that lead to ethanol production as well as to the reduction of branched-chain and aromatic aldehydes into fusel alcohols via the Ehrlich pathway (54). Concerning the biological function of yeast Aadp proteins, we note a lack of consensus as to whether they play a role in the production of aroma metabolites arising from the Ehrlich pathway. Dickinson et al. (55) found that none of the AAD genes could be implicated in fusel alcohol formation from amino acids, while Styger et al. attributed to AAD6 the aromatic profile peculiar to fermentation in the presence of excess branched amino acids: leucine, valine, and isoleucine (56). As the yeast strain used by Styger et al. (56) was isogenic to BY4741 but opposite in its mating type, their finding is inconsistent with our data, which indicate that AAD6 does not encode a functional enzyme.
The fact that few yeast Aad enzymes can reduce toxic aromatic aldehydes released by lignin depolymerization (23, 30, 31), coupled with the findings that yeast AAD genes are undergoing pseudogenization and that AAD orthologs are restricted to fungi in ligninolytic niches, leads us to hypothesize that Aad function originated among wood saprophytes. Consistent with this hypothesis, our phylogenetic analyses indicate that the orthologs most closely related to S. cerevisiae AAD are confined to the Basidiomycota and Ascomycota (Fig. 3). These phyla diverged about 400 mya (37), contemporaneous with the earliest fossil record of wood (56–58), suggesting that ancestral AAD genes arose about the same time that lignin became abundant in the terrestrial environment. The hemiascomyte clade that encompasses the saccharolytic yeasts (22, 59) emerged ∼150 mya, broadly coincidental with the rise of fruit-forming angiosperms (22, 60–63) (Fig. 4). We therefore propose that extant yeast Aad proteins are largely evolutionary relics, albeit ones that bear witness to fungal ancestors that invaded lignocellulosic niches created in the mid-Devonian with the rise of vascular plants on dry land.
Conclusion.
The role played by the subtelomeric AAD gene family in the yeast Saccharomyces cerevisiae has been obscure, as there is no obvious mutant phenotype and no documented enzymatic activity. Here, we report that of seven members of this family present in lab strain BY4741, only AAD4 and AAD14 encode proteins able to reduce aryl-aldehydes to their corresponding alcohols using NADPH as a cofactor. Among the five remaining putative AAD genes, aryl-aldehyde dehydrogenase activity could be resurrected in Aad10p after its N terminus was restored and in Aad6/Aad16p after two coding regions were fused. Aryl-aldehyde dehydrogenase activity could not be resurrected from either AAD3 or AAD15. Phylogenetic data suggest that AAD genes originated in wood-saprophytic fungi. The likely ancestral function of Aad proteins was to reductively detoxify aromatic aldehydes arising from lignin depolymerization. In contrast, the yeast ancestors of Saccharomyces cerevisiae adapted to sugar-rich niches that arose following the advent of fruit-bearing angiosperms. Ethanologenic fermentation favored the evolution of multiple alcohol dehydrogenases via duplication and neofunctionalization. Yeast ADH genes also encode aldehyde reductase activities, and these have played increasingly important roles in redox balance and detoxification, compensating for the loss of Aad activity through pseudogenization.
MATERIALS AND METHODS
Strains and media.
Unless otherwise stated, S. cerevisiae BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) was used as the host strain in all cloning, overexpression, and biochemical studies. The S. cerevisiae adh6 mutant (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Δadh6, BY4741 background) from the Yeast Knockout collection was included in detoxification assays. Yeast were cryopreserved as −80°C glycerol stocks and routinely propagated in either liquid or solid YPD medium (1% yeast extract, 2% peptone, 2% glucose) at 30°C. Experimental assays were performed using synthetic uracil drop-out (URA−) medium [1.7 g/liter yeast nitrogen base without amino acids, 5 g/liter (NH4)2SO4, 20 g/liter glucose, 1.92 g of uracil drop-out supplements (catalog no. Y1501; Sigma-Aldrich)] containing 76 mg/liter uracil (catalog no. U1128; Sigma-Aldrich) if required for growth. E. coli strain BL21 Star (catalog no. C6020-03; Invitrogen) was used to host heterologous expression and purification of recombinant Aad proteins. Luria broth and plate medium (10 g/liter tryptone, 5 g/liter yeast extract, 10 g/liter NaCl) was used for routine propagation of E. coli strains with 150 μg · ml−1 ampicillin, 50 μg · ml−1 kanamycin, or 34 μg · ml−1 chloramphenicol added for plasmid selection.
Bioinformatics. (i) Identification of ORF truncation and deletion.
Open reading frames plus 1-kb up- and downstream the sequences of the seven yeast AAD genes were retrieved from the Saccharomyces Genome Database (SGD), as well the PcAAD1 coding sequence; these were imported to VectorNTI Advanced 10.0 software (Invitrogen) for alignment. The inference of ORF truncation for ScAAD4, ScAAD10, ScAAD6, and ScAAD16 and the ORF deletion of ScAAD15 was based on the fact that (i) the out-of-frame nucleic acid sequences share ≥80% consensus positions (Fig. S1), (ii) a simulated single-base-pair mutation put all of the hypothetical coding sequences back in frame, (iii) in-frame sequences are similar in length to the coding sequence of the reference gene PcAAD1, and (iv) all hypothetical S. cerevisiae ORFs (ScORFs) encode comparably sized proteins that share ≥80% amino acid sequence similarity (Fig. S1).
(ii) Search for AAD homologs.
The SGD-retrieved ORFs of ScAAD3 and ScAAD14 and hypothetical ORFs of ScAAD4, ScAAD10, ScAAD6/16, and ScAAD15 were used as query sequences to perform a standard WU-BLASTN search against the genome data of Saccharomyces cerevisiae species (https://www.yeastgenome.org/blast-fungal), using default parameters. The retrieved subject sequences and their corresponding scaffold sequences were exported to VectorNTI Advanced 10.0 for further validation. AAD homologues were accepted as valid only if the chromosome numbers initially annotated by the sequencing project and the alignment between the exported scaffold and reference S288C sequences did not suggest any chromosome rearrangement. Nucleic acid and translated sequences of AAD homologues from 10 representative S. cerevisiae species were aligned with that of the reference S288C strain. Sequence alignments, oligonucleotide thermal properties, protein molecular weight (MW) calculation, and design of plasmids and primers were conducted using the software VectorNTI Advanced 10.0 (Invitrogen).
(iii) Search for AAD orthologs.
The ScAAD14 coding sequence was used as the query sequence to perform a BLASTn against nucleotide collection (nr/nt) genome databases (max target = 5,000, Exp. threshold = 10) at https://blast.ncbi.nlm.nih.gov/Blast.cgi. The matched AAD ortholog sequences were ranked by scores. For multiple orthologs from one species, only the first sequence (score, ≥60; length, >150 bp) was chosen to represent the corresponding species. A k-mer (k = 15)-based neighbor-joining tree construction was performed in the CLC Genomics Workbench 10 software.
(iv) Modeling of PcAad1p structure.
In silico modeling of PcAad1p structure was performed using the YASARA Structure software (YASARA Biosciences) with the resolved AKR11C1 structure as the template (64). Assessment of modeling quality was carried out using the SWISS-MODEL Web server (http://swissmodel.expasy.org/). The PcAad1p sequence with key amino acids and motifs thereby revealed was aligned with yeast Aadp proteins (Fig. 2).
Cloning, heterologous expression, and purification of recombinant yeast Aadp proteins.
PCR-based amplification of S. cerevisiae AAD genes was carried out on purified genomic BY4741 DNA using the forward and reverse primers described in Table 4. Fifty-microliter PCRs were performed using the Phusion PCR system (catalog no. F630S; Thermo Fisher; containing 100 ng of DNA, 0.2 mM dinucleoside triphosphate [dNTP], 1.5 mM MgCl2, 0.5 μM reverse and forward primers, 1 unit of neuraminidase [NA] polymerase), as follows: 1 cycle of 98°C for 30 s, 30 cycles of 98°C for 10 s, 65°C for 30 s, and 72°C for 45 s; and 1 cycle of 72°C for 7 min. The primers described in Table 4 generate PCR amplicons flanked by KpnI and NotI restriction sites at their 5′ and 3′ ends, respectively. Amplicons were A-tailed and ligated into pGEM-T Easy vectors (catalog no. A1360; Promega). Following transformation into E. coli DH5α and growth under ampicillin selection, positive clones were verified by double enzyme digestion and Sanger sequencing. KpnI-NotI fragments were then excised from the T-vectors and religated into plasmid pGS-21a (catalog no. SD0121; GenScript) previously linearized by KpnI-NotI digestion. Successful constructs were verified by plasmid sequencing and then transformed into E. coli BL21 Star as pGS-21a-ScAad3, pGS-21a-ScAad4, and pGS-21a-ScAad14. Expression and purification of recombinant yeast Aad proteins were carried out in parallel with reference protein PcAad1p, using procedures previously described for PcAad1p (10). Assays on reconstructed yeast ScAad3Cys73Tyrp, Aad6/16518Gp, and Aad10−35Cp proteins were carried out in parallel with a purified His6-GST tag.
TABLE 4.
Primers and plasmids for cloning, plasmid construction, and site-directed mutagenesis
| Primer | Primer sequencea | Purpose(s) | Yielded plasmid(s) |
|---|---|---|---|
| AAD3_pGS_F | GGTACCGACGACGACGACAAGATGATTGGGTCCGCGTCCG | ScAAD3 cloning to pGS-21a | pGS-21a-ScAad3 |
| AAD3_pGS_R | GCGGCCGCAACATTATTCGTACCATATTT | ||
| AAD4_pGS_F | GGTACCGACGACGACGACAAGATGGGCTCTATGAATAAGGAACA | ScAAD4 cloning to pGS-21a | pGS-21a-ScAad4 |
| AAD4_pGS_R | GCGGCCGCATCGAAGGAAATCTGCGCA | ||
| AAD14_pGS_F | GGTACCGACGACGACGACAAGATGACTGACTTGTTTAAACCTCT | ScAAD14 cloning to pGS-21a | pGS-21a-ScAad14 |
| AAD14_pGS_R | GCGGCCGCATTGTCAAAAGCTATCCTGGCA | ||
| PC_AAD_ORF1_F1_HR | GTAATTATCTACTTTTTACAACAAATATAAAACAAGATCTCGACTCTAGAGGATCCATGAACATCTGGGCACCCG | Subcloning PcAAD1 from pGS-21a-PcAad1 (Yang et al. [10]), for purpose of overexpression PcAAD1 in yeast BY4747 | YEplac195PGK/CYC1-JL52-URA3-PcAad1 |
| PC_AAD_ORF1_R1_HR | CCAAAGGCCATCTTGGTACCGGGCCCCCCCTCGAGGTCGACGGTATCGATAAGCTTCTACTTCTGGGGGCGGATAGC | ||
| SC_AAD3_F1_HR | GTAATTATCTACTTTTTACAACAAATATAAAACAAGATCTCGACTCTAGAGGATCCATGATTGGGTCCGCGTCCG | Subcloning ScAAD3 from pGS-21a-ScAad3, for purpose of overexpression ScAAD3 in yeast BY4747 | YEplac195PGK/CYC1-JL52-URA3-ScAad3 |
| SC_AAD3_R1_HR | CCAAAGGCCATCTTGGTACCGGGCCCCCCCTCGAGGTCGACGGTATCGATGCGGCCGCCTAAACATTATTCGTACCATATTT | ||
| SC_AAD4_F1_HR | GTAATTATCTACTTTTTACAACAAATATAAAACAAGATCTCGACTCTAGAGGATCCATGGGCTCTATGAATAAGGAACA | Subcloning ScAAD4 from pGS-21a-ScAad4, for purpose of overexpression ScAAD4 in yeast BY4747 | YEplac195PGK/CYC1-JL52-URA3-ScAad4 |
| SC_AAD4_R1_HR | CCAAAGGCCATCTTGGTACCGGGCCCCCCCTCGAGGTCGACGGTATCGATGCGGCCGCTTAATCGAAGGAAATCTGCGCA | ||
| SC_AAD14_F1_HR | GTAATTATCTACTTTTTACAACAAATATAAAACAAGATCTCGACTCTAGAGGATCCATGACTGACTTGTTTAAACCTCT | Subcloning ScAAD14 from pGS-21a-ScAad14, for purpose of overexpression ScAAD14 in yeast BY4747 | YEplac195PGK/CYC1-JL52-URA3-ScAad14 |
| SC_AAD14_R1_HR | CCAAAGGCCATCTTGGTACCGGGCCCCCCCTCGAGGTCGACGGTATCGATGCGGCCGCCTAATTGTCAAAAGCTATCCTGGCA | ||
| PcAad1Tyr76mut2Cys_F1 | AACTTCATTGATACCGCTAATGTCTGCCAAGACGAGACATCCGAGGAATTT | PcAad1p tyrosine73→cysteine73 mutagenesis | pGS-21a-PcAad1Tyr76Cys |
| PcAad1Tyr76mut2Cys_R1 | AAATTCCTCGGATGTCTCGTCTTGGCAGACATTAGCGGTATCAATGAAGTT | ||
| ScAad3MutCys2Tyr_BamHI_A1 | ATCGCGCGGATCCATGATTGGGTCCGCGTCCGACTCATCTAGC | ScAad3p cysteine73→tyrosine73 mutagenesis | pGS-21a-ScAad1Cys73 Tyr |
| ScAad3MutCys2Tyr_B1 | CCATTCTTCTGATTGCTCGTTTTGGTAGTTGTTTGCGGCATCAATGAAATT | ||
| ScAad3MutCys2Tyr_C1 | AATTTCATTGATGCCGCAAACAACTACCAAAACGAGCAATCAGAAGAATGG | ||
| ScAad3MutCys2Tyr_XhoI_D1 | ATCGCCGCTCGAGAACATTATTCGTACCATATTTTTGAGTCAAGG | ||
| ScAad6InserG_F1_A | ATCGATCGCGCGGATCCATGGCTGATTTATTTGCTCCTGCTCC | Fusion of ScAAD6-AAD16 | pGS-21a-ScAad6518G |
| ScAad6InserG_R1_B | AGACACACCCAAATAGAGGACCTTGCCCTGCTGCACTAGAATGTGTAAACT | ||
| ScAad6InserG_F2_C | AGTTTACACATTCTAGTGCAGCAGGGCAAGGTCCTCTATTTGGGTGTGTCT | ||
| ScAad6InserG_R2_D | ATCGATCGCCGCTCGAGTTAATCGAAGGAAATCTGCGCAGACATTGC | ||
| ScAad10Mute_F1_A | ATCGATCGCGCGGATCCATGTCTGAGGCTTTTGGACCTGCAC | ScAad10-35Cp N truncation repair | pGS-21a-ScAad10C-35 |
| ScAad10Mute_R1_B | ATCCAAGTCTCTGACTGCTCATACTGATAATTATTTGCAGTATCAATGAAATTTCC | ||
| ScAad10Mute_F2_C | GGAAATTTCATTGATACTGCAAATAATTATCAGTATGAGCAGTCAGAGACTTGGAT | ||
| ScAad10Mute_R2_D | ATCGATCGCCGCTCGAGCTAATCTTCGAAGCTAATCTTGGCA |
Italics indicate the enterokinase-coding sequence, boldface indicates start and stop codons, and underlining indicates restriction sites.
Subcloning of AAD into the YEplac195PGK/CYC1-JL52-URA3 yeast vector.
The coding sequence of PcAad1p was PCR amplified from plasmid pGS-21a-PcAad1 using primer set PC_AAD_ORF1_F1_HR and PC_AAD_ORF1_R1_HR in a 50-μl Phusion PCR (see Table 4 for primer sequences). This primer set generates amplicons flanked by 50-bp sequences homologous to the YEplac195PGK/CYC1-JL52-URA3 vector at multiple cloning sites. The purified PCR amplicon and BamHI- and HindIII-linearized YEplac195PGK/CYC1-JL52-URA3 were cotransformed to S. cerevisiae strain BY4741 using the standard lithium acetate protocol (65). Yeast transformants obtained on synthetic URA− plates were subjected to plasmid isolation and sequencing. The resulting plasmid was named YEplac195PGK/CYC1-JL52-URA3-PaAad1 (see Table 4). Yeast AAD genes were cloned into plasmid YEplac195PGK/CYC1-JL52-URA3 via homologous recombination using methods similar to those described above for PcAAD1. The primers used are described in Table 4.
Site-directed mutagenesis and reconstruction of hypothetical Aad ORFs. (i) Reconstruction of truncated ScAad6p and ScAad16p.
Bioinformatic analyses indicated that extant yeast genes AAD6 and AAD16 together once formed a functional open reading frame. To reconstruct this presumed ancestral AAD6/AAD16, we used a three-step PCR-based procedure. The first reaction uses S. cerevisiae BY4741 genomic DNA as the template with primers ScAad6InserG_F1_A and ScAad6InserG_R1_B, of which ScAad6InserG_R1_B carries a G insert at positions G517∼518. The second reaction uses the same template but with primer set ScAad6InserG_F2_C and ScAad6InserG_R2_D, which produces an amplicon that contains the G insert and also overlaps with the first amplicon. The two amplicons were then fused together by an overlapping PCR that uses primers ScAad6InserG_F1_A and ScAad6InserG_R2_D. The details for overlapping PCR and subsequent ligation are similar to those described above for ScAadC−35p. The resulting construct was verified by sequencing and termed pGS-21a-ScAad6518Gp.
(ii) Mutation of PcAad1Tyr76Cysp.
PCR-based site-directed mutagenesis (SDM) was performed on previously described plasmid pGS-21a-PcAad1 (10) to generate an A→G mutation that would replace the presumed catalytic tyrosine76 (coded by TAC) with a cysteine76 (coded by TGC). The PcAad1Tyr76mut2Cys_F1 and PcAad1Tyr76mut2Cys_R1 primers (0.5 μM working concentration each, both having the A→G mutation, as listed in Table 4) and 20 ng of plasmid were used in a 50-μl Phusion PCR. The PCR conditions consisted of one cycle of denaturation at 98°C for 60 s, followed by 20 cycles of amplification (98°C denaturation for 15 s, 50°C annealing for 30 s, and 72°C extension for 4 min), and one final extension at 72°C for 7 min. Two microliters of DpnI and 5 μl of 1× CutSmart buffer (catalog no. R0176S; New England BioLabs) were added to the completed reaction and incubated at 37°C for 1 h, after which an additional 2 μl of DpnI was added to completely digest the methylated template plasmid. The resulting reaction mixture was purified and then transformed to E. coli strain DH5α. Transformants were subjected to plasmid purification and sequencing to verify the presence of the mutation. The newly constructed plasmid was named pGS-21a-PcAad1Tyr76Cys.
(iii) Mutation of ScAad3p.
PCR-based site-direct mutagenesis was performed to mutate cystine73 (coded by TG218C) into a presumed functional tyrosine73 (coded by TAC) in a manner similar to that described above for PcAad1Cys76p, except that primer set ScAad3MutCys2Tyr_B1/ScAad3MutCys2Tyr_C1 was used against template vector pGS21a-ScAad3. The resulting constructs were screened by colony PCR using the primer set AAD3_pGS_F/AAD3_pGS_R, validated by Sanger sequencing, and then named pGS21a-ScAad3Cys73Tyr.
(iv) Repairing N-terminal truncation of ScAad10p.
Reconstruction of the presumed ScAad10p ancestor by repair of its N-terminal truncation was carried out using three rounds of PCR. The first reaction used S. cerevisiae genomic DNA as the template in conjunction with primers ScAad10Mute_F1_A and ScAad10Mute_R1_B, of which ScAad10Mute_R1_B carries a mutated C−35AG codon that replaces the presumed premature Stop codon T−35AG. The second reaction uses the same genomic template, but primer set ScAad10Mute_F2_C and ScAad10Mute_R2_D, which produces an amplicon with the corrected C−35AG codon and overlaps with the first amplicon. The third reaction is an overlapping PCR that uses purified amplicons from the first two rounds as templates in conjunction with primer set ScAad10Mute_F1_A and ScAad10Mute_R2_D. The first two reactions use standard the Phusion protocol described above but with annealing at 65°C. The overlapping Phusion PCR cycling conditions were 1 cycle at 95°C for 4 min, followed by 25 cycles of 95°C for 30 s, 68°C for 30 s, and 72°C for 3 min. The resulting amplicon was precipitated, digested with BamHI and Xhol, and then gel purified and ligated with similarly linearized pGS-21a. The new construct was verified by Sanger sequencing and termed pGS-21a-ScAad10C−35.
Analysis of AAD expression in response to aromatic aldehyde exposure.
Three independent colonies of S. cerevisiae strain BY4741 were picked from YPD agar, inoculated in 5 ml of YPD broth, and cultured overnight at 30°C, with shaking at 150 rpm. Half a milliliter of these overnight cultures was used to inoculate 180 ml of YPD broth in 1-liter Erlenmeyer flasks. Yeast cultures were grown in triplicate at 30°C at 150 rpm, and their optical density (OD) was recorded at λ = 600 nm. Aliquots of stock solutions of 3,4-dimethoxybenzaldehyde, hydroxymethylfurfural, 4-hydroxy-3-methoxybenzaldehyde, and trans-cinnamaldehyde were added to mid-log yeast cultures (OD, 0.8) to yield final concentrations of 25 mM, 25 mM, 25 mM, and 50 mM, respectively. Untreated cultures were included as a negative control. Cells were harvested 1 h and 2 h following treatment by centrifuging 45 ml of culture at 10,000 × g and 4°C for 5 min. The resulting pellets were immediately frozen in liquid nitrogen and stored at −80°C.
Yeast mRNA was extracted following the yeast RNA extraction protocol provided in the SV Total RNA isolation system kit (catalog no. Z3100; Promega). RNA quality was assessed via Bioanalyzer 2100 using the RNA 6000 Nano LabChip kit (Agilent Technologies, Massy, France) and quantified in a NanoDrop ND-1000 UV-visible light spectrophotometer (Fisher Scientific SAS, Illkirch, France). cDNA was synthesized from 1 μg of total RNA in 20-μl reaction mixtures using the iScript cDNA synthesis kit (catalog no. 1708891; Bio-Rad).
Due to high sequence homology among AAD family members, two sets of gene-specific primers (GSP) were designed for each AAD gene (Table 5). Housekeeping genes TAF10, TFC1, and UBC6 were used as internal controls for the normalization of expression data (66). The binding efficiencies of the primers were evaluated using the series dilution method (67). Real-time PCRs were carried out using a MyiQ single-color real-time PCR detection system (catalog no. 170-9740; Bio-Rad, France). Reactions were set up in triplicate for each of three biological replicates to ensure the reliability of the results. The reactions were performed in a 25-μl final reaction volume using iQ SYBR green Supermix (Bio-Rad), under previously described reaction conditions (10).
TABLE 5.
List of primers for quantitative PCR for expression studies
Plate assay of aldehyde-mediated growth inhibition.
Multicopy yeast expression vectors YEplac195PGK/CYC1-JL52-URA3 (harboring either ScAAD3, ScAAD4, ScAAD14, or PcAAD1) were transformed into S. cerevisiae strain BY4741 and the Yeast Knockout (YKO) adh6 (clone no. 6460, MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Δadh6) mutant using the standard lithium-acetate procedure (65). Three independent transformants were picked and used to inoculate replicate overnight cultures. The next day, 10 μl from these cultures was used to inoculate 5 ml of URA− medium, which was grown at 30°C and 150 rpm. At an OD at 600 nm (OD600) of 1, the culture was 105-fold diluted in URA− medium at 25°C. Two microliters of diluted cell suspensions was pipetted onto URA− agar containing one of the four aldehydes (3,4-dimethoxybenzaldehyde, hydroxymethylfurfural, 4-hydroxy-3-methoxybenzaldehyde, or trans-cinnamaldehyde) at concentrations of 0, 6.25, 12.5, or 50 mM. Colonies appearing on these plates were photographed every 12 h over the course of 72 h.
Analysis of AAD enzyme substrate specificity and enzymatic kinetics.
Heterologous expression, purification, and biochemical characterization of yeast Aad proteins were carried out in parallel, with the PcAad1p serving as a reference. PcAad1p and all recombinant yeast Aad proteins bear an N-terminal His6-GST and a C-terminal His6 tag and were purified following the GST-affinity batch purification (10). The activities of purified reference PcAad1p, yeast Aadp proteins, and mutated yeast Aad recombinant proteins were assayed against a panel of aliphatic/aromatic aldehydes and aryl-alcohols using both NAD(P)H and NAD(P)+ as reduction and oxidation cofactors (10). Reactions were quantified spectrophotometrically by following the consumption or production of cofactor NAD(P)+(H) at λ = 340 nm (ε340 = 6.2 mM−1 · cm−1) in 250-μl microplates containing 0.3 mM cofactor and 0.3 mM substrates (10) (microplate reader model 680XR; Bio-Rad). Kinetic parameters (Km and Vmax) quantifying NADP+(H) consumption/formation were assayed at 355 nm (ε355 = 5.12 mM−1 · cm−1) using a UV-visible spectrophotometer (Shimadzu UV1800) in 1-ml cuvettes, as described previously; the substrate absorption at this wavelength is negligible (10). For each recombinant protein, significant differences relative to a pGS-21a blank plasmid were calculated using a 2-tailed t test at a P value of <0.05.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Jean-Luc Parrou, Marie-Ange Teste, and Xingquan Liu for technical support, and to Emily Cook, Matt Herron, Pedram Samani, and Eugene Kroll for manuscript comments.
Work carried out at LISBP was in part supported by COST action FA0907 BIOFLAVOUR under the EU's Seventh Framework Programme for Research (FP7) to G.M.D.B., and by ANR grant BLAN07-2_200101 to J.-M.F. D.-D.Y. and J.-J.Z. were funded by Natural Science Foundation of China (NSFC 31301547), Ministry of Education (SRF for ROCS 2015), and ZAFU (2012FR066). F.R. and D.-D.Y. were funded by NASA NNX12AD87G and NIH R01HG003328.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01553-17.
REFERENCES
- 1.Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG. 1996. Life with 6000 genes. Science 274:546:563–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 2.Mackiewicz P, Kowalczuk M, Mackiewicz D, Nowicka A, Dudkiewicz M, Laszkiewicz A, Dudek MR, Cebrat S. 2002. How many protein-coding genes are there in the Saccharomyces cerevisiae genome? Yeast 19:619–629. doi: 10.1002/yea.865. [DOI] [PubMed] [Google Scholar]
- 3.Peña-Castillo L, Hughes TR. 2007. Why are there still over 1000 uncharacterized yeast genes? Genetics 176:7–14. doi: 10.1534/genetics.107.074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kowalczuk M, Mackiewicz P, Gierlik A, Dudek MR, Cebrat S. 1999. Total number of coding open reading frames in the yeast genome. Yeast 15:1031–1034. doi:. [DOI] [PubMed] [Google Scholar]
- 5.Lin D, Yin X, Wang X, Zhou P, Guo FB. 2013. Re-annotation of protein-coding genes in the genome of Saccharomyces cerevisiae based on support vector machines. PLoS One 8:e64477. doi: 10.1371/journal.pone.0064477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blandin G, Durrens P, Tekaia F, Aigle M, Bolotin-Fukuhara M, Bon E, Casaregola S, de Montigny J, Gaillardin C, Lepingle A, Llorente B, Malpertuy A, Neuveglise C, Ozier-Kalogeropoulos O, Perrin A, Potier S, Souciet J, Talla E, Toffano-Nioche C, Wesolowski-Louvel M, Marck C, Dujon B. 2000. Genomic exploration of the hemiascomycetous yeasts: 4. The genome of Saccharomyces cerevisiae revisited. FEBS Lett 487:31–36. [DOI] [PubMed] [Google Scholar]
- 7.Zhang CT, Wang J. 2000. Recognition of protein coding genes in the yeast genome at better than 95% accuracy based on the Z curve. Nucleic Acids Res 28:2804–2814. doi: 10.1093/nar/28.14.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muheim A, Waldner R, Sanglard D, Reiser J, Schoemaker HE, Leisola MS. 1991. Purification and properties of an aryl-alcohol dehydrogenase from the white-rot fungus Phanerochaete chrysosporium. Eur J Biochem 195:369–375. doi: 10.1111/j.1432-1033.1991.tb15715.x. [DOI] [PubMed] [Google Scholar]
- 9.Reiser J, Muheim A, Hardegger M, Frank G, Fiechter A. 1994. Aryl-alcohol dehydrogenase from the white-rot fungus Phanerochaete chrysosporium. Gene cloning, sequence analysis, expression, and purification of the recombinant enzyme. J Biol Chem 269:28152–28159. [PubMed] [Google Scholar]
- 10.Yang DD, Francois JM, de Billerbeck GM. 2012. Cloning, expression and characterization of an aryl-alcohol dehydrogenase from the white-rot fungus Phanerochaete chrysosporium strain BKM-F-1767. BMC Microbiol 12:126. doi: 10.1186/1471-2180-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penning TM. 2015. The aldo-keto reductases (AKRs): overview. Chem Biol Interact 234:236–246. doi: 10.1016/j.cbi.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delneri D, Gardner DC, Bruschi CV, Oliver SG. 1999. Disruption of seven hypothetical aryl alcohol dehydrogenase genes from Saccharomyces cerevisiae and construction of a multiple knock-out strain. Yeast 15:1681–1689. doi:. [DOI] [PubMed] [Google Scholar]
- 13.Delneri D, Gardner DC, Oliver SG. 1999. Analysis of the seven-member AAD gene set demonstrates that genetic redundancy in yeast may be more apparent than real. Genetics 153:1591–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen BA, Pilpel Y, Mitra RD, Church GM. 2002. Discrimination between paralogs using microarray analysis: Application to the Yap1p and Yap2p transcriptional networks. Mol Biol Cell 13:1608–1614. doi: 10.1091/mbc.01-10-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy S, Ihmels J, Carmi M, Weinberger A, Friedlander G, Barkai N. 2007. Strategy of transcription regulation in the budding yeast. PLoS One 2:e250. doi: 10.1371/journal.pone.0000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shapira M, Segal E, Botstein D. 2004. Disruption of yeast forkhead-associated cell cycle transcription by oxidative stress. Mol Biol Cell 15:5659–5669. doi: 10.1091/mbc.E04-04-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitagawa E, Takahashi J, Momose Y, Iwahashi H. 2002. Effects of the pesticide thiuram: genome-wide screening of indicator genes by yeast DNA microarray. Environ Sci Technol 36:3908–3915. doi: 10.1021/es015705v. [DOI] [PubMed] [Google Scholar]
- 19.Kitagawa E, Akama K, Iwahashi H. 2005. Effects of iodine on global gene expression in Saccharomyces cerevisiae. Biosci Biotechnol Biochem 69:2285–2293. doi: 10.1271/bbb.69.2285. [DOI] [PubMed] [Google Scholar]
- 20.Chechik G, Oh E, Rando O, Weissman J, Regev A, Koller D. 2008. Activity motifs reveal principles of timing in transcriptional control of the yeast metabolic network. Nat Biotechnol 26:1251–1259. doi: 10.1038/nbt.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwahashi H, Kitagawa E, Suzuki Y, Ueda Y, Ishizawa Y, Nobumasa H, Kuboki Y, Hosoda H, Iwahashi Y. 2007. Evaluation of toxicity of the mycotoxin citrinin using yeast ORF DNA microarray and Oligo DNA microarray. BMC Genomics 8:95. doi: 10.1186/1471-2164-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Connor ST, Lan J, North M, Loguinov A, Zhang L, Smith MT, Gu AZ, Vulpe C. 2012. Genome-wide functional and stress response profiling reveals toxic mechanism and genes required for tolerance to benzo[a]pyrene in S. cerevisiae. Front Genet 3:316. doi: 10.3389/fgene.2012.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dashko S, Zhou N, Compagno C, Piskur J. 2014. Why, when, and how did yeast evolve alcoholic fermentation? FEMS Yeast Res 14:826–832. doi: 10.1111/1567-1364.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs G, Boll M, Heider J. 2011. Microbial degradation of aromatic compounds–from one strategy to four. Nat Rev Microbiol 9:803–816. doi: 10.1038/nrmicro2652. [DOI] [PubMed] [Google Scholar]
- 25.Larroy C, Fernandez MR, Gonzalez E, Pares X, Biosca JA. 2002. Characterization of the Saccharomyces cerevisiae YMR318C (ADH6) gene product as a broad specificity NADPH-dependent alcohol dehydrogenase: relevance in aldehyde reduction. Biochem J 361:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larroy C, Pares X, Biosca JA. 2002. Characterization of a Saccharomyces cerevisiae NADP(H)-dependent alcohol dehydrogenase (ADHVII), a member of the cinnamyl alcohol dehydrogenase family. Eur J Biochem 269:5738–5745. doi: 10.1046/j.1432-1033.2002.03296.x. [DOI] [PubMed] [Google Scholar]
- 27.Ford G, Ellis EM. 2001. Three aldo-keto reductases of the yeast Saccharomyces cerevisiae. Chem Biol Interact 130–132:685–698. doi: 10.1016/S0009-2797(00)00259-3. [DOI] [PubMed] [Google Scholar]
- 28.Chang Q, Griest TA, Harter TM, Petrash JM. 2007. Functional studies of aldo-keto reductases in Saccharomyces cerevisiae. Biochim Biophys Acta 1773:321–329. doi: 10.1016/j.bbamcr.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ford G, Ellis EM. 2002. Characterization of Ypr1p from Saccharomyces cerevisiae as a 2-methylbutyraldehyde reductase. Yeast 19:1087–1096. doi: 10.1002/yea.899. [DOI] [PubMed] [Google Scholar]
- 30.Chandrasekharan UM, Sanker S, Glynias MJ, Karnik SS, Husain A. 1996. Angiotensin II-forming activity in a reconstructed ancestral chymase. Science 271:502–505. doi: 10.1126/science.271.5248.502. [DOI] [PubMed] [Google Scholar]
- 31.Schwab W, Davidovich-Rikanati R, Lewinsohn E. 2008. Biosynthesis of plant-derived flavor compounds. Plant J 54:712–732. doi: 10.1111/j.1365-313X.2008.03446.x. [DOI] [PubMed] [Google Scholar]
- 32.Kamimura N, Goto T, Takahashi K, Kasai D, Otsuka Y, Nakamura M, Katayama Y, Fukuda M, Masai E. 2017. A bacterial aromatic aldehyde dehydrogenase critical for the efficient catabolism of syringaldehyde. Sci Rep 7:44422. doi: 10.1038/srep44422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu ZL. 2011. Molecular mechanisms of yeast tolerance and in situ detoxification of lignocellulose hydrolysates. Appl Microbiol Biotechnol 90:809–825. doi: 10.1007/s00253-011-3167-9. [DOI] [PubMed] [Google Scholar]
- 34.Parawira W, Tekere M. 2011. Biotechnological strategies to overcome inhibitors in lignocellulose hydrolysates for ethanol production: review. Crit Rev Biotechnol 31:20–31. doi: 10.3109/07388551003757816. [DOI] [PubMed] [Google Scholar]
- 35.Larsson S, Quintana-Sainz A, Reimann A, Nilvebrant NO, Jönsson LJ. 2000. Influence of lignocellulose-derived aromatic compounds on oxygen-limited growth and ethanolic fermentation by Saccharomyces cerevisiae. Appl Biochem Biotechnol 84–86:617–632. doi: 10.1385/ABAB:84-86:1-9:617. [DOI] [PubMed] [Google Scholar]
- 36.Jönsson LJ, Martin C. 2016. Pretreatment of lignocellulose: formation of inhibitory by-products and strategies for minimizing their effects. Bioresour Technol 199:103–112. doi: 10.1016/j.biortech.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Petersson A, Almeida JR, Modig T, Karhumaa K, Hahn-Hagerdal B, Gorwa-Grauslund MF, Liden G. 2006. A 5-hydroxymethyl furfural reducing enzyme encoded by the Saccharomyces cerevisiae ADH6 gene conveys HMF tolerance. Yeast 23:455–464. doi: 10.1002/yea.1370. [DOI] [PubMed] [Google Scholar]
- 38.Guarro J, Gene J, Stchigel AM. 1999. Developments in fungal taxonomy. Clin Microbiol Rev 12:454–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Czemmel S, Galarneau ER, Travadon R, McElrone AJ, Cramer GR, Baumgartner K. 2015. Genes expressed in grapevine leaves reveal latent wood infection by the fungal pathogen Neofusicoccum parvum. PLoS One 10:e0121828. doi: 10.1371/journal.pone.0121828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D'Souza TM, Merritt CS, Reddy CA. 1999. Lignin-modifying enzymes of the white rot basidiomycete Ganoderma lucidum. Appl Environ Microbiol 65:5307–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bourbonnais R, Paice MG, Reid ID, Lanthier P, Yaguchi M. 1995. Lignin oxidation by laccase isozymes from Trametes versicolor and role of the mediator 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonate) in kraft lignin depolymerization. Appl Environ Microbiol 61:1876–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mäkelä MR, Sietio OM, de Vries RP, Timonen S, Hilden K. 2014. Oxalate-metabolising genes of the white-rot fungus Dichomitus squalens are differentially induced on wood and at high proton concentration. PLoS One 9:e87959. doi: 10.1371/journal.pone.0087959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffman CS, Wood V, Fantes PA. 2015. An ancient yeast for young geneticists: a primer on the Schizosaccharomyces pombe model system. Genetics 201:403–423. doi: 10.1534/genetics.115.181503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kodama K, Kyono T. 1974. Ascosporogenous yeasts isolated from tree exudates in japan. J Ferment Technol 52:605–613. [Google Scholar]
- 45.Di Rienzi SC, Lindstrom KC, Mann T, Noble WS, Raghuraman MK, Brewer BJ. 2012. Maintaining replication origins in the face of genomic change. Genome Res 22:1940–1952. doi: 10.1101/gr.138248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naumov GI, James SA, Naumova ES, Louis EJ, Roberts IN. 2000. Three new species in the Saccharomyces sensu stricto complex: Saccharomyces cariocanus, Saccharomyces kudriavzevii and Saccharomyces mikatae. Int J Syst Evol Microbiol 50:1931–1942. doi: 10.1099/00207713-50-5-1931. [DOI] [PubMed] [Google Scholar]
- 47.Charron G, Leducq JB, Bertin C, Dube AK, Landry CR. 2014. Exploring the northern limit of the distribution of Saccharomyces cerevisiae and Saccharomyces paradoxus in North America. FEMS Yeast Res 14:281–288. doi: 10.1111/1567-1364.12100. [DOI] [PubMed] [Google Scholar]
- 48.Hyma KE, Fay JC. 2013. Mixing of vineyard and oak-tree ecotypes of Saccharomyces cerevisiae in North American vineyards. Mol Ecol 22:2917–2930. doi: 10.1111/mec.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maganti H, Bartfai D, Xu JP. 2012. Ecological structuring of yeasts associated with trees around Hamilton, Ontario, Canada. FEMS Yeast Res 12:9–19. [DOI] [PubMed] [Google Scholar]
- 50.Sniegowski PD, Dombrowski PG, Fingerman E. 2002. Saccharomyces cerevisiae and Saccharomyces paradoxus coexist in a natural woodland site in North America and display different levels of reproductive isolation from European conspecifics. FEMS Yeast Res 1:299–306. doi: 10.1111/j.1567-1364.2002.tb00048.x. [DOI] [PubMed] [Google Scholar]
- 51.Kafri R, Springer M, Pilpel Y. 2009. Genetic redundancy: new tricks for old genes. Cell 136:389–392. doi: 10.1016/j.cell.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 52.Nowak MA, Boerlijst MC, Cooke J, Smith JM. 1997. Evolution of genetic redundancy. Nature 388:167–171. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- 53.Barton AB, Pekosz MR, Kurvathi RS, Kaback DB. 2008. Meiotic recombination at the ends of chromosomes in Saccharomyces cerevisiae. Genetics 179:1221–1235. doi: 10.1534/genetics.107.083493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown CA, Murray AW, Verstrepen KJ. 2010. Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr Biol 20:895–903. doi: 10.1016/j.cub.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dickinson JR, Salgado LE, Hewlins MJ. 2003. The catabolism of amino acids to long chain and complex alcohols in Saccharomyces cerevisiae. J Biol Chem 278:8028–8034. doi: 10.1074/jbc.M211914200. [DOI] [PubMed] [Google Scholar]
- 56.Styger G, Jacobson D, Prior BA, Bauer FF. 2013. Genetic analysis of the metabolic pathways responsible for aroma metabolite production by Saccharomyces cerevisiae. Appl Microbiol Biotechnol 97:4429–4442. doi: 10.1007/s00253-012-4522-1. [DOI] [PubMed] [Google Scholar]
- 57.Rothwell GW, Sanders H, Wyatt SE, Lev-Yadun S. 2008. A fossil record for growth regulation: the role of auxin in wood evolution. Ann Mo Bot Gard 95:121–134. doi: 10.3417/2006208. [DOI] [Google Scholar]
- 58.Gerrienne P, Gensel PG, Strullu-Derrien C, Lardeux H, Steemans P, Prestianni C. 2011. A simple type of wood in two Early Devonian plants. Science 333:837. doi: 10.1126/science.1208882. [DOI] [PubMed] [Google Scholar]
- 59.Weng JK, Chapple C. 2010. The origin and evolution of lignin biosynthesis. New Phytol 187:273–285. doi: 10.1111/j.1469-8137.2010.03327.x. [DOI] [PubMed] [Google Scholar]
- 60.Wang B, Zhang H, Jarzembowski EA. 2013. Early Cretaceous angiosperms and beetle evolution. Front Plant Sci 4:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soltis DE, Bell CD, Kim S, Soltis PS. 2008. Origin and early evolution of angiosperms. Ann N Y Acad Sci 1133:3–25. doi: 10.1196/annals.1438.005. [DOI] [PubMed] [Google Scholar]
- 62.Wikström N, Savolainen V, Chase MW. 2001. Evolution of the angiosperms: calibrating the family tree. Proc Biol Sci 268:2211–2220. doi: 10.1098/rspb.2001.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Piskur J, Rozpedowska E, Polakova S, Merico A, Compagno C. 2006. How did Saccharomyces evolve to become a good brewer? Trends Genet 22:183–186. doi: 10.1016/j.tig.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 64.Wilf P, Carvalho MR, Gandolfo MA, Cuneo NR. 2017. Eocene lantern fruits from Gondwanan Patagonia and the early origins of Solanaceae. Science 355:71–75. doi: 10.1126/science.aag2737. [DOI] [PubMed] [Google Scholar]
- 65.Marquardt T, Kostrewa D, Balakrishnan R, Gasperina A, Kambach C, Podjarny A, Winkler FK, Balendiran GK, Li XD. 2005. High-resolution crystal structure of AKR11C1 from Bacillus halodurans: an NADPH-dependent 4-hydroxy-2,3-trans-nonenal reductase. J Mol Biol 354:304–316. doi: 10.1016/j.jmb.2005.09.067. [DOI] [PubMed] [Google Scholar]
- 66.Gietz RD, Schiestl RH. 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 67.Teste MA, Duquenne M, Francois JM, Parrou JL. 2009. Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol Biol 10:99. doi: 10.1186/1471-2199-10-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tichopad A, Dilger M, Schwarz G, Pfaffl MW. 2003. Standardized determination of real-time PCR efficiency from a single reaction set-up. Nucleic Acids Res 31:e122. doi: 10.1093/nar/gng122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thomson JM, Gaucher EA, Burgan MF, De Kee DW, Li T, Aris JP, Benner SA. 2005. Resurrecting ancestral alcohol dehydrogenases from yeast. Nat Genet 37:630–635. doi: 10.1038/ng1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sugino RP, Innan H. 2005. Estimating the time to the whole-genome duplication and the duration of concerted evolution via gene conversion in yeast. Genetics 171:63–69. doi: 10.1534/genetics.105.043869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.