

Abstract
Many proteins contain intrinsically disordered regions (IDRs), functional polypeptide segments that in isolation adopt a highly flexible conformational ensemble instead of a single, well‐defined structure. Disorder prediction methods, which can discriminate ordered and disordered regions from the amino acid sequence, have contributed significantly to our current understanding of the distinct properties of intrinsically disordered proteins by enabling the characterization of individual examples as well as large‐scale analyses of these protein regions. One popular method, IUPred provides a robust prediction of protein disorder based on an energy estimation approach that captures the fundamental difference between the biophysical properties of ordered and disordered regions. This paper reviews the energy estimation method underlying IUPred and the basic properties of the web server. Through an example, it also illustrates how the prediction output can be interpreted in a more complex case by taking into account the heterogeneous nature of IDRs. Various applications that benefited from IUPred to provide improved disorder predictions, complementing domain annotations and aiding the identification of functional short linear motifs are also described here. IUPred is freely available for noncommercial users through the web server (http://iupred.enzim.hu and http://iupred.elte.hu) . The program can also be downloaded and installed locally for large‐scale analyses.
Keywords: intrinsically disordered proteins, globular domains, statistical potential, short linear motifs, sequence‐based prediction methods
Introduction
Understanding how the structural properties of proteins are linked to their function is an important challenge in protein science. For many decades it was assumed that the proper functioning of proteins requires the formation of a well‐folded three‐dimensional structure, motivating the determination and predictions of protein structures. However, this structure‐centric view had to be revisited to accommodate intrinsically disordered proteins and proteins regions (IDPs/IDRs), a novel class of proteins whose importance has been recognized only relatively recently.1 IDRs are polypeptide segments that function by relying on highly flexible conformational states instead of a single well‐defined structure. This can enable them to function by folding upon binding to their specific biological targets, forming flexible linkers, aiding the assembly of macromolecular complexes or organizing membrane‐less organelles through phase separation.2, 3, 4 IDPs/IDRs are involved in important biological functions and are particularly enriched in proteins implicated in cell signaling and regulation.5 Disordered regions are present in the proteome of any organisms but are most prevalent in eukaryotic sequences.6, 7, 8, 9 Further supporting their biomedical importance, disordered regions have been implicated in various diseases and can also hold a yet unexploited therapeutic potential.10, 11, 12
So far, several hundreds of disordered proteins have already been characterized experimentally. Many of these have been collected into the DisProt database which provides the largest collection of proteins with disordered regions.13 Another rich source of IDRs is the Protein Data Bank (PDB).14 While this database is primarily dedicated to structured regions, missing regions in X‐ray structures or highly mobile segments in nuclear magnetic resonance (NMR) ensembles are usually taken as an indirect evidence for the presence of disorder.15 It was suggested that the longer disordered segments, such as those collected in the Disprot database, which were largely studied for their biological importance, represent a different flavor of disorder compared to shorter segments that dominate IDRs collected from the PDB.3 However, these databases sample examples that are distinct from globular proteins, and both contributed to our understanding of the basic characteristics of disordered protein regions. They also served as the basis for developing a novel class of computational tools that aim to discriminate ordered and disordered segments based on their amino acid sequence, enabling the large‐scale characterization of IDPs and study of individual examples. Another important application of disorder prediction methods is aiding structure determination efforts by enabling construct optimization.16, 17
The group of Keith Dunker had pioneered the first disorder prediction method, even before the concept of protein disorder had become widely accepted. They collected a handful of examples of disordered protein regions and compared the amino acid composition of these proteins to that of globular proteins. They noted that disordered protein regions were generally enriched in polar and charged amino acids and depleted in hydrophobic amino acids.18 The pronounced differences indicated that protein disorder is encoded in the amino acid sequence, similar to the way the structure of ordered proteins is encoded in their sequence. Their first disorder prediction method utilized a neural network using various sequence attributes as inputs.19 Since then, more than 50 different disorder prediction methods have been developed by various research groups.20, 21 One class of methods relies on simple amino acid propensity scales, such as various types of hydrophobicity scales, the propensity to participate in regular secondary structure elements, or the combination of these factors. Another group of methods utilizes more sophisticated machine learning algorithms. Many of the more recent methods of protein disorder are meta‐predictors that achieve improved predictions by combining the output of multiple disorder prediction methods. Overall, disorder can be predicted from the amino acid sequence with around 80% accuracy by top performing methods.15, 22, 23, 24 However, this number can vary depending on the type of the dataset used for the evaluation. More specific methods have also been developed to aid the functional characterization of IDPs by predicting regions that are involved in binding to proteins partners or deoxyribose nucleic acid (DNA) or ribonucleic acid (RNA) molecules, or act as linkers.25 Overall, computational methods have contributed significantly to our understanding of biological properties of IDRs.2
The specific functional, evolutionary and system level properties of disordered proteins emerge due to their specific structural properties. From a biophysical point of view, however, it is not the existence of protein disorder that is really astonishing, rather the existence of protein order: that is, the ability of specific protein sequences to adopt a well‐defined three‐dimensional structure. To achieve this, globular structures have to form a large‐number of energetically favorable inter‐residue interactions in order to overcome the entropy penalty associated with the folding. A key element of folded structures is the burial of hydrophobic residues which form the hydrophobic core. But other physical forces also contribute to the stability of proteins, including hydrogen bonds, van der Waals interactions and electrostatic forces. The stability of some proteins relies on additional factors such as disulfide bonds or interactions with small ions. The over 100,000 structures collected in the PDB database provide different manifestations of the same principles to achieve a free energy state that favors a well‐folded structure. However, only specific amino acid sequences allow the formation of a well‐defined structure. Our basic assumption was that protein regions that contain amino acids which cannot form sufficient enough favorable interactions in an ideal compact state would be disordered.26
In theory, the contributions of the various energetic terms to the stability of a conformation can be characterized using the physical energies as calculated in molecular dynamics simulations. However, existing molecular force fields are still limited in their ability to accurately capture basic properties of IDPs due to problems with accuracy and speed.27 A more robust way to characterize protein structures relies on empirical or knowledge based force fields. Such functions use a coarse‐grained approach and transform the observed frequencies of amino acid pair interactions or other characteristics into energy‐like functions based on the Boltzmann statistics.28 One of the most problematic element of this model is the reference state which accounts for the expected number of interactions.29 An elegant solution that circumvents this problem was suggested by Ken Dill who proposed an iterative algorithm that avoids making direct assumptions about the reference state.30 Coarse‐grained models based on empirical force fields have been extremely useful in various areas of structure prediction.31 However, such applications were limited to cases when a structural model was available. To overcome this limitation, we developed an energy estimation method that opened up a new way to predict ordered and disordered protein regions from the amino acid sequence.26 This approach underlines the IUPred disorder prediction method as well as ANCHOR, a method that predicts specific regions located within IDRs involved in protein binding.26, 32
The Energy Estimation Method
The energy estimation method relies on a statistical pairwise potential that was derived using the approach suggested by Thomas and Dill.30 This choice of algorithm was shown to be crucial for the optimality of the energy estimation‐based disorder prediction method.26 For the calculations, a nonredundant dataset of globular protein structures was collected from the PDB with a resolution of 2.5 Å or better. The statistical potential, expressed as a 20 × 20 matrix, characterizes the general preference of each pair of amino acids to be in contact as observed in a dataset of globular proteins. From the coordinates of known structures, it can be specified which residues are in contact. Then, the energy of each residue is calculated by considering the number and types of the contacting amino acids and summing the appropriate elements of the statistical potential matrix.
| (1) |
where energy at position p of type i, M ij is the interaction energy between amino acid types i and j, and is the number of interactions of residue at position p with residues of type j in the given conformation. Variations in these energies arise depending on the type and number of contacting residues, with hydrophobic residues buried inside the protein core generally exhibiting more favorable energies.
To eliminate the need to know the precise arrangements of residues in the structure, an energy estimation method was developed that can be used to approximate the energies directly from the amino acid sequence. For this, a crude approximation was introduced assuming that energy of a given residue mostly depends on its own type and the types of the amino acids that surround it. The key component of the calculations is the energy estimation matrix, a 20 × 20 matrix that connects the elements of the amino acid composition vector to the energy of a given residue. The energy of a given residues can be obtained by multiplying the amino acid composition vector elements with the appropriate elements of this energy predictor matrix.
| (2) |
where is estimated energy at position p of type i, Pij is the ij element of the energy predictor matrix and is the jth element of the amino acid composition vector. This amino acid composition vector is specific for position p, as it calculated by considering only the local sequential environment within 2–100 residues in either direction. The choice of this range represents a trade‐off between the intention of covering most structured domains, but separating distinct domains in multidomain proteins.26 The matrix elements were optimized using least square fitting, to minimize the difference between energies estimated from the amino acid composition vector and the energies calculated from the known structure for each residue in the dataset of proteins. The correlation between the calculated and estimated energies was surprisingly strong at the level of complete proteins, confirming the validity of this approach.26
The energy estimation method enables the approximation of the pairwise energy for each residue without relying on the structure. Applying this energy estimation method for sequences of ordered and disordered proteins, a clear separation was observed between the two datasets in terms of their energy.26 However, this separation was less pronounced for shorter sequences, contributing to a wider twilight zone between ordered and disordered proteins with smaller length.33 Overall, the results confirmed that ordered residues can be discriminated from disordered ones based on their generally more favorable estimated energies. For the actual predictions, the position‐specific estimations of energies were averaged over a window of 21 residues and transformed into a score between 0 and 1, with the 0.5 score corresponding to a threshold where 5% of the positions of globular proteins were predicted as being disordered (false positive rate). Using this limit, 76% of positions of the IDP dataset were predicted to be disordered.26 This energy estimation approach is at the core of the IUPred disorder prediction method.26, 34 The key to the robustness of IUPred is that the main parameters of the method, the elements of the energy prediction matrix, were entirely derived from globular proteins, without relying on the collection of disordered proteins. This way, the pitfalls that originate from small and noisy datasets for disordered regions could be largely avoided.
Besides IUPred, the energy estimation approach is also applied in the ANCHOR method which was developed to predict regions that are disordered in isolation but can undergo disorder‐to‐order transition upon binding.32 Disordered binding have distinct properties compared to both globular proteins and disordered regions in general. ANCHOR aims to find these regions by the combination of three scores calculated based on the energy estimation approach. The first score corresponds to a smoothed IUPred score and ensures that a given residue belongs to a generally disordered region. The other two scores aim to capture the fundamental features of disordered binding regions by assuming that such regions cannot form enough favorable intrachain interactions to fold on their own but are likely to gain stabilizing energy by interacting with a globular protein partner. In accord, the estimated energy is calculated using two different amino acid composition vectors: the first one is calculated from the local sequential neighborhood, while the second one is calculated using to the average composition of globular proteins. The estimated energy calculated on this second amino acid composition vector is expected to yield more favorable energies in case of true binding regions. The overall tendency to be in a disordered binding region is obtained by a linear combination of the three terms, with the weights optimized on a small dataset of complexes formed between ordered and disordered protein segments. The developed method was shown to recognize disordered binding regions with almost 70% accuracy at the segment level tested on various datasets, largely independent of the secondary structure element adopted in the complex. In addition, disordered binding regions could also be discriminated from generally disordered regions, and the false positive rate on a dataset of globular proteins was below 5%. ANCHOR enabled the prediction of essential functional sites of disordered proteins engaged in specific protein binding, offering an additional way to use the energy estimation methods to characterize disordered regions.32, 35
The IUPred Web Server
The IUPred web server offers a per‐residue prediction of protein disorder.34 It takes a single amino acid sequence as an input either in plain or FASTA format. Alternatively, a UniProt identifier or accession numbers can also be submitted in which case the corresponding sequences are retrieved from the Uniprot web site. The results are returned in either text or graphical format, specifying the disorder tendency of each residue along the sequence. The core program to calculate the pairwise energy profile and disorder probability was written in C, the web server was written in PHP. The graphical output is generated on the fly using the JPGraph program (JpGraph, 2005, http://jpgraph.net/), producing a figure in PNG format. The default option for graphical/text output is automatically determined by the browser type, but it can be also changed by the user. The server can only take one sequence at a time. However, there is no limit on the number of requests per IP address, so predictions can be collected for large number of sequences via programmatic access. Alternatively, the IUPred program can be downloaded and installed locally, using the source C program.
IUPred offers three prediction types corresponding to long disorder, short disorder and structured domains. The long disorder option is the recommended choice for predicting biologically relevant disordered segments, such as those collected in the DisProt database. The short disorder option offers a variation of the algorithm that was optimized to predict missing residues in PDB structures. Such disordered regions are characteristically short and are most often located at the termini of the constructs.15 Correspondingly, one of the most obvious difference between short and long disorder predictions is that in the former case the terminal residues are generally predicted as more disordered. This is a common feature of many prediction methods aimed to perform well on CASP datasets and can be useful in terms of construct design. However, the biological relevance of these types of predictions is not well‐founded. The “structured domain” option was developed to delineate regions that are most likely to correspond to self‐contained globular domains from the relatively noisy prediction profiles by eliminating short disordered segments inserted within regions that are mostly predicted as ordered.
Thanks to the relative simplicity of algorithm and the advantage of C programming language, IUPred is impressively fast. In fact, it was shown that by taking into account the trade‐off between speed and accuracy, IUPred is a particularly good choice.36 While there are methods that achieve better performance by relying on multiple sequence alignments, these programs deliver predictions almost two orders of magnitude slower compared to IUPred. For this reason, IUPred is often the favored application for large‐scale prediction of protein disorder. Taking advantage of the graphical output, IUPred predictions can be used to explore the structural features of individual proteins as well.
The Interpretation of a Prediction Output
As most disorder prediction methods, IUPred provides a score which characterizes the disordered tendency of each position along the sequence. This score can take a value between 0 and 1. Residues with a predicted score above 0.5 are considered disordered, while residues with lower scores considered to be ordered. In theory, the location of ordered and disordered segments can be easily deciphered form such prediction profile. However, due to the complexity and heterogeneity of the protein disorder phenomenon as well as to noises in the predictions, the interpretation of the disorder profiles in many cases is more challenging. To illustrate how the prediction output can be interpreted in a more complex case, the human E3 ubiquitin‐protein ligase Mdm2 (MDM2) was selected as an example. The disorder prediction profile generated by IUPred for this protein is shown on Figure 1.
Figure 1.
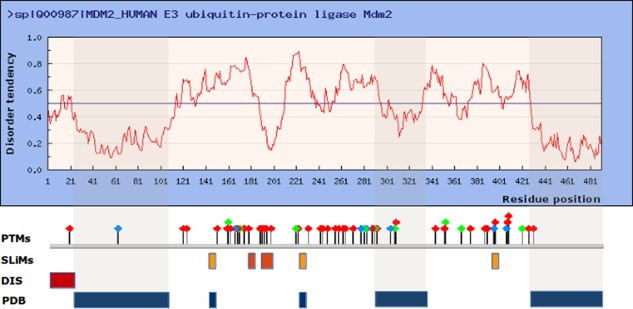
Prediction of protein disorder using the IUPred web server for the human E3 ubiquitin‐protein ligase Mdm2. The IUPred output was generated using the long option. Various structural and functional elements are shown below the prediction profile: PTMs plot various phosphorylation sites collected from PhosphositePlus,78 known linear motif sites (SLiMs) are indicated with lighter (USP7 binding motifs) and darker orange boxes (nuclear import and export signals), the experimentally verified disordered region (DIS) form the DisProt database is shown as a red box, while regions found in the PBD are indicated as blue boxes. The three regions corresponding to globular domains are shaded. Most of the phosphorylation sites and linear motifs are located outside these regions and are believed to be largely disordered, in agreement with the IUPred prediction.
MDM2 is an E3 ubiquitin ligase and one of the main cellular regulator of the p53 tumor suppressor by targeting it for proteasome‐mediated degradation through ubiquitination. MDM2 is a hub protein with over 100 protein and other macromolecular partners.37, 38 Based on known structures and domain assignments, three larger ordered segments have been located within this protein. The N‐terminal region (24–101) corresponds to the substrate binding SWIB domain, while the C‐terminal contains the RING domain (amino acid residues 430–480) that confers the catalytic ubiquitin ligase activity to the protein. MDM2 also contains a zing finger domain with unknown function (289–331). These regions are largely predicted as ordered. According to the DisProt database, the disordered nature of the first N‐terminal 24 residues was experimentally verified.39 At a closer look, this region is only partially disordered as region 18–24 forms a “lid” over part of the p53 peptide‐binding site and is displaced upon ligand binding. Regions with a similar dual character are often predicted by IUPred with scores hovering around the cutoff line. The remaining of the sequence is predicted largely disordered. These regions also mediate interactions with multiple partners, therefore contribute significantly to the large interaction capacity of MDM2.38 While there is no direct experimental evidence available to confirm the disordered status of this central region, the presence of a large number of PTMs and various short linear motifs (SLiM)37, 40—such as nuclear localization and export signals together with multiple binding sites for the USP7 (HAUSP) deubiquitinating enzyme—lends further support for this hypotheses, as such regions are usually located within IDRs. While the structure of two of the USP7 binding regions have been resolved by X‐ray crystallography,40 this cannot be taken as a proof of order, as the well‐defined conformation is only observed in a complex. The disorder tendency still shows variations within predicted IDRs indicating the presence of some locally more ordered segments. Such behavior is usually associated with binding sites that can undergo a disorder‐to‐order transition upon binding or with segments that adopt more compact but still flexible conformational states. One example for this can be seen around residue 200, showing a sharp dip in the IUPred prediction profile. The more ordered tendency of this region is also supported by multiple predictions collected in the MobiDB database.41 The actual segment was suggested to be involved in multiple protein interactions and overlaps with the nuclear export signal.38
When individual proteins are studied, the main strategy is to confirm disorder predictions by multiple methods and integrate information from the PDB and DisProt databases, domain and family annotations in addition to linear motif and PTM sites.42 As the presented example indicates, the disorder prediction provides a good starting point for exploring the main structural features of a protein, but many further studies are required to fully understand how regions with differing structural properties contribute to the molecular function of a protein.
IUPred Integrated Into Other Approaches
IUPred has inspired other methods of protein disorder predictions. Specifically, the UCON method follows similar principles compared to IUPred but it relies on a direct prediction of contacts based on a machine learning approach.43 A novel approach to estimate position specific estimated energy (PSEE) of a residue using contact energy and predicted relative solvent accessibility was also incorporated into the DisPredict2 method.44 IUPred is a frequent choice as one of contributing programs of various meta predictors of protein disorder,42 as it provides an orthogonal approach to most methods which were trained on either short or long disordered segments.45, 46, 47, 48, 49, 50 A particular application of disorder prediction methods is the optimization of construct design, for which IUPred has been used independently51 or as part of the DisMeta server.17 IUPred predictions have been also incorporated into novel databases that aim to provide genome level annotation of protein disorder. Among such databases, D2P2 collects predictions generated by multiple programs for a large number of available genome sequences.52 MobiDB provides disorder predictions for sequences available through the UniProt database and it is regularly updated to keep up with novel sequences.41 Both resources incorporate additional information, such as PTMs or known structures and provide their own consensus predictions.
By going beyond the general prediction of disordered regions, some methods aim to identify the specific functions these regions are involved in. Dedicated methods have been developed to predict linker regions, disordered DNA and RNA binding segments, and binding regions that undergo disorder‐to‐order transitions.53, 54, 55, 56 All these methods are based on machine learning approaches that use a combination of calculated and predicted sequence features, including disorder predictions generated by IUPred. The prediction of bioactive peptides—which can play important roles in signalling, regulation and immunity within an organism and also potentially carry therapeutic possibilities—represent an additional area where IUPred was found useful.57
Another major applications of disorder prediction methods, and IUPred in particular, is the identification of functionally relevant SLiM sites. SLiMs are generally defined by a specific sequence pattern—usually expressed as a regular expression—that contains the key amino acids required to bind to a given domain.58 The known motif can be used to find additional binding partners for a domain by scanning sequences of various proteins. However, due to the low information content of such motifs, matches can also occur purely by chance. Disorder prediction methods are used to reduce the number of potential false positive hits by ensuring that putative motifs hits reside within disordered regions, hence they are accessible for interactions and regulatory modifications.59 The Eukaryotic Linear Motif server, the largest collection of SLiMs and a motif prediction tool, enables the filtering of motif hits by providing the output of IUPred.60 Such filtering was also found useful in the interpretation of phosphorylation data extracted from the scientific literature and phosphoproteomic analyses.61 Various other motif‐centric tools, such as SLiMPrints or SLiMSearch, also rely on IUPred.62, 63 In these applications, a lower cutoff of 0.4 is suggested, to allow for a locally more ordered tendency that disordered binding regions often possess. ModPepInt, a method that offers predictions of binding partners for specific domains, such as SH3, SH2 and PDZ domains, also uses IUPred as a filter in a similar way.64
Disorder predictions are also be used to complement sequence family and domain annotations. Compared to globular domains that are usually evolutionary well conserved, IDRs in general can accommodate more sequence variations over time due to the lack of structural constraints.2 Conserved disordered regions have been observed in a relatively few cases, such as the kinase‐inhibitory domain of Cdk inhibitors or the Wiskott–Aldrich syndrome protein‐homology domain 2 of actin‐binding proteins.65 While it was recently suggested that protein disorder might not be the dominant factor behind the lack of sequence family annotation,66 the application of disorder prediction methods can direct the attention to ordered regions that are more likely to yield novel sequence families. The Pfam site, one of the most commonly used family annotation tool, uses IUPred as its disorder prediction method to complement its annotations together with prediction of coiled coil regions and low complexity segments.67 Similarly, the HMMer web server which generates the profile hidden markov models underlying Pfam annotations now also incorporates information about putative disordered regions according to IUPred.68 Besides speed, the other main requirements for such commonly used annotation tools, is low false positive rate. To improve on IUPred in this regard, it was combined with other tools to create a novel method, called MobiDB‐Lite.69 This fast and accurate method was integrated into the InterPro sequence family and domain annotation tool,70 and through this, it provides information for billions of sequences collected in the UniProt database with no additional annotation. The importance of combining sequence family annotation with predicted disorder is further underlined by the recent application of such information in the interpretation of cancer genome data. This type of information is used to visualize and analyze the structural properties of specific regions that are enriched in cancer mutations indicating critical functional sites targeted by the disease.71, 72 IUPred and ANCHOR predictions were also found to provide useful contribution to enhance the prediction of phenotype of single amino acid variants.73 The list of various approaches that take advantage of the energy estimation approach or directly the output of IUPred predictions is given in Table 1.
Table 1.
List of Prediction Tools Based on an Energy Estimation Approach or Using IUPred Predictions
Future Directions
Current prediction methods, including IUPred, are based on a binary classification of order and disorder. However, protein disorder is a heterogeneous phenomenon that encompasses various conformational states ranging from random coil‐like to molten globule‐like states with increasing transient secondary and tertiary elements.2 The length and position of discorded segments also vary, and include fully disordered proteins, longer disordered segments, domain linkers and loop regions. Disordered protein regions can be accurately described only in terms of conformational ensembles. In recent years, experimental methods have made significant advances to capture various properties of the conformational ensemble using small‐angle X‐ray scattering, fluorescence resonance energy transfer, and various types of NMR measurements. The experiments were complemented with various computational approaches to generate a set of conformations that conformed to the experimental constraints.74 The PED database was launched to systematically collect the generated conformational ensembles and to initiate their comparisons and analyses and to promote standardization and further improvements in this field.75 However, currently only a limited number of proteins have been characterized in such a detailed way. Computational methods that would enable the large‐scale characterization of the conformational ensembles of disordered proteins are lagging behind. One of the main challenges of future disorder prediction algorithms it to be able to recognize not only disordered regions in general but also their detailed properties, such as the presence of transient secondary structure elements or overall molecular dimension.76 Another frontier is the prediction of functionally important regions located within IDRs. While there are several methods that can recognize protein binding regions, DNA or RNA binding sites or linker regions,25 the performance of these methods falls behind that of general disorder prediction methods. A particularly challenging subset of IDPs corresponds to conditionally disordered segments: regions whose disordered status is regulated via specific physiological conditions such as pH, reducing agents or post‐translational modifications.77 Methods that would enable the characterization of the more detailed structural properties of IDPs would greatly advance our understanding of the functional properties of IDRs. In the future, the energy estimation method underlying IUPred might be able to address these problems as well.
Conflict of Interest
The author declares no conflict of interest.
References
- 1. Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: re‐assessing the protein structure‐function paradigm. J Mol Biol 293:321–331. [DOI] [PubMed] [Google Scholar]
- 2. van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM (2014) Classification of intrinsically disordered regions and proteins. Chem Rev 114:6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z (2001) Intrinsically disordered protein. J Mol Graph Model 19:26–59. [DOI] [PubMed] [Google Scholar]
- 4. Tompa P (2012) Intrinsically disordered proteins: a 10‐year recap. Trends Biochem Sci 37:509–516. [DOI] [PubMed] [Google Scholar]
- 5. Wright PE, Dyson HJ (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Romero P, Obradovic Z, Kissinger CR, Villafranca JE, Garner E, Guilliot S, Dunker AK (1998) Thousands of proteins likely to have long disordered regions. Pac Symp Biocomput 437–448. [PubMed] [Google Scholar]
- 7. Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 337:635–645. [DOI] [PubMed] [Google Scholar]
- 8. Peng Z, Yan J, Fan X, Mizianty MJ, Xue B, Wang K, Hu G, Uversky VN, Kurgan L (2015) Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol Life Sci 72:137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xue B, Dunker AK, Uversky VN (2012) Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn 30:137–149. [DOI] [PubMed] [Google Scholar]
- 10. Babu MM (2016) The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans 44:1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corbi‐Verge C, Kim PM (2016) Motif mediated protein‐protein interactions as drug targets. Cell Commun Signal 14:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu G, Wu Z, Wang K, Uversky VN, Kurgan L (2016) Untapped potential of disordered proteins in current druggable human proteome. Curr Drug Targets 17:1198–1205. [DOI] [PubMed] [Google Scholar]
- 13. Piovesan D, Tabaro F, Mičetić I, Necci M, Quaglia F, Oldfield CJ, Aspromonte MC, Davey NE, Davidović R, Dosztányi Z, Elofsson A, Gasparini A, Hatos A, Kajava AV, Kalmar L, Leonardi I, Lazar T, Macedo‐Ribeiro S, Macossay‐Castillo M, Meszaros A, Minervini G, Murvai N, Pujols J, Roche DB, Salladini E, Schad E, Schramm A, Szabo B, Tantos A, Tonello F, Tsirigos KD, Veljkovic N, Ventura S, Vranken W, Warholm P, Uversky VN, Dunker AK, Longhi S, Tompa P, Tosatto SCE (2017) DisProt 7.0: a major update of the database of disordered proteins. Nucleic Acids Res 45:D219–D227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rose PW, Prlić A, Altunkaya A, Bi C, Bradley AR, Christie CH, Costanzo LD, Duarte JM, Dutta S, Feng Z, Kramer Green R, Goodsell DS, Hudson B, Kairo T, Lowe R, Peisach E, Randle C, Rose AS, Shao C, Tao Y‐P, Valasatava Y, Voigt M, Westbrook JD, Woo J, Yang H, Young JY, Zardecki C, Berman HM, Burley SK (2017) The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res 45:D271–D281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Monastyrskyy B, Kryshtafovych A, Moult J, Tramontano A, Fidelis K (2014) Assessment of protein disorder region predictions in CASP10. Proteins 82:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oldfield CJ, Xue B, Van Y‐Y, Ulrich EL, Markley JL, Dunker AK, Uversky VN (2013) Utilization of protein intrinsic disorder knowledge in structural proteomics. Biochim Biophys Acta 1834:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang YJ, Acton TB, Montelione GT (2014) DisMeta: a meta server for construct design and optimization. Methods Mol Biol 1091:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xie Q, Arnold GE, Romero P, Obradovic Z, Garner E, Dunker AK (1998) The sequence attribute method for determining relationships between sequence and protein disorder. Genome Inform Ser Workshop Genome Inform 9:193–200. [PubMed] [Google Scholar]
- 19. Garner E, Cannon P, Romero P, Obradovic Z, Dunker AK (1998) Predicting disordered regions from amino acid sequence: Common themes despite differing structural characterization. Genome Inform Ser Workshop Genome Inform 9:201–213. [PubMed] [Google Scholar]
- 20. He B, Wang K, Liu Y, Xue B, Uversky VN, Dunker AK (2009) Predicting intrinsic disorder in proteins: an overview. Cell Res 19:929–949. [DOI] [PubMed] [Google Scholar]
- 21. Dosztányi Z, Mészáros B, Simon I (2010) Bioinformatical approaches to characterize intrinsically disordered/unstructured proteins. Brief Bioinform 11:225–243. [DOI] [PubMed] [Google Scholar]
- 22. Peng Z‐L, Kurgan L (2012) Comprehensive comparative assessment of in‐silico predictors of disordered regions. Curr Protein Pept Sci 13:6–18. [DOI] [PubMed] [Google Scholar]
- 23. Walsh I, Giollo M, Di Domenico T, Ferrari C, Zimmermann O, Tosatto SCE (2015) Comprehensive large‐scale assessment of intrinsic protein disorder. Bioinformatics 31:201–208. [DOI] [PubMed] [Google Scholar]
- 24. Necci M, Piovesan D, Dosztanyi Z, Tompa P, Tosatto SCE (2017) A comprehensive assessment of long intrinsic protein disorder from the DisProt database. Bioinformatics. Available from: https://doi.org/10.1093/bioinformatics/btx590 [DOI] [PubMed] [Google Scholar]
- 25. Meng F, Uversky VN, Kurgan L (2017) Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell Mol Life Sci 74:3069–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dosztányi Z, Csizmók V, Tompa P, Simon I (2005) The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol 347:827–839. [DOI] [PubMed] [Google Scholar]
- 27. Piana S, Donchev AG, Robustelli P, Shaw DE (2015) Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J Phys Chem B 119:5113–5123. [DOI] [PubMed] [Google Scholar]
- 28. Koppensteiner WA, Sippl MJ (1998) Knowledge‐based potentials–back to the roots. Biochemistry 63:247–252. [PubMed] [Google Scholar]
- 29. Thomas PD, Dill KA (1996) Statistical potentials extracted from protein structures: how accurate are they?. J Mol Biol 257:457–469. [DOI] [PubMed] [Google Scholar]
- 30. Thomas PD, Dill KA (1996) An iterative method for extracting energy‐like quantities from protein structures. Proc Natl Acad Sci USA 93:11628–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kmiecik S, Gront D, Kolinski M, Wieteska L, Dawid AE, Kolinski A (2016) Coarse‐grained protein models and their applications. Chem Rev 116:7898–7936. [DOI] [PubMed] [Google Scholar]
- 32. Mészáros B, Simon I, Dosztányi Z (2009) Prediction of protein binding regions in disordered proteins. PLoS Comput Biol 5:e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Szilágyi A, Györffy D, Závodszky P (2008) The twilight zone between protein order and disorder. Biophys J 95:1612–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dosztányi Z, Csizmok V, Tompa P, Simon I (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21:3433–3434. [DOI] [PubMed] [Google Scholar]
- 35. Dosztányi Z, Mészáros B, Simon I (2009) ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics 25:2745–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walsh I, Martin AJM, Di Domenico T, Tosatto SCE (2012) ESpritz: accurate and fast prediction of protein disorder. Bioinformatics 28:503–509. [DOI] [PubMed] [Google Scholar]
- 37. Meek DW, Knippschild U (2003) Posttranslational modification of MDM2. Mol Cancer Res 1:1017–1026. [PubMed] [Google Scholar]
- 38. Fåhraeus R, Olivares‐Illana V (2014) MDM2's social network. Oncogene 33:4365–4376. [DOI] [PubMed] [Google Scholar]
- 39. Uhrinova S, Uhrin D, Powers H, Watt K, Zheleva D, Fischer P, McInnes C, Barlow PN (2005) Structure of free MDM2 N‐terminal domain reveals conformational adjustments that accompany p53‐binding. J Mol Biol 350:587–598. [DOI] [PubMed] [Google Scholar]
- 40. Sarkari F, La Delfa A, Arrowsmith CH, Frappier L, Sheng Y, Saridakis V (2010) Further insight into substrate recognition by USP7: structural and biochemical analysis of the HdmX and Hdm2 interactions with USP7. J Mol Biol 402:825–837. [DOI] [PubMed] [Google Scholar]
- 41. Potenza E, Di Domenico T, Walsh I, Tosatto SCE (2015) MobiDB 2.0: an improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res 43:D315–D320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Punta M, Simon I, Dosztányi Z (2015) Prediction and analysis of intrinsically disordered proteins. Methods Mol Biol 1261:35–59. [DOI] [PubMed] [Google Scholar]
- 43. Schlessinger A, Punta M, Rost B (2007) Natively unstructured regions in proteins identified from contact predictions. Bioinformatics 23:2376–2384. [DOI] [PubMed] [Google Scholar]
- 44. Iqbal S, Hoque MT (2016) Estimation of position specific energy as a feature of protein residues from sequence alone for structural classification. PLoS One 11:e0161452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schlessinger A, Punta M, Yachdav G, Kajan L, Rost B (2009) Improved disorder prediction by combination of orthogonal approaches. PLoS One 4:e4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mizianty MJ, Peng Z, Kurgan L (2013) MFDp2: Accurate predictor of disorder in proteins by fusion of disorder probabilities, content and profiles. Intrinsically Disord Proteins 1:e24428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kozlowski LP, Bujnicki JM (2012) MetaDisorder: a meta‐server for the prediction of intrinsic disorder in proteins. BMC Bioinformatics 13:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mizianty MJ, Stach W, Chen K, Kedarisetti KD, Disfani FM, Kurgan L (2010) Improved sequence‐based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 26:i489–i496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lieutaud P, Canard B, Longhi S (2008) MeDor: a metaserver for predicting protein disorder. BMC Genomics 9:S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ishida T, Kinoshita K (2008) Prediction of disordered regions in proteins based on the meta approach. Bioinformatics 24:1344–1348. [DOI] [PubMed] [Google Scholar]
- 51. Punta M, Love J, Handelman S, Hunt JF, Shapiro L, Hendrickson WA, Rost B (2009) Structural genomics target selection for the New York consortium on membrane protein structure. J Struct Funct Genomics 10:255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker AK, Gough J (2013) D2P2: database of disordered protein predictions. Nucleic Acids Res 41:D508–D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meng F, Kurgan L (2016) DFLpred: High‐throughput prediction of disordered flexible linker regions in protein sequences. Bioinformatics 32:i341–i350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Peng Z, Kurgan L (2015) High‐throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res 43:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yan J, Dunker AK, Uversky VN, Kurgan L (2016) Molecular recognition features (MoRFs) in three domains of life. Mol. Biosyst 12:697–710. [DOI] [PubMed] [Google Scholar]
- 56. Disfani FM, Hsu W‐L, Mizianty MJ, Oldfield CJ, Xue B, Dunker AK, Uversky VN, Kurgan L (2012) MoRFpred, a computational tool for sequence‐based prediction and characterization of short disorder‐to‐order transitioning binding regions in proteins. Bioinformatics 28:i75–i83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mooney C, Haslam NJ, Holton TA, Pollastri G, Shields DC (2013) PeptideLocator: prediction of bioactive peptides in protein sequences. Bioinformatics 29:1120–1126. [DOI] [PubMed] [Google Scholar]
- 58. Van Roey K, Uyar B, Weatheritt RJ, Dinkel H, Seiler M, Budd A, Gibson TJ, Davey NE (2014) Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem Rev 114:6733–6778. [DOI] [PubMed] [Google Scholar]
- 59. Gibson TJ, Dinkel H, Van Roey K, Diella F (2015) Experimental detection of short regulatory motifs in eukaryotic proteins: tips for good practice as well as for bad. Cell Commun Signal 13:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dinkel H, Van Roey K, Michael S, Kumar M, Uyar B, Altenberg B, Milchevskaya V, Schneider M, Kühn H, Behrendt A, Dahl SL, Damerell V, Diebel S, Kalman S, Klein S, Knudsen AC, Mader C, Merrill S, Staudt A, Thiel V, Welti L, Davey NE, Diella F, Gibson TJ (2016) ELM 2016–data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res 44:D294–D300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F (2011) Phospho.ELM: a database of phosphorylation sites–update 2011. Nucleic Acids Res 39:D261–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Krystkowiak I, Davey NE (2017) SLiMSearch: a framework for proteome‐wide discovery and annotation of functional modules in intrinsically disordered regions. Nucleic Acids Res. Available from: https://doi.org/10.1093/nar/gkx238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Davey NE, Cowan JL, Shields DC, Gibson TJ, Coldwell MJ, Edwards RJ (2012) SLiMPrints: conservation‐based discovery of functional motif fingerprints in intrinsically disordered protein regions. Nucleic Acids Res 40:10628–10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kundu K, Mann M, Costa F, Backofen R (2014) MoDPepInt: an interactive web server for prediction of modular domain‐peptide interactions. Bioinformatics 30:2668–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tompa P, Fuxreiter M, Oldfield CJ, Simon I, Dunker AK, Uversky VN (2009) Close encounters of the third kind: disordered domains and the interactions of proteins. Bioessays 31:328–335. [DOI] [PubMed] [Google Scholar]
- 66. Perdigão N, Heinrich J, Stolte C, Sabir KS, Buckley MJ, Tabor B, Signal B, Gloss BS, Hammang CJ, Rost B, Schafferhans A, O'Donoghue SI (2015) Unexpected features of the dark proteome. Proc Natl Acad Sci USA 112:15898–15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador‐Vegas A, Salazar GA, Tate J, Bateman A (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR (2015) HMMer web server: 2015 update. Nucleic Acids Res 43:W30–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Necci M, Piovesan D, Dosztányi Z, Tosatto SCE (2017) MobiDB‐lite: fast and highly specific consensus prediction of intrinsic disorder in proteins. Bioinformatics 33:1402–1404. [DOI] [PubMed] [Google Scholar]
- 70. Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, Chang H‐Y, Dosztányi Z, El‐Gebali S, Fraser M, Gough J, Haft D, Holliday GL, Huang H, Huang X, Letunic I, Lopez R, Lu S, Marchler‐Bauer A, Mi H, Mistry J, Natale DA, Necci M, Nuka G, Orengo CA, Park Y, Pesseat S, Piovesan D, Potter SC, Rawlings ND, Redaschi N, Richardson L, Rivoire C, Sangrador‐Vegas A, Sigrist C, Sillitoe I, Smithers B, Squizzato S, Sutton G, Thanki N, Thomas PD, Tosatto SCE, Wu CH, Xenarios I, Yeh L‐S, Young S‐Y, Mitchell AL (2017) InterPro in 2017‐beyond protein family and domain annotations. Nucleic Acids Res 45:D190–D199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, Jia M, Jubb H, Sondka Z, Thompson S, De T, Campbell PJ (2017) COSMIC: somatic cancer genetics at high‐resolution. Nucleic Acids Res 45:D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mészáros B, Zeke A, Reményi A, Simon I, Dosztányi Z (2016) Systematic analysis of somatic mutations driving cancer: uncovering functional protein regions in disease development. Biol Direct 11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yates CM, Filippis I, Kelley LA, Sternberg MJE (2014) SuSPect: enhanced prediction of single amino acid variant (SAV) phenotype using network features. J Mol Biol 426:2692–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fisher CK, Stultz CM (2011) Constructing ensembles for intrinsically disordered proteins. Curr Opin Struct Biol 21:426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Varadi M, Tompa P (2015) The Protein Ensemble Database. Adv Exp Med Biol 870:335–349. [DOI] [PubMed] [Google Scholar]
- 76. Sormanni P, Piovesan D, Heller GT, Bonomi M, Kukic P, Camilloni C, Fuxreiter M, Dosztanyi Z, Pappu RV, Babu MM, Longhi S, Tompa P, Dunker AK, Uversky VN, Tosatto SCE, Vendruscolo M (2017) Simultaneous quantification of protein order and disorder. Nat Chem Biol 13:339–342. [DOI] [PubMed] [Google Scholar]
- 77. Jakob U, Kriwacki R, Uversky VN (2014) Conditionally and transiently disordered proteins: awakening cryptic disorder to regulate protein function. Chem Rev 114:6779–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43:D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]