

Abstract
Our understanding of the world of biomolecular structures is based upon the interpretation of macromolecular models, of which ∼90% are an interpretation of electron density maps. This structural information guides scientific progress and exploration in many biomedical disciplines. The Protein Data Bank's web portals have made these structures available for mass scientific consumption and greatly broaden the scope of information presented in scientific publications. The portals provide numerous quality metrics; however, the portion of the structure that is most vital for interpretation of the function may have the most difficult to interpret electron density and this ambiguity is not reflected by any single metric. The possible consequences of basing research on suboptimal models make it imperative to inspect the agreement of a model with its experimental evidence. Molstack, a web‐based interactive publishing platform for structural data, allows users to present density maps and structural models by displaying a collection of maps and models, including different interpretation of one's own data, re‐refinements, and corrections of existing structures. Molstack organizes the sharing and dissemination of these structural models along with their experimental evidence as an interactive session. Molstack was designed with three groups of users in mind; researchers can present the evidence of their interpretation, reviewers and readers can independently judge the experimental evidence of the authors' conclusions, and other researchers can present or even publish their new hypotheses in the context of prior results. The server is available at http://molstack.bioreproducibility.org.
Keywords: macromolecular crystallography, electron density interpretation, reproducibility, structural biology, web server
Introduction
Both the most prominent (X‐ray crystallography) and the fastest growing (cryo‐EM) techniques for determining the atomic structures of macromolecules generate electron density or Coulomb potential maps (respectively) which must be interpreted to generate a structural model. Macromolecular crystallography remains the most widely used technique for determining atomic structures of proteins and their complexes, which ultimately expands our understanding of biological processes in a unique and invaluable way. Extensive efforts have resulted in easy‐to‐use, sophisticated software that aids researchers during most of the major phases in processing crystallographic data: data reduction, structure determination, model building, and refinement.1, 2, 3, 4, 5, 6, 7 These semiautomated software pipelines have not only made crystallography as a tool accessible to researchers from many fields, but also enabled a surge in the number of high‐quality structures from individual labs and structural genomics efforts alike.8 The difficult and sometimes most time‐consuming step in structural analysis of the crystallographic data is the iterative model alteration and validation process. Recent advances in cryo‐EM have greatly increased the rate of structure determination at relatively low resolutions, which increases the level of uncertainty and ambiguity in structure refinement/validation and renders structure interpretation more challenging.
The Protein Data Bank (PDB)9 is not only a repository of atomic coordinates, but serves as a portal for the macromolecular community who subsequently use structural information to design individual experiments and research projects.10 The increasing number of structures has promoted the development of new structural bioinformatics and data mining algorithms, which have resulted in knowledge‐based refinement and validation methods that have improved the quality of subsequent structures. As a result, the average quality of structures deposited in the PDB is steadily improving over time11 (Fig. 1). Some of these methods for structure validation have been standardized and applied to the entire PDB repository.12, 13 Many journals have implemented policies requiring that the PDBs structural validation reports be provided to reviewers,14 although reviewers are not usually provided with models and corresponding experimental data. Moreover, old structures are not updated to reflect changes in validation standards, and occasionally even new deposits are of suboptimal quality in terms of model correctness and model‐to‐data correspondence.15, 16 Furthermore, even those who are aware that the structures in PDB contain local errors do not have access to an easy‐to‐use tool that will permit them to verify the integrity of specific regions of structures.
Figure 1.
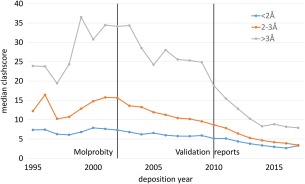
The quality of the structures in the PDB coincides with the introduction of modern validation tools and change in mandatory policies. The quality metrics of structures (as represented by clashscore calculated with MolProbity) have evolved over time. Since the introduction of MolProbity, the quality of high (blue), medium (orange) and low (gray) resolution structures have steadily improved year‐to‐year. However, the largest improvement in overall quality for low resolution structures can be observed for 2010 when the validation reports introduced in 2009 were made mandatory by journals for structural publication. Although the improvement of the structural quality is likely due to improvement of many computer programs used in macromolecular crystallography and increased awareness and experience of the community, this coincidence nicely illustrates evolution of the quality of the structures in context of Validation software and policy development.
Structural information provides the basis for scientific hypotheses regarding the function of macromolecules, and for that reason, any mistake may have tremendous ripple effect.17 For researchers interested in particular macromolecules, whether they are involved in functional, structural, or computational research, it is important that the feature that interests them is accurately modeled. Therefore, it is imperative for consumers of the PDB to validate the overall and local quality of all relevant structures early in the analysis process to reduce potential future frustration. For example, the diffraction data may not support the interpretation that a protein binds the ligand modeled into its structure. Other errors of structural interpretation are errors in ligand identification,15 peptide modelling18 and misinterpretation of metal ions, which can be difficult to correctly identify and to accurately model.19 In principle, some validation reports provide metrics designed to identify regions of poorly modeled density, such as local ligand density fit (LLDF), real‐space R‐value Z‐score (RSRZ), and clash score parameters. While they provide an indication that a region is suboptimally modeled, the nature of the potential problem is still undisclosed. In addition, some of these metrics are not necessarily calculated with high reliability (Fig. 2).
Figure 2.
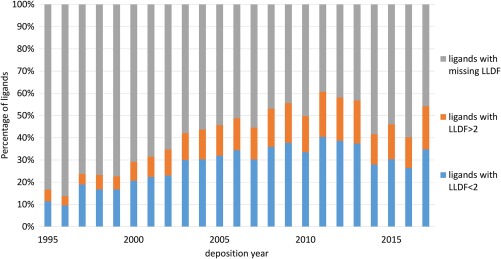
The presence of LLDF parameters for all ligands in the PDB. A large portion of ligands in the PDB does not have LLDF calculated (shown in gray, ligands with missing LLDF) and therefore LLDF cannot be used as a quality metric for those ligands.
Researchers conducting data mining experiments face similar problems, only on a larger scale. Low quality model fragments increase the noise within datasets and can complicate tasks like classification,20 derivation of expected distributions,21 and machine learning. For example, one of the training sets for ligand docking to proteins contained models that were not well supported by experimental data, which may have reduced the accuracy of computational ligand screening based on these sets.22 The presence of “bad apples” necessitates creating strict filtering criteria for methods development and selection of high‐quality reference structures.23 Overall structure quality metrics, such as R factors, resolution, RMSD, etc. are not always sufficient for this type of research. It is imperative, therefore, that the quality of the PDB be maintained by carefully reviewing the structures and not relying entirely on post‐factum automatic re‐refinements like PDB_REDO.
The uses of protein structure validation methods generally fall into two categories: those that are used by crystallographers while modeling/refining the structure, and those that report structure metrics to the general public after the structure has been released by the PDB. Validation methods used during structure modeling and refinement help crystallographers interpret the experimental data in the most consistent and reproducible manner currently available. The methods that are applied to improve the quality of the models include validation methods that spot the problems during refinement24, 25 and methods that attempt to eliminate problems automatically during structure refinement.26, 27 This is in contrast to general validation methods, which are primarily passive reports about overall quality mostly limited to mandatory validation carried out by PDB.13 General validation methods will check for errors/inconsistencies, yet will not attempt to substantially update a model or, more importantly, will not answer the question of whether a model of suboptimal quality is the result of noisy, poor resolution electron density maps and/or a poor interpretation of them. For example, a low RSRZ shows that the data do not justify the local conformation of a model, but does not necessarily answer the question about how (or whether) the model may be improved.
The iterative process of using interactive validation tools to guide manual changes is now deeply entrenched in the crystallographic mindset. Various tools have been integrated into different pipelines and different software, but these are not necessarily tightly integrated. The usual approach is to iteratively repeat the process of manual model building, refinement, validation, and model rebuilding to satisfy different validation metrics. The quality of the structures produced by this process greatly depends on the existing model building and validation technology that assists the researcher in interpreting the structure. Refining structural models to produce high quality structures is often a tedious task, which seems to obey the 90–90 rule from computer programing: “The first 90% of the code accounts for the first 90% of the development time. The remaining 10% of the code accounts for the other 90% of the development time.”28 That is why even structures deposited by the most experienced crystallographers may sometimes contain suboptimal fragments that may affect the interpretation. Additionally, in general, software improvements are not easily applied to older structures, and therefore only newly determined structures benefit from new software developments (Fig. 1).
The best way to ensure that the region of interest is of the best quality is not only to inspect the validation reports, but to examine the model in the context of the electron density and to evaluate whether the region is optimal, taking into account current knowledge, tools, and experience.
The community has already benefited from the mandatory deposition of structure factors17 and would likely benefit from deposition of raw experimental data (metadata and diffraction images).10 Thanks to these raw data, most macromolecular models can be re‐reduced and re‐refined, and in questionable cases, can be reinterpreted globally or locally.29 This is usually done by individual researchers on a structure‐by‐structure basis. In most cases, both of the approaches require specialized software for re‐interpretation.
Considering the decreasing cost per unit of computational power and advancements in automated model building, it seems plausible in the near future to create a structural repository that will provide structural models that are semiautomatically and continuously improved using state‐of‐the‐art software. Automated model building and rebuilding may ensure that in the future all the models are interpreted in an uniform (standardized) manner and that any ambiguity (such as disorder) of the models reflects the inherent dynamic properties of the studied system, rather than the imagination (or lack thereof) of the person interpreting experimental data. PDB_REDO30 is an excellent example how experimental data can be improved just by taking advantage of software improvements that have occurred since the original structure determination. However, automatic re‐refinement algorithms like PDB_REDO are not yet powerful enough to reinterpret ligand‐protein complexes.22 In any case, the final assessment of the model validity should be done on a case‐by‐case basis, regardless of the level of automation of data interpretation.
Summarizing, the majority of structural models are interpretations of the electron density maps. Due to various software limitations, the quality of data, and human expectation bias, some regions may be suboptimally interpreted. Some of the inconsistencies will likely be flagged by validation software, but many will remain unnoticed. While experienced crystallographers are able to remediate the problems using specialized software, many users do not have access to these tools. Therefore, researchers and reviewers that use and evaluate structural data should expect strong experimental evidence to support the interpreted model and any conclusions based on it. Traditionally such evidence has been provided in the form of static figures that show appropriate electron density maps that substantiate the claim. However, the authors' assertions can be put under scrutiny only after the model and experimental data are released to the public, which is usually done after publication. In many cases, the release of experimental data has led to models being examined by interested parties and specific claims being called into question, sometimes resulting in the retraction of publications, sometimes not. In any case, the publication of a re‐refined, corrected model is sometimes a complicated ordeal.17 In addition, it has also been argued by some authors that the different interpretations stem from the inability of the researchers to reproduce the same electron density maps that were used by the original authors.31
In our opinion, the process of “self‐correction” in structural biology can be significantly improved by enhancing two elements: presentation of the experimental evidence, and the possibility to present different interpretations based on the same experimental data. We hope that our new server Molstack will be used to enhance these two elements.
Results
Molstack is a cloud‐based tool that allows the visualization and analysis of multiple sets of coordinate and electron density data in stacks in dual, synchronized side‐by‐side windows for easy comparison. Each stack can be used to present and analyze various data, including (1) atomic coordinates, 2mFc‐DFo maps, and an omit map for small molecule ligands and its surrounding; (2) alternative ligands or ligands in alternative conformations that may be used to interpret the experimental evidences for the same site; (3) a collection of structures such as structures with different ligand, mutants of the same protein, or homologous structures from different organisms; (4) re‐interpretation, re‐refinements, or corrections of existing structures; or (5) supporting data like anomalous difference maps. Molstack organizes the sharing and dissemination of these structural models along with their experimental evidence as an interactive session.
The technologies we have used allow interactive scenes, including electron density maps, to display in modern desktop and mobile browsers. The stack author can predefine views, but users can manually navigate the scene using controls similar to those used by COOT.32
Molstack operates in two distinct modes, viewing and editing. Anyone can view publicly available projects, but users who wish to create stacks must register and log in. The editing mode comprises two components: creating stacks (Fig. 3) and annotating views (Fig. 4). As mentioned above, we define a stack as a collection of maps and models that will be displayed together. More than one view can be created to focus attention on different areas within a stack.
Figure 3.
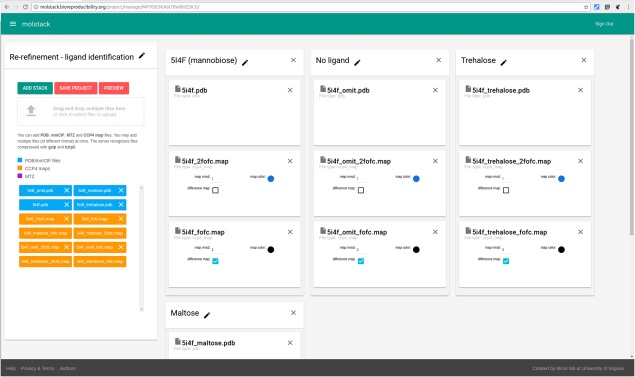
Project editing—creating stacks. The interface allows upload of multiple files (left panel) that can be assigned to stacks (center part).
Figure 4.
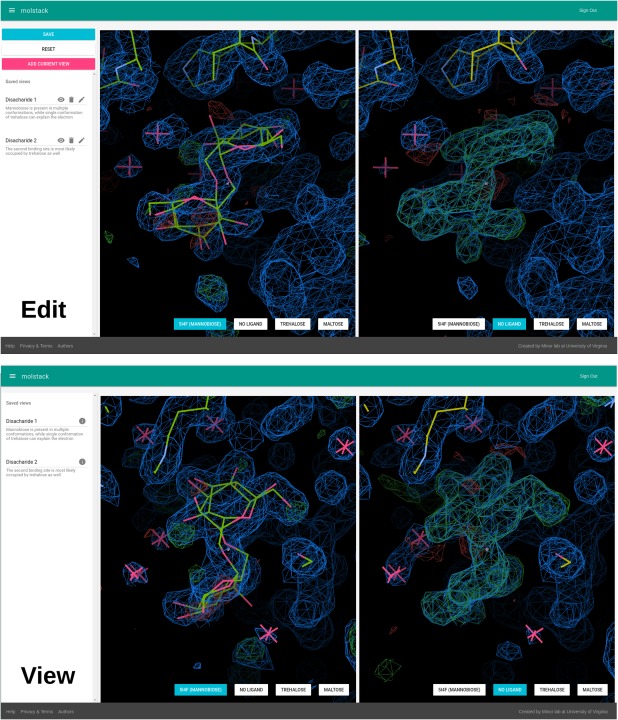
Project editing—creating views and project viewing. Using the preview mode user can view the stacks and navigate the scenes. The particular view can be then saved and a name and description of the view can be added using the left panel. These views will be then accessible to other users when viewing the project.
The first step of creating a stack is to upload the necessary files, which can be accomplished by dragging and dropping the files into the file “drop zone” or by browsing for the selected file. Molstack currently supports uploading models in PDB format, structure factors in MTZ format, and maps in CCP4 format. Uploaded files will appear in the available file list on the left. The user can create multiple stacks (currently up to six, but each stack can display multiple files) by dragging and dropping files from the available file zone to a respective stack. The structural models will be displayed as uploaded. The user may specify the color of the map, default map RMSD (sigma) level, and indicate whether the map should be treated as a difference map (both positive and negative densities will be displayed). When an MTZ file is added to the stack, a map creation dialog is opened with radio buttons allowing the stack author to select the amplitudes and phases that should be used for Fourier synthesis. Anomalous amplitudes (DANO) can be used to generate anomalous difference maps. Each stack can be given a descriptive name and annotated with a short description that will be displayed together with the scenes by clicking the edit icon (a pencil).
The second element of editing a project is annotating the views. Once the components of the stacks are defined, the user can preview the resulting scenes, select views to display, and add annotations to them. The user can navigate to any part of the model and map and save the particular view using the “Add Current View” button. The views can be used to highlight particular fragments of interest, such as ligands, binding sites, or unusual features. Names and descriptions of the views can be added by clicking the edit icon.
The viewing mode is used to present a project to other users (Fig. 4). It has a simplified interface compared to the project editing preview. The view displays two synchronized scenes that can be switched to a different stack using buttons at the bottom of each scene. The description of each stack can be displayed after hovering over the name of a stack. On the left side of the display there is a list of views that were predefined by the author of the project. The user can click on a view name or its description—that will switch both views to a predefined position. It has to be noted that we decided not to tie a particular stack with a particular view, as this provides greater interactivity. The user can select a view and a stack at their discretion, analyze the descriptions, and make their own conclusions about the presented models and data.
Accounts, Security, and Data Privacy
To create a new project, a user needs to create an account. The account can be created using an email address, or by using existing ORCID or LinkedIn accounts to authenticate.
By default, each project is assigned with a randomized link that can be used to share the resulting presentation with the public. In the current version of Molstack, users have the opportunity to control if the project is public (accessible by everyone). Until it is made public it will remain accessible only to the owner of the project. Once made public the project is permanent—it cannot be removed. This allows for persistence of the published projects, which makes it possible to assign a DOI. Currently published projects do not have a DOI assigned but this feature will be implemented in the future.
Currently each project that is made public is displayed on the list of public projects. In the near future Molstack will implement “semi‐private” projects, which will be accessible by only those with a link to the specific project. The random nature of the link will make possible to share a project (presentation) with collaborators, journals, or reviewers without exposing the project to the general public. This link sharing mechanism is similar to those implemented in Dropbox, Google Drive, or OneDrive.
Discussion
Applications and examples
Molstack was designed to be a flexible tool for presenting and comparing structural models and various types of density maps; therefore, no assumptions are made about the coordinate and electron density data used to create stacks. We do, however, have several future applications of Molstack in mind, and have designed the core of Molstack to accommodate these enhancements or to serve as a module that could be utilized by others. Currently, Molstack is optimized to show the structural consequences of altering some parameters. One of the most important applications is the presentation of a model in the presence of various maps. For example, a ligand and its binding environment can be shown in the presence of an omit map for that ligand. This is a crucial comparison for evaluating a ligand's reliability, but usually not readily available to peer reviewers and almost never shown in the main text of publications in high impact journals. Traditionally, if such evidence is presented in a manuscript, it is in the form of a two‐dimensional figure of one view of the ligand in question.
Molstack can present a stack of the model with refined density side by side with a stack containing the omit map with an initial view that encompasses the ligand in question. The 2mFo‐DFc and mFo‐DFc electron density maps calculated with a ligand present in the model will be displayed as one stack on one side while 2mFo‐DFc and mFo‐DFc “omit maps” will be displayed side‐by‐side on the other side. This approach not only creates a more interactive experience for the reader, but also allows the authors to display the actual maps that they were looking at when they proposed their model. This is important because even with all the data submitted to the PDB and data archives such as proteindiffraction.org,33 maps generated subsequently will be biased by the choice of software and various other parameters. Choices about refinement steps and parameters are almost never accessible. Molstack allows authors to present the maps they based their decisions on. Moreover, Molstack permits authors to present information by a means that is not possible in a two‐dimensional environment, such as anomalous difference maps, Polder maps,34 or kick maps,35 etc.
Additionally, Molstack provides a means to present less “formal” map interpretations than the models deposited to the PDB. For example, one can provide several interpretations (in forms of different stacks) of density that are likely to correspond to a ligand bound during the purification process, but cannot reliably be identified as a single compound. Molstack would also be an appropriate platform to display ligands that are “negative hypotheses,” or to show how multiple conformations could account for a disordered fragment. Moreover, Molstack is a practical tool to demonstrate ligand disintegration resulting from radiation decay during an experiment.
The ability to display multiple stacks makes Molstack a platform well suited to present different re‐refinement and re‐interpretation efforts. The ability to present several models (original interpretation, new interpretations, negative interpretations, etc.) and supporting maps simplifies the visualization of disputed regions, making it easier for others to make their own informed decisions. All the potential alternative hypotheses mentioned above are caused by the nature of potential ambiguity in the interpretation of electron density maps and the choices made when generating the maps. Publication of multiple stacks in Molstack can present such potential alternative interpretations of experimental evidence more flexibly than the single interpretation allowed in a PDB deposit.
The capability of visualizing multiple structures also makes Molstack a good tool to compare related structures. For example, Molstack can present several structures of the same protein with different ligands or several different protein mutants. Future versions will allow the comparison of homologous proteins from different organisms without manually superposing them, allowing one to visualize and analyze structural commonalities and differences between them. Moreover, all models can be displayed along with the electron density maps as experimental evidences for the analyzed features.
Finally, Molstack can allow peer reviewers access to the most critical parts of structure when the entire PDB deposit is still not disclosed. Project authors have the possibility to upload only a fragment of a structure and corresponding region of a map.
To demonstrate these applications, we have created four example Molstack projects: re‐refinement of a ligand (Wlodawer et al, to be published) (http://molstack.bioreproducibility.org/project/view/MPYO83KA6I78W8HZSIC0/), identification of the type of a metal ion based on anomalous difference maps36 (http://molstack.bioreproducibility.org/project/view/LBJOZYOTCIWN8DBL8JDI/), comparison of an apo‐enzyme with its complexes with cofactor and substrate analogs37 (http://molstack.bioreproducibility.org/project/view/JJISBFEC2K6OU51GOETK/) and identification of the unexpected ligand (http://molstack.bioreproducibility.org/project/view/1AG4O774JD7Y1SAFQ9CR/).
Conclusions
In our experience, the interpretation of electron density regions that correspond to well‐ordered fragments of macromolecule is relatively straightforward. These regions can be interpreted by almost anyone given the proper tools (such as COOT32 and KING38) and some simple training. These observations are similar to the experience from FoldIt,39 where the complex scientific problem of protein folding has been re‐framed as a puzzle game.40 This shows that even problems that were once hard to solve, can be handled by virtually anyone with the proper tools. On the other hand, ligand binding sites can be dynamic or exhibit static disorder, and therefore require more experience and insight. These regions are also the fragments least likely to be modeled (or modeled well) by any of the sophisticated programs that have removed the tedium of manually tracing well‐ordered electron density.41, 42 As a result, the interpretation of such regions may be more subjective.
These regions are often the very regions of highest interest to individual academic researchers and industry alike. Avenues of research are sometimes based on unique structures, and an unexpected binding mode or binding site is certainly unique for a ligand. In the case of structural based drug discovery, many conclusions are drawn and hypotheses formed based on small portions of a structure.
Additionally, crystallographers have never held the PDB deposits up as infallible because they know that this collection is imperfect and contains some suboptimally modeled fragments. Moreover, models themselves are just the interpretation of the underlying experimental density that can quite often have alternative interpretations. Therefore, many researchers regularly validate and reinterpret previous data aiding in the process of “self‐curation” of the science.
Unfortunately, it has been inherently difficult to present additional experimental evidence, different hypotheses and alternative interpretations of data. We hope that Molstack will ease the burden of presenting this information and allow for better, more reproducible science by streamlining the presentation of various maps and their interpretations.
The current version of Molstack can be accessed via http://molstack.bioreproducibility.org. Apart from the applications as a cloud‐based tool discussed above, we also see the potential use of Molstack in other projects that will benefit from comparison of different maps and their interpretations—for example, examining structural variability, evaluating radiation damage, or comparing maps after automated reprocessing. We are sure that the flexible nature of Molstack will allow the community to envision many more applications than are presented here.
Materials and Methods
The server has been implemented using the Django framework with the PostgreSQL database backend. The user authentication with external services (ORCID and LinkedIn) is based on the OAuth2 protocol. Electron density maps are generated from structure factors using FFT program from CCP4 using grid sampled at 1/2.5 of the maximum resolution.
The interactive parts of the website (project editing and viewing) are build using the React JavaScript framework. The current version uses the Uglymol viewer (https://uglymol.github.io/) for displaying models and electron densities, but Molstack is designed to be flexible and will accommodate different viewers in the future. We plan to add an NGL Viewer43 as an alternative visualization tool.
Molstack has been tested on current versions of Google Chrome (on Linux, Windows, macOS, and Android), Mozilla Firefox (on Linux, Windows, MacOS), Microsoft Edge (on Windows), and Apple Safari (on MacOS).
Acknowledgments
We thank Matthew Zimmerman and Ivan Shabalin for valuable discussion.
References
- 1. Kabsch W (2010) Xds. Acta Crystallogr Sect D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 3. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr Sect D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adams PD, Baker D, Brunger AT, Das R, DiMaio F, Read RJ, Richardson DC, Richardson JS, Terwilliger TC (2013) Advances, interactions, and future developments in the CNS, Phenix, and Rosetta structural biology software systems. Annu Rev Biophys 42:265–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA (2005) Auto‐rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X‐ray diffraction experiment. Acta Crystallogr Sect D 61:449–457. [DOI] [PubMed] [Google Scholar]
- 6. Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr Sect A 64:112–122. [DOI] [PubMed] [Google Scholar]
- 7. Minor W, Cymborowski M, Otwinowski Z, Chruszcz M (2006) HKL‐3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr Sect D 62:859–866. [DOI] [PubMed] [Google Scholar]
- 8. Grabowski M, Niedzialkowska E, Zimmerman MD, Minor W (2016) The impact of structural genomics: the first quindecennial. J Struct Funct Genomics 17:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rupp B, Wlodawer A, Minor W, Helliwell JR, Jaskolski M (2016) Correcting the record of structural publications requires joint effort of the community and journal editors. FEBS J 283:4452–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shao C, Yang H, Westbrook JD, Young JY, Zardecki C, Burley SK (2017) Multivariate analyses of quality metrics for crystal structures in the PDB archive. Structure 25:458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Read RJ, Adams PD, Arendall WB, III , Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lutteke T, Otwinowski Z, Perrakis A, Richardson JS, Sheffler WH, Smith JL, Tickle IJ, Vriend G, Zwart PH (2011) A new generation of crystallographic validation tools for the protein data bank. Structure 19:1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gore S, Velankar S, Kleywegt GJ (2012) Implementing an X‐ray validation pipeline for the Protein Data Bank. Acta Crystallogr Sect D 68:478–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baker EN, Dauter Z, Einspahr H, Weiss MS (2010) In defence of our science ‐ validation now! Acta Crystallogr Sect F 66:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weichenberger CX, Pozharski E, Rupp B (2013) Visualizing ligand molecules in Twilight electron density. Acta Crystallogr Sect F69:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cooper DR, Porebski PJ, Chruszcz M, Minor W (2011) X‐ray crystallography: assessment and validation of protein‐small molecule complexes for drug discovery. Expert Opin Drug Discov 6:771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Minor W, Dauter Z, Helliwell JR, Jaskolski M, Wlodawer A (2016) Safeguarding structural data repositories against bad apples. Structure 24:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weichenberger CX, Pozharski E, Rupp B (2017) Twilight reloaded: the peptide experience. Acta Crystallogr Sect D 73:211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng H, Chruszcz M, Lasota P, Lebioda L, Minor W (2008) Data mining of metal ion environments present in protein structures. J Inorg Biochem 102:1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zheng H, Shabalin IG, Handing KB, Bujnicki JM, Minor W (2015) Magnesium‐binding architectures in RNA crystal structures: validation, binding preferences, classification and motif detection. Nucleic Acids Res 43:3789–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lovell SC, Davis IW, Arendall WB, 3rd , de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins 50:437–450. [DOI] [PubMed] [Google Scholar]
- 22. Chruszcz M, Domagalski M, Osinski T, Wlodawer A, Minor W (2010) Unmet challenges of structural genomics. Curr Opin Struct Biol 20:587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leaver‐Fay A, O'Meara MJ, Tyka M, Jacak R, Song Y, Kellogg EH, Thompson J, Davis IW, Pache RA, Lyskov S, Gray JJ, Kortemme T, Richardson JS, Havranek JJ, Snoeyink J, Baker D, Kuhlman B (2013) Scientific benchmarks for guiding macromolecular energy function improvement. Methods Enzymol 523:109–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cymborowski M, Klimecka M, Chruszcz M, Zimmerman MD, Shumilin IA, Borek D, Lazarski K, Joachimiak A, Otwinowski Z, Anderson W, Minor W (2010) To automate or not to automate: this is the question. J Struct Funct Genomics 11:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Urzhumtseva L, Afonine PV, Adams PD, Urzhumtsev A (2009) Crystallographic model quality at a glance. Acta Crystallogr Sect D 65:297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bell JA, Ho KL, Farid R (2012) Significant reduction in errors associated with nonbonded contacts in protein crystal structures: automated all‐atom refinement with PrimeX. Acta Crystallogr Sect D 68:935–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Headd JJ, Immormino RM, Keedy DA, Emsley P, Richardson DC, Richardson JS (2009) Autofix for backward‐fit sidechains: using MolProbity and real‐space refinement to put misfits in their place. J Struct Funct Genomics 10:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bentley J (1985) Programmimg pearls. Commun ACM 28:896–901. [Google Scholar]
- 29. Shabalin I, Dauter Z, Jaskolski M, Minor W, Wlodawer A (2015) Crystallography and chemistry should always go together: a cautionary tale of protein complexes with cisplatin and carboplatin. Acta Crystallogr Sect D 71:1965–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Joosten RP, Salzemann J, Bloch V, Stockinger H, Berglund AC, Blanchet C, Bongcam‐Rudloff E, Combet C, Da Costa AL, Deleage G, Diarena M, Fabbretti R, Fettahi G, Flegel V, Gisel A, Kasam V, Kervinen T, Korpelainen E, Mattila K, Pagni M, Reichstadt M, Breton V, Tickle IJ, Vriend G (2009) PDB_REDO: automated re‐refinement of X‐ray structure models in the PDB. J Appl Crystallogr 42:376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Salunke DM, Nair DT (2017) Macromolecular structures: quality assessment and biological interpretation. IUBMB Life 69:563–571. [DOI] [PubMed] [Google Scholar]
- 32. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr Sect D 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grabowski M, Langner KM, Cymborowski M, Porebski PJ, Sroka P, Zheng H, Cooper DR, Zimmerman MD, Elsliger MA, Burley SK, Minor W (2016) A public database of macromolecular diffraction experiments. Acta Crystallogr Sect D 72:1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, Adams PD (2017) Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr Sect D 73:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Praznikar J, Afonine PV, Guncar G, Adams PD, Turk D (2009) Averaged kick maps: less noise, more signal… and probably less bias. Acta Crystallogr Sect D 65:921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Handing KB, Shabalin IG, Kassaar O, Khazaipoul S, Blindauer CA, Stewart AJ, Chruszcz M, Minor W (2016) Circulatory zinc transport is controlled by distinct interdomain sites on mammalian albumins. Chem Sci 7:6635–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Porebski PJ, Klimecka M, Chruszcz M, Nicholls RA, Murzyn K, Cuff ME, Xu X, Cymborowski M, Murshudov GN, Savchenko A, Edwards A, Minor W (2012) Structural characterization of Helicobacter pylori dethiobiotin synthetase reveals differences between family members. FEBS J 279:1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen VB, Davis IW, Richardson DC (2009) KING (Kinemage, Next Generation): a versatile interactive molecular and scientific visualization program. Protein Sci 18:2403–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cooper S, Khatib F, Treuille A, Barbero J, Lee J, Beenen M, Leaver‐Fay A, Baker D, Popovic Z, Players F (2010) Predicting protein structures with a multiplayer online game. Nature 466:756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khatib F, DiMaio F, Cooper S, Kazmierczyk M, Gilski M, Krzywda S, Zabranska H, Pichova I, Thompson J, Popovic Z, Jaskolski M, Baker D (2011) Crystal structure of a monomeric retroviral protease solved by protein folding game players. Nat Struct Mol Biol 18:1175–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langer G, Cohen SX, Lamzin VS, Perrakis A (2008) Automated macromolecular model building for X‐ray crystallography using ARP/wARP version 7. Nat Protoc 3:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cowtan K (2012) Completion of autobuilt protein models using a database of protein fragments. Acta Crystallogr Sect D 68:328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rose AS, Hildebrand PW (2015) NGL Viewer: a web application for molecular visualization. Nucleic Acids Res 43:W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]