

SUMMARY
The central mechanism by which neurotensin (Nts) potentiates weight loss has remained elusive. We leveraged chemogenetics to reveal that Nts-expressing neurons of the lateral hypothalamic area (LHA) promote weight loss in mice by increasing volitional activity and restraining food intake. Intriguingly, these dual weight loss behaviors are mediated by distinct signaling pathways: Nts action via NtsR1 is essential for the anorectic effect of the LHA Nts circuit, but not for regulation of locomotor or drinking behavior. Furthermore, although LHA Nts neurons cannot reduce intake of freely available obesogenic foods, they effectively restrain motivated feeding in hungry, weight-restricted animals. LHA Nts neurons are thus vital mediators of central Nts action, particularly in the face of negative energy balance. Enhanced action via LHA Nts neurons may therefore be useful to suppress the increased appetitive drive that occurs after lifestyle-mediated weight loss, and hence to prevent weight regain.
Keywords: neurotensin, neurotensin receptor, lateral hypothalamic area, feeding, locomotor activity, drinking, obesity, energy balance
Graphical abstract
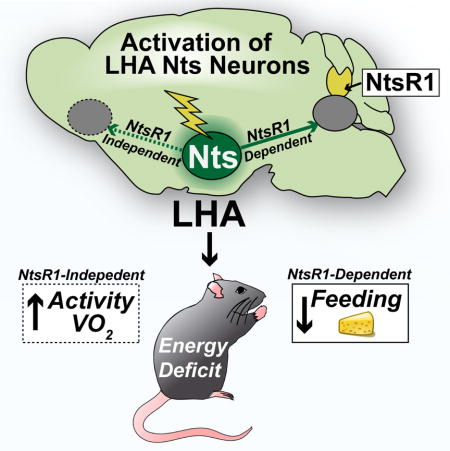
Obesity is a formidable health concern yet non-surgical treatments including lifestyle modification and pharmacotherapy provide limited long-term weight loss (Khera et al., 2016). Individuals who lose weight experience compensatory increases in appetite and diminished metabolic rate, and as a result most regain weight (Sumithran et al., 2011; Wing and Hill, 2001). Understanding how the brain coordinates feeding and energy expenditure is therefore crucial to identify strategies to support sustained weight loss.
The neuropeptide neurotensin (Nts) acts centrally to suppress feeding in hungry and obese rodents, and holds potential to counteract increased appetitive drive that thwarts sustained weight loss (Boules et al., 2000; Cooke et al., 2009; Feifel et al., 2010; Hawkins, 1986b; Kim et al., 2008; Luttinger et al., 1982). Nts can signal via the G-protein-coupled Nts receptors−1 and −2 (NtsR1 and NtsR2) and NtsR1 has been specifically implicated in mediating anorectic effects (Kim et al., 2008; Remaury et al., 2002). However, Nts and NtsR1 are broadly distributed throughout the brain and also regulate analgesia, thermoregulation and blood pressure (Geisler et al., 2006), and it remains unclear which Nts neurons specifically modify energy balance. For example, Nts in the nucleus accumbens blunts physical activity with no effect on feeding or body weight (Ervin et al., 1981; Kalivas et al., 1984) while Nts in the ventral tegmental area (VTA) limits feeding and promotes locomotor activity, dual behaviors that could support weight loss (Cador et al., 1986; Hawkins, 1986a; Stanley et al., 1983). These data indicate that specific, yet to be defined Nts neurons promote weight loss behaviors, in part via Nts release to the VTA. Establishing the precise Nts population that modifies energy balance, and the requirement for signaling via NtsR1, is necessary to design therapies to support weight loss without disrupting other Nts-mediated physiology.
Nts-expressing neurons within the lateral hypothalamic area (LHA) are positioned to potentiate weight loss, consistent with the role of the LHA in coordinating ingestive and locomotor behaviors (Brown et al., 2015; Opland et al., 2013). The large population of LHA Nts neurons is distinct from orexigenic melanin-concentrating hormone or orexin/hypocretin neurons (Brown et al., 2017), and although some LHA Nts neurons co-express GABA (Jennings et al., 2015; Patterson et al., 2015), they but do not provoke the voracious feeding response that occurs with activation of all LHA GABA neurons (Jennings et al., 2013; Jennings et al., 2015; Navarro et al., 2016; Nieh et al., 2015). Instead, LHA Nts neurons are physiologically activated by signals that suppress feeding, such as dehydration anorexia (Watts and Sanchez-Watts, 2007) or the appetite-suppressing hormone, leptin (Leinninger et al., 2011). However, only 15% of LHA Nts neurons mediate leptin action (Brown et al., 2017; Leinninger et al., 2011), yet the overall role of the large population of LHA Nts neurons, and Nts signaling from them, is unclear. Since LHA Nts neurons release Nts to the VTA, where NtsR1-expressing dopamine (DA) neurons have been implicated in coordinating feeding and locomotor activity (Opland et al., 2013; Patterson et al., 2015; Woodworth et al., 2017), LHA Nts neurons might support NtsR1 and/or DA-mediated weight loss behaviors. Indeed, experimental activation of LHA Nts neurons increases DA-dependent locomotor activity (Patterson et al., 2015) but the potential anorectic role of LHA Nts neurons and NtsR1 was not examined. We therefore used chemogenetics to activate LHA Nts neurons in the presence and absence of NtsR1 to determine if they mediate the known anorectic and locomotor effects of Nts, and their potential for stimulating weight loss behaviors.
RESULTS
Models to Define the Role of LHA Nts signaling via NtsR1
To experimentally activate LHA Nts neurons and simultaneously determine the requirement for Nts signaling via NtsR1, we crossed NtsCre mice to an NtsR1 knockout line, generating NtsCre mice with intact NtsR1 (NtsCre;NtsR1++) or lacking NtsR1 (NtsCre;NtsR1KOKO mice). Adult males were injected with cre-inducible AAV-hM3Dq-mCherry in the LHA to express excitatory DREADD receptors selectively in LHA Nts neurons (Fig. 1A). Treating mice with CNO induced cFos in mCherry-labeled LHA Nts neurons, verifying that this method activates LHA Nts neurons (Fig. 1B). We next confirmed that developmental deletion of NtsR1 does not impair the structure of LHA Nts neurons; indeed, NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice have similar amounts of mCherry-labeled Nts cell bodies in the LHA and terminals in the VTA, a projection site of LHA Nts neurons (Fig. 1 C, D). Since both NtsR1 and NtsR2 are expressed in the VTA (Fig. 1E and (Woodworth et al., 2017), we used this region to verify that NtsCre;NtsR1KOKO mice specifically lack Ntsr1 gene expression, without any compensatory increase in Ntsr2 (Fig. 1F). Thus, activating the intact LHA Nts neurons in NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice will reveal their physiologic contributions to energy balance and the specific role of Nts action via NtsR1.
Figure 1. Examination of the LHA Nts➔VTA circuit in NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice.
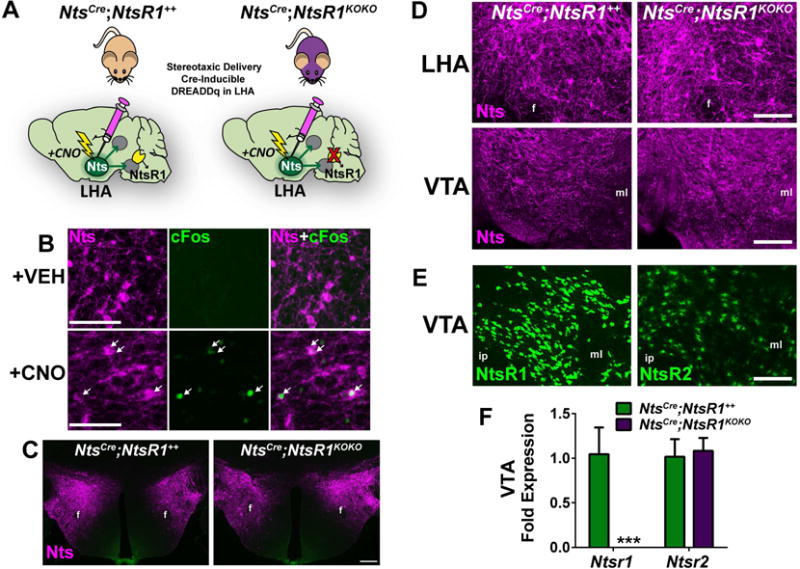
A) NtsCre mice with either intact or developmentally deleted NtsR1 (NtsCre;NtsR1++ or NtsCre;NtsR1KOKO mice) were injected bilaterally in the LHA with cre-inducible AAV-hm3Dq-mCherry, permitting chemoogenetic activation of LHA Nts neurons by CNO injection. B) i.p CNO treatment induces cFos expression in LHA Nts neurons (white arrows). Scale bar=50μM C) LHA Nts neurons expressing hM3Dq-mCherry in NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice (scale bar=200μm). D) hM3Dq-mCherrry-labeled LHA Nts cell bodies in the LHA and terminals in the VTA of NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice. E) Both NtsRs are expressed in the VTA, visualized using Ntsr1Cre;GFP and Ntsr2Cre;GFP mice. F) Ntsr1 and Ntsr2 mRNA expression in the VTA of NtsCre;NtsR1++ mice and NtsCre;NtsR1KOKO mice. Data represent mean ± SEM, n=10–11 per group. Data were analyzed by unpaired t-test for each gene, ***p<0.001. For D and E, scale bar=100μM. Abbreviations: f=fornix, ml=medial lemniscus, ip=interpeduncular nucleus.
Acute Activation of LHA Nts Neurons Promotes Activity and Suppresses Feeding
We first examined how activation of LHA Nts neurons alters energy balance by analyzing NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice in metabolic chambers after single injections of VEH or CNO. LHA Nts activation increased water intake over 8 hr in NtsCre;NtsR1++ mice and to a lesser extent in NtsCre;NtsR1KOKO mice, suggesting that NtsR1 may contribute to, but is not essential for LHA Nts-induced drinking (Fig. 2A,B). Activating LHA Nts neurons increased locomotor activity and energy expenditure in NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice in a similar time course (Fig. 2C–F), indicating that the physical activity and caloric usage occurs in an NtsR1-independent manner. While CNO-treated NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice were less active during the dark cycle, presumably to compensate for increased light cycle activity (Fig. S1A–C), their modest elevation in energy expenditure persisted over 24 hr (Fig. S1D–E). Given the increased energy demands induced by activation of LHA Nts neurons, we examined whether NtsCre;NtsR1++ or NtsCre;NtsR1KOKO mice consumed more calories to maintain energy balance. Interestingly, NtsCre;NtsR1++ mice did not increase feeding after CNO injection when they are also most active (Fig. 2G vs. C), and reduced feeding during the dark cycle despite their activity-induced caloric deficit, resulting in decreased total food intake over 24 hr (Fig 2I and s). By contrast, NtsCre;NtsR1KOKO mice increased food intake immediately after CNO injection with only modest reductions in the dark cycle, resulting in no feeding difference over 24 hr compared to VEH-treated mice (Fig 2I and S1F,G). Together, the restrained feeding and elevated locomotor behaviors induced by activating LHA Nts neurons in NtsCre;NtsR1++ mice resulted in weight loss, but NtsCre;NtsR1KOKO mice maintained their weight, likely due to their compensatory food intake (Fig. 2I, J). These data suggest that activation of LHA Nts neurons promotes locomotor activity, energy expenditure and NtsR1-dependent suppression of feeding to promote weight loss.
Figure 2. Acute activation of LHA Nts neurons promotes energy expenditure and suppresses feeding.
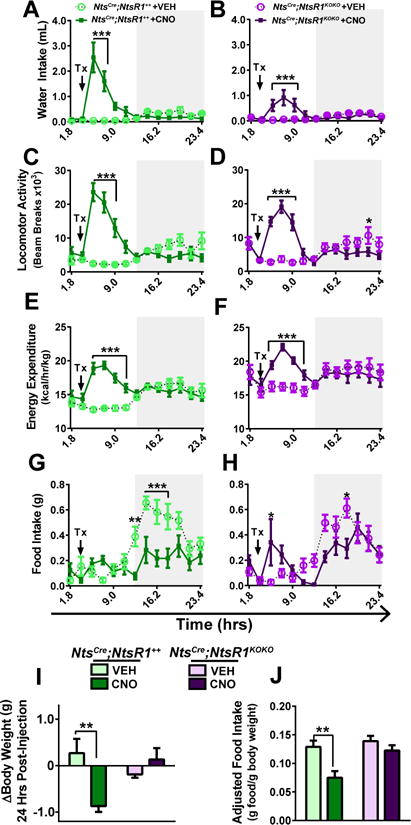
VEH or CNO-treated NtsCre;NtsR1++and NtsCre;NtsR1KOKO mice were analyzed in TSE metabolic cages. In A–H, each point represents 108 minutes and gray boxes denote the dark cycle. A, B) water intake, C, D) locomotor activity, E, F) energy expenditure, and G, H) feeding in NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice. I) total change in body weight 24 hours post-injection. J) Total weight-adjusted food intake over 24 hours. Data were analyzed by repeated measures two-way ANOVA with Sidak post-tests, and graphed data represent mean ± SEM. **p < 0.01, ***p < 0.001. NtsCre;NtsR1++ n=10–11; NtsCre;NtsR1KOKO n=9–10.
To verify the requirement for NtsR1 in LHA Nts-induced anorectic behavior we pretreated NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice with the NtsR1 antagonist SR48692 (0.3 mg/kg, i.p.), then administered VEH or CNO to activate LHA Nts neurons. CNO-induced hyperdipsia appeared less robust in NtsCre;NtsR1KOKO mice compared to NtsCre;NtsR1++ mice, but the NtsR1 antagonist had no effect on water intake in either group (Fig. 3A, D), suggesting that LHA Nts-mediated hyperdipsia does not entirely depend on NtsR1. Treatment with the NtsR1 antagonist did not diminish the total CNO-induced locomotor activity of NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice over 24 hr (Fig. 3B,E, and consistent with Supp Fig 1A–C). By contrast, the LHA Nts-mediated reduction of chow intake over 24 hr was abolished in NtsCre;NtsR1++ mice pre-treated with the NtsR1 antagonist, with no effect in NtsCre;NtsR1KOKO mice (Fig. 3C,F). Indeed, activation of LHA Nts neurons while NtsR1 signaling was pharmacologically or genetically disrupted resulted in increased feeding during the light cycle (Fig. S2, Fig. 2H), which likely represents a compensatory response to the increased energy expenditure that occurs concomitantly with LHA Nts neuron activation. These data suggest that Nts signaling via NtsR1 limits compensatory feeding in response to energy deficit, such as may be induced by exercise.
Figure 3. Acute NtsR1 or D1R blockade abolishes LHA Nts-induced suppression of feeding.
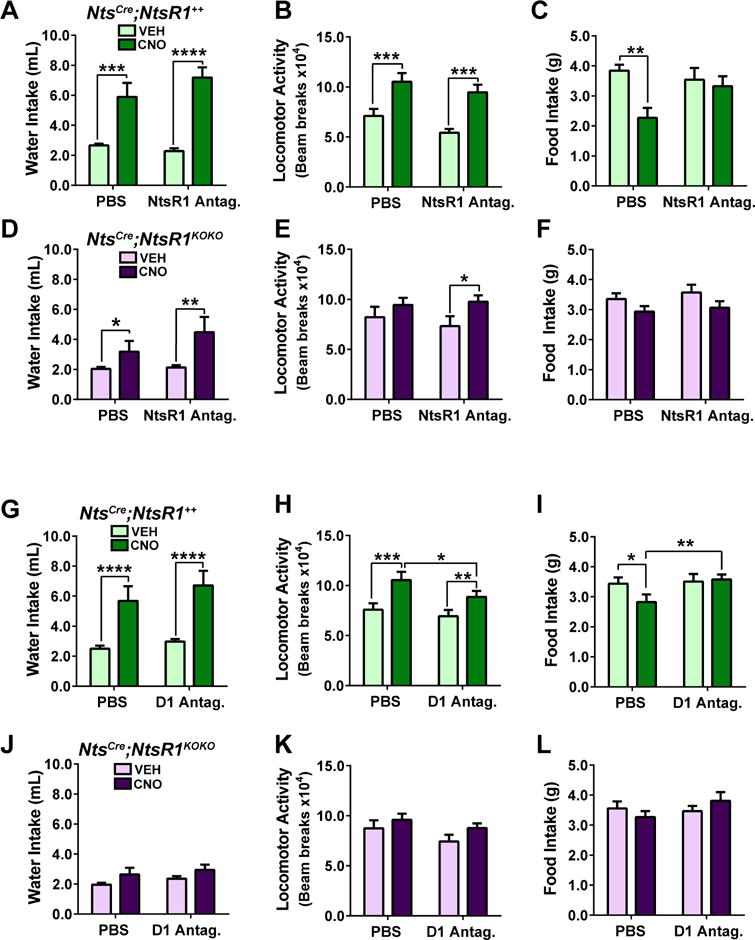
Mice were pretreated with PBS, SR48692 (NtsR1 antagonist) or SCH23390 (D1R antagonist) 30 min. prior to VEH or CNO injection. Graphed data represent mean ± SEM over 24 hours post VEH or CNO injection. A–C) water intake, locomotor activity and feeding in NtsCre;NtsR1++ mice pretreated with the NtsR1 antagonist. D–F) water intake, locomotor activity and feeding in NtsCre;NtsR1KOKO mice pretreated with the NtsR1 antagonist. G–L) same parameters in NtsCre;NtsR1++ or NtsCre;NtsR1KOKO mice pretreated with a D1R antagonist (SCH23390). Data were analyzed by repeated measures two-way ANOVA with Sidak post-tests, *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001. NtsCre;NtsR1++ n=10–11; NtsCre;NtsR1KOKO n=9–10.
Since activation of LHA Nts neurons promotes DA release and D1R-mediated locomotor activity (Patterson et al., 2015), we next investigated whether D1R is necessary for all LHA Nts-induced weight-loss behaviors. Pre-treatment with the D1R antagonist SCH23390 (0.1 mg/kg i.p.) prior to activating LHA Nts neurons did not block drinking behavior in NtsCre;NtsR1++ or NtsCre;NtsR1KOKO mice (Fig. 3G, J), suggesting that D1R signaling is not required for LHA Nts-induced water intake. CNO-induced drinking behavior over 24 hours in NtsCre;NtsR1KOKO mice was generally blunted during this D1R antagonist experiment (Fig. 3J). We speculate that this may be due to delayed acclimation of NtsCre;NtsR1KOKO mice to the novel environment of the metabolic cages and water bottle lixits, consistent with the increased susceptibility to anxiety reported in NtsR1KOKO mice (Fitzpatrick et al., 2012). However, such a phenotype is mild, and the NtsCre;NtsR1KOKO mice did ultimately acclimate, demonstrated by their significantly induced drinking during subsequent experimental treatment with NtsR1 antagonist (Fig. 3D). D1R antagonism blunted LHA Nts-induced locomotor activity in NtsCre;NtsR1++ mice, consistent with previous studies (Patterson et al., 2015), and also in NtsCre;NtsR1KOKO mice (Fig. 3H, K, p=0.06 for NtsCre;NtsR1KOKO mice). D1R blockade also abolished the ability of LHA Nts neurons to suppress feeding in NtsCre;NtsR1++ mice, similar to the effect of disrupting NtsR1 signaling (Fig. 3I, Fig. S2C,D). D1R blockade did not alter feeding in NtsCre;NtsR1KOKO mice, consistent with the lack of NtsR1 impeding DA release to the NA (Patterson et al., 2015). In sum, these data indicate that NtsR1 and D1R signaling are required for LHA Nts neurons to acutely restrain compensatory food intake, but they are not essential for inducing drinking or locomotor activity.
LHA Nts Action Promotes Sustained Weight Loss
LHA Nts neuron activation promotes weight loss acutely over 24 hours, but to determine if LHA Nts action could sustain weight loss, we activated the LHA Nts neurons of NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice for 5 consecutive days in their home cages. While the temporally-precise, automated measurements obtained via metabolic cages revealed the modest anorectic effect of activating LHA Nts neurons in normal weight mice (Fig. 2G), we did not observe a significant reduction in feeding in NtsCre;NtsR1++ mice (Fig. 4A). This is likely due to the increased error of hand-weighing chow at the beginning and end of the light cycle each day, along with the modest anorectic effect of LHA Nts neurons in ad lib-fed mice (Fig 2G, J). Despite the lack of significant feeding differences, NtsCre;NtsR1++ mice lost weight after the first injection of CNO and maintained that reduced body weight throughout the study (Fig. 4B). Since acute activation of LHA Nts neurons promoted weight loss via suppressing feeding and increasing physical activity in metabolic chambers (Fig. 2), we measured physical activity in locomotor chambers for 1 hr after the final VEH or CNO treatment on day 5. Although this method provided a snapshot of CNO-induced locomotor activity compared to the continuous monitoring provided by the metabolic cages (Fig. 2C), the trend towards increased activity in CNO-treated mice (Fig. 4C) suggests that repeated activation of LHA Nts neurons maintains elevated locomotor activity and presumably increased energy expenditure. Chronic activation of LHA Nts neurons also sustained vigorous water consumption throughout the study (Fig. 4D). Importantly, CNO treatment had no effect on the behavior or body weight of NtsCre;NtsR1++ mice expressing channel rhodopsin (ChR) in LHA Nts neurons instead of hM3Dq-mCherry (Fig. S3), confirming that our results are not due to off-target effects of CNO or its metabolite clozapine (Gomez et al., 2017). Interestingly, activation of LHA Nts neurons in NtsCre;NtsR1KOKO mice also induced sustained weight loss over 5 days (Fig. 4F), which conflicts with our observed lack of weight loss in metabolic cages due to compensatory feeding (Fig. 2I). Taken together, these data suggest that chronic activation of LHA Nts neurons can promote weight loss in ad lib-fed animals in home cages via an NtsR1-independent mechanism. These data further support our hypothesis that NtsR1KOKO mice may have been susceptible to novel-environment anxiety during testing in metabolic chambers that altered their intake behavior, and highlight the importance of testing mice in both novel and home cage conditions. Indeed, while CNO-induced water drinking was blunted in NtsCre; NtsR1KOKO mice in metabolic chambers, drinking behavior was similar to NtsCre;NtsR1++ mice in home cages with familiar water bottles (Fig. 4D, H), suggesting that NtsR1 is not required for LHA Nts-induced drinking behavior.
Figure 4. Chronic activation of LHA Nts neurons induces mild weight loss in chow-fed lean mice.
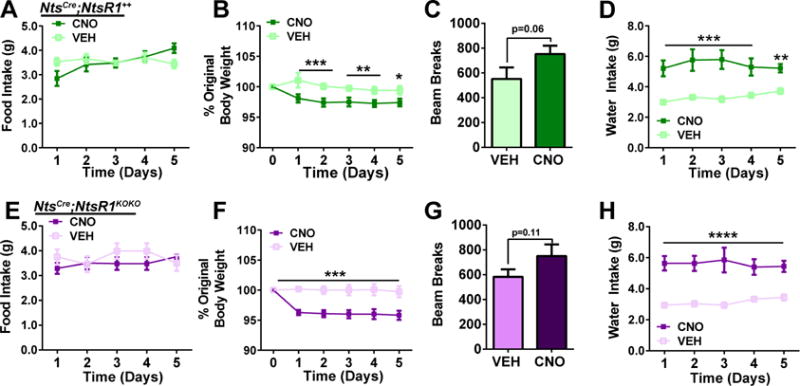
NtsCre;NtsR1++and NtsCre;NtsR1KOKO mice were treated once daily with VEH or CNO for five consecutive days. A) chow intake, B) body weight, C) 1 hr locomotor activity, and D) water intake in NtsCre;NtsR1++ mice (n=11). E) chow consumption, F) body weight, G) 1 hr locomotor activity, and H) water intake in NtsCre;NtsR1KOKO mice (n=11). Data were analyzed by repeated-measures two-way ANOVA with Sidak post-tests, except for locomotor activity which was analyzed by paired t-test. Graphed data represent mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ****p<0.0001.
Role of LHA Nts Neurons in Palatable Food Intake
Next, we investigated whether activating LHA Nts neurons could support weight loss in mice made obese by free access to a palatable high fat (45%), high sucrose diet (HF diet). Obese NtsCre;NtsR1++ mice expressing hM3Dq-mCherry in LHA Nts neurons were given daily treatments of VEH or CNO while in metabolic chambers. As in normal weight mice, chronic activation of LHA Nts neurons promoted locomotor activity immediately after CNO injection, followed by a compensatory decrease in activity during the dark cycle (Fig. 5A, B). However, chronic activation of LHA Nts neurons did not significantly increase energy expenditure in obese mice as observed in lean animals (Fig. 5C vs. Fig S1D). Whereas activating LHA Nts neurons suppressed feeding in normal weight mice, activation in obese mice increased intake of the palatable HF diet during the light cycle with no subsequent suppression of feeding during the dark phase (Fig. 5D, E). As a result, obese CNO-treated mice maintained their high body weight, while obese VEH-treated controls slightly lost weight during the experiment, likely due to the stress of repeated injections (Fig. 5F). Thus, although activation of LHA Nts neurons promoted locomotor activity in obese mice, it was insufficient to offset the consumption of freely available palatable food necessary to support weight loss.
Figure 5. Repeated activation of LHA Nts neurons does not restrain ad lib palatable food intake in obese or lean mice.
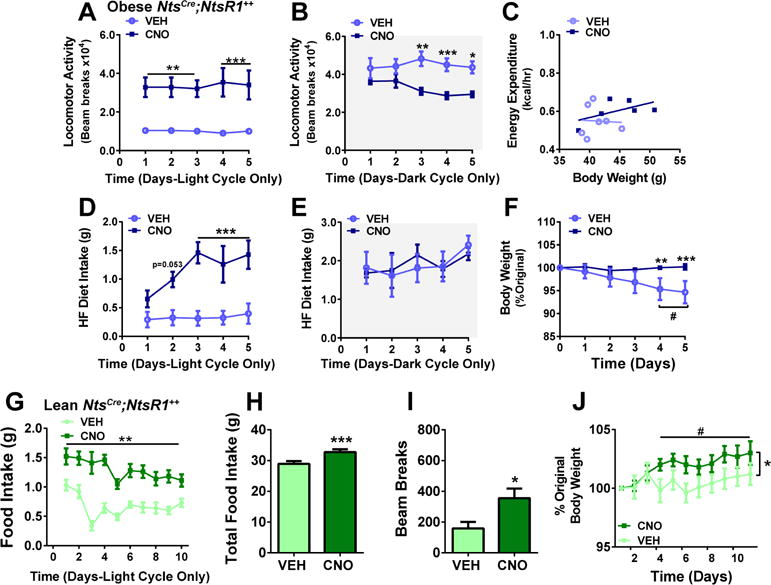
LHA Nts neurons of diet-induced obese mice were activated for 5 days. A) daily locomotor activity during the light cycle vs. B) dark cycle, C) plot showing average rate of energy expenditure vs. body weight (p=0.44), D) food intake during light cycle compared to E) dark cycle, F) percent original body weight over five days of chronic VEH or CNO injections. VEH n=6, CNO n=5–6. Data were analyzed by standard two-way ANOVA except for c which was analyzed by ANCOVA. Asterisks indicate significant difference between VEH and CNO at given time point, *p < 0.05, **p < 0.01, ***p < 0.001. In f, (#) represents significant difference in body weight at days 4–5 compared to day 0 in the VEH group, #p <0.05. For g-j, lean NtsCre;NtsR1++ mice were injected each morning on VEH or CNO for 10 days with ad lib access to HF diet in home cages. G) HF diet intake during the light cycle, H) total food consumed over 10 days, I) 1 hour locomotor activity, and J) body weight in lean NtsCre;NtsR1++ mice (n=10). *p<0.05, **p<0.01, ***p<0.001 between VEH and CNO from Days 4–10. #p<0.05 for CNO time point compared to Day 1. Daily food intake and body weight were analyzed by repeated-measures two-way ANOVA with Sidak post-tests. Total food intake and locomotor activity was analyzed by paired t-test. Graphed data represent mean ± SEM.
Prolonged obesity disrupts neural circuitry that regulate feeding and motivation (Johnson and Kenny, 2010; Stice et al., 2008), hence activation of LHA Nts neurons during established obesity may no longer be able to promote weight loss behaviors. We therefore tested whether LHA Nts neurons could prevent normal weight mice from HF diet-induced weight gain. Lean chow-fed NtsCre;NtsR1++ mice were switched to ad lib palatable HF diet followed by daily VEH or CNO treatments to activate LHA Nts neurons. Similar to obese mice, activation of LHA Nts neurons caused lean NtsCre;NtsR1++ mice to consume significantly more palatable HF diet than VEH-treated controls (Fig. 5G, H). Although activation of LHA Nts neurons also increased locomotor activity (Fig. 5I, measured on day 5), this was counteracted by increased food intake, leading to weight gain (Fig. 5J). Thus, while activation of LHA Nts neurons mildly suppresses chow intake, it cannot restrain consumption of freely accessible palatable foods that promote obesity.
LHA Nts Neurons Suppress Fasting-Induced Refeeding
LHA Nts neurons release Nts to target regions including the VTA, where VTA NtsR1 neurons contain DA (Brown et al., 2017; Patterson et al., 2015; Woodworth et al., 2017). Since VTA DA signaling modifies the motivation to work for food rewards (e.g. “wanting”), but not their hedonic value (“liking”) or ad lib food consumption (Berridge et al., 2010; Salamone and Correa, 2012), we speculated that LHA Nts neurons might selectively modulate working for food and/or feeding in an energy-restricted state (Ahn and Phillips, 1999; Branch et al., 2013). To test this, we fasted NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice overnight and activated LHA Nts neurons upon restoration of chow the following morning. CNO-mediated activation of LHA Nts neurons restrained re-feeding, which persisted over 24 hr in NtsCre;NtsR1++ mice (Fig. 6A, B) and resulted in less weight regain compared to VEH-treated controls (Fig. 6C). Although LHA Nts-mediated suppression of re-feeding in NtsCre;NtsR1KOKO mice was non-significant, one explanation for their failure to re-gain body weight could be that increased LHA Nts-induced activity and energy expenditure, which occurs independently of NtsR1 (Fig. 2), is sufficient to maintain negative energy balance in a fasted state. These data demonstrate that LHA Nts neurons can suppress chow feeding and subsequent weight gain even under conditions that increase the drive to feed, such as fasting-induced weight loss (Ahima et al., 1996).
Figure 6. Activation of LHA Nts neurons suppresses fasting-induced ad lib and motivated food intake.
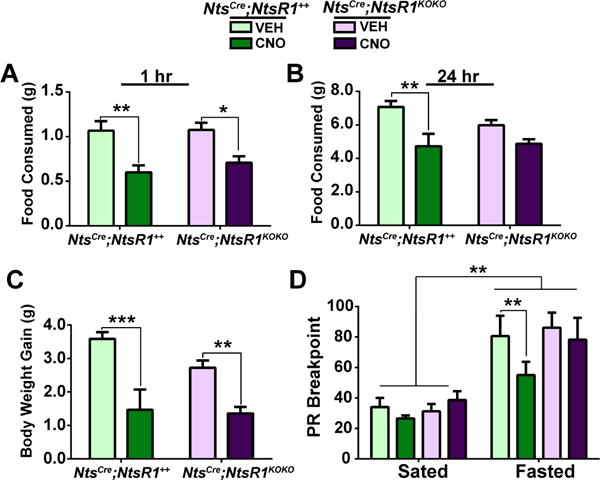
NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice were food-deprived overnight and received VEH or CNO with food restoration the following morning. A, B) Ad lib chow re-feeding 1 and 24 hours after food restoration. C) Body weight gain during 24 hours of ad lib re-feeding after overnight food-deprivation. D) PR breakpoint for sucrose pellets after VEH or CNO injection in both fed and fasted states. NtsCre;NtsR1++ n=9, NtsCre;NtsR1KOKO n=12, data were analyzed by repeated measures two-way ANOVA with Sidak post-tests, *p < 0.05, **p < 0.01, ***p < 0.001. Graphed data represent mean ± SEM.
Given the established role of mesolimbic DA signaling in progressive-ratio (PR) responding for food (Salamone and Correa, 2012; Salamone et al., 2007), we next investigated if LHA Nts neurons alter motivation to work for sucrose. NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice were trained to nose-poke (e.g. work) for sucrose pellets, and both groups learned the task equally well (Fig. S4A–D). When tested on a PR schedule (Sharma et al., 2012), activation of LHA Nts neurons did not alter PR breakpoints in ad lib fed (Sated) NtsCre;NtsR1++ or NtsCre;NtsR1KOKO mice (Fig. 6D). Thus, activation of LHA Nts neurons in an energy-replete context does not suppress freely accessible (Fig. 5) or motivated intake of palatable food. By contrast, activation of LHA Nts neurons after overnight fasting suppressed motivated sucrose responding in NtsCre;NtsR1++ but not NtsCre;NtsR1KOKO mice (Fig. 6D), suggesting that LHA Nts action via NtsR1 reduces motivated sucrose “wanting” in the face of physiologic energy deficit. Importantly, the increased locomotor activity of mice upon activation of LHA Nts neurons did not impair their ability to obtain sucrose, as magazine entries were unaffected by CNO treatment (Fig. S4E). Together, these data provide evidence that enhanced LHA Nts neuronal activity counteracts the drive to consume palatable food in an energy-restricted state, similar to what occurs after diet-induced weight loss (Sumithran et al., 2011; van der Plasse et al., 2015).
Aversive stimuli or stress also suppress feeding (Jennings et al., 2015; Stamatakis et al., 2013), thus we investigated whether activation of LHA Nts neurons promotes these conditions. First, mice were tested in conditioned place-preference (CPP) chambers paired with either VEH or CNO. NtsCre;NtsR1++ mice demonstrated neither aversion nor preference for the CNO-paired chamber, but NtsCre;NtsR1KOKO mice significantly preferred the chamber associated with CNO-mediated activation of LHA Nts neurons (Fig. 7A). These behaviors are not attributable to nonspecific CNO effects, as CNO failed to modify place preference in mice expressing ChR in LHA Nts neurons instead of hM3Dq-mCherry (Fig. 7B). Together, these data suggest that activating LHA Nts neurons does not suppress feeding due to aversion, but lack of NtsR1 might positively shift incentive valence and counteract feeding suppression via these neurons. Activation of LHA Nts neurons did not increase anxiety-like behavior as measured via elevated plus maze, thus activation of the neurons does not cause stress-induced suppression of feeding (Fig. 7C–D). Lastly, because we anecdotally observed behaviors such as gnawing, grooming, and digging in CNO-treated mice, we examined if these were directed at procuring food or toward any object, in this case a nestlet placed in the cage. Activation of LHA Nts neurons significantly increased nestlet-shredding in both NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice (Fig. 7e-f), suggesting that LHA Nts neurons increases behaviors directed at objects other than food, which may contribute to their increased activity and energy expenditure (Fig. 1). Collectively, these data indicate that LHA Nts-mediated suppression of food intake is not confounded by aversion or anxiety, and is associated with behaviors directed at objects other than food.
Figure 7. Assessment of reward, anxiety, and repetitive behaviors associated with LHA Nts➔NtsR1 circuit activation.
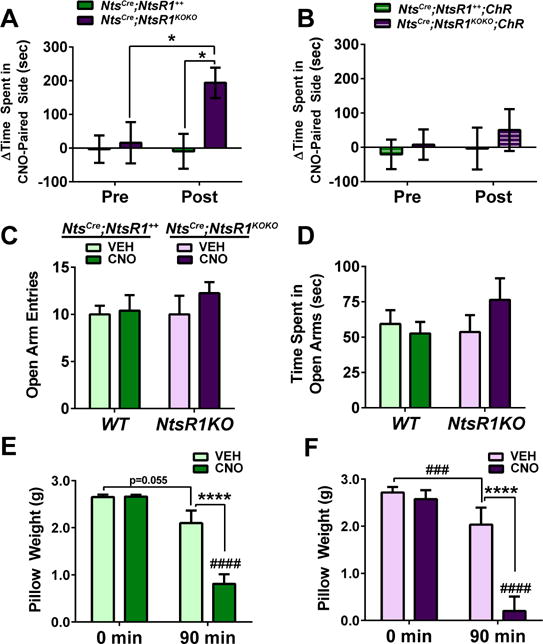
CPP was used assess whether activation of LHA Nts neurons is positively reinforcing or aversive. A) Time-spent in the CNO and VEH-paired sides during the pre- and post-tests reveals that activation of LHA Nts neurons is rewarding in NtsCre;NtsR1KOKO (n=15) but not NtsCre;NtsR1++ (n=17) mice. B) No differences were detected in NtsChR (n=12) and NtsCre;NtsR1KOKO ChR (n=10) controls. Anxiety-like behavior was assessed via EPM and no differences were detected in open arm C) entries or D) time spent (NtsCre;NtsR1+++VEH n=10, NtsCre;NtsR1+++CNO n=10, NtsCre;NtsR1KOKO +VEH n=6, NtsCre;NtsR1KOKO +CNO n=8). E, F) nestlet weight 90 minutes after VEH or CNO injection in NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice (NtsCre;NtsR1+++VEH n=8, NtsCre;NtsR1+++CNO n=10, NtsCre;NtsR1KOKO +VEH n=6, NtsCre;NtsR1KOKO +CNO n=8). Nestlet-shredding and EPM data were analyzed by standard two-way ANOVA while CPP was analyzed by repeated-measures two-way ANOVA to compare pretest to post-test, with Sidak post-tests in both analyses. Graphed data represent mean ± SEM. Significant differences between VEH and CNO-treated mice: *p < 0.05, **p < 0.01, ***p < 0.001. Significant differences between genotypes with the same treatment: ###p < 0.001, ####p < 0.0001.
DISCUSSION
We demonstrate that activation of LHA Nts neurons increases volitional activity and energy expenditure while suppressing food intake, hence promoting dual behaviors to support weight loss. LHA Nts neurons modulate these behaviors via distinct mechanisms: Nts-mediated signaling via NtsR1 is required for reduction of motivated feeding in the face of exercise- or fasting-induced energy deficit, while the locomotor behavior occurs via an NtsR1-independent mechanism. Although LHA Nts neurons cannot suppress intake of freely accessible palatable foods that promote weight gain, they do restrain feeding in fasted, weight-reduced mice that are highly motivated to eat. Taken together, these data suggest that activation of the LHA Nts population might be useful to maintain weight loss in the face of negative energy balance. For example, restraining increased appetite that occurs after an initial bout of weight loss may prevent weight regain, and support further weight loss via continued diet and exercise.
Previous work implicated central Nts in regulating feeding and locomotor activity (Cador et al., 1986; Feifel et al., 2010; Kelley et al., 1989) yet the Nts populations mediating these effects were unknown. Similar to the effects of pharmacologic Nts or NtsR1 agonists in the VTA (Cador et al., 1986; Kalivas et al., 1983; Kelley et al., 1989; St-Gelais et al., 2004), we demonstrate that activation of LHA Nts neurons (known to project to the VTA) increased physical activity while suppressing food intake in energy replete mice. However, because LHA Nts-induced locomotor activity did not depend on intact NtsR1 and was only mildly blunted by D1R antagonism, it is likely controlled by a non-Nts signal released from LHA Nts neurons. By contrast, both acute NtsR1 and D1R antagonism abolished LHA Nts-induced suppression of feeding in NtsCre;NtsR1++ mice, implying obligatory, overlapping roles for Nts and DA in this process. While we observed ad lib feeding and weight loss behavior that differed with cage environment in NtsCre;NtsR1KOKO mice, possibly due to their susceptibility to anxiety or developmental differences and future work employing methods to disrupt NtsR1 signaling in normal adult mice will be helpful to decipher the precise role for NtsR1 in LHA Nts-mediated weight loss behaviors. Since LHA Nts neurons directly engage VTA NtsR1 neurons to promote DA release (Opland et al., 2013; Patterson et al., 2015), it is possible that LHA Nts neurons modulate VTA DA signaling to potentiate weight loss. Going forward it will also be important to determine if LHA Nts-induced NtsR1 signaling may be a specific pathway to engage mesolimbic DA circuits that suppress appetite.
LHA Nts neurons do not provide an absolute brake on feeding, as their activation only suppressed ad lib intake of chow but not HF diet. A possible explanation for this discrepancy is that the palatability of HF diet overrides the ability of LHA Nts neurons to restrain intake. Mechanistically, this suggests that LHA Nts neurons do not modify opioid signaling systems that encode food “liking” that can drive overconsumption (Berridge et al., 2010). LHA Nts neurons are, however, linked with the VTA DA system (Patterson et al., 2015) that modifies the motivation to obtain food rewards (Berridge et al., 2010) and food necessary for survival. For example, the changes in circulating ghrelin and leptin during food-deprivation modulate VTA DA signaling to increase the motivation to eat and restore energy balance (Branch et al., 2013; Larder and O’Rahilly, 2012; Wilson et al., 1995). Anorectic signals thus more profoundly diminish feeding in fasted animals with an elevated drive to eat; for example, exogenous leptin treatment more effectively reduces food intake in fasted vs. fed rodents (Ahima et al., 1996; Sharma et al., 2012). Similarly, LHA Nts neurons robustly suppress fasting-induced chow intake and weight regain, and may in fact contribute to leptin-mediated suppression of motivated feeding (Leinninger et al., 2011). Interestingly, activation of LHA Nts neurons only suppressed DA-dependent operant responding for palatable food in fasted NtsCre;NtsR1++ mice, but not sated mice. Since feeding suppression is more easily detected when levels of food “wanting” are high (as during fasting), our finding is consistent with the possibility that LHA Nts neurons suppress feeding by reducing the “wanting” of food. The failure of LHA Nts neurons to suppress operant palatable intake in the absence of NtsR1, however, supports an essential role for Nts action via NtsR1 in limiting operant-reinforced feeding.
Some LHA Nts-mediated behaviors, such as locomotor activity and drinking, were increased both in NtsCre;NtsR1++ and NtsCre;NtsR1KOKO mice, hence do not depend on NtsR1. NtsCre;NtsR1KOKO mice appeared to have blunted hyperdipsia compared to NtsCre;NtsR1++ mice after acute activation of LHA Nts neurons in metabolic cages (Fig 2A,B and Fig. 3A, D), but this did not reach significance. In contrast, both groups exhibited similar increases in water intake during 5 days of daily activation in home cages (Fig. 4D, H). Since NtsR1KOKO mice are mildly anxious when placed in novel open field environments (Fitzpatrick et al., 2012), it is possible that the modestly-stressful environment of metabolic cages deterred NtsCre;NtsR1KOKO mice from drinking, while this was not apparent in non-anxiety producing home cages. Altogether, we cannot rule out a contribution of NtsR1 in drinking behavior, though it does not appear to be essential for it. Nts may still contribute to locomotor and drinking behaviors via NtsR2, which is predominantly expressed by astrocytes in the VTA (Woodworth et al., 2017)). Alternately, signals other than Nts that are released from LHA Nts neurons might mediate drinking and locomotor activity. For example, some LHA Nts neurons contain GABA (Jennings et al., 2015), which is presumably co-released with Nts. Similar to LHA Nts action, optogenetic activation of LHA GABA neurons increases NA DA release (Nieh et al., 2016), water intake, and non- appetitive gnawing (Navarro et al., 2016; Nieh et al., 2015). Conversely, activation of LHA GABA neurons increases feeding and either does not affect or suppresses locomotor activity (Jennings et al., 2015; Navarro et al., 2016; Nieh et al., 2015), whereas LHA Nts activation suppresses chow feeding and increases locomotor activity. Furthermore, LHA GABA neurons that project to the VTA promote conditioned reward (Nieh et al., 2016) whereas LHA Nts neurons only induce such reward in the absence of NtsR1. These discrepancies suggest that LHA Nts neurons do not fully overlap with the population of LHA GABA neurons, and that perhaps only a subset of LHA Nts neurons are also GABAergic. Within this putative subset of LHA Nts-GABA neurons, the loss of Nts signaling via NtsR1 might bias GABAergic actions, and could explain our observation that activation of LHA Nts neurons in NtsCre;NtsR1KOKO mice causes conditioned place-preference similar to that induced by the activation of LHA GABA neurons. Since hypothalamic Nts is reduced in obesity (Beck et al., 1990), this might also favor GABAergic signaling that potentiates feeding (Jennings et al., 2015; Navarro et al., 2016; Nieh et al., 2015). Collectively, we hypothesize that Nts is co-expressed on subsets of GABAergic LHA neurons, and stimulation of the LHA Nts population as a whole promotes a repertoire of beneficial behaviors that may promote weight loss.
LHA Nts neurons may also differentially control locomotor, drinking and feeding behaviors via distinct projections. Since LHA Nts neurons robustly project to the VTA and SN, where NtsR1 is expressed almost exclusively on DA neurons, the NtsR1-dependent suppression of feeding might be mediated via these LHA Nts afferents. Determining whether VTA-projecting LHA Nts neurons co-release GABA may provide insight into the control of feeding behaviors. For instance, LHA Nts neurons might co-release GABA onto VTA GABA interneurons, thereby disinhibiting DA neurons and promoting DA signaling (Nieh et al., 2016). LHA Nts neurons also project locally and inhibit neighboring hypocretin/orexin neurons (Goforth et al., 2014; Leinninger et al., 2011), which could decrease food intake, especially in a food-restricted state. Our data thus reveal that LHA Nts neurons are important mediators of central anorectic Nts action via NtsR1. Future work will be important to define the additional signaling mechanisms and projections by which LHA Nts neurons support weight loss behaviors.
EXPERIMENTAL PROCEDURES
Animals
Mice were bred and housed in a 12h light/12h dark cycle and cared for by Campus Animal Resources (CAR) at Michigan State University. Animals had ad lib access to chow (Teklad 7913) and water unless otherwise noted. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Michigan State University in accordance with Association for Assessment and Accreditation of Laboratory Animal Care and National Institutes of Health guidelines.
Surgery
At 8–12 weeks of age, male NtsCre;++ and NtsCre:NtsR1KOKO mice underwent stereotaxic surgery (as described in Woodworth et al., 2017), and were injected bilaterally with either 300 uL of AAV-hM3Dq-mCherry or AAV-ChR-mCherry (UNC vector core) into the LHA (AP: −1.34, ML: - 1.05, DV: −5.2) per the mouse brain atlas of Paxinos and Watson. Analysis began 1–2 weeks after recovery. Mice were only included in the final study data if injections were confined to the LHA on both sides. Approximately 90% of animals included in the study were bilaterally targeted; however in 10% of cases, animals with robust unilateral targeting were included in the study if CNO injection induced >1mL of water consumption, as analysis of several cohorts revealed this as a reliable indicator of LHA Nts targeting. To visualize NtsR1 neurons, adult male and female NtsR1Cre;GFP mice were bilaterally injected with 1uL FlpO adenovirus (Vector Biolabs) into the lateral ventricles (A/P: −0.22, M/L: +/− 1.0, D/V: −2.0). Mice were perfused 10 days after surgery to permit sufficient time for FlpO-mediated excision of the frt-flanked Neo cassette and GFP expression.
Metabolic Analysis
At 1–2 weeks post-surgery, mice were placed in TSE cages for metabolic phenotyping (PhenoMaster, TSE Systems). After 24–48 hours of acclimation, mice were i.p. injected 1–2 hr after the onset of the light cycle with PBS, SR48692 (0.3 mg/kg), or SCH23390 (0.1mg/kg), followed by VEH or CNO (0.3 mg/kg) 30 min later. Mice were given a 24 hr washout between PBS and antagonist injections. They were continuously monitored for food and water intake, locomotor activity, and energy expenditure. Ambient temperature was maintained at 20–23°C and the airflow rate through the chambers was adjusted to maintain an oxygen differential around 0.3% at resting conditions. Metabolic parameters including VO2, respiratory exchange ratio, and energy expenditure were assessed via indirect calorimetry by comparing O2 and CO2 concentrations relative to a reference cage.
Chronic Activation in Lean Mice
Chow-Experiments
Mice were injected with VEH or CNO between 8–9AM once daily for five consecutive days in home cages while fed ad lib standard chow (Harlan Teklad 7913). Food, water, and body weight were weighed prior to the morning injection and again between 5–6pm. A cross-over study design was used, thus each animal received both VEH and CNO over the course of two separate experiments, with 24 hr between studies. On day 5, locomotor activity was assessed in CPP chambers described below. Animals were randomly assigned to one side of the chamber and were allowed to acclimate for 30 min. Following acclimation, they received VEH or CNO and locomotor activity was measured by laser beam breaks for 1 hr. HF-Diet Experiments: As above, mice were injected once daily with VEH or CNO between 8–9AM in home cages with ad lib access to high fat, high sugar diet (HF diet, D12451, Research Diets). Mice received 24 hr of access to HF diet prior to the study. On day 5, locomotor activity was assessed as described above, and injections were given for 10 consecutive days to allow time for weight gain. These experiments also used a cross-over design, with 2 wk between experiments during which mice only had access to standard chow.
Activation of LHA Nts Circuit in Obese Mice
Four week old male NtsCre;NtsR1++ mice were weaned onto 45% high fat, high sucrose diet (HF diet, D12451, Research Diets) to induce obesity. After 6–8 months of HF diet consumption, AAV-hM3Dq-mCherry was injected into the LHA as described above. After 2 wk, obese mice were acclimated in TSE metabolic cages for 2 days, then were divided into separate treatment groups and injected with VEH or CNO for 5 consecutive days in TSE cages at 8AM each morning.
Fasting-Induced Re-feeding
Chow was removed from home cages ~5PM and mice were given a clean cage bottom. Mice had ad lib access to water during food-deprivation. The following morning between 8AM–9AM, fasted mice were given ip VEH or CNO and chow pellets were returned to the feeder. Food intake, water intake, and body weight was measured at 1 hr and 24 hr after injection. The study was performed using a cross-over design, such that half the mice received VEH or CNO, and after 3 full days of recovery from fasting, the experiment was repeated with the opposite treatment, allowing each animal to serve as its own control.
Gene Expression, Immunostaining and Other Behavioral Testing
See supplemental information.
Statistics
Student’s t-tests and 2-way ANOVA were calculated using Prism 6 (GraphPad). Repeated measures two-way ANOVA with Sidak post-tests was used when each animal was given both VEH and CNO, and when data from the same animals were collected at different time points. For energy expenditure data, analysis of covariance (ANCOVA) was computed in SPSS 22 (IBM). Body weight was analyzed as a covariate to correct for any inherent differences it may have on metabolism (Tschop et al., 2012). Data were tested for homogeneity of regression, independence of the covariate (body weight), and linearity of regression prior to running the ANCOVA. For all data, *p<0.05, **p<0.01 and ***p<0.001. (#) is used to indicate comparison to starting time line within a given group.
Supplementary Material
Acknowledgments
We thank the University of Michigan Transgenic Animal Model Core and the Van Andel Research Institute Transgenic and Targeting Core for assistance in generating NtsR1IRES-Cre mice, and Sandra O’Reilly for assistance with mouse metabolic phenotyping. We also thank Andrew Eagle, A.J. Robison, and Michelle Mazei-Robison for guidance regarding the conditioned place preference experiments. This research was supported by grants from the NIH to HLW (F30-DK107163) and GML (R01-DK103808).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
HLW and GML designed the experiments and wrote the manuscript. HLW performed the experiments and analyzed the data. BGB and HMB assisted with operant and metabolism experiments. RB and PPB performed gene expression and helped generate obese mice, while LES assisted with perfusions.
References
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- Ahn S, Phillips AG. Dopaminergic correlates of sensory-specific satiety in the medial prefrontal cortex and nucleus accumbens of the rat. J Neurosci. 1999;19:RC29. doi: 10.1523/JNEUROSCI.19-19-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck B, Burlet A, Nicolas JP, Burlet C. Hyperphagia in obesity is associated with a central peptidergic dysregulation in rats. J Nutr. 1990;120:806–811. doi: 10.1093/jn/120.7.806. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Ho CY, Richard JM, DiFeliceantonio AG. The tempted brain eats: pleasure and desire circuits in obesity and eating disorders. Brain Res. 2010;1350:43–64. doi: 10.1016/j.brainres.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boules M, Cusack B, Zhao L, Fauq A, McCormick DJ, Richelson E. A novel neurotensin peptide analog given extracranially decreases food intake and weight in rodents. Brain Res. 2000;865:35–44. doi: 10.1016/s0006-8993(00)02187-9. [DOI] [PubMed] [Google Scholar]
- Branch SY, Goertz RB, Sharpe AL, Pierce J, Roy S, Ko D, Paladini CA, Beckstead MJ. Food restriction increases glutamate receptor-mediated burst firing of dopamine neurons. J Neurosci. 2013;33:13861–13872. doi: 10.1523/JNEUROSCI.5099-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Bugescu R, Mayer T, Gata-Garcia A, Kurt G, Woodworth HL, Leinninger GM. Loss of Action via Neurotensin-Leptin Receptor Neurons Disrupts Leptin and Ghrelin-Mediated Control of Energy Balance. Endocrinology. 2017 doi: 10.1210/en.2017-00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Woodworth HL, Leinninger GM. To ingest or rest? Specialized roles of lateral hypothalamic area neurons in coordinating energy balance. Front Syst Neurosci. 2015;9:9. doi: 10.3389/fnsys.2015.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cador M, Kelley AE, Le Moal M, Stinus L. Ventral tegmental area infusion of substance P, neurotensin and enkephalin: differential effects on feeding behavior. Neuroscience. 1986;18:659–669. doi: 10.1016/0306-4522(86)90061-8. [DOI] [PubMed] [Google Scholar]
- Cooke JH, Patterson M, Patel SR, Smith KL, Ghatei MA, Bloom SR, Murphy KG. Peripheral and central administration of xenin and neurotensin suppress food intake in rodents. Obesity (Silver Spring) 2009;17:1135–1143. doi: 10.1038/oby.2008.652. [DOI] [PubMed] [Google Scholar]
- Ervin GN, Birkemo LS, Nemeroff CB, Prange AJ., Jr Neurotensin blocks certain amphetamine-induced behaviours. Nature. 1981;291:73–76. doi: 10.1038/291073a0. [DOI] [PubMed] [Google Scholar]
- Feifel D, Goldenberg J, Melendez G, Shilling PD. The acute and subchronic effects of a brain-penetrating, neurotensin-1 receptor agonist on feeding, body weight and temperature. Neuropharmacology. 2010;58:195–198. doi: 10.1016/j.neuropharm.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick K, Winrow CJ, Gotter AL, Millstein J, Arbuzova J, Brunner J, Kasarskis A, Vitaterna MH, Renger JJ, Turek FW. Altered sleep and affect in the neurotensin receptor 1 knockout mouse. Sleep. 2012;35:949–956. doi: 10.5665/sleep.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Berod A, Zahm DS, Rostene W. Brain neurotensin, psychostimulants, and stress–emphasis on neuroanatomical substrates. Peptides. 2006;27:2364–2384. doi: 10.1016/j.peptides.2006.03.037. [DOI] [PubMed] [Google Scholar]
- Goforth PB, Leinninger GM, Patterson CM, Satin LS, Myers MG., Jr Leptin acts via lateral hypothalamic area neurotensin neurons to inhibit orexin neurons by multiple GABA-independent mechanisms. J Neurosci. 2014;34:11405–11415. doi: 10.1523/JNEUROSCI.5167-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, Ellis RJ, Richie CT, Harvey BK, Dannals RF, et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science. 2017;357:503–507. doi: 10.1126/science.aan2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins MF. Aphagia in the rat following microinjection of neurotensin into the ventral tegmental area. Life Sci. 1986a;38:2383–2388. doi: 10.1016/0024-3205(86)90606-5. [DOI] [PubMed] [Google Scholar]
- Hawkins MF. Central nervous system neurotensin and feeding. Physiol Behav. 1986b;36:1–8. doi: 10.1016/0031-9384(86)90064-8. [DOI] [PubMed] [Google Scholar]
- Jennings JH, Rizzi G, Stamatakis AM, Ung RL, Stuber GD. The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science. 2013;341:1517–1521. doi: 10.1126/science.1241812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JH, Ung RL, Resendez SL, Stamatakis AM, Taylor JG, Huang J, Veleta K, Kantak PA, Aita M, Shilling-Scrivo K, et al. Visualizing hypothalamic network dynamics for appetitive and consummatory behaviors. Cell. 2015;160:516–527. doi: 10.1016/j.cell.2014.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci. 2010;13:635–641. doi: 10.1038/nn.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Burgess SK, Nemeroff CB, Prange AJ., Jr Behavioral and neurochemical effects of neurotensin microinjection into the ventral tegmental area of the rat. Neuroscience. 1983;8:495–505. doi: 10.1016/0306-4522(83)90195-1. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Nemeroff CB, Prange AJ., Jr Neurotensin microinjection into the nucleus accumbens antagonizes dopamine-induced increase in locomotion and rearing. Neuroscience. 1984;11:919–930. doi: 10.1016/0306-4522(84)90203-3. [DOI] [PubMed] [Google Scholar]
- Kelley AE, Cador M, Stinus L, Le Moal M. Neurotensin, substance P, neurokinin-alpha, and enkephalin: injection into ventral tegmental area in the rat produces differential effects on operant responding. Psychopharmacology (Berl) 1989;97:243–252. doi: 10.1007/BF00442258. [DOI] [PubMed] [Google Scholar]
- Khera R, Murad MH, Chandar AK, Dulai PS, Wang Z, Prokop LJ, Loomba R, Camilleri M, Singh S. Association of Pharmacological Treatments for Obesity With Weight Loss and Adverse Events: A Systematic Review and Meta-analysis. JAMA. 2016;315:2424–2434. doi: 10.1001/jama.2016.7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ER, Leckstrom A, Mizuno TM. Impaired anorectic effect of leptin in neurotensin receptor 1-deficient mice. Behav Brain Res. 2008;194:66–71. doi: 10.1016/j.bbr.2008.06.024. [DOI] [PubMed] [Google Scholar]
- Larder R, O’Rahilly S. Shedding pounds after going under the knife: guts over glory-why diets fail. Nat Med. 2012;18:666–667. doi: 10.1038/nm.2747. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Opland DM, Jo YH, Faouzi M, Christensen L, Cappellucci LA, Rhodes CJ, Gnegy ME, Becker JB, Pothos EN, et al. Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell Metab. 2011;14:313–323. doi: 10.1016/j.cmet.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttinger D, King RA, Sheppard D, Strupp J, Nemeroff CB, Prange AJ., Jr The effect of neurotensin on food consumption in the rat. Eur J Pharmacol. 1982;81:499–503. doi: 10.1016/0014-2999(82)90116-9. [DOI] [PubMed] [Google Scholar]
- Navarro M, Olney JJ, Burnham NW, Mazzone CM, Lowery-Gionta EG, Pleil KE, Kash TL, Thiele TE. Lateral Hypothalamus GABAergic Neurons Modulate Consummatory Behaviors Regardless of the Caloric Content or Biological Relevance of the Consumed Stimuli. Neuropsychopharmacology. 2016;41:1505–1512. doi: 10.1038/npp.2015.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieh EH, Matthews GA, Allsop SA, Presbrey KN, Leppla CA, Wichmann R, Neve R, Wildes CP, Tye KM. Decoding neural circuits that control compulsive sucrose seeking. Cell. 2015;160:528–541. doi: 10.1016/j.cell.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieh EH, Vander Weele CM, Matthews GA, Presbrey KN, Wichmann R, Leppla CA, Izadmehr EM, Tye KM. Inhibitory Input from the Lateral Hypothalamus to the Ventral Tegmental Area Disinhibits Dopamine Neurons and Promotes Behavioral Activation. Neuron. 2016;90:1286–1298. doi: 10.1016/j.neuron.2016.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opland D, Sutton A, Woodworth H, Brown J, Bugescu R, Garcia A, Christensen L, Rhodes C, Myers M, Jr, Leinninger G. Loss of neurotensin receptor-1 disrupts the control of the mesolimbic dopamine system by leptin and promotes hedonic feeding and obesity. Mol Metab. 2013;2:423–434. doi: 10.1016/j.molmet.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson CM, Wong JM, Leinninger GM, Allison MB, Mabrouk OS, Kasper CL, Gonzalez IE, Mackenzie A, Jones JC, Kennedy RT, et al. Ventral tegmental area neurotensin signaling links the lateral hypothalamus to locomotor activity and striatal dopamine efflux in male mice. Endocrinology. 2015;156:1692–1700. doi: 10.1210/en.2014-1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remaury A, Vita N, Gendreau S, Jung M, Arnone M, Poncelet M, Culouscou JM, Le Fur G, Soubrie P, Caput D, et al. Targeted inactivation of the neurotensin type 1 receptor reveals its role in body temperature control and feeding behavior but not in analgesia. Brain Res. 2002;953:63–72. doi: 10.1016/s0006-8993(02)03271-7. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012;76:470–485. doi: 10.1016/j.neuron.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone JD, Correa M, Farrar A, Mingote SM. Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology (Berl) 2007;191:461–482. doi: 10.1007/s00213-006-0668-9. [DOI] [PubMed] [Google Scholar]
- Sharma S, Hryhorczuk C, Fulton S. Progressive-ratio responding for palatable high-fat and high-sugar food in mice. J Vis Exp. 2012:e3754. doi: 10.3791/3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Gelais F, Legault M, Bourque MJ, Rompre PP, Trudeau LE. Role of calcium in neurotensin-evoked enhancement in firing in mesencephalic dopamine neurons. J Neurosci. 2004;24:2566–2574. doi: 10.1523/JNEUROSCI.5376-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis AM, Jennings JH, Ung RL, Blair GA, Weinberg RJ, Neve RL, Boyce F, Mattis J, Ramakrishnan C, Deisseroth K, et al. A unique population of ventral tegmental area neurons inhibits the lateral habenula to promote reward. Neuron. 2013;80:1039–1053. doi: 10.1016/j.neuron.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley BG, Hoebel BG, Leibowitz SF. Neurotensin: effects of hypothalamic and intravenous injections on eating and drinking in rats. Peptides. 1983;4:493–500. doi: 10.1016/0196-9781(83)90054-2. [DOI] [PubMed] [Google Scholar]
- Stice E, Spoor S, Bohon C, Small DM. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science. 2008;322:449–452. doi: 10.1126/science.1161550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumithran P, Prendergast LA, Delbridge E, Purcell K, Shulkes A, Kriketos A, Proietto J. Long-term persistence of hormonal adaptations to weight loss. N Engl J Med. 2011;365:1597–1604. doi: 10.1056/NEJMoa1105816. [DOI] [PubMed] [Google Scholar]
- Tschop MH, Speakman JR, Arch JR, Auwerx J, Bruning JC, Chan L, Eckel RH, Farese RV, Jr, Galgani JE, Hambly C, et al. A guide to analysis of mouse energy metabolism. Nat Methods. 2012;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Plasse G, van Zessen R, Luijendijk MC, Erkan H, Stuber GD, Ramakers GM, Adan RA. Modulation of cue-induced firing of ventral tegmental area dopamine neurons by leptin and ghrelin. Int J Obes (Lond) 2015;39:1742–1749. doi: 10.1038/ijo.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AG, Sanchez-Watts G. Rapid and preferential activation of Fos protein in hypocretin/orexin neurons following the reversal of dehydration-anorexia. J Comp Neurol. 2007;502:768–782. doi: 10.1002/cne.21316. [DOI] [PubMed] [Google Scholar]
- Wilson C, Nomikos GG, Collu M, Fibiger HC. Dopaminergic correlates of motivated behavior: importance of drive. J Neurosci. 1995;15:5169–5178. doi: 10.1523/JNEUROSCI.15-07-05169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing RR, Hill JO. Successful weight loss maintenance. Annu Rev Nutr. 2001;21:323–341. doi: 10.1146/annurev.nutr.21.1.323. [DOI] [PubMed] [Google Scholar]
- Woodworth HL, Batchelor HM, Beekly BG, Bugescu R, Brown JA, Kurt G, Leinninger GM. Neurotensin receptor-1 identifies a subset of ventral tegmental dopamine neurons that coordinate energy balance. Cell Rep. 2017;20:1881–1892. doi: 10.1016/j.celrep.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.