

Abstract
PixelDB, the Peptide Exosite Location Database, compiles 1966 non‐redundant, high‐resolution structures of protein–peptide complexes filtered to minimize the impact of crystal packing on peptide conformation. The database is organized to facilitate study of structurally conserved versus non‐conserved elements of protein–peptide engagement. PixelDB clusters complexes based on the structural similarity of the peptide‐binding protein, and by comparing complexes within a cluster highlights examples of domains that engage peptides using more than one binding mode. PixelDB also identifies conserved peptide core structural motifs characteristic of each binding mode. Peptide regions that flank core motifs often make non‐structurally conserved interactions with the protein surface in regions we call exosites. Many examples establish that exosite contacts can be important for enhancing protein binding and interaction specificity. PixelDB provides a resource for computational and structural biologists to study, model, and predict core‐motif and exosite‐contacting peptide interactions. PixelDB is available to the community without restriction in a convenient flat‐file format with accompanying visualization tools.
Keywords: protein–peptide interactions, exosites, structural biology database, peptide binding motifs, peptide docking
Abbreviations
- CSV
comma separated values
- ECR
exosite contacting region
- ELM
eukaryotic linear motif
- NISR
non‐interacting surface residues
- PDB
protein data bank
- TM
template modeling
Introduction
Protein–peptide interactions govern many cellular processes and can be important for structural scaffolding and complex assembly, signal transduction, transcriptional regulation, intracellular localization, and enzyme‐substrate recognition.1 In many protein–peptide complexes, a protein engages a relatively short, conserved motif in an interaction partner. Experimentally determined sequence motifs for numerous peptide‐binding proteins are documented in databases such as the eukaryotic linear motif resource (ELM),2 and for many complexes there are high‐resolution structures in the protein data bank (PDB) that provide insight into binding mechanism. Several previous studies have used structures of proteins bound to peptides, or knowledge of peptide binding motifs, as the starting point to design reagents that can block the formation of protein–protein complexes.3, 4, 5, 6, 7, 8
Despite the central importance of peptide motifs for protein–peptide interactions, conserved motifs may not contain all of the information needed to fully determine the affinity and specificity of a protein–peptide complex. The interface formed with many conserved motifs is small, and provides few enthalpically favorable interactions compared to the formation of larger protein–protein complexes.9 At the same time, flexible peptide ligands have to overcome entropy loss upon adopting a bound conformation, which contributes to making many protein–peptide interactions quite weak. Furthermore, the amount of information encoded in a short motif is limited, and may not be sufficient to establish binding preferences among related proteins. Unsurprisingly, given these considerations, studies of various protein–peptide complexes have provided evidence that sequence regions flanking core motifs, that is, sequences N‐ or C‐terminal to the motif, can in many cases make additional interactions with a protein binding partner to enhance affinity and/or specificity.10, 11, 12, 13, 14 We refer to the region of a protein surface contacted by core‐flanking sequence as an exosite, and the peptide region that contacts an exosite as an exosite contacting region (ECR).
Exosites, which are important for natural protein–peptide complexes, also offer opportunities to protein designers. For example, to compete with an endogenous interaction, a peptide inhibitor could be designed to make affinity‐enhancing interactions with exosite regions. This could be achieved by designing an ECR using structure‐based computational methods.15 However, we do not currently know much about ECR interactions. Incomplete understanding of ECR structural determinants was highlighted in the latest CAPRI challenge, where participants were tasked with predicting the binding mode of a peptide to a known partner structure. For CAPRI targets 60–64, most groups excelled at predicting the correct core binding mode, but none were able to correctly predict the flanking structure.16
Many databases have compiled examples of protein–peptide complexes and extracted interesting structural trends.17 Some examples are mentioned here and summarized in Table 1 to provide context for our new database. Although existing databases provide sets of protein–peptide complexes that are useful for analysis and for benchmarking docking algorithms, none of these databases has been analyzed for structural conservation of peptide binding modes, or to provide examples of exosites. Closer to our current focus is the work of Stein and Aloy, who compiled structures of protein domains bound to peptides containing motifs documented in the 2007 version of the ELM database.13 The resulting collection of 390 complex structures covers 30 different domains and provides examples of the roles of both ELM residues and surrounding “context” residues in binding. The authors used FoldX to estimate the relative roles of motif versus context residues in binding, and found an average contribution of 21% of the binding energy from the context. Analysis also revealed that variation in the conformation of context residues was significantly greater than variation of the motif residues, when comparing complexes involving the same family of binding protein.
Table 1.
Protein–Peptide Databases Compared with PixelDB
| Database name | Number of complexes | Peptide definition | Quality filter | Availability |
|---|---|---|---|---|
| PepBind18 | 5314 | <35 AA, MeSH definition and manual curation | None | Webserver |
| DOMMINO 2.019 | 13,592 | <20 AA and no SCOP | None | Webserver |
| PepX20 | 1431 | 5–35 AA | Resolution < 2.5 Å | Webserver |
| Receptor > 35 AA | ||||
| Removed sequence redundancy >40% | ||||
| PeptiSite21 | 650 | 5–50 AA, bound to receptor that has Uniprot identifier, >25% surface buried upon binding | None | Webserver and ICM format |
| peptiDB9 | 103 | 5–15 AA | Resolution < 2.0 Å | List of PDB IDs |
| Removed sequence redundancy > 70% | ||||
| PixelDB | 1966 | Length cutoffs and machine learning | Resolution < 2.5 Å | Github |
| Crystal packing |
To expand the amount of data available for analysis, and to provide a resource useful for exploring the roles of peptide core versus flanking regions in protein–peptide molecular recognition, we built PixelDB (Peptide Exosite Location Database). The database is a highly curated compendium of protein complexes that provides many examples of both structurally conserved and non‐conserved interactions. To define core regions in structures without any prior knowledge of core sequence motifs (and therefore no reliance on databases such as ELM), we developed a structural definition of such sites. In our scheme, we defined a peptide structural core motif as the part of a peptide that adopts a conserved binding mode in different structures of related complexes. The core region is an estimate of the minimum peptide structure required for binding. Non‐structurally conserved peptide elements in complexes, including elements that are observed only in some structures and thus are probably not required for binding, were considered as potential ECRs. To obtain as many examples as possible, we implemented automated search protocols to find protein–peptide complexes in the PDB and annotate them with structurally conserved and non‐conserved peptide positions.
This article presents the methods used to construct PixelDB, including the processing used to select examples, our clustering of complexes, and initial analysis of the resulting data. We also include a description of the files and scripts that we are releasing with the database, which is freely available on line. We expect that PixelDB will find multiple uses in the protein modeling and structural biology communities, both for studying protein–peptide molecular recognition and for testing new modeling methods. We have annotated the incorporated data with labels that will help users extract subsets of examples appropriate for different uses. We look forward to new insights into protein–peptide recognition that may emerge from use of PixelDB.
Methods
Database building
PixelDB was constructed by analyzing structures only, without reference to sequence or sequence conservation, except that very high sequence similarity was used to limit redundancy. An overview of the process is shown in Figure 1(A). The PDB (09/01/16) was queried for protein–peptide complexes using the following criteria: asymmetric unit with two or more chains, X‐ray structure with resolution of 2.5 Å or better, a chain of at least 25 amino acids (candidate receptor), and no DNA or RNA molecules. A second filter was applied such that all retained structures included a chain of 5–80 amino acids (candidate peptide). Missing side chain atoms were added using SCWRL4.22 Structures were standardized by removing heteroatoms and hydrogens, renumbering residues for each chain, and inserting a gap in residue numbering when two consecutive alpha carbons were more than 4 Å apart. Processed PDB‐format files are available as part of the PixelDB distribution.
Figure 1.
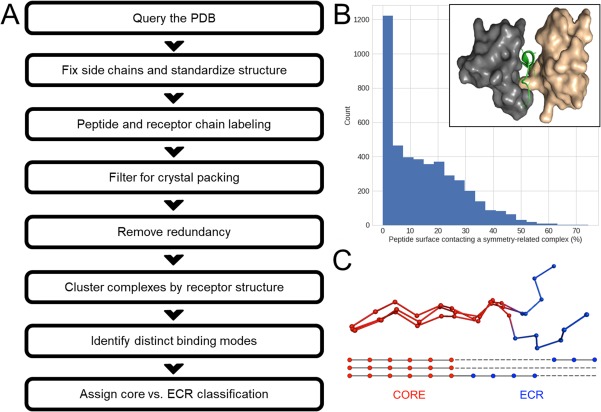
Overview of the construction of PixelDB. (A) Steps used to assemble the database, see Methods for details. (B) Histogram showing the distribution of peptide contacts with symmetry‐related complexes among 4385 structures considered for PixelDB. Complexes with >20% surface contacts were filtered out. The inset shows an example of a protein–peptide complex (PDB ID 4N7H 34) in which the peptide (green) makes extensive contacts with both the WW domain receptor (gray) and a symmetry mate of the receptor (tan). (C) Cartoon illustrating the definition of core versus ECR regions based on structural conservation across complexes. The alignment at the bottom of the panel reflects a structure‐based alignment, not a sequence alignment.
To assign one chain in each complex as peptide, and others as receptor, we used length cutoffs, manual curation and automated assignment by machine learning. Every chain of 5–25 residues without a gap in the sequence was classified as peptide, and every chain of 50 residues or more was classified as receptor. Also, chains with length >25% of all residues in the asymmetric unit were classified as receptor. Remaining chains of 25–50 residues were classified as receptor or peptide either by using a nearest‐neighbors classification algorithm, or by manual assignment. To train the nearest‐neighbors classifier we used three features: length of the chain to be classified in comparison to the summed lengths of all chains, secondary structure content determined from the structure using STRIDE,23 and number of chains in the crystal structure. Complexes with classifications assigned by chain‐length cutoffs, as mentioned above, were used as the training data. Training was performed using Scikit‐learn,24 and then applied to classify remaining complexes. Many examples, including the smallest classified receptor and longest classified peptide (Supporting Information, Fig. S1), were manually inspected to validate the classification results.
Crystallographic symmetry mates were obtained using Pymol,25 and the contacting surface area between each chain in the asymmetric unit and all symmetry‐related complexes was obtained using SurfNet.26 Peptides with more than 20% of their surface area, in the context of the complex, in contact with a symmetry‐related complex were filtered out, because their binding conformation might be affected by crystal lattice packing [Fig. 1(B)]. The binding partner chain(s) of a peptide were defined as those classified as receptor that contacted at least 10% of the peptide surface. Peptides with <40% surface burial with a binding partner were removed from the database. Complexes involving interactions between chains classified as peptide were removed. Therefore, each PixelDB entry represents the structure of a single chain assigned as peptide bound to one or more receptor chains.
Receptor‐based clustering and identification of binding modes and exosites
A graph was built in which each node was a receptor structure and an edge was added between nodes for which the template modeling (TM) score from DeepAlign27 was higher than 0.8. The TM score used here was the number of aligned residues within 4 Å of each other, divided by the length of the longer chain. The largest cliques in this network were iteratively found, identified as receptor clusters, and removed from the graph using the NetworkX python package.28 Each cluster therefore represents a set of structures in which the receptors all share a TM score above 0.8. The restrictive threshold and stringent clustering algorithm were used to ensure that variation in peptide conformation within clusters is not due to receptor structure differences. Complexes within each cluster that contained at least two members were structurally aligned based on the receptor chain using 3DCOMB.29
Within a cluster, peptide sequences were aligned based on their binding geometry (i.e. based on their positioning relative to the receptor, not sequence). Dynamic programming was used to generate a pairwise peptide alignment, with similarity based on differences of the Cartesian coordinates of alpha carbons in receptor‐aligned structures, with a gap penalty of 2.5 Å. Pairs of complexes that shared more than 99% peptide sequence identity and 95% receptor sequence identity were considered redundant, and one of the two was removed.
For each receptor cluster, a second graph was built in which each node represented four consecutive peptide residues and, using the pairwise peptide alignment previously obtained, an edge between two nodes was created if all four residues in the peptide segments from two different structures were structurally aligned. Maximum cliques were iteratively found, identified as binding modes, and all nodes from those complexes were removed from the graph. Therefore, each binding mode cluster is composed of complexes that share at least four structurally conserved residue positions. Finally, using dynamic programming, all peptides within a binding mode were aligned using their Cartesian positions, adding peptides progressively to a growing alignment. For each position in the alignment, a conservation score was calculated as the percent of aligned examples that conserve that alpha carbon position.
To avoid double‐counting symmetry related binding sites, for each receptor–peptide complex we tested for high structural similarity to all other complexes by identifying those with TM score >0.9. When symmetry related complexes were identified, we included both examples in clustering to identify binding modes. In cases where the peptide/receptor had near‐identical sequence similarity, one example of the complex was removed using the redundancy filter described above.
Peptide residues with alpha carbon positions that were conserved in 80% or more of the examples in the binding mode were classified as core and are labeled with “c” in the database. We imposed a binary classification such that the rest of the peptide was classified as (potential) exosite contacting region (ECR, marked with “e” in the database) [Fig. 1(C)]. To smooth the core/ECR partitioning, single residues of one classification (core vs. ECR) that were flanked on either side by residues of the opposite classification were re‐assigned (e.g. a three‐residue assignment of core–ECR–core would be re‐coded as core–core–core). Residues designated as ECR were not required to contact the receptor, although most do (see Results and Discussion). In a binding mode containing only one complex, all residues were classified as core. Finally, continuous core and continuous ECR regions were defined as regions with at least four contiguous core or ECR residues. On the receptor side of the interface, core‐binding sites and exosites were defined as residues that had a least one atom within 4.5 Å of any atom of a peptide core or ECR residue. Continuous core‐binding sites and continuous exosites had to be within 4.5 Å of a continuous core or continuous exosite residue, respectively. Surface residues that were classified as both core‐binding and exosite were labeled as core‐binding sites. Receptor residues not in contact with any peptide but with at least 25% surface exposed to solvent were classified as non‐interacting surface residues (NISR), and other receptor residues were classified as interior residues. For receptors with more than one binding mode, residues designated as ECR with respect to a first binding mode can also be core residues for a second binding mode. To avoid such residues distorting the ECR statistics we report here, we introduced a dual category. Residues designated as dual are indicated in the database with “d,” and complexes including peptides with dual residues were not included when compiling statistics reported in the results. The criteria for a residue to be marked as dual was that it be labeled as ECR, yet have its alpha carbon positioned within 2.5 Å of a core residue alpha carbon from another binding mode.
Database statistics and organization
Each PixelDB entry was characterized using multiple descriptors and is reported in a comma separated values (CSV) file in which each entry corresponds to a unique protein–peptide structure. Each entry contains the PDB ID, the PDB title, publication Pubmed ID, crystal structure resolution, receptor structural classification according to CATH,30 sequence domain annotation using PFAM,31 and gene annotation using Uniprot32 obtained from SIFTS.33 The peptide–protein complex structures are described by the chain IDs and lengths of the receptor chains, and by the chain ID and the sequence of the crystalized peptide as recorded in the PDB SEQRES field. The part of the peptide sequence that is resolved in the structure, which is sometimes shorter, is also given. The assigned receptor cluster and unique binding mode ID are included. Each peptide residue was given a STRIDE secondary structure assignment (Bridge, Coil, Strand, Helix310, AlphaHelix, Turn), a binned value between 0 and 9 reflecting the percent of surface exposed to solvent (9 fully exposed; 0 fully buried) and a core, ECR or dual classification; capital letters are used to denote a continuous region. Finally, b‐factor values, normalized for each complex, are reported for each peptide residue using a value between 0 and 9, where 9 represents 3 standard deviations or more above the mean, and 0 represents 3 standard deviations or more below the mean. In order to simplify b‐factor representation in the database, values were rounded to the nearest integer. The length of the longest continuous peptide core or ECR region is reported. Receptor surface descriptors include designation of residues as core‐binding site or exosite, and receptor secondary structure content and amino acid composition.
PixelDB is organized around the principle of receptor conformation clustering and peptide binding mode. It is therefore structured in a series of directories, where each directory bears a receptor cluster number and contains: all the protein–peptide complexes in that cluster in PDB format (re‐processed), CSV files that describe the cluster and the pairwise receptor sequence identities, and a PML file that allows easy color‐based PyMol visualization of the core binding sites and exosites of each binding mode. PDB files are named as: PDB ID followed by the receptor chain(s), peptide chain, cluster number, and binding mode number. For example, 4H26_DE_F_6_1.pdb comes from PDB 4H26, and is part of cluster 6 and binding mode 1; chains D and E are receptor chains, and chain F is the peptide. All files constituting the database and its analysis can be downloaded from a GitHub repository: https://github.com/KeatingLab/PixelDB under the MIT license.
Statistical analysis
We analyzed the distribution of amino acids, solvent‐exposed surface area, secondary structure content, and normalized b‐factors for categories of residues as follows: peptide ECR, peptide core, receptor core‐binding site, receptor exosite, receptor NISR, and receptor interior. Values were obtained for each binding mode and a bootstrapped average was reported (from 10,000 iterations of sampling with replacement from the binding modes). As indicated above, complexes with residues designated as dual were removed for this analysis; the observed general trends were not affected by this additional filtering step. Full computational details of the analysis are provided in a Jupyter (http://jupyter.org) notebook file included in the GitHub repository.
Results and Discussion
Protein complex identification and annotation
An overview of the steps used to create PixelDB is shown in Figure 1(A). The initial query and filtering of the PDB returned 3548 structures that encompassed 4832 potential peptide chains with fewer than 50 residues. Assigning each chain as peptide or receptor/protein binding partner is non‐trivial. In previous studies this was done using manual assignment or chain‐length cutoffs (see Table 1). We used machine learning to assist with the classification, as described in the Methods section, leading to identification of 4385 complexes of a peptide bound to a least one receptor chain. The use of machine learning led to the inclusion of longer peptides and shorter receptors than would be obtained by simple chain‐length criteria. For example, the longest chain assigned as peptide in PixelDB has 50 residues, and the shortest receptor has 36 residues (Supplementary Information, Fig. S1). Including longer peptides increases the coverage of exosite‐binding peptides, which on average have a length of 19.9 residues (Supplementary Information, Fig. S2).
Among the 4385 candidate complexes, 1424 peptides had more than 20% of their surface area in contact with a crystallographic symmetry mate of the complex and were therefore filtered out. An additional 194 peptides were removed because they had minimal surface area buried with the receptor chain(s), and 100 were removed because they engaged in peptide–peptide interactions. Finally, 658 sequence‐redundant complexes were removed. The 1966 remaining entries in PixelDB were grouped into 486 receptor clusters. The average pairwise sequence identity within a cluster is, on average, 77% (see distribution in Supplementary Information, Fig. S3). 687 unique binding modes were identified.
275 of these binding modes, represented by 1271 complexes, contained at least two structures. 122 binding modes had at least one entry with both continuous ECR and continuous core of length four or more residues, for a total of 321 complexes. 198 of these complexes contained ECR sites that were reclassified as “dual” and were therefore not included in analyses described below. Including versus removing complexes with dual sites did not affect the general trends identified in the results. A final subset of 123 complexes and 76 unique binding modes was used for analysis of core, core‐binding site, exosite, and ECR properties. Examples of complexes with peptide core and ECR regions, and of receptor clusters that contain multiple binding modes, are shown in Figures 2 and 3.
Figure 2.
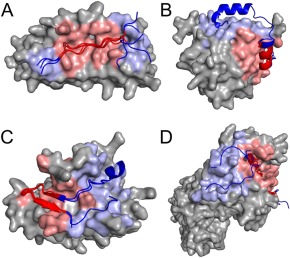
Examples of peptide binding modes containing one or more ECRs and exosite(s). (A) Cluster 28 binding mode 2 (ankyrin repeat), (B) Cluster 25 binding mode 1 (eukaryotic initiation factor 4E), (C) Cluster 111 binding mode 1 (apical membrane antigen 1), and (D) Cluster 9 binding mode 1 (major histocompatibility complex). Peptides are shown in cartoon representation and one receptor of each cluster is shown using a surface representation. Core residues and core‐binding sites are shown in red and residues in ECRs or exosites are shown in blue. Representative PDB IDs for each binding mode are, respectively: 3UXG,35 5ABY,36 3RSI 37 and 4H25.38
Figure 3.
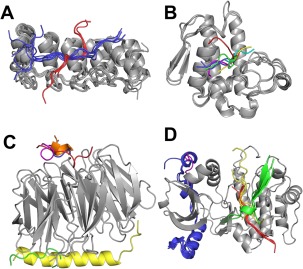
Examples of different binding modes within one receptor cluster. (A) Cluster 28 (tankyrase and ankyrin), (B) Cluster 53 (phospholipase A2), (C) Cluster 11 (WD‐repeat), and (D) Cluster 13 (protein kinase). Receptors (gray) are bound to different peptides in different binding modes. Peptides within the same binding mode are shown using the same color. To simplify representation, only one peptide per binding mode and one receptor chain is shown in C and D. Representative PDB IDs for each receptor are, respectively: 3TWR,39 3JTI, 4J81,40 and 1NVS.41
Our structural definitions mean that residues can potentially be designated as ECR based on decisions made by crystallographers. For example, given the same diffraction data, one author might decide to model only the best‐resolved part of a peptide while another author might decide to model most of it, perhaps with high b‐factors. In such a case, the uncertain part will be identified as an ECR in our analysis. If this type of distinction were prevalent in PixelDB, we might expect to see greater discrepancies between the lengths of crystallized peptides versus resolved peptides in complexes that contain ECR. However, this is not the case. The difference between the length of the crystallized versus resolved peptides is not significantly different for entries that contain versus do not contain continuous ECR (T‐test P‐value = 0.88). After adjusting for binding mode size, resolved segments in peptides that contain vs. do not contain ECR are, respectively, 6.5 and 6.3 residues shorter than the length of the peptide used for crystallization.
The influence of the crystal lattice is a significant concern when using crystal structures to study the conformation of peptides bound to proteins. If a peptide contacts adjacent complexes in the crystal, this could perturb its conformation compared to the interactions expected in solution. To our knowledge, none of the previously published protein–peptide databases have controlled systematically for crystal packing artifacts, even though these may have large effects using observed binding poses. Our database‐building process revealed that such contacts are abundant, and parameterization of computational methods using observed binding poses, without taking crystal packing into account, is likely to introduce biases. One approach to this problem would be to include the contacting chain in the symmetry mate as part of the complex. But because such contacts are not likely to be biologically relevant, we chose to remove them instead. Even a moderate threshold for removal, for example removal of all peptides that bury more than 20% surface area with symmetry mates, resulted in removing almost a third of the initial database. A more stringent threshold of 5% surface‐area burial would have removed more than two‐thirds of the initial database (2991 entries out of 4385 initial peptide‐protein complexes) [Fig. 1(B)]. Ultimately, we used the 20% cutoff as a compromise. This analysis illustrates some of the challenges associated with using X‐ray structures to define peptide binding geometries.
The contents of PixelDB are described in Table 2. The database has relatively low coverage of known domains. It includes proteins with 377 unique PFAM IDs, 758 unique Uniprot IDs, and 328 unique CATH superfamily assignments. These represent, respectively, 5.0%, 2.3%, and 4.6% of the total unique entries found in the PDB. The coverage is even lower in complexes that provide examples of exosites, which represent 93 (1.2%), 168 (0.5%), and 87 (1.2%) unique PFAM, Uniprot, and CATH entries, respectively. These results are consistent with the observation of Kuang et al.,19 who noted that protein–peptide interactions are generally underrepresented in the PDB. PixelDB coverage of structural diversity is lower than that of some other databases, due to the more stringent cutoffs applied that decrease the number of examples but increase the quality of complexes.
Table 2.
Features of PixelDB
| Feature | Mean | Median | Min:Max |
|---|---|---|---|
| No. of structures per receptor cluster/binding mode | 4.0/2.7 | 1/1 | 1:291/1:171 |
| No. of binding modes per receptor cluster | 1.5 | 1 | 1:45 |
| No. of unique CATH superfamilies per receptor cluster/binding mode | 1.3/1.3 | 1/1 | 0:7/0:7 |
| No. of unique PFAM families per receptor cluster/binding mode | 1.2/1.1 | 1/1 | 0:10/0:10 |
| No. of unique Uniprot IDs per receptor cluster/binding mode | 1.8/1.4 | 1/1 | 0:46/0:31 |
| Avg. pairwise sequence identity per receptor cluster/binding mode (%) | 75/83 | 89/95 | 13:100/14:100 |
| Length of peptide (AA) | 14.1 | 11 | 5:50 |
| Length of receptor (AA) | 317.1 | 266 | 36:1360 |
| Longest continuous core segment (AA) | 12.9 | 10 | 3:50 |
| Longest continuous ECR segment (AA) | 0.86 | 0 | 0:35 |
Protein interaction domains with multiple peptide binding modes
Clustering protein complexes by receptor structure allowed us to identify instances in which the same receptor fold engaged peptide ligands differently. A total of 93 out of the 486 receptor clusters had more than one binding mode. By our definition, a different binding mode does not require that a peptide interact in a different pocket or change binding orientation, although we do include 21 cases in which the same receptor (same PDB ID and receptor chain) engages different peptides in different regions on its surface. The stringent definition of binding mode (four consecutive residues that are structurally conserved) means that we include examples in which two or more peptides engage the same binding pocket but with a conformation that is detectably, yet perhaps modestly, shifted or rotated.
Because receptors were clustered based on structural, not sequence, similarity, we investigated whether the proteins that use different binding modes diverge in sequence. In 37 receptor clusters that had at least two binding modes with more than two complexes per binding mode, average sequence identity of receptors was compared within or between binding modes. We found that receptors engaging peptides in the same binding mode were 80% identical in sequence (on average, corrected for cluster size), and there was on average 56% identity between receptors that bind peptides differently (Supporting Information, Fig. S3). We found many examples of protein receptors in the same family engaging peptides using different binding modes. Using PFAM ID, Uniprot ID, or CATH superfamily classification to reflect evolutionary relationships, we found that, on average, 97% of CATH homologous superfamilies, 44% of Uniprot IDs and 67% of PFAM family examples found in one binding mode were also found in at least one other. Within PixelDB, the CATH superfamilies with the most unique binding modes are the immunoglobulins (CATH 2.60.40.10), with 32 different binding modes, followed by quinoprotein amine dehydrogenase (CATH 2.130.10.10), and trypsin‐like serine proteases (CATH 2.40.10.10), each with 10 binding modes.
Features of core versus exosite interactions
Our definition of core versus ECR regions initially used a binary classification, so every peptide residue was placed in one category or the other. Residues that had properties of both core and ECR were subsequently designated as dual, as described above. Consequently, not all ECR residues contact the receptor, although most do: 91% of ECR residues have solvent‐accessible surface area <40%. Similarly, not all ECRs flank core sequences. We uncovered interesting examples, such as MHC complexes, in which peptides are anchored by N‐ and C‐terminally conserved interaction modes with more variability in the middle of the peptide. Nevertheless, most segments classified as ECR lie at the N‐ or C‐terminus of the peptide in the solved structure and thus do flank core regions. Of 123 complexes that have a continuous ECR, 117 (95%) have an N‐terminal and/or C‐terminal ECR.
We investigated whether protein–peptide contacts involving continuous core versus continuous ECR had different characteristics, because differences might be important for interpreting structural roles and could facilitate automated identification of different types of sites for protein design. First, we calculated the frequencies of different amino acid types at continuous receptor core‐binding sites versus continuous exosites versus NISR versus interior sites, which encompass respectively 2883, 643, 6365, and 27529 residues (Fig. 4). Core‐binding sites and exosites are different in composition: exosites tend to be more charged, with a higher proportion of Asp, Lys, Arg, and His. Core‐binding sites have more hydrophobic residues Leu, Met, and Phe. Compared to the amino‐acid distribution of NISRs, the composition of exosites is similar, but includes a higher fraction of Leu, Ile, and Tyr, and somewhat fewer polar residues (especially Thr, Gln). Core‐binding sites and residues in the protein interior shared similar profiles, consistent with previously published findings.42 Peptide chain differences in the amino‐acid composition of core versus ECR regions were similar to receptor core‐binding site versus exosite amino‐acid compositions (Fig. 4). Core residues are slightly more hydrophobic than ECRs (e.g. more Leu and Ile), consistent with peptide cores forming hydrophobic interactions with a hydrophobic core‐binding site. ECR in our dataset have a notably higher content of serine (ECR vs. core content of 13% vs. 5%), which we cannot explain at this time.
Figure 4.
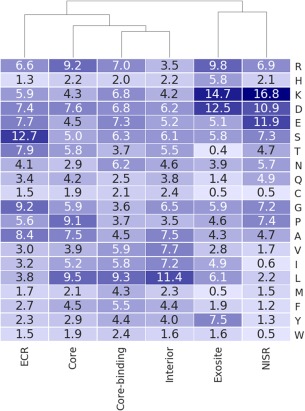
Amino‐acid composition (%) of different peptide and receptor regions. See text for definitions of different types of sites. Types of sites are clustered by compositional similarity.
When we compared the solvent accessibility of core versus ECR residues, we found a strong tendency for the core residues to be more buried and the ECR residues more exposed (Supporting Information, Table S1). This was true for residues in these regions regardless of amino‐acid type (Supporting Information, Fig. S4). Unsurprisingly, given their greater exposure, ECR tend to have higher normalized b‐factors than core residues (Supporting Information, Table S2). The b‐factors for peptide residues in PixelDB complexes are correlated well with solvent accessible surface area (Pearson correlation of 0.45), independent of their annotation (core vs. ECR). We observed that ECR regions have less helix and beta‐strand secondary structure than the core regions, and more coil (Supporting Information, Table S3). It is possible that ECR may be more flexible. However, the X‐ray structures analyzed here do not provide reliable information about flexibility and dynamics, and beyond reporting normalized b‐factors, we have not investigated that further at this time.
Conclusions
PixelDB is a database of high‐quality protein–peptide complex structures that we designed as a resource to study the roles of core‐motif flanking residues in peptide binding. Compared to complexes in other such compendia, PixelDB peptide conformations are less affected by crystal contacts. The clustering scheme that we used facilitates the identification of domains that can engage peptides in different binding geometries and provides examples where a core motif is structurally conserved but other parts of the peptide sequence make variable interactions with a receptor. We anticipate that PixelDB will find use as a benchmark for peptide binding site prediction and for peptide docking. Examples where a single type of receptor binds to peptides using different sites may provide particularly interesting and challenging tests for predictors. PixelDB will also advance work on the difficult problem of predicting and designing the structures of flexible peptide tails.15 ,43
In this work, we applied a binary classification that assigned all peptide residues as core versus ECR (although those that met both criteria were subsequently re‐labeled as dual), and all peptide‐binding regions on the protein as core‐binding or exosite regions. It is likely that ECR regions in some complexes may be flexible and dynamic, whereas in other complexes the ECR may engage the protein binding partner in a well‐defined conformation. Nevertheless, many of the extra‐motif interactions that we capture in PixelDB are likely to be important for modulating protein–peptide affinity and specificity. It is notable that Stein and Aloy estimated that as much as 88% of peptide binding energy can come from non‐motif residues in examples where a protein domain binds to a short linear motif.13 Although the crystal structures used to build PixelDB do not readily allow us to further subdivide complexes based on flexibility or dynamics, computational analysis of the energy landscapes surrounding the observed structures could be a useful way to make further distinctions.
We urge prospective users to think carefully about what types of complexes they wish to analyze when using PixelDB, and to take advantage of the annotations we provide to identify those examples that best suit their purposes. We do not wish to imply that our classification of residues as core versus ECR is absolute, nor do we intend to ascribe special roles to cores or ECRs as we have labeled them here. We do, however, hope that the availability of a large number of annotated example complexes in PixelDB will propel further investigation of the roles of structurally conserved versus non‐conserved peptide residues in biomolecular recognition.
Supporting information
Supporting Information 1
Supporting Information 2
Supporting Information 3
Acknowledgments
The authors thank NSERC and FRQNT for postdoctoral funding to V.F., and Gina De Felice and Robert M. Lefkowitz (1975) for funding to M.D. We thank all Keating lab members for insightful discussions. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Short Statement: The protein data bank (PDB) contains >100,000 solved structures and is a rich source of information about how proteins execute their functions. To provide a resource for scientists studying how proteins bind to peptides, the database PixelDB compiles 1966 high‐quality structures of protein–peptide complexes and organizes them into related clusters. The database annotates structurally conserved and non‐conserved elements in interaction interfaces and can be used to study determinants of peptide binding affinity and specificity.
References
- 1. Wright PE, Dyson HJ (2014) Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dinkel H, Van Roey K, Michael S, Kumar M, Uyar B, Altenberg B, Milchevskaya V, Schneider M, Kühn H, Behrendt A, Dahl SL, Damerell V, Diebel S, Kalman S, Klein S, Knudsen AC, Mäder C, Merrill S, Staudt A, Thiel V, Welti L, Davey NE, Diella F, Gibson TJ (2016) ELM 2016 ‐ data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res 44:D294–D300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cushing PR, Fellows A, Villone D, Boisguérin P, Madden DR (2008) The relative binding affinities of PDZ partners for CFTR: a biochemical basis for efficient endocytic recycling. Biochemistry 47:10084–10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, Sawyer TK (2013) Stapled α‐helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53‐dependent cancer therapy. Proc Natl Acad Sci USA 110:E3445–E3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Golemi‐Kotra D, Mahaffy R, Footer MJ, Holtzman JH, Pollard TD, Theriot JA, Schepartz A (2004) High affinity, paralog‐specific recognition of the Mena EVH1 domain by a miniature protein. J Am Chem Soc 126:4–5. [DOI] [PubMed] [Google Scholar]
- 6. Foight GW, Ryan JA, Gullá SV, Letai A, Keating AE (2014) Designed BH3 peptides with high affinity and specificity for targeting Mcl‐1 in cells. ACS Chem Biol 9:1962–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jenson JM, Ryan JA, Grant RA, Letai A, Keating AE (2017) Epistatic mutations in PUMA BH3 drive an alternate binding mode to potently and selectively inhibit anti‐apoptotic Bfl‐1. Elife 6:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y, Ho TG, Bertinetti D, Neddermann M, Franz E, Mo GC, Schendowich LP, Sukhu A, Spelts RC, Zhang J, Herberg FW, Kennedy EJ (2014) Isoform‐selective disruption of AKAP‐localized PKA using hydrocarbon stapled peptides. ACS Chem Biol 9:635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. London N, Movshovitz‐Attias D, Schueler‐Furman O (2010) The structural basis of peptide–protein binding strategies. Structure 18:188–199. [DOI] [PubMed] [Google Scholar]
- 10. Procko E, Berguig GY, Shen BW, Song Y, Frayo S, Convertine AJ, Margineantu D, Booth G, Correia BE, Cheng Y, Schief WR, Hockenbery DM, Press OW, Stoddard BL, Stayton PS, Baker D (2014) A computationally designed inhibitor of an Epstein‐Barr viral Bcl‐2 protein induces apoptosis in infected cells. Cell 157:1644–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Godkin AJ, Smith KJ, Willis A, Tejada‐Simon MV, Zhang J, Elliott T, Hill AVS (2001) Naturally processed HLA class II peptides reveal highly conserved immunogenic flanking region sequence preferences that reflect antigen processing rather than peptide‐MHC interactions. J Immunol 166:6720–6727. [DOI] [PubMed] [Google Scholar]
- 12. Chang CW, Counago RM, Williams SJ, Boden M, Kobe B (2013) Distinctive Conformation of minor site‐specific nuclear localization signals bound to importin‐a. Traffic 14:1144–1154. [DOI] [PubMed] [Google Scholar]
- 13. Stein A, Aloy P (2008) Contextual specificity in peptide‐mediated protein interactions. PLoS One 3:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ball LJ, Kühne R, Hoffmann B, Häfner A, Schmieder P, Volkmer‐Engert R, Hof M, Wahl M, Schneider‐Mergener J, Walter U, Oschkinat H, Jarchau T (2000) Dual epitope recognition by the VASP EVH1 domain modulates polyproline ligand specificity and binding affinity. EMBO J 19:4903–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sood VD, Baker D (2006) Recapitulation and design of protein binding peptide structures and sequences. J Mol Biol 357:917–927. [DOI] [PubMed] [Google Scholar]
- 16. Lensink MF, Velankar S, Wodak SJ (2017) Modeling protein–protein and protein–peptide complexes: CAPRI 6th edition. Proteins 85:359–377. [DOI] [PubMed] [Google Scholar]
- 17. Abriata LA (2016) Structural database resources for biological macromolecules. Brief Bioinform 18(4):659–669. [DOI] [PubMed] [Google Scholar]
- 18. Krivov GG, Shapovalov MV, Dunbrack RL (2009) Improved prediction of protein side‐chain conformations with SCWRL4. Proteins 77:778–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frishman D, Argos P (1995) Knowledge‐based protein secondary structure assignment. Proteins 23:566–579. [DOI] [PubMed] [Google Scholar]
- 20. Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E (2011) Scikit‐learn: machine learning in {P}ython. J Mach Learn Res 12:2825–2830. [Google Scholar]
- 21. Schrödinger LLC (2010) The {PyMOL} molecular graphics system, version 1.8r4.
- 22. Laskowski RA (1995) SURFNET: a program for visualizing molecular surfaces, cavities, and intermolecular interactions. J Mol Graph 13:323–330. [DOI] [PubMed] [Google Scholar]
- 23. Wang S, Ma J, Peng J, Xu J (2013) Protein structure alignment beyond spatial proximity. Sci Rep 3:1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hagberg A, Swart PJ, Schult DA, Exploring network structure, dynamics, and function using NetworkX. In: Varoquaux G, Vaught T, Millman J, Eds. (2008) Proceedings of the 7th Python in science conference (SciPy2008), Vol. 2008. Pasadena, CA USA, pp. 11–15.
- 25. Wang S, Peng J, Xu J (2011) Alignment of distantly related protein structures: algorithm, bound and implications to homology modeling. Bioinformatics 27:2537–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Das S, Lee D, Sillitoe I, Dawson NL, Lees JG, Orengo CA (2014) Functional classification of CATH superfamilies: a domain‐based approach for protein function annotation. Bioinformatics 31:3460–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pundir S, Magrane M, Martin MJ, O'Donovan C (2015) Searching and navigating UniProt databases. Curr Protoc Bioinforma 50:1.27.1–1.27.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Velankar S, Dana JM, Jacobsen J, Van Ginkel G, Gane PJ, Luo J, Oldfield TJ, O'Donovan C, Martin MJ, Kleywegt GJ (2013) SIFTS: structure integration with function, taxonomy and sequences resource. Nucleic Acids Res 41:D483–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuang X, Dhroso A, Han JG, Shyu CR, Korkin D (2016) DOMMINO 2.0: integrating structurally resolved protein‐, RNA‐, and DNA‐mediated macromolecular interactions. Database 2016:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Levy ED (2010) A simple definition of structural regions in proteins and its use in analyzing interface evolution. J Mol Biol 403:660–670. [DOI] [PubMed] [Google Scholar]
- 32. Kleiger G, Saha A, Lewis S, Kuhlman B, Deshaies RJ (2009) Rapid E2‐E3 assembly and disassembly enable processive ubiquitylation of Cullin‐RING ubiquitin ligase substrates. Cell 139:957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O'Hayre M, Gutkind JS, Hurley JH (2014) Structural and biochemical basis for ubiquitin ligase recruitment by arrestin‐related domain‐containing protein‐3 (ARRDC3) shiqian qi1. J Biol Chem 289:4743–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Das AA, Sharma OP, Kumar MS, Krishna R, Mathur PP (2013) PepBind: a comprehensive database and computational tool for analysis of protein–peptide interactions. Genomics, Proteomics Bioinforma 11:241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vanhee P, Reumers J, Stricher F, Baeten L, Serrano L, Schymkowitz J, Rousseau F (2009) PepX: a structural database of non‐redundant protein peptide complexes. Nucleic Acids Res 38:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Acharya C, Kufareva I, Ilatovskiy AV, Abagyan R (2014) PeptiSite: a structural database of peptide binding sites in 4D. Biochem Biophys Res Commun 445:717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information 1
Supporting Information 2
Supporting Information 3