

Abstract
Characterization of life processes at the molecular level requires structural details of protein interactions. The number of experimentally determined structures of protein–protein complexes accounts only for a fraction of known protein interactions. This gap in structural description of the interactome has to be bridged by modeling. An essential part of the development of structural modeling/docking techniques for protein interactions is databases of protein–protein complexes. They are necessary for studying protein interfaces, providing a knowledge base for docking algorithms, and developing intermolecular potentials, search procedures, and scoring functions. Development of protein–protein docking techniques requires thorough benchmarking of different parts of the docking protocols on carefully curated sets of protein–protein complexes. We present a comprehensive description of the Dockground resource (http://dockground.compbio.ku.edu) for structural modeling of protein interactions, including previously unpublished unbound docking benchmark set 4, and the X‐ray docking decoy set 2. The resource offers a variety of interconnected datasets of protein–protein complexes and other data for the development and testing of different aspects of protein docking methodologies. Based on protein–protein complexes extracted from the PDB biounit files, Dockground offers sets of X‐ray unbound, simulated unbound, model, and docking decoy structures. All datasets are freely available for download, as a whole or selecting specific structures, through a user‐friendly interface on one integrated website.
Keywords: protein recognition, protein–protein interactions, structure prediction, benchmark sets
Introduction
Protein interactions are the key part of molecular mechanisms in living systems. Because of the limitations of experimental techniques, only a fraction of known protein–protein interactions has experimentally resolved structures.1, 2 Thus, computational modeling is essential for our ability to understand and manipulate biological processes at the molecular level.3 Modeling structures of protein–protein complexes (protein docking) aims at determining a mutual arrangement of the proteins, given the structure (experimentally determined or modeled) of the interactors.4 Docking methodologies can be roughly divided into: template free, where sampling of the binding modes is performed with no prior knowledge of similar experimentally determined structures of the complex, and template‐based or comparative, where such similar complexes (templates) determine the docking predictions. Both types of docking protocols usually consist of the scan (global search for an approximate structure of the complex) with a coarse‐grained, computationally inexpensive objective function, followed by scoring/refinement of the putative matches, using more accurate functions.4, 5 Adequate scoring should capture relevant aspects of the protein structure/function relationships. Such scoring can be either based on the general physical principles or derived from empirical concepts, based on known protein structures (knowledge‐based, or statistical, potentials). Statistical potentials provide balance between accuracy and computational efficiency, and thus are successfully applied to protein–protein docking.6, 7, 8, 9
A number of databases of protein–protein complexes have been compiled and used to investigate physicochemical and structural preferences at protein–protein interfaces.10, 11, 12, 13, 14, 15, 16 Docking methodologies are evaluated by community‐wide blind assessment CAPRI,17 and by benchmarking on pre‐compiled protein–protein sets. Such benchmark sets are based on bound and unbound X‐ray protein structures,16, 18, 19, 20 and protein models.21, 22, 23 Docking has to distinguish correct matches from false‐positives. Thus, an important part in developing intermolecular potentials and scoring functions is docking decoy sets (scoring benchmarks), where near‐native matches are paired with incorrect docking predictions (decoys).19, 24, 25, 26
Comparative docking relies on target/template relationships based on sequence27 sequence/structure (threading), and structure similarity27, 28, 29, 30, 31 with the latter showing a great promise in terms of availability of the templates.32 Evolutionary conserved surface patches may yield similar binding modes for otherwise dissimilar proteins,33, 34 which implies that docking can also be performed by the structure alignment between the target proteins and the interface parts of the templates. The key element in the template‐based docking success is the quality (diversity, non‐redundancy, and completeness of PDB structures) of the template libraries. Obviously, simply selecting all pairwise protein–protein complexes from PDB would produce the complete set of currently known structures. However, utilizing such a brute‐force set would tremendously increase computation time due to the presence of many identical or highly similar complexes. The set will also contain erroneous, low‐quality, and biologically irrelevant structures.15, 35 Thus, groups working on structure alignment docking typically generate their own template libraries by filtering PDB in order to retain only the relevant interactions.36, 37, 38, 39, 40, 41, 42
The uniqueness of the Dockground (http://dockground.compbio.ku.edu) resource is that it offers various datasets of X‐ray and modeled structures, suitable for testing most aspects of protein docking. Currently, it consists of five integrated databases of protein–protein complexes: (i) bound, (ii) unbound, (iii) models, (iv) docking decoys, and (v) docking templates. The first part is the basis for the generation of the other four databases (Fig. 1). The user‐friendly Web interface provides easy download of the current and previous versions of the pre‐compiled datasets and advanced generation of custom datasets. The paper presents, for the first time, a comprehensive description of the Dockground resource in one place, including previously unpublished recent additions and developments.
Figure 1.
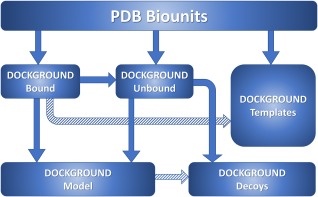
Schematic representation of interconnectivity between different Dockground modules and their relation to the external PDB. Stroked arrows represent future developments.
Database Content and Description
X‐ray bound structures
Details of the initial procedure for generating the bound–bound part of Dockground were published elsewhere,15 and here we provide only a brief summary. A relational PostgreSQL database of annotated structures is generated from the non‐obsolete PDB biological unit files of X‐ray structures (Fig. 1). During the update procedure, in‐house programs automatically exclude undesirable complexes (chains with <30 amino acids, interwoven/tangled chains, and disordered termini at the interface), characterize the entries, chains, and pairwise complexes by several attributes (presence of a ligand, ions, RNA or DNA at the interface, disulfide bridge across the interface, membrane‐associated proteins, etc., see Table 1) and extract a downloadable set of representative structures based on most common parameters (for the full list, see Selection criteria under Bound → Build Database tab on the Dockground web page, Fig. 2). Redundancy between representative structures is removed at 30% sequence identity level, for at least one of the interacting monomers.
Table 1.
Statistics on protein interfaces in Dockground annotated by various attributes
| Attribute | Number of entries | Fraction of entriesa |
|---|---|---|
| Disulfide bond at interface | 6938 | 0.032 |
| DNA/RNA | 32,669 | 0.152 |
| Membrane | 18,784 | 0.087 |
| Disordered | 635 | 0.003 |
| Tangled | 2317 | 0.011 |
| Ligand at interface | 64,013b | 0.297 |
With respect to the total number of interfaces (215,363) stored in the PostgreSQL database.
For 15,820 different ligands.
Figure 2.
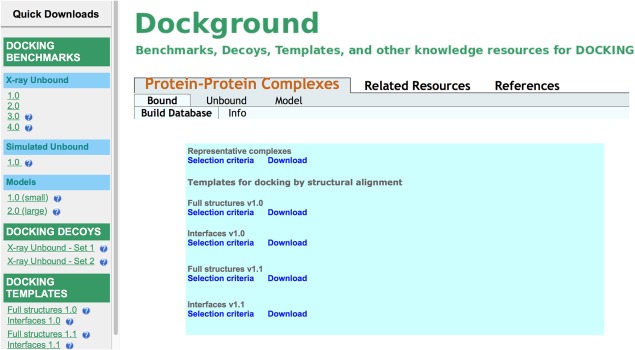
Dockground bound page, along with the Quick Downloads. The detailed search part is shown in Figure 3.
In addition, the bound part of Dockground has an option of generating custom datasets from the main database content, based on a number of parameters (Fig. 3, top). Results are provided as downloadable Excel‐readable table (Fig. 3, bottom) and redundancy can be removed based on the user‐defined sequence identity level.
Figure 3.

Dockground search screen and fragment of the corresponding search results.
The bound–bound Dockground database currently contains 215,363 pairwise complexes (149,416 of which are homo‐dimers) formed by 171,948 polypeptide chains, extracted from 50,779 PDB biounit files. Statistics of several properties of the complexes are in Figure 4. An automated representative set of structures contains 3171 pairwise complexes.
Figure 4.
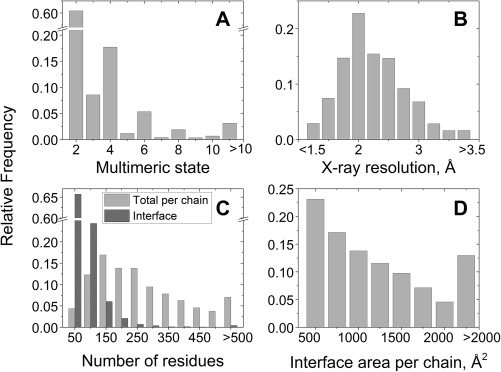
Statistics on the basic (bound) part of Dockground. Normalization of data is with respect to the total number of PDB biounit files (A, B), the total number of chains (C), and the total number of pairwise complexes (C, D) in the database.
X‐ray unbound structures
Proteins undergo conformational changes upon binding. Modeling such changes is a challenge due to the very large number of internal degrees of freedom determining protein conformation. Thus, an accurate prediction of protein complexes from the unbound components poses a big problem for the docking algorithms. Because of that, the datasets of unbound structures corresponding to the co‐crystallized complexes16, 18, 19, 20 are essential for the development and validation of docking approaches. The Dockground unbound set distinguishes itself by being an integral part of a large resource and the basis for other datasets (Fig. 1), for example, allowing a straightforward comparison of docking performance for unbound and model structures. Dockground has three legacy datasets of unbound structures, described earlier.16 In this paper, we present for the first time the most recent docking benchmark set 4.
The initial step in compiling the dataset was performed using ProPairs software20 run on the entire PDB with the default values of the parameters. This resulted in 1020 binary complexes, each having both interactors in the unbound form (unbound/bound sequence identity >70%). Applying more stringent criterion for the sequence identity between bound and unbound structures for both interactors (96% identity, 80% coverage, as in Weng's benchmark 519) reduced this number to 427. Additional purging of the dataset by the structural similarity between bound complexes (the threshold for TM‐scores of the structural alignments between both pairs of interactors set to 0.8) yielded the final dataset of 396 complexes, out of which 223 structures have single‐chain monomers only, and the rest have one or both interacting subunits consisting of two or more polypeptide chains. This number is significantly larger than the number of entries in the most recent Weng's benchmark 5 (230 complexes)19 with only 77 complexes shared by both datasets (at least one PDB code is the same for bound or unbound structures). This is due to the different ways of generating the sets and removing redundancies. Weng's benchmark 5 was generated by adding newer PDB structures to the previous benchmark 4,43 while we started from scratch. We also used a different definition of the redundancy between bound complexes (sequence ID at the interface >40% in the ProPairs algorithm versus belonging to the same SCOP domain family in the Weng's benchmarks).
Among the 319 unique complexes (not shared with the Weng's benchmark set 5) in our dataset, only 39 do not have structural or sequence similarity to complexes in the Weng's set (in pairwise comparison of the bound proteins, the maximal TM‐score <0.6, and the maximal sequence identity <26%); and 34 unique complexes have an almost identical analog in the Weng's set (the minimal TM‐score >0.9 and the minimal sequence identity >95%). The rest of the unique structures have at least one complex in the Weng's set that shares the same fold (the minimal TM‐score >0.6 and the maximal sequence identity <60%).
The Weng's benchmark has relatively more easy docking (rigid‐body) cases, whereas our dataset is more balanced between easy and medium difficulty cases [Fig. 5(A)]. However, the 39 truly unique complexes (no structural or sequence similarity to complexes in the Weng's set) are also biased toward the easy cases (12, 4, and 12 complexes in the rigid‐body, medium, and difficult docking categories, respectively). Compared to the Weng's set, our dataset contains more “Others, miscellaneous” complexes, and a similar distribution of other functional categories [Fig. 5(B)]. The 39 truly unique complexes are distributed only over four functional categories (2 – E, 10 – OG, 2 – OR, and 25 – OX, see legend to Fig. 5B).
Figure 5.
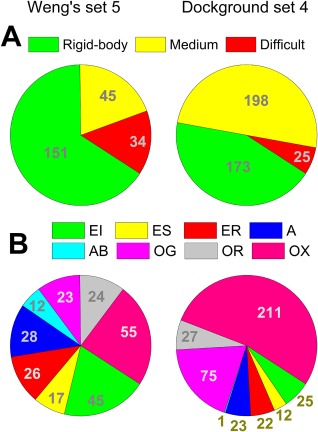
Comparison of complexes in the Weng's benchmark set 5 and the Dockground set 4. (A) Docking difficulty level (adopted from Ref. 19). Rigid‐body (or easy) cases correspond to the protein complexes with bound/unbound interface Cα RMSD (i‐RMSD) ≤ 1.5 Å; medium difficulty cases correspond to 1.5 < i‐RMSD < 2.2 Å; and difficult cases correspond to i‐RMSD >2.2 Å. (B) Functional categories of complexes, as in Ref. 19: EI, Enzyme‐Inhibitor; ES, Enzyme‐Substrate; ER, Enzyme complex with a regulatory or accessory chain; A, Antibody‐Antigen; AB, Antigen‐Bound Antibody; OG, Other, G‐protein containing; OR, Other, Receptor containing; and OX, Other, miscellaneous. The figure shows the absolute number of complexes in each category.
Each structure in the dataset is represented by four PDB‐formatted files: XXXX_b1.pdb, XXXX_b2.pdb, XXXX_u1.pdb, and XXXX_u2.pdb, where XXXX is PDB code for the bound structure and b1, u1, b2, and u2 are first bound, first unbound, second bound, and second unbound partners, respectively. For user convenience, files for the unbound structures contain coordinates, transformed by structural alignment of the original unbound PDB structures onto the corresponding bound structures. Also, residues in the unbound files are renumbered to match the residue numbering in the PDB files for bound structures. Mapping alignments by BLAST44 are provided in the REMARK section of the unbound files. The text file LIST.txt provides the original PDB codes and the chain IDs for the bound and unbound structures, along with the bound/unbound Cα RMSD, TM‐scores, and sequence identities. In the case of multichain interactors comparison was made for concatenated chains. The file FORMAT.txt contains legends for the columns in LIST.txt. Files are available for download as a zipped archive under the “Unbound → Build Database” tab, and as a Quick Download link.
Simulated unbound structures
Sets of protein structures determined in both bound and unbound states are essential for benchmarking of the docking procedures. However, the number of such proteins in PDB is relatively small (see above). A radical expansion of such sets is possible if the unbound structures are computationally simulated. Dockground provides a large (3205 single chains from 1918 complexes) dataset of such simulated unbound structures. Protein complexes were selected from the bound part of Dockground with the following criteria: mean buried surface area ≥500 Å2, include alternative binding modes, homo/hetero n‐mers, and oligomers, and the sequence redundancy cutoff of 97%. Proteins were separated from the interacting partner and subjected to 1 ns Langevin dynamics simulation in CHARMM (CHARMM2245 force field), with electrostatics by Generalized Born approximation. The simulated unbound structures were selected according to criteria from the systematic comparison of experimentally determined bound and unbound structures (details in Ref. 46).
The PDB‐formatted files of the simulated unbound structures are available under the “Unbound → Build Database” tab, and as a Quick Download link. Users can download either the entire set or any combination of the available subsets. In addition to the obligate and/or non‐obligate complexes, the interface has an option to download structures, for which simulated unbound structures were generated for both monomers in the complex or for one only. Users can also include simulated unbound structures, for which corresponding X‐ray unbound structure exists in the Dockground unbound docking benchmark 3. The names of the files, similarly to the X‐ray unbound sets, start with the PDB code of the initial bound structure, followed by _u1 or _u2 for the first and second chain in the initial complex, respectively.
Model‐model complexes
In the structural reconstruction of protein interaction networks, most individual protein structures will be models of limited accuracy (due to high‐throughput modeling, lack of homologs, structural flexibility, and such ). However, docking techniques have been tested largely on the datasets of the X‐ray structures only, mainly due to the lack of adequate benchmark sets of models. Dockground provides two such carefully curated sets of representative modeled structures of individual proteins with controlled levels of inaccuracy. The first, smaller set is based on Dockground unbound benchmark 3 and consists of arrays of six models with 1, 2, … 6 Å model‐to‐native Cα RMSD for each of the proteins from 63 binary complexes. The models were built either by simple single‐template modeling, or by the Nudged Elastic Band method (details in Ref. 21). The benchmark can be downloaded either as a whole set through the Quick Downloads link or as a customizable set under “Model → Build Database → Select protein from set 1.0” tab.
The second, larger set was constructed from 165 protein complexes generated by the built‐in engine of the Dockground bound part. For all proteins in the set, arrays of six models with the 1, 2, … 6 Å model‐to‐native Cα RMSD were selected from the modeling trajectories generated by I‐TASSER47, 48 (details in Ref. 22). This set is available for download via the Quick Downloads link. While the first set allows direct comparison of docking methodologies performance on X‐ray unbound and modeled structures, the second set ensures statistical significance of the benchmarking. Also, all models in the second set are “genuine” (from an actual structural modeling protocol), rather than “simulated” by extra‐ or interpolation from such models, as in a significant part of the first set.
X‐ray docking decoys
Testing of the scoring functions for protein–protein docking requires a set of docking poses for a number of protein–protein complexes. Among these poses, one or several matches per complex have to be close to the native structure, preferably within the binding funnel,49 while the rest have to be false‐positive ones (decoys). Ideally, the decoys should be spread in space, to avoid bias in testing of the scoring functions due to clustering. Dockground provides two sets of docking decoys (scoring benchmarks). The first set consists of 100 non‐native and 1 near‐native (ligand RMSD to the native structure <5 Å) generated by GRAMM‐X50 for 61 unbound complexes from the Dockground benchmark 2 (for details, see Ref. 25). However, due to the use of the unbound structures in the docking procedure, the 100 top wrong docking solutions have better shape complementarity then near‐native match, which may lead to a bias in testing of the scoring function (i.e., one may detect the near‐native match by requiring a function to favor worse complementarity).
Recently, we generated a new larger decoy set (presented here for the first time), which addresses this problem. For each of the 396 unbound–unbound complexes from Dockground benchmark 4, we generated 300,000 low‐resolution docking solutions by GRAMM51, 52 with the grid step 3.5 Å and 10° angular interval. To avoid interference of different scoring schemes, the matches are unscored/unrefined, ranked by the shape complementarity alone, as implemented in the GRAMM scan stage. Thus, most near‐native matches, in acceptable or better category, according to the CAPRI criteria53 (201 complexes, 50.8% of the set), were ranked outside the top 100,000 in the prediction list. The unbound structures for 43 complexes did not yield near‐native matches. These complexes were excluded from the final dataset.
The 99 incorrect docking matches, with ranking similar to the near‐native one, were selected around the near‐native structure with a maximally spread spatial distribution (Fig. 6). The selection was done by an automated iterative procedure that utilizes the angles between vectors connecting centers of mass of the receptor and the ligand in the predicted poses. In the first iteration, the incorrect models were selected from the ranked matches sublist of ±50 positions around the near‐native match. The vectors of the selected matches had to form ≥5° angle with the vectors of any other selected matches. If 99 incorrect matches were not selected in the first iteration, in the second iteration, the sublist around the near‐native match was expanded by 50 positions in either direction (i.e., to contain ±100 positions around the near‐native match); and the minimum allowed angle between the vectors was halved. The procedure iterated until all 99 incorrect models were selected. The maximum number of iterations needed was 5, for five complexes. For 323 complexes, the full set of incorrect matches was selected in the first iteration.
Figure 6.
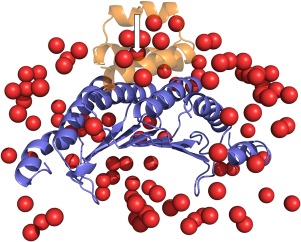
Example of docking decoys. False positive matches are shown by the ligand (smaller protein) center of mass (red) for 1f93 complex. The near‐native match is indicated by an arrow. The native structure of the complex is in cartoon representation.
Both decoy sets are available for download as zipped archives from the Quick Downloads links. In addition, decoys generated for each complex, are available for download separately under the “Unbound → Docking Decoys” tab.
Docking templates
Search for the templates in comparative docking can be done either by sequence or structural similarity with the latter gaining increasing popularity due to increasing availability of the template structures.32 Detection of the structurally similar templates can be carried out either by the entire structures or by the interface.54 The key element in successful application of any template‐based docking is the quality (diversity, non‐redundancy, and structure completeness) of the template libraries. Simply selecting all pairwise complexes from PDB would result in many identical or highly similar complexes, leading to a very significant slow‐down of docking. The set would also contain erroneous, low‐quality, and non‐biological structures.15, 35 Also, “static” sets of target and template structures would allow consistent comparison of the template‐based docking methodologies developed at different times, since comparing their performance on the evolving PDB may produce confusing results.
Dockground offers a carefully curated, non‐redundant library of templates containing 4950 full structures of binary complexes, and 5936 protein–protein interfaces extracted from the full structures at 12 Å distance cutoff.55 Redundancy was removed by clustering based on structural similarity. High structural quality of the template interfaces was achieved by automated procedures and manual curation (details in Ref. 56). The library is available for download from Dockground under Quick Downloads links as sets 1.1. Initially, the datasets were generated using a less sophisticated clustering algorithm, resulting in a larger number of structures (5050 full structures and 7107 interfaces). Because of the high demand from the scientific community, these datasets were posted online well ahead of the corresponding publication56 and are kept in Dockground as legacy sets 1.0.
In addition to the template structures, a set of carefully selected targets is also provided in the downloadable zip archives. Each archive contains the folders templates, targets, and info. The templates folder contains two PDB‐formatted files of atomic coordinates per library entry. The files are named by the original PDB file, from which the entry was extracted, where is the four‐symbol PDB code, and are the model numbers (as in PDB biounit file), and are the chain identifiers for the first and the second component of the complex, and N = 1 and 2 identifies the component. Separation of library entries into two files makes it easier to use the set in the docking programs. However, simple merging of the two files (e.g., by cat Linux command) produces the complex or interface structure without geometrical clashes and distinct chain identifiers. The targets folder (in both full‐structure and interface archives) consists of 2 × 293 similarly named PDB‐formatted files for the full structures of the targets. The info folder contains two text files per structure in the target set (named similar to the files in the target folder, but with the extension .txt) with information on all statistically significant structural alignments (TM‐scores >0.4) of the targets to full‐structures or interfaces in the template set. The folder also contains a text file with information on the resulting template‐based docking predictions.
These sets can be used either for modeling of new protein complexes by full or interface alignment (using files in the templates folder only), and for benchmarking of docking techniques (using both target and template folders and comparing results with the data in the info folder).
Concluding Remarks and Future Development
We present the comprehensive, all‐in‐one description of our Dockground data resource for modeling of protein complexes, containing a variety of inter‐connected datasets for development and testing of different aspects of protein docking methodologies. The current data include co‐crystallized (bound) protein complexes, unbound X‐ray and simulated docking benchmark sets, model–model docking benchmark sets, scoring benchmarks (docking decoys), and templates for comparative docking. The datasets are available for download from the single site through a user‐friendly Web interface.
The central part of Dockground is based on the entire PDB and is currently updated by a semiautomated procedure, whereas the rest of the datasets are updated less frequently, with little or no automation. Incorporation of all the update procedures into a single automated (or at least, semi‐automated) pipeline is a major future development of the resource. We also plan to implement interfaces for generating custom sets, similar to one for the bound part, for all other Dockground parts. In the bound part, we will implement removal of the redundancy based on structure. We will generate docking decoys for models of the individual proteins, and an automated pipeline for generating template libraries from the bound Dockground part. We will also add new datasets for modeling of protein complexes, such as libraries of rotamers and rotamer–rotamer transitions for flexible docking.57
Acknowledgments
A number of people have contributed over the years to the development of Dockground. Dominique Douguet designed and generated the first bound database, Ying Gao built the original X‐ray unbound sets, Shiyong Liu generated the first set of docking decoys, and Anatoly Ruvinsky and Tatsiana Kirys created the simulated unbound set, with the help of Deepak Singla. Andrey Tovchigrechko designed and implemented the original set of simulated protein models, which eventually led to the development of the Dockground current model–model sets. He also wrote GRAMM‐X code used to build the original docking decoys.
Contributor Information
Petras J. Kundrotas, Email: pkundro@ku.edu
Ilya A. Vakser, Email: vakser@ku.edu
References
- 1. Petrey D, Honig B (2014) Structural bioinformatics of the interactome. Ann Rev Bioph 43:193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mosca R, Pons T, Ceol A, Valencia A, Aloy P (2013) Towards a detailed atlas of protein–protein interactions. Curr Opin Struct Biol 23:929–940. [DOI] [PubMed] [Google Scholar]
- 3. Vakser IA (2013) Low‐resolution structural modeling of protein interactome. Curr Opin Struct Biol 23:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vakser IA (2014) Protein–protein docking: from interaction to interactome. Biophys J 107:1785–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moal IH, Moretti R, Baker D, Fernandez‐Recio J (2013) Scoring functions for protein–protein interactions. Curr Opin Struct Biol 23:862–867. [DOI] [PubMed] [Google Scholar]
- 6. Mintseris J, Weng Z (2003) Atomic contact vectors in protein–protein recognition. Proteins 53:629–639. [DOI] [PubMed] [Google Scholar]
- 7. Chuang GY, Kozakov D, Brenke R, Comeau SR, Vajda S (2008) DARS (Decoys As the Reference State) potentials for protein–protein docking. Biophys J 95:4217–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu SY, Vakser IA (2011) DECK: distance and environment‐dependent, coarse‐grained, knowledge‐based potentials for protein–protein docking. BMC Bioinformatics 12:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moal IH, Torchala M, Bates PA, Fernandez‐Recio J (2012) The scoring of poses in protein–protein docking: current capabilities and future directions. BMC Bioinformatics 14:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keskin O, Tsai CJ, Wolfson H, Nussinov R (2004) A new, structurally nonredundant, diverse data set of protein–protein interfaces and its implications. Protein Sci 13:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davis FP, Sali A (2005) PIBASE: a comprehensive database of structurally defined protein interfaces. Bioinformatics 21:1901–1907. [DOI] [PubMed] [Google Scholar]
- 12. Teyra J, Doms A, Schroeder M, Pisabarro MT (2006) SCOWLP: a web‐based database for detailed characterization and visualization of protein interfaces. BMC Bioinformatics 7:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jefferson ER, Walsh TP, Roberts TJ, Barton GJ (2007) SNAPPI‐DB: a database and API of structures, iNterfaces and alignments for protein–protein interactions. Nucleic Acids Res 35:D580–D589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kundrotas PJ, Alexov E (2007) PROTCOM: searchable database of protein complexes enhanced with domain–domain structures. Nucleic Acids Res 35:D575–D579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Douguet D, Chen HC, Tovchigrechko A, Vakser IA (2006) DOCKGROUND resource for studying protein–protein interfaces. Bioinformatics 22:2612–2618. [DOI] [PubMed] [Google Scholar]
- 16. Gao Y, Douguet D, Tovchigrechko A, Vakser IA (2007) DOCKGROUND system of databases for protein recognition studies: unbound structures for docking. Proteins 69:845–851. [DOI] [PubMed] [Google Scholar]
- 17. Lensink MF, Velankar S, Wodak SJ (2017) Modeling protein–protein and protein–peptide complexes: CAPRI 6th edition. Proteins 85:359–377. [DOI] [PubMed] [Google Scholar]
- 18. Chen R, Mintseris J, Janin J, Weng Z (2003) A protein–protein docking benchmark. Proteins 52:88–91. [DOI] [PubMed] [Google Scholar]
- 19. Vreven T, Moal IH, Vangone A, Pierce BG, Kastritis PL, Torchala M, Chaleil R, Jimenez‐Garcia B, Bates PA, Fernandez‐Recio J, Bonvin AMJJ, Weng Z (2015) Updates to the integrated protein–protein interaction benchmarks: Docking Benchmark Version 5 and Affinity Benchmark Version 2. J Mol Biol 427:3031–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krull F, Korff G, Elghobashi‐Meinhardt N, Knapp EW (2015) ProPairs: a data set for protein−protein docking. J Chem Inf Model 55:1485–1507. [DOI] [PubMed] [Google Scholar]
- 21. Anishchenko I, Kundrotas PJ, Tuzikov AV, Vakser IA (2014) Protein models: the Grand Challenge of protein docking. Proteins 82:278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anishchenko I, Kundrotas PJ, Tuzikov AV, Vakser IA (2015) Protein models docking benchmark 2. Proteins 83:891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bohnuud T, Luo L, Wodak SJ, Bonvin AMJJ, Weng Z, Vajda S, Schueler‐Furman O, Kozakov D (2017) A benchmark testing ground for integrating homology modeling and protein docking. Proteins 85:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gray JJ, Moughon S, Wang C, Schueler‐Furman O, Kuhlman B, Rohl CA, Baker D (2003) Protein–protein docking with simultaneous optimization of rigid‐body displacement and side‐chain conformations. J Mol Biol 331:281–299. [DOI] [PubMed] [Google Scholar]
- 25. Liu S, Gao Y, Vakser IA (2008) DOCKGROUND protein–protein docking decoy set. Bioinformatics 24:2634–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lensink MF, Wodak SJ (2014) Score_set: a CAPRI benchmark for scoring protein complexes. Proteins 82:3163–3169. [DOI] [PubMed] [Google Scholar]
- 27. Aloy P, Pichaud M, Russell RB (2005) Protein complexes: structure prediction challenges for the 21st century. Curr Opin Struct Biol 15:15–22. [DOI] [PubMed] [Google Scholar]
- 28. Szilagyi A, Zhang Y (2014) Template‐based structure modeling of protein–protein interactions. Curr Opin Struct Biol 24:10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dey F, Zhang QC, Petrey D, Honig B (2013) Toward a ‘‘structural BLAST’’: using structural relationships to infer function. Protein Sci 22:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuzu G, Keskin O, Gursoy A, Nussinov R (2012) Constructing structural networks of signaling pathways on the proteome scale. Curr Opin Struct Biol 22:367–377. [DOI] [PubMed] [Google Scholar]
- 31. Szilagyi A, Grimm V, Arakaki AK, Skolnick J (2005) Prediction of physical protein–protein interactions. Phys Biol 2:S1–S16. [DOI] [PubMed] [Google Scholar]
- 32. Kundrotas PJ, Zhu Z, Janin J, Vakser IA (2012) Templates are available to model nearly all complexes of structurally characterized proteins. Proc Natl Acad Sci USA 109:9438–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keskin O, Nussinov R (2005) Favorable scaffolds: proteins with different sequence, structure and function may associate in similar ways. Protein Eng 18:11–24. [DOI] [PubMed] [Google Scholar]
- 34. Zhang QC, Petrey D, Norel R, Honig BH (2010) Protein interface conservation across structure space. Proc Natl Acad Sci USA 107:10896–10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kundrotas PJ, Vakser IA, Janin J (2013) Structural templates for modeling homodimers. Protein Sci 22:1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, Maniatis T, Califano A, Honig B (2012) Structure‐based prediction of protein–protein interactions on a genome‐wide scale. Nature 490:556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Henrick K, Thornton JM (1998) PQS: a protein quaternary structure file server. Trends Biochem Sci 23:358–361. [DOI] [PubMed] [Google Scholar]
- 38. Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. [DOI] [PubMed] [Google Scholar]
- 39. Tuncbag N, Gursoy A, Nussinov R, Keskin O (2011) Predicting protein–protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat Protoc 6:1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tuncbag N, Gursoy A, Guney E, Nussinov R, Keskin O (2008) Architectures and functional coverage of protein–protein interfaces. J Mol Biol 381:785–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu H, Domingues FS, Sommer I, Lengauer T (2006) NOXclass: prediction of protein–protein interaction types. BMC Bioinformatics 7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cukuroglu E, Gursoy A, Nussinov R, Keskin O (2014) Non‐redundant unique interface structures as templates for modeling protein interactions. PloS One 9:e86738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hwang H, Vreven T, Janin J, Weng Z (2010) Protein–protein docking benchmark version 4.0. Proteins 78:3111–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph‐McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz‐Kuczera J, Yin D, Karplus M (1998) All‐atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 46. Kirys T, Ruvinsky AM, Singla D, Tuzikov AV, Kundrotas PJ, Vakser IA (2015) Simulated unbound structures for benchmarking of protein docking in the DOCKGROUND resource. BMC Bioinformatics 16:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roy A, Kucukural A, Zhang Y (2010) I‐TASSER: a unified platform for automated protein structure and function prediction. Nat Protocols 5:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y (2008) I‐TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hunjan J, Tovchigrechko A, Gao Y, Vakser IA (2008) The size of the intermolecular energy funnel in protein–protein interactions. Proteins 72:344–352. [DOI] [PubMed] [Google Scholar]
- 50. Tovchigrechko A, Vakser IA (2006) GRAMM‐X public web server for protein–protein docking. Nucleic Acids Res 34:W310–W314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Katchalski‐Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA (1992) Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci USA 89:2195–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vakser IA (1995) Protein docking for low‐resolution structures. Protein Eng 8:371–377. [DOI] [PubMed] [Google Scholar]
- 53. Mendez R, Leplae R, De Maria L, Wodak SJ (2003) Assessment of blind predictions of protein–protein interactions: current status of docking methods. Proteins 52:51–67. [DOI] [PubMed] [Google Scholar]
- 54. Kundrotas PJ, Vakser IA (2013) Global and local structural similarity in protein–protein complexes: implications for template‐based docking. Proteins 81:2137–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sinha R, Kundrotas PJ, Vakser IA (2012) Protein docking by the interface structure similarity: how much structure is needed?. PLoS One 7:e31349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Anishchenko I, Kundrotas PJ, Tuzikov AV, Vakser IA (2015) Structural templates for comparative protein docking. Proteins 83:1563–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kirys T, Ruvinsky A, Tuzikov AV, Vakser IA (2012) Rotamer libraries and probabilities of transition between rotamers for the side chains in protein–protein binding. Proteins 80:2089–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]