

ABSTRACT
Sulfolobus islandicus is rapidly emerging as a model system for studying the biology and evolution within the TACK lineage of the archaeal domain. As the tree of life grows, identifying the cellular functions of genes within this lineage will have significant impacts on our understanding of the evolution of the last archaeal eukaryote common ancestor (LEACA) and the differentiation of archaea from eukaryotes during the evolution of the modern-day cell. To increase our understanding of this key archaeal organism, we report a novel high-throughput method for targeted gene inactivation in S. islandicus through one-step microhomology-directed homologous recombination (HR). We validated the efficacy of this approach by systematically deleting 21 individual toxin-antitoxin gene pairs and its application to delete chromosomal regions as large as 50 kb. Sequence analysis of 96 ArgD+ transformants showed that S. islandicus can effectively incorporate donor markers as short segments through HR in a continuous or discontinuous manner. We determined that the minimal size of homology allowing native argD marker replacement was as few as 10 bp, whereas argD marker replacement was frequently observed when increasing the size of homology to 30 to 50 bp. The microhomology-mediated gene inactivation system developed here will greatly facilitate isolation of S. islandicus gene deletion strains, making generation of a collection of genome-wide targeted mutants feasible and providing a tool to investigate homologous recombination in this organism.
IMPORTANCE Current procedures for the construction of deletion mutants of S. islandicus are still tedious and time-consuming. We developed a novel procedure based on microhomology-mediated HR, allowing for rapid and efficient removal for genetic regions as large as 50 kb. Our work will greatly facilitate functional genomic studies in this promising model organism. Additionally, we developed a quantitative genetic assay to measure HR properties in S. islandicus, providing evidence that the ability to incorporate short, mismatched donor DNA into the genome through HR was probably a common trait for members of the Sulfolobus genus that are recombinogenic.
KEYWORDS: genetics, Sulfolobus islandicus, gene inactivation, homologous recombination, hyperthermophilic crenarchaea
INTRODUCTION
Sulfolobus islandicus has been rapidly emerging as a model system for studying the unique biology of the crenarchaeal division of the archaeal domain (1–3). Most notably, many different strains of S. islandicus have been investigated as unique models for studying virus-host interactions and CRISPR (clustered regularly interspaced short palindromic repeats) biology (4–10). In comparison, the other crenarchaeal model, Sulfolobus acidocaldarius, is a single strain (11, 12) and appears to be less susceptible to infection by mobile genetic elements (13).
In the past 8 years, genetic toolboxes in diverse S. islandicus strains have been greatly augmented by the addition of multiple selectable/counterselectable markers, versatile gene deletion methodologies, gene silencing, and genome editing approaches relying on their endogenous CRISPR-Cas systems (14–21). A current limitation in S. islandicus genetics is lack of high-throughput genetic tools to rapidly generate mutant strains on a genome-wide scale. Current procedures for deleting a gene of interest are time-consuming and tedious, because the construction of knockout plasmids generally requires cloning of two or three homologous arms (22), which greatly impedes the progress in functional genomic studies for the slow-growing hyperthermophilic crenarchaeon S. islandicus.
In addition to S. islandicus, homologous recombination (HR)-based chromosomal gene inactivation systems have also been reported in other members of Sulfolobales, including Metallosphaera sedula (23) and two well-studied organisms belonging to the Sulfolobus genus, i.e., S. acidocaldarius (12) and S. solfataricus (24). HR has been investigated in some detail in S. acidocaldarius, in which a quantitative genetic assay to characterize functional properties of HR showed that single or multiple donor tracts could be incorporated into the recipient chromosome as short segments (25). Moreover, it has also been reported that small deletions of the pyrE gene in the S. acidocaldarius chromosome could be effectively replaced by electroporating linear DNA fragments harboring a wild-type pyrE gene sequence flanked with 10 to 30 bp of homology (26). The ability to incorporate exogenous donor DNA as short segments via HR in S. acidocaldarius allowed the successful establishment of a PCR-mediated gene targeting technique used to disrupt two individual loci involved in UV photoproduct repair (27). To date, it remains to be tested whether these HR properties are S. acidocaldarius specific or are ubiquitous in Sulfolobus species. Although the minimal length of homology required for sufficient HR in S. islandicus has not yet been determined, it has been previously reported that HR could occur efficiently for 34-bp or 50-bp repeats when using a marker insertion and target gene deletion (MID) strategy for genetic analysis of proliferating cell nuclear antigen (PCNA) genes (19). This raised an important question as to whether microhomology could mediate HR for gene targeting in S. islandicus.
In this study, by using stringent agmatine prototrophy selection (16) in an argD-deficient strain of S. islandicus M.16.4, we asked whether microhomology-mediated gene targeting systems could be adapted to S. islandicus to develop a high-throughput knockout system for this model crenarchaeon. This method was validated by rapidly deleting 21 toxin-antitoxin (TA) gene pairs as well as large chromosomal DNA fragments of up to 50 kb. In addition, we investigated the functional properties of HR and tested the HR frequencies in S. islandicus. We envision that this novel approach will enable us to construct a collection of individual deletion mutants for all of the nonessential genes in S. islandicus, greatly facilitating gene function studies in this organism.
RESULTS AND DISCUSSION
S. islandicus can incorporate exogenous homoeologous (nonidentical) donor DNA into its chromosome via HR.
Previously, when conducting gene knockout experiments using the marker replacement strategy in S. islandicus M.16.4, we occasionally found that the heterogenous SsoargD marker (a 755-bp homoeologous donor DNA that consisted of the coding sequence, native promoter, and terminator region of the argD gene from S. solfataricus P2) was not integrated into the position of the target gene in some ArgD+ transformants (data not shown), raising the question whether unintended recombination occurred between the SsoargD marker cassette and the argD deletion allele in the chromosome of S. islandicus. To investigate the recombination events in detail, we designed a genetic assay in which a PCR-generated SsoargD marker cassette (linear donor DNA with 755 bp) (Fig. 1A) was electroporated into an argD-deficient strain, RJW008 (recipient). RJW008 harbored 381 bp of in-frame deletions within the argD gene (405 bp in total), with a KpnI site (GGTACC) introduced in the argD deletion allele (Fig. 1A). We found that ArgD+ colonies could be consistently obtained when using more than 250 ng of donor DNA, and a positive correlation was observed between the transformation efficiency and amount of donor DNA (Fig. 1B). As shown in our previous study, agmatine prototrophy selection was extremely stringent, and no background colonies grew on agmatine-free plates (16); therefore, the only way to generate ArgD+ transformants was through HR between the short homologous DNA segments of the marker cassette and the chromosome, transferring the donor sequences and thereby recovering a functional argD allele.
FIG 1.
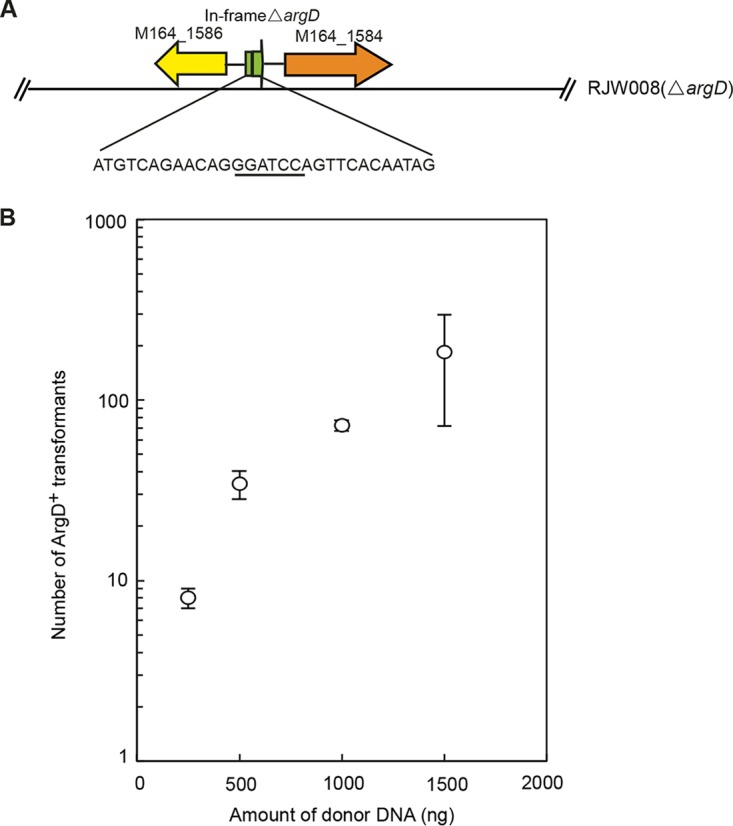
Genetic assay of HR in S. islandicus using a heterogenous donor DNA. (A) Genomic context of the argD mutant allele in S. islandicus RJW008. The nucleotide sequences in the argD mutant allele are indicated, and the introduced KpnI site is underlined. (B) Transformation efficiency using the PCR-generated SsoargD marker cassette as a donor DNA. Amounts of 250 ng, 500 ng, 1,000 ng, and 1,500 ng of donor DNA were transformed into the recipient strain RJW008 via electroporation, selecting agmatine-prototrophic colonies on selective plates after 10 to 12 days of incubation at 76 to 78°C. Each transformation was performed three times.
Analysis of recombinant genotypes reveals highly diverse HR events in S. islandicus.
We then compared the nucleotide sequence variations between the heterogenous SsoargD marker cassette (755 bp) and the regions (378 bp) surrounding the argD mutant allele in the chromosome of recipient strain RJW008. As shown in Fig. 2 and summarized in Table S1 in the supplemental material, 26 mismatches and 4 gaps were found between the recipient and the donor DNAs in total. To better characterize these variations, we choose to use “donor marker” and “recipient marker” to define them, as Grogan and Rockwood have previously done in S. acidocaldarius (25). Two nucleotides, at positions 14 and 15 and positions 40 and 41, respectively, were linked together; therefore, they were considered one single donor or recipient marker. Similarly, two continuous gaps at nucleotides (nt) 278 and 279 in the recipient, corresponding to nucleotides (CT) from donor DNA, were defined as a single marker. In addition, one gap followed by a mismatch between recipient and donor DNAs linked together at nt 328 and 329 and nt 329, and they were used as single marker. Notably, the 380-bp donor DNA that corresponded to the argD deletion allele in the recipient (Fig. 3A) was expected to transfer as a whole and was defined as a “selectable donor marker.” Thus, the 255-bp left flanking regions of the argD deletion allele contained 17 markers, leading to 18 intervals with homology ranging in length from 2 to 54 bp (Table S1). The 118-bp right flanking regions of the argD deletion allele contained 9 markers, generating 10 intervals with homology from 2 to 31 bp in length (Table S1). The HR events that occurred at these short homologous segments spaced by donor markers allow the selectable donor marker to replace the argD-deleted portion from the recipient chromosome, generating ArgD+ recombinants.
FIG 2.

Alignment between the donor DNA and the sequences surrounding the argD mutant allele in the recipient chromosome. Sequences highlighted with red boxes represent the donor marker. The KpnI site (GGTACC) that was introduced into the argD mutant allele in recipient is highlighted in bold.
FIG 3.

Molecular analysis of recombinants derived from the HR assay using the SsoargD marker cassette as the donor DNA. (A) Sequence features of the argD deletion locus in the chromosome of recipient S. islandicus RJW008 and donor DNA (SsoargD, 755 bp). Open and filled circles denote the recipient and donor markers, respectively. Open and filled triangles represent 1-nt gaps in the recipient and donor DNAs in BLAST analysis, respectively. RJW008 contained a 381-bp deletion within the argD gene, while a KpnI site (GGTACC) was introduced when constructing the in-frame argD deletion strain. Note that the first nucleotide (G) of the KpnI site had a match with that from donor DNA and thus was not included into the argD deletion allele (indicated by the bracket). The 380-bp region from donor DNA, corresponding to the regions surrounding the argD deletion allele, was defined as the “selectable donor marker.” The primer set UR-F/R was used to amplify the argD locus in recombinants. (B) Recombinants that incorporated a single donor tract. (C) Recombinants that incorporated two donor tracts. The number of recombinants with an identical genotype is indicated.
To characterize HR events in detail, we amplified the argD locus from 96 independent ArgD+ transformants by PCR and then sequenced the amplicons. The frequency of donor marker transfer was plotted against the given position of donor markers, and it was shown that HR frequency was dependent on the distance away from the selectable donor marker (see Fig. S1 in the supplemental material). This result was consistent with prior studies using a genetic assay of HR performed in S. acidocaldarius, in which a pyrE gene fragment with synonymous substitutions was electroporated into an 18-bp pyrE-deficient recipient (25). In addition, we found that donor markers with a 2- or 3-bp separation did not segregate from each other. For example, the donor markers positioned at nt 66, 69, and 72 that were separated by 2 bp (Fig. 2) were always incorporated into the recipient chromosome as a whole. Similarly, the two donor markers at nt 253 and 263 that distributed at the left and right of the selectable donor marker, respectively (2-bp distance), were always found in the argD locus of all 96 ArgD+ transformants. In addition, the donor markers at nt 41 and 45 that were separated by 3 bp were always linked together as well.
For donor markers that were separated by 4 bp or more, segregation from each other was readily observed. The distinct donor or recipient markers provided high resolution to trace the HR events. Sequence analysis of the argD locus from 96 individual transformants allowed us to identify 42 distinct genotypes of recombinants, as schematically visualized in Fig. 3B and C. In summary, these recombinants were grouped into two categories, i.e., the recombinants that harbored a single donor tract (Fig. 3B) (91 recombinants belonging to 37 distinct genotypes) and the recombinants containing two donor tracts (Fig. 3C) (5 recombinants with 5 distinct genotypes). However, no recombinants that incorporated three or more donor marker tracts were detected among 96 transformants. This observation was inconsistent with the results reported by Grogan and Rockwood for S. acidocaldarius (25), probably because of the varied complexity of donor DNAs that were used in the genetic assay of HR. Taken together, our results showed that in S. islandicus, donor sequences could also be transferred into the recipient chromosome as short segments through double- or multiple-crossover HR events. Nevertheless, it remained unclear whether this HR trait was universal for all of the Sulfolobus genus. At least in S. solfataricus, the HR capacity seemed to be divergent due to the fact that HR had not been detected in two type strains, i.e., S. solfataricus P2 and S. solfataricus P1, whereas it proved to be active in a closely related strain S. solfataricus 98/2 (24, 28). Although it was difficult to study functional properties or accurately assess the frequency of HR in S. solfataricus 98/2 due to the lack of stringent selection systems, single or multiple linear DNA fragments have been successfully used for chromosomal gene disruptions via double-crossover HR. Thus, we reasoned that the ability to incorporate exogenous donor DNA as short segments by discontinuous or continuous HR was a common trait for Sulfolobus species that were highly recombinogenic.
Ten base pairs of homology is sufficient to allow DNA integration into the S. islandicus chromosome via double-crossover HR.
Having demonstrated that HR is efficient in S. islandicus, we next investigated how the length of homology affected the HR frequency. To this end, donor DNAs encompassing regions that consisted of the deletion portion (381 bp) of the argD gene and its flanking sequences with different sizes (0 to 1,500 bp) were generated by PCR and then electroporated into recipient cells (Fig. 4A). Since only colonies that completely recovered a wild-type argD allele through double-crossover HR could grow on agmatine-free plates, the efficiency of HR was calculated as the number of ArgD+ transformants divided by amount of donor DNA. As shown in Fig. 4B, homology arms as short as 10 to 20 bp allowed the selection for the restored argD gene through double-crossover HR. Although the HR events occurred very rarely, the recombinants were still detectable in transformation experiments (1 ± 1 CFU and 6 ± 2 CFU per 1.5 μg DNA for 10-bp and 20-bp homologous flanking regions, respectively). However, when the size of flanking regions reached 30 bp, the number of recombinants (184 CFU/1.5 μg DNA) significantly increased. Additionally, a strong positive correlation was observed between HR efficiency and the length of the homology arms (Fig. 4B). The HR efficiency was comparable when the size of flanking regions was in the range of 750 to 1,500 bp, and the highest HR efficiency was obtained when 750-bp homology arms were used (2.41 × 104 CFU/1.5 μg DNA). Given the transformation efficiency (5.32 × 104 CFU/1.5 μg DNA) estimated by electroporating the replicative plasmid pSeSd-StoargD into strain RJW008, there was an HR event within approximately half of the transformed cells.
FIG 4.
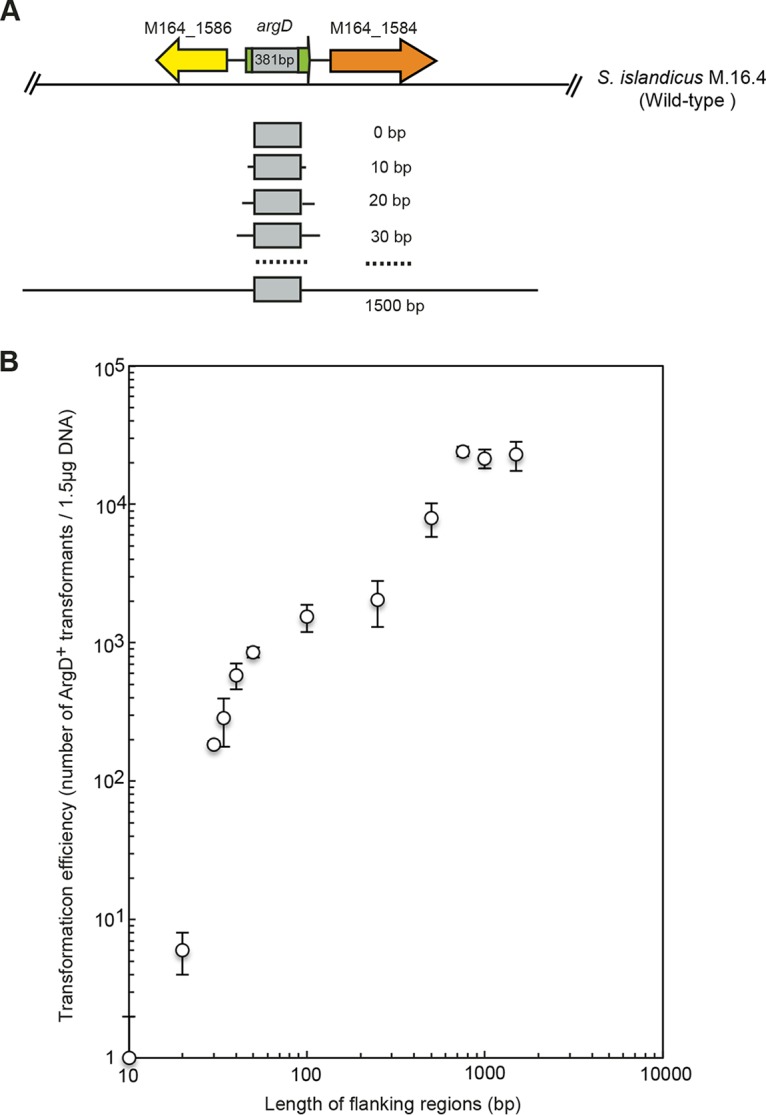
Genetic assay of HR in S. islandicus using a homologous donor DNA. (A) Generation of homologous donor DNAs with flanking regions of various lengths. A wild-type argD region (381 bp), corresponding to the sequences that have been deleted from the argD gene, and its flanking regions varying from 0 to 1,500 bp were amplified from the genome of wild-type S. islandicus M.16.4. Gray boxes indicate the deletion in the argD gene. (B) Impact of homology size on transformation efficiency. Purified PCR products (1.5 μg) were used to transform RJW008 via electroporation. Each transformation was performed three times.
The impact of homology size on HR efficiency using linear DNA has rarely been systematically investigated in archaea. Recently, Farkas et al. performed an HR assay using uracil selection in Pyrococcus furiosus COM1, a pyrF-deficient strain derived from P. furiosus DSM3638 that could naturally uptake foreign DNA (29). The HR frequency reported in our study was approximately 5-fold higher than that for P. furiosus COM1 (2.15 × 104 CFU/1.5 μg DNA versus 2.90 × 103 CFU/μg DNA when 1,000-bp homologous flanking regions were used). Taking the results together, although the frequency of double-crossover HR events correlated positively to the length of the homology arms, 30 to 50 bp of microhomology was sufficient to enable the occurrence of double-crossover HR in S. islandicus, suggesting that S. islandicus was a promising model to develop a microhomology-mediated gene inaction system. This system has been extensively used in Saccharomyces cerevisiae and Escherichia coli (30, 31) and also was recently reported for a related species, S. acidocaldarius (27).
Building a one-step, microhomology-mediated gene inactivation system in S. islandicus.
Based on the efficient HR with short regions of homology that we demonstrated above, we sought to develop a one-step, microhomology-mediated gene inactivation system in S. islandicus. As a proof of concept, we chose the lacS gene, encoding a β-glycosidase, for targeting, as the LacS− and LacS+ phenotypes could be readily distinguished by blue/white X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining. To determine the impact of homology length on gene deletion efficiency, DNA fragments containing a StoargD marker cassette (a 740-bp marker cassette containing the argD gene from Sulfolobus tokodaii under the control of its native promoter and terminator regions) flanked by 0- to 40-bp homologous regions of the lacS gene were created (see Fig. S2 in the supplemental material). The primers used to generate lacS disruption cassettes contained sequences homologous to the StoargD marker and 0, 10, 15, 20, 25, 30, 35, or 40 bp of homology to the lacS gene. As shown in Table 1, agmatine-prototrophic colonies were detectable, even at very low frequencies, when 25-bp homology arms were used. However, when the length of the homology arms reached 30 to 40 bp, we routinely obtained 18 to 35 transformants per 1.5 μg DNA. Significantly, the transformation efficiency jumped to ∼103 transformants per 1.5 μg DNA after extending the length of homology to 650 to 750 bp. Phenotype examination of the ArgD+ transformant by X-Gal staining further confirmed the 100% accuracy of lacS gene targeting (Table 1).
TABLE 1.
Influence of length of homology on lacS gene targeting efficiency
| Length of homology to lacS (bp) | Avg no. of ArgD+ transformants/1.5 μg DNAb | % of ArgD+ transformants with correct lacS disruptionc |
|---|---|---|
| 0, 10, 15, 20 | 0 | 0 |
| 25 | 5 | 100 |
| 30 | 18 | 100 |
| 35 | 22 | 100 |
| 40 | 35 | 100 |
| 739/652a | 1,275 | 100 |
The lacS gene disruption scheme was PCR amplified from the nonreplicative plasmid pKlacS-StoargD. The length of homology was 739 bp upstream of the lacS gene and 652 bp downstream of the lacS gene.
Electroporation-mediated transformation was conducted at least four times.
Phenotypically screened via blue/white X-Gal staining.
Homologous regions of 30 to 40 bp were sufficient to effectively generate deletions of a gene of interest (GOI) in S. islandicus. We thus proposed a microhomology-mediated gene inactivation system in this organism. In this procedure, a gene deletion cassette consisting of an agmatine-positive selection marker flanked by 30 to 40 bp of homology of the GOI was created by one-step PCR amplification and then transformed into an argD-deficient strain via electroporation, selecting ArgD+ transformants on agmatine-free plates. Regarding this system, there were several concerns that needed to be addressed. First, as described previously, HR events occurred frequently between the heterogenous SsoargD marker and the argD mutant allele in the chromosome of the S. islandicus genetic host, thereby generating false-positive transformants in gene deletion studies. To circumvent the occurrences of these unintended HR events, a more divergent argD marker cassette (StoargD) derived from the phylogenetically distantly related species S. tokodaii was used. Second, given that the expense of synthesizing oligonucleotides longer than 59 nt increased dramatically, primers used for generating deletion cassettes of the GOI were uniformly designed to be 59 nt in length. Thus, the primers for generating the gene deletion cassette were typically designed to contain 19-nt and 20-nt priming sequences to the 5′ and 3′ regions of the StoargD marker cassette and a 40-nt or 39-nt homology extension. Finally, we previously showed that the introduction of a 50-bp or 34-bp repeated sequence within the pcna1 and pcna2 targeting constructs, respectively, allowed the marker gene cassette to pop out from the chromosome via a single-crossover HR event, with a frequency of 10−6 to 10−7 (19). Thus, to achieve markerless gene deletions, we can modify the one-step, microhomology-mediated gene inactivation system by repurposing a marker insertion and target gene deletion (MID) strategy that we previously developed in S. islandicus REY15A (19) as follows. (i) The double marker genes StoargD and SsopyrEF were used when creating gene deletion cassettes via PCR, which enables agmatine-positive selection and 5-fluoroorotic acid (5-FOA) counterselection, respectively. Correspondingly, RJW007 (ΔpyrEF ΔargD) was used as a genetic host. (ii) The length of the three homology arms in MID, i.e., the In-arms, Out-arms, and Tg-arms (19), could be shortened to 34 to 40 bp for double- or single-crossover HR. Therefore, by introducing sequences of these short homology arms into the 5′ or 3′ ends of primers used to amplify the dual marker genes, we can easily create unmarked gene deletion cassettes via one-step PCR.
High-throughput, systematic gene deletion in S. islandicus.
Next, we tested whether the microhomology-mediated gene inactivation system represented a general approach for genetic manipulations in S. islandicus. Thus, we systematically disrupted 22 toxin-antitoxin (TA) gene pairs, belonging to the VapBC (virulence-associated proteins B and C) type, in S. islandicus RJW008 (Fig. 5A; see Table S2 in the supplemental material). The primers used for generating TA deletion cassettes were designed to be 59 nt in length (Table 2) and consisted of 19-nt and 20-nt priming sequences to the 5′ and 3′ regions of the StoargD marker cassette and a 40-nt or 39-nt homology extension (Fig. 5B). Again, 7 to 198 transformants per 1.5 μg DNA were routinely produced on agmatine-deficient plates. PCR verifications of ArgD+ transformants revealed that 21 of the 22 TA deletion strains were successfully created individually, with the exception being TA5 (Fig. 5C). Phenotypic characterization of TA mutants through drop dilution assays demonstrated that all individual TA mutants almost exhibited equivalent fitness in dextrin-tryptone plates, which was also comparable to that of the parental strain (Fig. 5D). Further investigation was required to decipher the physiological roles of TA systems in S. islandicus by constructing mutants lacking multiple TA modules.
FIG 5.

Systematic disruption of toxin-antitoxin (TA) gene pairs via a one-step, microhomology-mediated gene inactivation system. (A) Distribution of 22 vapBC TA loci across the chromosome of wild-type S. islandicus M.16.4. Three replicative origins (OriC-1, -2, and -3) are labeled. (B) Schematic diagram showing the deletion of TA loci via the one-step, microhomology-mediated gene inactivation approach. White and hatched boxes denote 5′ and 3′ flanking regions of the target TA loci, which were 40 and 39 bp in length, respectively. (C) Verification of TA deletion strains by PCR analysis. The TA loci were amplified from representative TA knockout strains (Δ) and the genetic host RJW008 (+) using primer sets annealing outside the 5′ and 3′ homologous regions for HR. The expected lengths of PCR products amplified from the genetic host and TA knockout strains are summarized in Table S3 in the supplemental material. The TA5 deletion strain was not obtained (see Fig. S3 in the supplemental material). Lanes L, GeneRuler Express DNA ladder (Thermo Fisher Scientific, USA). (D) Drop dilution assay of individual TA mutants. Ten microliters of serial dilutions of culture samples were spotted on nutrient-rich medium plates and then incubated for 10 days at 76 to 78°C.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′→3′)a |
|---|---|
| StoargD-F1 | TCACCCCGGGGGATATGTGCTTTCATATGTTAAA |
| StoargD-R1 | ACGCCCCGGGGTCTATGGATTCATAGACTTTTGTC |
| StoargD-F2 | CATGCCATGGGGATATGTGCTTTCATATGTTAAA |
| StoargD-R2 | ACTAACGCGTGTCTATGGATTCATAGACTTTTGTC |
| SsoargD-F | ATTCTCCAATATATGGGGTTT |
| SsoargD-R | TACTTTCTTACTGCTTTGATCAA |
| SispyrEF-F | CGCTACTCTATAGCCTTCACTTC |
| SispyrEF-R | AGATGATGAGGTGGACAGGTT |
| UR-F | GAGAACGCTATCACTTCGTAAC |
| UR-R | CCTCTACTTTTATTCTATATCTTGGA |
| argD-1500bp-F | CATTGACCGAGAGGAGTATTA |
| argD-1500bp-R | AACTCCTATTATTAGAATTGAATTTA |
| argD-1000bp-F | CTGAGCTACGGCGGCAC |
| argD-1000bp-R | AACCAATATCCTTTCACCAGG |
| argD-750bp-F | TTGGTTACCCAATGGCGAT |
| argD-750bp-R | TGGAAATATGCTGGTTCTCCA |
| argD-500bp-F | ACTAATACTGTAAACTTCTCTATCTCTG |
| argD-500bp-R | AAAACGAAACTAGACGCTAATC |
| argD-250bp-F | TCCAATATAGCGGATTTATTGA |
| argD-250bp-R | AGGAACTGACTTTTCAGTTAATCC |
| argD-100bp-F | TTATAACGTTTATCCTAAGGTTTATG |
| argD-100bp-R | ATCAAATATAAGGTTTTCGATTTT |
| argD-50bp-F | ATATTGTTGAGAATGAGATGGG |
| argD-50bp-R | GAATGAATTAAAAAACAAATAGAGA |
| argD-40bp-F | GAATGAGATGGGGGCATAAG |
| argD-40bp-R | AAAAACAAATAGAGATTAATATAAAAAACT |
| argD-34bp-F | GATGGGGGCATAAGAAAGA |
| argD-34bp-R | AAATAGAGATTAATATAAAAAACTATTGT |
| argD-30bp-F | GGGGCATAAGAAAGAAAGAT |
| argD-30bp-R | AGAGATTAATATAAAAAACTATTGTGA |
| argD-20bp-F | AAAGAAAGATGTCAGAACAGG |
| argD-20bp-R | ATAAAAAACTATTGTGAACTCCTA |
| argD-10bp-F | GTCAGAACAGGAGGTATTACAAAA |
| argD-10bp-R | ATTGTGAACTCCTATCGGCAT |
| argD-0bp-F | GAGGTATTACAAAAAAATAATTCC |
| argD-0bp-R | CCTATCGGCATAAAACATTT |
| lacS-DR(40)-F | gtctcaagccggattccaatctgaaatgggcacaccagggGGATATGTGCTTTCATATGTTAAA |
| lacS-DR(40)-R | ctgaaggtctccagtacagtcttttagtaccataatccacGTCTATGGATTCATAGACTTTTGTC |
| lacS-DR(35)-F | aagccggattccaatctgaaatgggcacaccagggGGATATGTGCTTTCATATGTTAAA |
| lacS-DR(35)-R | ggtctccagtacagtcttttagtaccataatccacGTCTATGGATTCATAGACTTTTGTC |
| lacS-DR(30)-F | ggattccaatctgaaatgggcacaccagggGGATATGTGCTTTCATATGTTAAA |
| lacS-DR(30)-R | ccagtacagtcttttagtaccataatccacGTCTATGGATTCATAGACTTTTGTC |
| lacS-DR(25)-F | ccaatctgaaatgggcacaccagggGGATATGTGCTTTCATATGTTAAA |
| lacS-DR(25)-R | acagtcttttagtaccataatccacGTCTATGGATTCATAGACTTTTGTC |
| lacS-DR(20)-F | ctgaaatgggcacaccagggGGATATGTGCTTTCATATGTTAAA |
| lacS-DR(20)-R | cttttagtaccataatccacGTCTATGGATTCATAGACTTTTGTC |
| lacS-DR(15)-F | atgggcacaccagggGGATATGTGCTTTCATATGTTAAA |
| lacS-DR(15)-R | agtaccataatccacGTCTATGGATTCATAGACTTTTGTC |
| lacS-DR(10)-F | cacaccagggGGATATGTGCTTTCATATGTTAAA |
| lacS-DR(10)-R | cataatccacGTCTATGGATTCATAGACTTTTGTC |
| lacS-DR(0)-F | GGATATGTGCTTTCATATGTTAAA |
| lacS-DR(0)-R | GTCTATGGATTCATAGACTTTTGTC |
| lacS-DR(739)-F | CCTTATCTCTGGATTGATGCTT |
| lacS-DR(652)-R | TTACTAAACAGAGGAGTTAACTTACTG |
| TA1-DR-F | cggaaagttaatttatgatgattctacccatttattgaatGGATATGTGCTTTCATATG |
| TA1-DR-R | taattatgagtagaaaacttacaacgatttccatttctgGTCTATGGATTCATAGACTT |
| TA2-DR-F | ttcttctttaattcttcgtcgttcgatacaatctctaatcGGATATGTGCTTTCATATG |
| TA2-DR-R | gggttattaaatgggtatatgtgggcaaggaatttatatGTCTATGGATTCATAGACTT |
| TA3-DR-F | tctgcacaatttaggtcttagtctaaaataattccgagtcGGATATGTGCTTTCATATG |
| TA3-DR-R | tacgacataaagtggaatgaagagatagagggtttcataGTCTATGGATTCATAGACTT |
| TA4-DR-F | cttaacccttaatactcatataaatattgcttcattcgttGGATATGTGCTTTCATATG |
| TA4-DR-R | aaacttttaagtggaaagtagaaagtatacatatgcaagGTCTATGGATTCATAGACTT |
| TA5-DR-F | gtatgtatataggatatatgaaaacaataatgataagggaGGATATGTGCTTTCATATG |
| TA5-DR-R | aattagaattagatttagttcgtcaaattcttctttaacGTCTATGGATTCATAGACTT |
| TA6-DR-F | agttaataatctcggggatttataatataaataatggcatGGATATGTGCTTTCATATG |
| TA6-DR-R | ctttgtcaaagatgtgatctgtggaaattattatcttatGTCTATGGATTCATAGACTT |
| TA7-DR-F | aatattcttatgggatatatagttacagttgatgaaagagGGATATGTGCTTTCATATG |
| TA7-DR-R | agaattctctcttaacttgaagtaaatcaaaatctgtatGTCTATGGATTCATAGACTT |
| TA8-DR-F | ttcatatgtcagacgtaataagcgtaagagtgaagaaagaGGATATGTGCTTTCATATG |
| TA8-DR-R | tttcacatactttttatcaaatcctttagcgaaatggcaGTCTATGGATTCATAGACTT |
| TA9-DR-F | attgtatttttatatactctttcaatctcagagtccgtacGGATATGTGCTTTCATATG |
| TA9-DR-R | tcatggaggtaaaagttcacaagaaagggattatagttaGTCTATGGATTCATAGACTT |
| TA10-DR-F | tactctgctactatagctcagaaacagatatcttaacaatGGATATGTGCTTTCATATG |
| TA10-DR-R | tatcatatgtcagtagtaataacgattaaagtggacaagGTCTATGGATTCATAGACTT |
| TA11-DR-F | aagtaaaggttactagaaactttcaagttactataccctcGGATATGTGCTTTCATATG |
| TA11-DR-R | attattcttacaccatgtttttcagcaattttccttaatGTCTATGGATTCATAGACTT |
| TA12-DR-F | aaacaatgtggtaagcactgttatatcaataagagttgacGGATATGTGCTTTCATATG |
| TA12-DR-R | aaggcgataaaaaatactttgaaattcctggataatttcGTCTATGGATTCATAGACTT |
| TA13-DR-F | taaactcctgtgaaaaatactatagtatgagaatagtaacGGATATGTGCTTTCATATG |
| TA13-DR-R | caaactatttgtcatgaatccttcagcttctctttaacaGTCTATGGATTCATAGACTT |
| TA14-DR-F | catcggtttaacttatattatatggtttatcaaataaagtGGATATGTGCTTTCATATG |
| TA14-DR-R | atcaatgttagttctcatatctcgtcaataaaactaataGTCTATGGATTCATAGACTT |
| TA15-DR-F | gctaaatttgtccttaattttaggccacactttcaatattGGATATGTGCTTTCATATG |
| TA15-DR-R | ccttcccctgatcatgatagaacagtttaaatatttaagGTCTATGGATTCATAGACTT |
| TA16-DR-F | aatatgccaataattagtataaggattgatgatgagttgaGGATATGTGCTTTCATATG |
| TA16-DR-R | tacaaatagtatgtaggctcattaaattctgaaatctctGTCTATGGATTCATAGACTT |
| TA17-DR-F | tcagacaaatcttatggataggatcataaggataggtaaaGGATATGTGCTTTCATATG |
| TA17-DR-R | gttatccaggttgaaagtatccccattaaagcaatatgaGTCTATGGATTCATAGACTT |
| TA18-DR-F | attccaaagaaatatcgtagacgaatttgattaagagcacGGATATGTGCTTTCATATG |
| TA18-DR-R | tagcaaacttacggagtacttctttgctatctttgagagGTCTATGGATTCATAGACTT |
| TA19-DR-F | taatatacgaattctacttttacatatgccagtgattagcGGATATGTGCTTTCATATG |
| TA19-DR-R | atgtgaattatgaaatgtaagtctttcacttttctaagtGTCTATGGATTCATAGACTT |
| TA20-DR-F | aattaccaaataatttctatccctaacttcctcgcaactaGGATATGTGCTTTCATATG |
| TA20-DR-R | tgagttaaaattatggaagttagggtaaaggttaataagGTCTATGGATTCATAGACTT |
| TA21-DR-F | ttgcatattcctatcattgctaattaactcaccaatctttGGATATGTGCTTTCATATG |
| TA21-DR-R | attaagttcaatataaatagtagtgggaaagcaagtgctGTCTATGGATTCATAGACTT |
| TA22-DR-F | ttctataggtataaattcaacatcataatcttttgccgttGGATATGTGCTTTCATATG |
| TA22-DR-R | gagttaaactattatttatgatggtcacaaaggtacacaGTCTATGGATTCATAGACTT |
| upsAB-DR-F | cgatgttctctgagtaagataaattttgatatagtgagagGGATATGTGCTTTCATATG |
| upsAB-DR-R | ctgatccaaggttaaaatagtagtagtttccattaatctGTCTATGGATTCATAGACTT |
| 23/35kb-DR-Fb | cctagatttagatttcaaacaagtcatagaatatagtataGGATATGTGCTTTCATATG |
| 23kb-DR-R | gaaatcatattttgtgaccatgggatttaatgaaactttGTCTATGGATTCATAGACTT |
| 35/50kb-DR-Rc | taaacttctatcagataattaacgttgcgttagtataatGTCTATGGATTCATAGACTT |
| 50kb-DR-F | aagtcataaacttaataactggcataattaccattatataGGATATGTGCTTTCATATG |
| TA1-flankP-F | CAAAGTCGGTATGGTGGATT |
| TA1-flankP-R | GAAATAGTCTTTGTTTCCTTAGGA |
| TA2-flankP-F | TAGAAGTGCTATTTGATCCTTACC |
| TA2-flankP-R | ATCAACAAAGCAACACAAAAGA |
| TA3-flankP-F | AACGCATGTTTCTGGTCAATA |
| TA3-flankP-R | AAAAGTACGACATAAAGTGGAATG |
| TA4-flankP-F | TAATATGTTGGCCTACATGATAAT |
| TA4-flankP-R | CGTCTATAACATCAATACCTTCG |
| TA5-flankP-F | CTCTTCTCTAATCTCCTTCGGT |
| TA5-flankP-R | GGAGGCAAAGGATATTAAAACT |
| TA6-flankP-F | ACAAATCTGCATAAGTGGGGA |
| TA6-flankP-R | CACGGGTAAACACTATATCCTTCT |
| TA7-flankP-F | AGAGTTATACACGTCGTCCCTT |
| TA7-flankP-R | GATGGCTAAATATTGACTTAATTATTAT |
| TA8-flankP-F | GGAGAGAAGACTAATCCCAGAAT |
| TA8-flankP-R | AACAACTGCTGCTCTTAAAATAAA |
| TA9-flankP-F | AATCAGGGAGTTTAAATGGTACA |
| TA9-flankP-R | ATTACTTTTCCTTTTCCGAGAC |
| TA10-flankP-F | ATACAAGGAGTCAAGTACCGCT |
| TA10-flankP-R | TAGTAATAACGATTAAAGTGGACAAG |
| TA11-flankP-F | ATCGGATTTCTACTGAGTCGTAG |
| TA11-flankP-R | TTCAATTATTTGCATTGGGATA |
| TA12-flankP-F | GGTTTGAAGTGAACAAGCTCC |
| TA12-flankP-R | CAATTCCGATAGATCCAAAATC |
| TA13-flankP-F | CAAACTTGAGGTTCAAGGAGAT |
| TA13-flankP-R | GCAAACTATTTGTCATGAATCCT |
| TA14-flankP-F | GCTTCAGCAACAATCCTCATAAC |
| TA14-flankP-R | AGAGTAGTTAGCCTTCTTTGTAAGAGAA |
| TA15-flankP-F | GTGAAACGACAGTCAGTTCTTTAG |
| TA15-flankP-R | AACTTCTTAATGACAGCCTTCCT |
| TA16-flankP-F | TTTTGGACTCTAAAAACGCATT |
| TA16-flankP-R | CTAGCTTATGATTAGGAAATGCTCT |
| TA17-flankP-F | TCAAAAATACTACCGTGAATCGT |
| TA17-flankP-R | CACACAATCCAGCACTGAACA |
| TA18-flankP-F | TACTGTACTCTTTGGCGGAAC |
| TA18-flankP-R | CATAACTGCATGAGAACACCC |
| TA19-flankP-F | AAAATGTGTCTCGTGAAAGTCA |
| TA19-flankP-R | TATGTCGAATTTGACGAGGG |
| TA20-flankP-F | CTTATAGAATATAAATCTTGCCCTT |
| TA20-flankP-R | CTGTTATGCCTCTTAAGGTACG |
| TA21-flankP-F | TCATCATGGCTAGAATCTAGCG |
| TA21-flankP-R | AGATGTTCCAATACCCTTCTCTG |
| TA22-flankP-F | AGTACAGAGACACCTTTATTAGGAA |
| TA22-flankP-R | ATTGATCGTAAAAGATGGGTTAT |
| upsAB-flankP-F | ACTTGATACTGATACTATTTTTCGGA |
| upsAB-flankP-R | TTCGTAAAACCTAATCTGATCCA |
| 23/35kb-flankP-Fd | GTTAGCGGAAGTGAGGTTATG |
| 23kb-flankP-R | GGGACATTCCATTATTTCTCAG |
| 35kb-flankP-R | TTAATCTGCTTGATCCCTGTAG |
| 50kb-flankP-F | TCCTCCTCACTTAGGCTCAGA |
| 50kb-flankP-R | GTCAGGGATTTGGGTGATAAG |
Lowercase sequences are homologous regions of the gene(s) or DNA fragments of interest, and added restriction sites are underlined.
The forward primers that are used to create the deletion cassette of 23-kb and 35-kb DNA fragments have identical sequences.
The reverse primers that are used to create the deletion cassette of 35-kb and 50-kb DNA fragments have identical sequences.
The forward primers that are used to verify the 23-kb and 35-kb deletion mutants have identical sequences.
Notably, when transforming the TA5 deletion cassette into the argD-deficient strain RJW008, we occasionally observed very few agmatine-prototrophic colonies (1 to 3 CFU/1.5 μg DNA), although in most genetic transformations, no colonies were produced. A total of 10 colonies, collected from multiple electroporation experiments, were then assessed for the correct gene deletion. Amplification using primers (TA5-FlankP-F/R) complementary to sequences flanking TA5 loci generated amplicons of the same size from both ArgD+ transformants (TA5-T1-T10) and genetic host RJW008, suggesting that the StoargD marker was not correctly integrated into the genome at the TA5 loci (see Fig. S3B in the supplemental material). In addition, unintended recombination events between the StoargD marker cassette and the argD deletion allele in RJW008 were also excluded because all of the ArgD+ transformants still harbored an argD deletion allele in their genomes (Fig. S3C). Furthermore, amplification using StoargD-specific primers (StoargD-F1/R1) revealed that the StoargD marker cassette was indeed present in the genomes of all the ArgD+ transformants but was absent in RJW008 (Fig. S3D), confirming a successful integration of the StoargD marker cassette into the genome of RJW008. However, the integration did not occur at the expected TA5 loci. Partial sequence matches between the 40 or 39 bp of homology located at 5′ and 3′ flanking regions of the TA5 deletion cassette and the genome of RJW008 were frequently observed when using the BLASTN program (optimized for somewhat similar sequences). We reasoned that if recombination occurred at these sites, then the StoargD marker cassette could have integrated into the RJW008 genome, resulting in agmatine-prototrophic colonies. This phenomenon was not unusual; as demonstrated in our previous HR assay, recombination breakpoints could be readily generated even within internal sequences as short as 4 bp separated by two donor markers. Additional attempts to delete TA5 using different homology arms were still unsuccessful, suggesting that TA5 was essential for S. islandicus cell survival.
We also validated applications of the microhomology-mediated gene inactivation strategy in another genetically tractable strain, S. islandicus REY15A (22), by efficiently deleting two prepilin subunit-encoding genes (upsAB) in the ups (UV-inducible pilus of Sulfolobales) operon (see Fig. S4 in the supplemental material). Phase-contrast microscopy analysis showed that the UV-induced cellular aggregation in the genetic host S. islandicus E235 (ΔpyrEF ΔlacS ΔargD; derived from the original isolate S. islandicus REY15A) was very obvious, whereas no cellular aggregation was observed in the upsAB deletion mutant (see Fig. S5 in the supplemental material). These results further supported a view that UpsA and UpsB are indispensable for the formation of UV-induced cellular aggregation in S. acidocaldarius (32).
Taken together, these proof-of-concept experiments showed that the microhomology-mediated gene deletion system was a powerful and reliable approach to rapidly delete nonessential genes in S. islandicus. Genome-wide targeted mutant collections have been a valuable resource for functional genomic studies; however, for a long time, this kind of mutant collection was available only for E. coli and S. cerevisiae and was unavailable for any archaeal organisms. As the construction of a gene deletion cassette could be easily realized via one-step PCR amplification, it is now feasible to systematically inactivate each nonessential gene and construct genome-wide targeted mutant collections in S. islandicus, which will make this organism as an excellent paradigm to study archaeal biology.
Microhomology can mediate deletion of large chromosomal DNA fragments up to 50 kb through HR in S. islandicus.
To further explore whether the microhomology-mediated gene inactivation system could be utilized to create large deletions from the chromosome, a variable region, positioned at nt 822574 to 872690 of the S. islandicus M.16.4 genome, was selected for DNA targeting tests. This region contained two repeat-spacer CRISPR arrays (CRISPR-A1 and CRISPR-A2), type IIIB Cmr-α, and type IA modules of a CRISPR-Cas system, as well as an integrated virus-like element that interrupted the transcriptional regulator csa3 gene (4). There were 39 putative open reading frames (ORFs) within this 50-kb region, and no genes were considered to be essential by transposon sequencing (Tn-seq) analysis in the same strain (Changyi Zhang, Alex Phillips, Gary J. Olsen, Kira S. Makarova, and Rachel J. Whitaker, unpublished data).
The DNA targeting efficiency was tested by the construction of three different mutants, with 23-kb, 35-kb, and 50-kb regions from the chromosome of the genetic host RJW008 replaced by the StoargD marker cassette via HR (Fig. 6A). Electroporations of these three DNA fragment deletion cassettes were almost equally efficient in producing agmatine-auxotrophic colonies, and 3 to 8 ArgD+ transformants were generally observed per 1.5 μg DNA. In each DNA fragment deletion experiment, six individual transformants were verified by PCR analysis using the primers that annealed outside the 5′- and 3′-homologous regions for HR, and amplicons of the expected sizes were observed (Fig. 6B). Sanger sequencing analysis of the resulting PCR products further confirmed precise integration of the StoargD marker cassette at the locus of target DNA via double-crossover HR events (Fig. 6C). These results showed that the microhomology-mediated gene deletion system can also effectively create large-scale chromosomal deletions in S. islandicus, and it could be expanded to other archaea that encode similar homologous recombination machinery.
FIG 6.

Large-scale deletion of chromosomal DNA fragments via a one-step, microhomology-mediated gene inactivation system. (A) Schematic representation of the selected variable regions that are located at nt 822574 to 872690 in the chromosome of S. islandicus. Gene clusters highlighted in red and pink are the type IIIB Cmr-α and type IA modules, respectively. Gene cassettes in blue are the adaptation modules of the CRISPR-Cas system. The transcriptional regulator csa3 gene (yellow) was interrupted by an integrated virus-like element containing 13 putative ORFs. Chromosomal deletions of 23-, 35-, and 50-kb regions are indicated. (B) PCR analysis of 23-, 35-, and 50-kb deletion strains. Six agmatine-prototrophic transformants (designated 23kb-T, 35kb-T, and 50kb-T; lanes 2 to 7) were analyzed by PCR using the primer sets annealing outside the 5′ and 3′ homologous regions for HR. PCR bands of 1,573, 1,325, and 1,136 bp were obtained from 23kb-T, 35kb-T, and 50kb-T, respectively, corresponding to the designed deletion regions. In the genetic host RJW008, bands were not observed (lane 1) due to the difficulty in amplifying very long PCR products (24,042, 35,293, and 50,513 bp, respectively). (C) Confirmation of chromosomal deletions by Sanger sequencing. The resulting PCR products amplified from representative 23kb-T, 35kb-T, and 50kb-T using different primer combinations (relative positions are labeled in panel A) were sequenced, and the 5′ and 3′ junction sequences between the chromosomal and StoargD marker gene are indicated.
Conclusions.
In this study, we revealed that S. islandicus was able to efficiently incorporate short, multiply mismatched donor DNA in a continuous or discontinuous manner by diverse HR events. Importantly, the highly efficient HR in S. islandicus was further validated by the observation that 30 to 50 bp of microhomology was sufficient to allow marker replacement in both an HR assay and a lacS gene targeting test. The apparent ease of recombination enables us to develop a one-step, microhomology-mediated gene inactivation system in two genetically tractable strains, i.e., S. islandicus M.16.4 and REY15A. Given that the gene deletion cassette did not require cloning and could be easily obtained by one-step PCR amplification, we envision that the high-throughput approach reported here, in combination with the nonessential gene profiles, could be applied to create genome-wide targeted gene deletions. This will revolutionize genetics and functional genomics studies for hyperthermophilic crenarchaea. Furthermore, a similar approach could be applied to other hyperthermophilic archaea that are equipped with a functional HR system.
MATERIALS AND METHODS
Sulfolobus strains and growth conditions.
The strains used in this work are described in Table 3. Unless otherwise stated, all Sulfolobus strains were aerobically cultivated in 75-cm2 U-shape canted-neck cell culture flasks (Corning, USA) containing a dextrin-tryptone (DY) liquid medium (pH 3.5) at 76 to 78°C without shaking. The DY liquid medium was composed of 3.0 g liter−1 of K2SO4, 0.5 g liter−1 of NaH2PO4, 0.145 g liter−1 of MgSO4, 0.1 g liter−1 of CaCl2 · 2H2O, 2 g liter−1 of dextrin from maize starch (Sigma-Aldrich, USA), and 1 g liter−1 of tryptone (Bacto, BD Biosciences, USA) and supplemented with 20 μl liter−1 of trace mineral solution. The trace mineral solution was prepared by dissolving 3 g of FeCl3, 0.5 g of CoCl2 · 6H2O, 0.5 g of MnCl2 · 4H2O, 0.5 g of ZnCl2, and 0.5 g of CuCl2 · 2H2O in 100 ml of 1 N HCl. When appropriate, uracil (20 μg/ml), agmatine (50 μg/ml), or both were added to DY to make DYU, DYA, or DYUA medium, respectively. Plate medium was solidified with 0.7% (wt/vol) (final concentration) gellan gum (Gelzan CM; Sigma-Aldrich, USA) and supplemented with 0.96 g liter−1 of CaCl2 · 2H2O and 2.805 g liter−1 of MgSO4, formulated as described previously (16). Growth of Sulfolobus cells was monitored by measurement of optical density at 600 nm (OD600) using a CO8000 cell density meter (WPA, Cambridge, United Kingdom).
TABLE 3.
Strains and plasmids used in this study
| Strain or plasmid | Genotype and/or descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| S. tokodaii | Wild type | DSMZ |
| S. solfataricus P2 | Wild type | DSMZ |
| S. islandicus | ||
| M.16.4 | Wild type | 2 |
| RJW002 | ΔpyrEF; derived from S. islandicus M.16.4 | 15 |
| RJW007 | ΔpyrEF ΔargD; derived from RJW002 | This study |
| RJW008 | ΔpyrEF::pyrEF ΔargD; derived from RJW007 | This study |
| ΔlacS | ΔargD ΔlacS::StoargD; derived from RJW008 | This study |
| ΔTA1 | ΔargD ΔTA1::StoargD; derived from RJW008 | This study |
| ΔTA2 | ΔargD ΔTA2::StoargD; derived from RJW008 | This study |
| ΔTA3 | ΔargD ΔTA3::StoargD; derived from RJW008 | This study |
| ΔTA4 | ΔargD ΔTA4::StoargD; derived from RJW008 | This study |
| ΔTA6 | ΔargD ΔTA6::StoargD; derived from RJW008 | This study |
| ΔTA7 | ΔargD ΔTA7::StoargD; derived from RJW008 | This study |
| ΔTA8 | ΔargD ΔTA8::StoargD; derived from RJW008 | This study |
| ΔTA9 | ΔargD ΔTA9::StoargD; derived from RJW008 | This study |
| ΔTA10 | ΔargD ΔTA10::StoargD; derived from RJW008 | This study |
| ΔTA11 | ΔargD ΔTA11::StoargD; derived from RJW008 | This study |
| ΔTA12 | ΔargD ΔTA12::StoargD; derived from RJW008 | This study |
| ΔTA13 | ΔargD ΔTA13::StoargD; derived from RJW008 | This study |
| ΔTA14 | ΔargD ΔTA14::StoargD; derived from RJW008 | This study |
| ΔTA15 | ΔargD ΔTA15::StoargD; derived from RJW008 | This study |
| ΔTA16 | ΔargD ΔTA16::StoargD; derived from RJW008 | This study |
| ΔTA17 | ΔargD ΔTA17::StoargD; derived from RJW008 | This study |
| ΔTA18 | ΔargD ΔTA18::StoargD; derived from RJW008 | This study |
| ΔTA19 | ΔargD ΔTA19::StoargD; derived from RJW008 | This study |
| ΔTA20 | ΔargD ΔTA20::StoargD; derived from RJW008 | This study |
| ΔTA21 | ΔargD ΔTA21::StoargD; derived from RJW008 | This study |
| ΔTA22 | ΔargD ΔTA22::StoargD; derived from RJW008 | This study |
| Δ23kb | ΔargD Δ23kb::StoargD; derived from RJW008 | This study |
| Δ35kb | ΔargD Δ35kb::StoargD; derived from RJW008 | This study |
| Δ50kb | ΔargD Δ50kb::StoargD; derived from RJW008 | This study |
| E235 | ΔpyrEF ΔlacS ΔargD; S. islandicus REY15A carrying a triple deletion of pyrEF, lacS, and argD | Gift from Q. She's lab |
| ΔupsAB | ΔpyrEF ΔlacS ΔargD ΔupsAB::StoargD; derived from S. islandicus E235 | This study |
| Plasmids | ||
| pRJW1 | pUC19 carrying Simr | 15 |
| pRJW10 | Simr was replaced with the argD expression cassette (StoargD) amplified from S. tokodaii at NcoI-MluI sites | This study |
| pKlacS-Simr | pUC19 carrying the Up-arm and Dn-arm of lacS and Simr; lacS knockout plasmid | 15 |
| pKlacS-StoargD | Simr in pKlacS-Simr was replaced with StoargD at NcoI-MluI sites; lacS knockout plasmid | This study |
| pSeSd-StoargD | StoargD was cloned into Sulfolobus-E. coli shuttle vector pSeSd at XmaI site | This study |
| pPIS-argD | pUC19 carrying the hybrid marker SsopyrEF-lacS and Up-arm and Dn-arm of argD; argD knockout plasmid | 16 |
Simr, simvastatin resistance marker cassette.
Construction of plasmids harboring an agmatine selectable marker (StoargD).
The plasmids used in this work are shown in Table 3.
A 740-bp StoargD marker cassette containing the argD gene under the control of its native promoter and terminator regions was PCR amplified from genomic DNA of S. tokodaii using primers StoargD-F1/R1 or StoargD-F2/R2 (Table 2), with XmaI/XmaI or NcoI/MluI sites introduced, respectively, in the resulting PCR products. To generate pSeSd-StoargD, the StoargD cassette was cloned into the XmaI site of Sulfolobus-E. coli shuttle vector pSeSd (33). The replicative plasmid pSeSd-StoargD was used as a positive control to assess the Sulfolobus transformation efficiency on agmatine-free plates.
For construction of pRJW10, the NcoI/MluI-digested StoargD cassette was cloned into pRJW1 by replacing a simvastatin resistance (Simr) marker at the corresponding sites (15). To generate pKlacS-StoargD, the Simr marker was replaced by the StoargD cassette at the NcoI and MluI sites of plasmid pKlacS-Simr (15).
Generation of donor DNA used for genetic assay of HR in S. islandicus.
A total of 755 bp of homoeologous donor DNA (SsoargD) that consisted of the coding sequence, native promoter, and terminator region of the argD gene was PCR amplified from the S. solfataricus P2 genome using primers SsoargD-F/R (Table 2). Nucleotide sequence variations between SsoargD and the DNA regions surrounding the argD deletion allele in the chromosome of the genetic host S. islandicus RJW008 were compared by BLAST analysis with SnapGene software (Fig. 2) and are shown in Table S1 in the supplemental material. The homologous donor DNA used in the HR assay was PCR amplified from the genome of S. islandicus M.16.4 using the following primer sets: argD-0bp-F/R, argD-10bp-F/R, argD-20bp-F/R, argD-30bp-F/R, argD-34bp-F/R, argD-40bp-F/R, argD-50bp-F/R, argD-100bp-F/R, argD-250bp-F/R, argD-500bp-F/R, argD-750bp-F/R, argD-1000bp-F/R, and argD-1500bp-F/R (Table 2). The resulting PCR products encompassed the in-frame deleted region (381 bp) of the wild-type argD gene as well as its 5′ and 3′ flanking regions with sizes ranging from 0 to 1,500 bp. Each PCR amplification was completed with Phusion high-fidelity DNA polymerase (New England BioLabs, USA). The donor DNA fragments generated by PCR were highly concentrated (>300 ng/μl) with a DNA Clean & Concentrator-25 column (Zymo Research, USA) and eluted with molecular biology-grade water (Corning, USA). For the homoeologous (nonidentical) donor DNA (SsoargD), 250 ng, 500 ng, 1,000 ng, or 1,500 ng of purified PCR products was electroporated into the genetic host RJW008, whereas for the homologous donor DNA, 1,500 ng of purified PCR products was used.
The ArgD+ colonies were selected on DY plates lacking agmatine after 8 to 12 days of incubation at 76 to 78°C.
Molecular analysis of recombinants.
Independent ArgD+ transformants originating from the HR assay using the SsoargD marker cassette as donor DNA were analyzed according to a well-established colony PCR procedure (16) using primers UR-F/R (Table 2). The resulting PCR products (the accurate size of the amplicon was dependent on the donor sequence tracts), encompassing the argD allele generated through HR between donor DNA and the chromosome of the recipient strain, were treated with ExoSAP-IT for the PCR product cleanup kit (Affymetrix, USA) and then sequenced to confirm the genotypes of recombinants.
Generation of gene deletion cassettes via one-step PCR.
The lacS deletion cassette containing the StoargD marker flanked by 0- to 40-bp homology arms was amplified from plasmid pKlacS-StoargD via one-step PCR with primer sets as lacS-DR(0)-F/R, lacS-DR(10)-F/R, lacS-DR(15)-F/R, lacS-DR(20)-F/R, lacS-DR(25)-F/R, lacS-DR(30)-F/R, lacS-DR(35)-F/R, and lacS-DR(40)-F/R (Table 2). Additionally, a 2.1-kb linear lacS deletion cassette that contained the StoargD marker and 739-bp upstream and 652-bp downstream flanking regions of lacS gene was PCR amplified from plasmid pKlacS-StoargD using primer sets lacS-DR (739)-F and lacS-DR (652)-R (Table 2). For the gene deletion cassettes that were used to target 22 individual TA loci, upsAB, or large-scale chromosomal regions (23-, 35-, and 50-kb DNA fragments), 39-bp and 40-bp homology arms were introduced at 5′ and 3′ ends of the StoargD marker, respectively, via one-step PCR with primers listed in Table 2. The one-step PCR mixture was prepared as follows: 0.1 to 1 ng of nonreplicative plasmid pKlacS-StoargD was added to 50-μl reaction mixtures containing 1× Phusion HF buffer, 0.5 μM forward/reverse primers, 200 μM deoxynucleoside triphosphates (dNTPs), and 1.0 unit of Phusion high-fidelity DNA polymerase. The PCR amplification was performed in a Bio-Rad T100 thermal cycler with the following parameters: 98°C for 2 min, followed by 35 cycles of 98°C for 10 s, 56 to 59°C for 30 s, and 72°C for 30 s and a final extension at 72°C for 10 min. To obtain highly concentrated PCR products (>300 ng/μl) for S. islandicus transformation, multiple PCRs using 50-μl mixtures were performed, and the products were combined and then purified with a DNA Clean & Concentrator-25 column (Zymo, USA). The PCR products were finally eluted from column with 25 μl of molecular biology-grade water. The quality of purified PCR products was determined by electrophoresis on a 1% agarose gel, and the DNA concentration was measured with a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA). For gene disruption, 1,500 ng of gene deletion cassette was electroporated into the genetic host S. islandicus RJW008 or S. islandicus E235 (Table 3), selecting ArgD+ colonies on DY or DYU plates lacking agmatine after 8 to 12 days of incubation at 76 to 78°C.
Genetic manipulation of S. islandicus.
S. islandicus RJW008 and S. islandicus E235 were grown in Falcon tissue culture-treated flasks (capacity, 750 ml; Thermo Fisher Scientific, USA) containing 150 ml DYA and DYUA liquid medium, respectively, without shaking. Cell cultures were harvested at an OD600 of 0.18 to 0.25 (early exponential growth phase) and then centrifuged at 4,000 × g (5810 R centrifuge; Eppendorf) for 12 min at room temperature. The supernatant was carefully removed, and the pellet was washed with 50 ml 20 mM sucrose three times under the same centrifugation conditions as described above. The pellet was resuspended with an appropriate volume of 20 mM sucrose, adjusted to a final OD600 of 12 to 15, and then aliquoted in 50-μl portions. Highly concentrated PCR products or circular replicative plasmid DNA (>300 ng/μl; 1 to 5 μl) was mixed with 50 μl competent cells in microcentrifuge tubes and incubated at room temperature for 5 min, and then the mixture was transferred into a Gene Pulser cuvette (0.1-cm gap; Bio-Rad). Electroporation was carried out at 1.2 kV, 25 μF, and 600 Ω using a Gene Pulser II electroporation system (Bio-Rad). After electroporation, 800 μl of prewarmed β-alanine–malate buffer (34) (1% sucrose, 20 mM β-alanine, 1.5 mM malate, and 10 mM MgSO4, pH 4.5) was immediately added to the transformed cells and incubated at 76°C for 60 min without shaking. To obtain single Ura+ colonies, transformed cells were grown in liquid DT medium (0.2% [vol/wt] dextrin, 0.1% [wt/vol] EZMix-N-Z-amine A) lacking uracil for approximately 14 days prior to plating on selective DT plates. ArgD+ colonies were directly selected by plating transformed cells onto nutrient-rich plates lacking agmatine. S. islandicus RJW007 with a double deletion of pyrEF and argD was constructed essentially as described previously by employing a so-called plasmid integration and segregation (PIS) methodology (16), but RJW002 (ΔpyrEF) was used as the genetic host in this study. To generate RJW008 (ΔargD), a 3.45-kb PCR product amplified from the S. islandicus M.16.4 genome with primers SispyrEF-F/R (Table 2), containing wild-type SispyrEF genes as well as their flanking regions (1.1 kb each for upstream and downstream of SispyrEF), was transformed into RJW007 by electroporation, restoring the pyrEF deletion allele to a wild-type allele. RJW008 was used as a recipient strain in the HR assay or as a genetic host for the disruption of lacS, TA gene pairs, and large-scale chromosomal regions. S. islandicus E235 (ΔpyrEF ΔlacS ΔargD), derived from the original isolate S. islandicus REY15A (35), was used as the genetic host to construct the upsAB disruption mutant.
Drop dilution assay of TA disruption mutants.
Mid-log-phase (OD600 = 0.4 to 0.6) cultures of genetic host RJW008 and toxin-antitoxin locus (TA) mutant strains were normalized to a final OD600 of 0.5 by using fresh DY medium. The cultures were serially diluted 10-fold with DY, and 10-μl serial dilutions (100, 10−1, 10−2, 10−3, and 10−4) were spotted on DY plates containing agmatine. Cell growth was examined by incubating the plates at 76 to 78°C for 10 days, and images were digitally captured.
UV treatment and phase-contrast microscopy analysis of S. islandicus cells.
UV treatment of S. islandicus E235 and ΔupsAB mutant cells was conducted as described previously (36) with minor modifications. Briefly, 12 ml of fresh S. islandicus cells (OD600 = 0.2 to 0.3) were transferred into a plastic petri dish (100 by 15 mm; Fisher Scientific, USA), and then irradiated with a UV dose of 75 J/m2 (UVP CL-1000 UV cross-linker; λ = 254 nm). Subsequently, the cells were transferred into an Erlenmeyer flask with a screw cap, stored in the dark for 15 min at room temperature, and then cultivated with shaking (150 rpm) at 76°C. The control cells were prepared following exactly the same procedure except that UV irradiation was not conducted. After 6 h of incubation, 5 μl of cells was taken and dropped onto frosted microscope slides (76 by 25.4 by 1.0 mm; Fisher Scientific, USA) covered by 500 μl of 1.2% agarose in 1× basal salt solution (3.0 g liter−1 of K2SO4, 0.5 g liter−1 of NaH2PO4, 0.145 g liter−1 of MgSO4, 0.1 g liter−1 of CaCl2 · 2H2O). Afterwards, a microscope cover glass (22 by 22 mm; thickness, 0.13 to 0.17 mm; Fisher Scientific, USA) was immediately placed on the top of slides. Cells were observed with a phase-contrast microscope (Zeiss, USA), and images were recorded using ZEN imaging software.
Supplementary Material
ACKNOWLEDGMENTS
We thank Melinda E. Baughman and Yuan Li for experimental assistance. We especially thank Qunxin She and Mingxia Feng for providing the S. islandicus E235 strain. We also thank Ken Ringwald and Matthew Pauly for critical reading of the manuscript.
This work was supported mainly by a grant from the Division of Environmental Biology (DEB) (1355171 to R.J.W.), U.S. National Science Foundation. The work was also partially supported by a grant from the National Aeronautics and Space Administration (NASA) through the NASA Astrobiology Institute under cooperative agreement no. NNA13AA91A, issued through the Science Mission Directorate.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02167-17.
REFERENCES
- 1.Whitaker RJ, Grogan DW, Taylor JW. 2003. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 301:976–978. doi: 10.1126/science.1086909. [DOI] [PubMed] [Google Scholar]
- 2.Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ. 2009. Biogeography of the Sulfolobus islandicus pan-genome. Proc Natl Acad Sci U S A 106:8605–8610. doi: 10.1073/pnas.0808945106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cadillo-Quiroz H, Didelot X, Held NL, Herrera A, Darling A, Reno ML, Krause DJ, Whitaker RJ. 2012. Patterns of gene flow define species of thermophilic Archaea. PLoS Biol 10:e1001265. doi: 10.1371/journal.pbio.1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bautista MA, Zhang C, Whitaker RJ. 2015. Virus-induced dormancy in the archaeon Sulfolobus islandicus. mBio 6:e02565-14. doi: 10.1128/mBio.02565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bautista MA, Black JA, Youngblut ND, Whitaker RJ. 2017. Differentiation and structure in Sulfolobus islandicus rod-shaped virus populations. Viruses 9:E120. doi: 10.3390/v9050120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Held NL, Herrera A, Whitaker RJ. 2013. Reassortment of CRISPR repeat-spacer loci in Sulfolobus islandicus. Environ Microbiol 15:3065–3076. doi: 10.1111/1462-2920.12146. [DOI] [PubMed] [Google Scholar]
- 7.Uldahl KB, Jensen SB, Bhoobalan-Chitty Y, Martinez-Alvarez L, Papathanasiou P, Peng X. 2016. Life cycle characterization of Sulfolobus monocaudavirus 1, an extremophilic spindle-shaped virus with extracellular tail development. J Virol 90:5693–5699. doi: 10.1128/JVI.00075-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leon-Sobrino C, Kot WP, Garrett RA. 2016. Transcriptome changes in STSV2-infected Sulfolobus islandicus REY15A undergoing continuous CRISPR spacer acquisition. Mol Microbiol 99:719–728. doi: 10.1111/mmi.13263. [DOI] [PubMed] [Google Scholar]
- 9.Erdmann S, Le Moine Bauer S, Garrett RA. 2014. Inter-viral conflicts that exploit host CRISPR immune systems of Sulfolobus. Mol Microbiol 91:900–917. doi: 10.1111/mmi.12503. [DOI] [PubMed] [Google Scholar]
- 10.Quax TE, Voet M, Sismeiro O, Dillies MA, Jagla B, Coppee JY, Sezonov G, Forterre P, van der Oost J, Lavigne R, Prangishvili D. 2013. Massive activation of archaeal defense genes during viral infection. J Virol 87:8419–8428. doi: 10.1128/JVI.01020-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen L, Brugger K, Skovgaard M, Redder P, She Q, Torarinsson E, Greve B, Awayez M, Zibat A, Klenk HP, Garrett RA. 2005. The genome of Sulfolobus acidocaldarius, a model organism of the Crenarchaeota. J Bacteriol 187:4992–4999. doi: 10.1128/JB.187.14.4992-4999.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner M, van Wolferen M, Wagner A, Lassak K, Meyer BH, Reimann J, Albers SV. 2012. Versatile genetic tool box for the crenarchaeote Sulfolobus acidocaldarius. Front Microbiol 3:214. doi: 10.3389/fmicb.2012.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson RE, Kouris A, Seward CH, Campbell KM, Whitaker RJ. 2017. Structured populations of Sulfolobus acidocaldarius with susceptibility to mobile genetic elements. Genome Biol Evol doi: 10.1093/gbe/evx104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng L, Zhu H, Chen Z, Liang YX, She Q. 2009. Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles 13:735–746. doi: 10.1007/s00792-009-0254-2. [DOI] [PubMed] [Google Scholar]
- 15.Zhang C, Whitaker RJ. 2012. A broadly applicable gene knockout system for the thermoacidophilic archaeon Sulfolobus islandicus based on simvastatin selection. Microbiology 158:1513–1522. doi: 10.1099/mic.0.058289-0. [DOI] [PubMed] [Google Scholar]
- 16.Zhang C, Cooper TE, Krause DJ, Whitaker RJ. 2013. Augmenting the genetic toolbox for Sulfolobus islandicus with a stringent positive selectable marker for agmatine prototrophy. Appl Environ Microbiol 79:5539–5549. doi: 10.1128/AEM.01608-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C, She Q, Bi H, Whitaker RJ. 2016. The apt/6-methylpurine counterselection system and its applications in genetic studies of the hyperthermophilic archaeon Sulfolobus islandicus. Appl Environ Microbiol 82:3070–3081. doi: 10.1128/AEM.00455-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaubert C, Danioux C, Oberto J, Cortez D, Bize A, Krupovic M, She Q, Forterre P, Prangishvili D, Sezonov G. 2013. Genomics and genetics of Sulfolobus islandicus LAL14/1, a model hyperthermophilic archaeon. Open Biol 3:130010. doi: 10.1098/rsob.130010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C, Guo L, Deng L, Wu Y, Liang Y, Huang L, She Q. 2010. Revealing the essentiality of multiple archaeal pcna genes using a mutant propagation assay based on an improved knockout method. Microbiology 156:3386–3397. doi: 10.1099/mic.0.042523-0. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Pan S, Zhang Y, Ren M, Feng M, Peng N, Chen L, Liang YX, She Q. 2016. Harnessing type I and Type III CRISPR-Cas systems for genome editing. Nucleic Acids Res 44:e34. doi: 10.1093/nar/gkv1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng W, Feng M, Feng X, Liang YX, She Q. 2015. An archaeal CRISPR type III-B system exhibiting distinctive RNA targeting features and mediating dual RNA and DNA interference. Nucleic Acids Res 43:406–417. doi: 10.1093/nar/gku1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng N, Han W, Li Y, Liang Y, She Q. 2017. Genetic technologies for extremely thermophilic microorganisms of Sulfolobus, the only genetically tractable genus of crenarchaea. Sci China Life Sci 60:370–385. doi: 10.1007/s11427-016-0355-8. [DOI] [PubMed] [Google Scholar]
- 23.Maezato Y, Johnson T, McCarthy S, Dana K, Blum P. 2012. Metal resistance and lithoautotrophy in the extreme thermoacidophile Metallosphaera sedula. J Bacteriol 194:6856–6863. doi: 10.1128/JB.01413-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Worthington P, Hoang V, Perez-Pomares F, Blum P. 2003. Targeted disruption of the alpha-amylase gene in the hyperthermophilic archaeon Sulfolobus solfataricus. J Bacteriol 185:482–488. doi: 10.1128/JB.185.2.482-488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grogan DW, Rockwood J. 2010. Discontinuity and limited linkage in the homologous recombination system of a hyperthermophilic archaeon. J Bacteriol 192:4660–4668. doi: 10.1128/JB.00447-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurosawa N, Grogan DW. 2005. Homologous recombination of exogenous DNA with the Sulfolobus acidocaldarius genome: properties and uses. FEMS Microbiol Lett 253:141–149. doi: 10.1016/j.femsle.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 27.Sakofsky CJ, Runck LA, Grogan DW. 2011. Sulfolobus mutants, generated via PCR products, which lack putative enzymes of UV photoproduct repair. Archaea 2011:864015. doi: 10.1155/2011/864015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jonuscheit M, Martusewitsch E, Stedman KM, Schleper C. 2003. A reporter gene system for the hyperthermophilic archaeon Sulfolobus solfataricus based on a selectable and integrative shuttle vector. Mol Microbiol 48:1241–1252. doi: 10.1046/j.1365-2958.2003.03509.x. [DOI] [PubMed] [Google Scholar]
- 29.Farkas J, Stirrett K, Lipscomb GL, Nixon W, Scott RA, Adams MW, Westpheling J. 2012. Recombinogenic properties of Pyrococcus furiosus strain COM1 enable rapid selection of targeted mutants. Appl Environ Microbiol 78:4669–4676. doi: 10.1128/AEM.00936-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. 1993. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res 21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Wolferen M, Ajon M, Driessen AJ, Albers SV. 2013. Molecular analysis of the UV-inducible pili operon from Sulfolobus acidocaldarius. Microbiologyopen 2:928–937. doi: 10.1002/mbo3.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng N, Deng L, Mei Y, Jiang D, Hu Y, Awayez M, Liang Y, She Q. 2012. A synthetic arabinose-inducible promoter confers high levels of recombinant protein expression in hyperthermophilic archaeon Sulfolobus islandicus. Appl Environ Microbiol 78:5630–5637. doi: 10.1128/AEM.00855-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berkner S, Grogan D, Albers SV, Lipps G. 2007. Small multicopy, non-integrative shuttle vectors based on the plasmid pRN1 for Sulfolobus acidocaldarius and Sulfolobus solfataricus, model organisms of the (cren-)archaea. Nucleic Acids Res 35:e88. doi: 10.1093/nar/gkm449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo L, Brugger K, Liu C, Shah SA, Zheng H, Zhu Y, Wang S, Lillestol RK, Chen L, Frank J, Prangishvili D, Paulin L, She Q, Huang L, Garrett RA. 2011. Genome analyses of Icelandic strains of Sulfolobus islandicus, model organisms for genetic and virus-host interaction studies. J Bacteriol 193:1672–1680. doi: 10.1128/JB.01487-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frols S, Gordon PM, Panlilio MA, Duggin IG, Bell SD, Sensen CW, Schleper C. 2007. Response of the hyperthermophilic archaeon Sulfolobus solfataricus to UV damage. J Bacteriol 189:8708–8718. doi: 10.1128/JB.01016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.