

Abstract
Cryo‐electron microscopy (cryoEM) is becoming popular as a tool to solve biomolecular structures with the recent availability of direct electron detectors allowing automated acquisition of high resolution data. The Bsoft software package, developed over 20 years for analyzing electron micrographs, offers a full workflow for validated single particle analysis with extensive functionality, enabling customization for specific cases. With the increasing use of cryoEM and its automation, proper validation of the results is a bigger concern. The three major validation approaches, independent data sets, resolution‐limited processing, and coherence testing, can be incorporated into any Bsoft workflow. Here, the main workflow is divided into four phases: (i) micrograph preprocessing, (ii) particle picking, (iii) particle alignment and reconstruction, and (iv) interpretation. Each of these phases represents a conceptual unit that can be automated, followed by a check point to assess the results. The aim in the first three phases is to reconstruct one or more validated maps at the best resolution possible. Map interpretation then involves identification of components, segmentation, quantification, and modeling. The algorithms in Bsoft are well established, with future plans focused on ease of use, automation and institutionalizing validation.
Keywords: Micrograph frame alignment, contrast transfer function, particle picking, 3D reconstruction, cryo‐electron microscopy, micrograph processing, segmentation
Abbreviations
- 2D
Two‐dimensional
- 3D
three‐dimensional
- Cryo‐EM
cryo‐electron microscopy
- CTF
contrast transfer function
- DED
direct electron detector
- EM
electron microscopy
- EMX
electron microscopy exchange format
- FSC
fourier shell correlation
- MPA
multiple particle analysis
- MRC
Medical Research Council image format
- SPA
single particle analysis
- SNR
signal to noise ratio
- STAR
self‐defining text archiving and retrieval
- XML
extensible markup language
Introduction
The 3D electron microscopy (3DEM) field evolved through numerous hardware and software advances in single particle analysis (SPA), reaching near atomic resolution recently.1 As a longstanding package in the 3DEM community, the evolution of Bsoft reflects these changes, incorporating new algorithms to handle better quality data. The key events in hardware development that enabled rapid improvement in structural analysis are: the first functional microscope in 1931,2 the field emission gun in 1968,3 and most recently, direct electron detectors (DEDs) (original chip in 19984, 5). Similarly, the software to process electron micrographs for 3D reconstruction also went through three major jumps in achievable resolution: the first 3D reconstruction in 1968,6 understanding the contrast transfer function (CTF) initially noted by Thon in 19667 and subsequently corrected in 1975,8 and compensating for specimen movement from dose‐fractionated DED micrographs in 2013.9, 10
Bsoft is a general data processing package (freely available from http://bsoft.ws) intended mainly for the electron microscopy community (see previous papers11, 12, 13). It has multiple capabilities, with three main themes: single particle analysis (SPA), tomography, and interpretation of 3D reconstructions. Previous versions of Bsoft offered a whole SPA workflow including CTF adjustment and reconstruction.11, 12 Bsoft now includes the ability to use dose‐fractionated micrographs for solving biomolecular structures to high resolution. The aim is to aid the user in processing electron microscopy data through easy image manipulation and automating as much of the workflow as possible. The danger is of course that such freedom can lead to inappropriate processing, invalidating the results. The current effort in improving Bsoft is therefore to (i) institutionalize validation, and (ii) accelerate processing.
I organized the text in the following manner: First, I explain the approaches for doing validated SPA. Next, I present the considerations required to do SPA in Bsoft, dividing it into four phases as it relates to automation. Finally, I discuss several case studies to illustrate the different ways Bsoft has been used to answer biological questions. Throughout I explore the developments required to automate SPA with validation.
Validation
Despite the advances in SPA, it remains an ill‐defined problem with many pitfalls.14, 15, 16 The decisions of the user on how to pick particles and how to align them are crucial elements in producing a justifiable reconstruction. Although validation has recently risen to a more visible topic of discussion,17 it has always been an issue of consideration in SPA. Ultimate validation is through independent methods, such as the agreement between EM‐derived maps and data from X‐ray crystallography or NMR. Here, I concentrate on internal validation, i.e., using only electron micrographs. These can be applied at any resolution, and is as suitable for assessing initial maps from small numbers of particles as it is for high‐resolution maps.
The principle in internal validation is based on coherency between the particle images. There are two approaches to establish coherency in the data: (i) cross‐validation and (ii) direct demonstration of coherency. In cross‐validation, only a fraction of the data is used for processing, while the remainder is used for validation. In the second approach, the fact that there is coherency between the particle images should lead to better reconstructions than from aligned random data. The assessment of coherence is based on interpreting a measure such as the Fourier shell correlation (FSC) curve.
Independent data sets
The idea of independent data sets is simply assessing robustness of the results under repetition. In the 3D reconstruction field, processing two data sets independently and comparing their output has been proposed very early.18, 19 Grigorieff revisited the issue20 and the term “gold standard”1 was later attached.17, 21 An example of a comparison by FSC is shown in Figure 1(A) (black curve). To estimate the resolution, Rosenthal and Henderson24 made an argument to use a cutoff of 0.143 on the curve, currently widely adopted [the bottom horizontal dashed line in Fig. 1(A)]. The analysis can be readily extended to more than two data sets.
Figure 1.
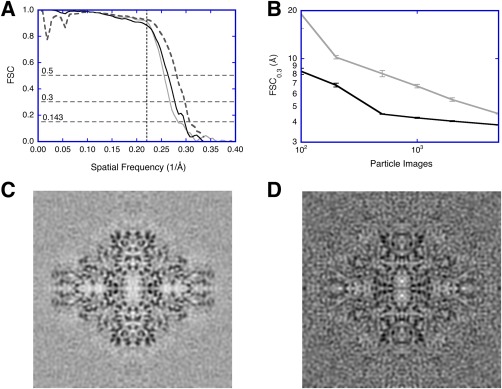
Validation issues in SPA using a public data set of ß‐galactosidase micrographs (https://www.ebi.ac.uk/pdbe/emdb/empiar/entry/10013/).22 The micrographs were split into two independent sets. Within each set, particles were aligned with resolution limits, and two half‐maps calculated to assess resolution. The full maps from the two independent sets were also compared to assess resolution (see Fig. 2 for workflows). (A) FSC curves for the final reconstructions from resolution‐limited processing of one micrograph set (gray), from the two independent data sets (black), and compared to the published map (EMD 5995)22 as repetition with different software packages (dashed). The horizontal dashed lines indicate the different cutoffs used (0.143, 0.3, and 0.5). The vertical dashed line at 4.5 Å (spatial frequency 0.222/Å) indicates the resolution limit used in the alignment. (B) Test for coherence:23 The resolution (FSC0.3) of reconstructions calculated from aligned noise images (gray) compared to those from real images (black). Each point is the average and standard deviation of ten assessments done with random selection of particle or noise images. (C) Reconstruction from 5000 real images. (D) Reconstruction from 5000 aligned noise images
Resolution limited alignment
In many studies in the past the particle images have been aligned to a common reference. This was then followed by reconstructing two maps from mutually exclusive sets of images and comparing them by FSC [using a conservative cutoff of 0.5 or 0.3 to estimate the resolution, the upper two horizontal dashed lines in Fig. 1(A)]. Grigorieff showed that the use of a common reference could lead to aligned noise, producing an invalid reconstruction.20 However, when the SNR is high enough, the signal in the images constrains the alignment to be more representative of the particle. Therefore, restricting the alignment to lower frequencies where the SNR is high then allows the use of higher frequencies as validation. This is similar to the R‐free measure used in X‐ray crystallography,25 in the sense that part of the data is used for fitting/alignment, and an independent part is used to evaluate how good the fit/alignment is. In this approach, the projection‐matching during alignment is limited in resolution to those frequencies the user has good faith in (i.e., scored high by FSC). Information in resolution shells recovered beyond this limit in resolution is then taken as validation that the particles exhibit coherent information. Similarly, Shaihk et al.26, 27 implemented a method where a certain range of frequencies were excluded during alignment, subsequently used for validation. In a case with good data and appropriate processing, the FSC curve is typically close to that for independent data sets [Fig. 1(A), compare the black and gray curves].
Chen et al. proposed a related validation method.28 The particle images are aligned using all the data, accompanied by a second version of the particles with noise substituted beyond a user‐defined limit. Two FSC curves are then produced: One for the original set, and one for the noise‐substituted set. The difference beyond the chosen resolution limit then indicates coherence. This approach requires twice the amount of processing but achieves the same end as resolution‐limited alignment.
Tilt‐pair analysis
The 2D nature of an electron micrograph means that any handedness (chiral) information has been lost. Belnap et al.29 developed a method based on tilt‐pair micrographs to establish the hand of a reconstruction (program dhand in Bsoft). In a further application, Rosenthal and Henderson proposed the use of tilt‐pair micrographs for validation.24 In their method, the known difference in tilt angle between the micrographs is compared to the difference in orientations determined for the particle images (a form of cross‐validation). A small angular error is taken as indicating a valid reconstruction, and in this respect, differs from other methods based on the FSC curve.
Demonstration of particle coherence
It is well known that SPA processing has enough freedom to allow alignment of pure noise to a reference such that the reference can be reproduced.14, 15 This is inherently related to the SNR, where at low SNR, alignment to noise dominates, while at high SNR, alignment to the signal dominates.20 The signal in SPA is effectively the coherence between the particles that should give a measure better than noise. One way to determine whether the alignment targets a coherent signal, is to compare the results from a set of particles to that of an equal number of noise images.23 With a relatively small number of images (∼1000 asymmetric units), one can already distinguish coherency in the data [Fig. 1(B)]. This is useful in assessing whether the micrographs contain particles that can be processed at an early stage in data acquisition. Furthermore, experienced image processors know that the quality of the background noise in reconstructions is an indicator of whether the images contain a coherent signal [Fig. 1(C)], or noise [Fig. 1(D)].
In a similar vein, Vargas et al.30 proposed a method to assess the reliability of particle orientations. They make use of the tendency for high‐scoring projection‐matching results to cluster around the appropriate orientation for a real particle, compared to random orientations for noise images.
Single Particle Work Flow in Bsoft: Validation and Automation
The current state of SPA allows for automation of parts of the workflow, with particular points at which the user needs to assess progress and deal with problematic issues. I therefore divided the work flow into phases relating to checkpoints in automation (Fig. 2). Figure 2(A) shows what is typically understood as SPA, as the first three phases. The first phase is to prepare the micrograph images into a suitable form with associated metadata. The second phase involves particle picking, the crucial step in determining eventual success in reconstruction. The third phase entails iterative alignment and reconstruction, with appropriate validation and resolution assessment. Figure 2(B) lays out the work flow for separate data sets [each done as in Fig. 1(A)], culminating in a comparison of independently reconstructed maps. The final phase is the interpretation of the reconstruction, covering aspects of annotation, segmentation, quantitation, and modeling [Fig. 2(C)].
Figure 2.
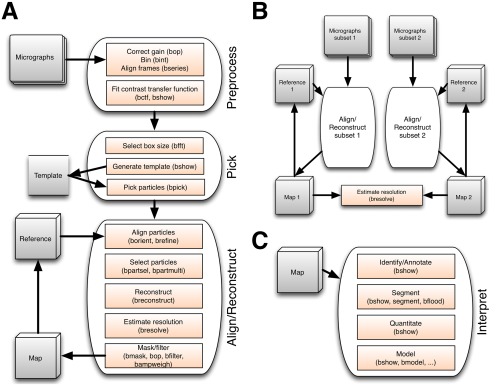
(A) Diagram outlining the first three phases of single particle analysis in Bsoft. The preprocessing of the micrographs can be done as the data is acquired on the microscope, including determining CTF parameters. The CTF parameters are then checked manually, the particle template produced, and particles picked. An initial, low‐resolution reference map is produced and the particles aligned and reconstructed iteratively until the resolution stops improving. (B) For validation by independent sets, the micrographs are split into two subsets and processed separately. The two maps are then compared by Fourier shell correlation to assess the resolution and whether the processing is diverging. (C) The final phase, interpretation, requires extensive case‐specific input from the user for identification and annotation of components, segmentation into regions‐of‐interest, quantitation of parts, and building detailed or coarse‐grained models
Bsoft provides an array of tools to do diverse types of image processing, including SPA. One of these is the graphical program, bshow, with extensive functionality to manage workflows (Supporting Information Fig. S1), while most of the other programs are run from the command line. Examples are given in the supplementary material.
Phase 1: Micrograph Preprocessing and CTF Fitting
The preprocessing details depend on how the micrographs were acquired. The goal is to set up the micrographs with associated parameters suitable for particle picking, alignment and reconstruction. The user can combine all of the preprocessing operations in a script for automation.
Gain correction
The microscope acquisition software usually gain‐correct the micrographs taken on CCD or DED cameras during imaging. However, to ease the burden on the microscope computer, the user can choose to postpone gain correction and write non‐corrected files in a compressed format (such as LZW compression of TIF files. The micrographs are then decompressed and corrected by multiplication with the gain reference (program bop).
Binning
Micrographs can be taken at high magnification (or in super‐resolution mode), and afterwards binned (program bint). The advantage is that it averages some of the noise, increasing the SNR and aiding further processing. The amount of binning should be chosen to ensure that the expected resolution is about 2/3 of Nyquist to avoid significant loss of information (see Supporting Information Fig. S2 for loss of information due to binning). Alternatively, the user can rescale the micrographs in frequency space using the program bftr. It is possible to bin aggressively to obtain an initial low resolution map, and return later to refine the map to higher resolution from the original data.
Frame alignment
A key operation in SPA is to compensate for specimen movement during data acquisition.9 The DEDs are fast enough to allow capturing series of images (frames or dose‐fractionated micrographs, also called “movie‐mode”). The frames are then aligned and averaged to obtain an average image that is further processed in the same way as a micrograph taken on film or with a CCD camera.
The Bsoft algorithm (program bseries) is a full‐frame alignment composed of two parts: (i) progressive and (ii) iterative. In the progressive part, a reference frame is selected. The next frame is aligned against it and the two averaged. Every successive frame is then aligned against the average of the previous frames, giving a progressively better reference. The final average is then used to start the second part, an iterative alignment, stopping when the change in shifts or change in correlation coefficients become negligible. Good alignment depends on the dose (typically ∼2 e‐/frame pixel), the choice of the initial reference frame and the resolution limits. The user should choose an initial reference a few frames from the beginning because the specimen often moves more during the initial exposure to the electron beam.9 Cross‐correlation is typically limited to resolution shells with high amplitudes, often within the first zero of the CTF for the most defocused micrographs (∼ 20–30 Å). The quality of frame alignment can be assessed from the isotropy of the Thon rings of the power spectrum of the frame average (Fig. 3).
Figure 3.

Improvement in power spectrum on frame alignment with the program bseries: (A) Power spectrum of the frame average before alignment shows suppression of Thon rings in one direction indicating severe drift. (B) Power spectrum of the frame average after alignment shows good isotropy.
DEDs offer a direct count of the electrons impacting them. The frame averaging in bseries has therefore been designed to preserve the count sum per pixel, allowing the user to retain accumulated dose information. The frames can be dose‐weighted based on the method from Grant and Grigorieff.31 The averaging suppresses the variance of the noise, effectively increasing the SNR as a function of the number of frames. Note that the signal here is any object in the view, including carbon film, aggregates, etc.
Power spectrum calculation and determining CTF parameters
The power spectrum calculation and CTF fitting can be done interactively in bshow (Supporting Information Fig. S3), or automatically with bctf (see Supplementary Material for details). A good way to calculate the power spectrum for a noisy image is to divide it into tiles, Fourier transform all the tiles, and average their power spectra.32, 33 The size of the tiles is important: too large and the average power spectrum is noisy, too small and the high frequency oscillations are not adequately sampled. To ensure that the CTF minima are separated by at least two pixels at about half‐Nyquist, the tile edge size should be:
| (1) |
where λ is the electron wavelength, Δf is the defocus, and u is the pixel size (all in Å) (see Supplementary Material for a detailed derivation). The power spectrum in Bsoft can be calculated from a micrograph, frames or picked particles.
For tilted specimens, the defocus changes perpendicular to the tilt axis. If the tilt axis and tilt angles are known, the power spectra of the tiles can be rescaled to agree with the defocus in the middle of the micrograph. The scaling at a distance, d t, from the tilt axis for a tilt angle, α, is given by:
| (2) |
where s 0 is the spatial frequency at the origin (middle of the image), and Δf 0 is the defocus at the origin [derived from the second term in Supporting Information Eq. (S2)]. In bshow, the user can enter an approximate average defocus and tilt parameters when calculating the power spectrum. On the command line, the tilt angle and axis must be specified for bctf, either as options or in a parameter file.
The CTF parameters are determined by dividing the problem into parts, avoiding refining too many parameters at once. In bshow the fitting buttons are at the bottom of the CTF dialog window (Supporting Information Fig. S3). The first one is a quick fit that ignores astigmatism and search for a defocus in the range indicated on the right. The next four buttons are for refinement of different sets of parameters [see Supporting Information Eq. (S4)]: the background (it fits to the minima or zeroes), the envelope (its fits to the maxima), the defocus, and astigmatism parameters (defocus deviation and astigmatism angle). The background is modeled as one of six possible curves that should approximately pass through the radial power spectrum minima (Supporting Information Table S2). The last three curves are duplicates of the first three, except for a Gaussian peak to fit the strong water signal at 3.8 Å that is often present in DED micrographs [Supporting Information Fig. S3(A)]. The CTF can also be fitted to micrographs taken with a phase plate, adjusting the amplitude contrast close to one.
Phase 2: Particle Picking
In the initial stages of a project on a new specimen and with small numbers of micrographs, manual picking is still preferred. However, when an initial reference map is well established and the goal is higher resolution, automated picking becomes a necessity. The results must still be checked in bshow to make sure that the number of poorly selected particles are not excessive (preferably below 20%).
Selecting particles
The selection of particles from micrographs is a segmentation problem ‐ it suffers from the same issues as all segmentation approaches. It is inherently subjective ‐ the user must select what signal is desired. Past efforts attempted to pick particles using a synthetic, idealized reference, or to use specific properties thought to distinguish signal from noise (such as variance). However, the most effective method turns out to be based on what the user recognizes as desired particles. The user therefore selects representative particles and an average is calculated as a template (in bshow). Once a suitable template is generated (Fig. 4), the particles can be selected and extracted automatically (program bpick). The automated picking is effective to a large degree, but the inclusion of undesirable objects, such as ice and carbon support remains a complicating issue. The user can then check the quality of picking in bshow, and improve it manually where necessary.
Figure 4.
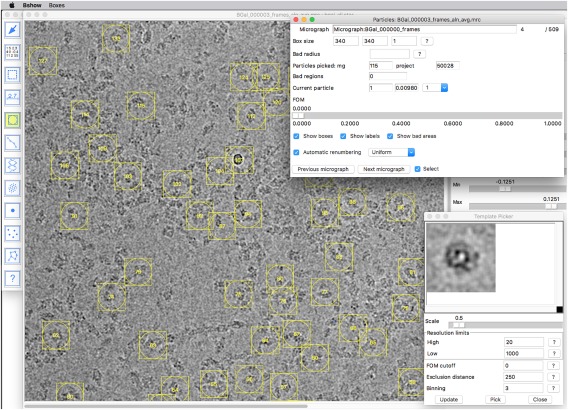
Particle picking: First a template is generated from a small set of hand‐picked particles (lower right). Then a larger set is picked automatically, pruned by removing badly picked particles, and a new template generated. This can be iterated until the user is satisfied with the result. The template is subsequently used to automatically pick particles from multiple micrographs
The choice and consequences of the box size
The choice of box size for particle picking is an important consideration, determining the limit of resolution achievable and the cost (time) of processing. The sampled nature of the data implies hard limits for the accuracy of alignment. In practice, the maximum amount of workable information extends up to about two thirds of Nyquist frequency (i.e., about three times the pixel size). At this distance from the origin, the angle that is subtended by one pixel separation (in frequency space) gives the limit for angular accuracy (in radians):
| (3) |
where r lim is the limiting (high) resolution (Å), D is the physical box size (Å), and n e is the box edge size in pixels (see Supporting Information Fig. S4 for example curves based on equation (3)).
The cost of a global search for orientations depends on the number of images, n i, the box size, and the number of orientations (or views). The latter is given by the number of projections calculated from the 3D reference map that covers the surface of the unit sphere within the asymmetric unit. For an angular step size of Δα, the number of projections is:
| (4) |
where n s is the number of asymmetric units for a given symmetry. The cost is therefore on the order of , or , where n i is the number of images. Considering the angular limit in equation (3) and calculating the number of projections from it according to equation (4), the upper limit for the cost is a fourth order of the box size, , and the memory required to hold the projections is . The box size chosen is therefore important in determining the speed of processing. This is also the reason that the global search is usually done with a coarse angular step size (program borient), followed by a refinement of orientation that is not coupled to calculating a large number of projections (program brefine).
Another consideration is to make sure a box size is chosen that can be Fourier transformed fast (use bfft with the –test option to find a suitable size). Suggestions range from a tight fit (∼20% bigger than the particle size) to a more generous size (3 times the particle diameter). The former ensures little extraneous interference, while the latter allows better angular accuracy in the final refinement (D in equation (3) is larger, therefore Δαlim is smaller). A possible reconciliation is to use a soft circular mask to eliminate background with a larger box size.
Phase 3: Particle Alignment, Reconstruction and Validation
The core steps and most time‐consuming part of SPA is particle alignment and reconstruction. Careful choice of a starting reference and parameters ensures efficient processing and obtaining a valid result. This phase can be fully automated for a simple SPA run. For more complicated cases, different levels of user intervention will be necessary, depending on the details and goals. Where conformational or compositional variability is expected, multiple reference maps must be generated and each used to align the particles. The particles are then classified based on the best fit to a reference map as claculated during alignment.
The initial 3D reference/template
The alignment of the particle images in Bsoft is reference‐based. To start processing, a reference must be produced with low frequency features similar to those of the particles. This map must be a cube with the edge equal to the edge of a particle image. The user must also ensure that the sampling (pixel size) is the same as the particle images, and that the origin is appropriately specified (usually in the center of the box).
There are three different ways in which a reference map can be constructed in Bsoft. The first is creating a synthetic image with some features resembling the particle. The program beditimg creates images from scratch, or modify existing images. The user can create a simple reference map by placing a few spherical densities in appropriate locations within a volume. Symmetry can be applied using the program bsym (see Supporting Information Fig. S5 for an example).
Alternatively, an existing map from the EMDB can be used, or calculated from an atomic structure from the PDB. The programs bsf and bgex convert atomic coordinates into 3D density suitable for a reference map.
Finally, random orientations can be assigned to the particle images (program emgrand), and an initial map reconstructed from them. This works better for particles with high symmetry than with asymmetric particles.34
The program borient can also use a set of 2D images as reference. The alignment can then be done using the program borient as described in the next session, with subsequent averaging to produce class averages (Supporting Information Fig. S6).
Alignment
The user typically starts an alignment iteration with a global search for orientations within the asymmetric unit using a coarse angular step size in the program borient. This can then be followed by subsequent iterations using borient with smaller angular step sizes, or using brefine to improve orientations from previous iterations. In all cases, low and high resolution limits are typically used: the former to avoid the strong low frequency components with little orientational information, and the latter to eliminate noise.35
In both borient and brefine, projections from the 3D reference are compared to the particle images to determine the best views. In both programs, central sections (the frequency space equivalent of projections) are calculated using an interpolation developed for the Radon package (Supporting Information Fig. S7).36, 37 The CTF is imposed on each reference projection to generate a more appropriate comparison with the particle image. The projection‐matching algorithm used in borient is based on the polar Fourier transform approach35 (see Supporting Information Fig. S8 for flow diagrams). The second program, brefine, does a frequency space refinement of orientation, magnification and defocus parameters per particle (see Supporting Information Fig. S9 for a flow diagram).
In borient the angular step size determines the number of projections. These are then matched to particle images using the low and high resolution limits chosen by the user. The angular step size is related to the high resolution limit as indicated in equation (3), so that the user only needs to select the latter. In brefine, the angular accuracy specified for the grid search option is similarly linked to the high resolution limit chosen. In both cases the recovery of information beyond the high resolution limit is taken as a measure of coherence, i.e. validation [Fig. 1(A)].
Particle selection
Once a set of particles have been aligned, they can be selected in many different ways based on figures‐of‐merit (FOMs) using the program bpartsel. Two FOMs are calculated in borient or brefine runs: a cross‐correlation coefficient (CCC) within the resolution limits set by the user, and a cross‐validation coefficient (CVC) outside those limits. For borient, the latter is calculated for the excluded low resolution shells, and can be used to eliminate some noise and junk images. For brefine, the CVC covers shells just beyond the high resolution limit as an assessment of recovering coherence. Because the defocus affects contrast and thus also the magnitude of the FOMs, they can be scaled to the average defocus. Another option selects a maximum number of particles per view to counter the effects of preferred orientations.
The alignment can be run on the same set of particles with different reference maps, each resulting in a parameter file. These parameter files can be compared with the program bpartmulti, and the particles assigned a selection number indicating the best match to a reference. These selection numbers are then used to calculate multiple reconstructions.
Reconstruction
The main reconstruction program in Bsoft, breconstruct, does a frequency space nearest neighbor integration with oversampling. During reconstruction in breconstruct, the particle images can be adjusted for the CTF (using any of the options shown in Supporting Information Table S1). All symmetry‐related views are incorporated during the integration. A resolution limit for integration is set to avoid high‐frequency noise.
Multiple reconstructions can be calculated. Typically, two halfmaps (from alternating particle selections) are calculated for resolution estimation, as well as a full map. If the program bpartmulti has been used to generate multiple selections, all the corresponding reconstructions can be calculated (limited only by the available computer memory).
Resolution estimation
The program bresolve calculates a Fourier shell correlation curve between any two given maps of the same size and sampling. The maps can be masked, but care must be taken to use an appropriate mask (see below). The resolution is estimated using user‐provided cutoffs: for independent data sets the resolution is estimated at 0.143,21 while for resolution‐limited alignment, a conservative cutoff of 0.3 is typically used.
The local resolution can be calculated from two maps using the program blocres.38 The key is to use a large enough box size (typically 5–7 times the global resolution) to avoid artificially good resolution estimates. The output is a map of local resolution estimates that can be used to color a map (Fig. 5). For further examples using blocres, see.39, 40, 41, 42 The local resolution map can then be used to filter different parts of a map to appropriate resolution limits with the program blocfilt.38, 43
Figure 5.
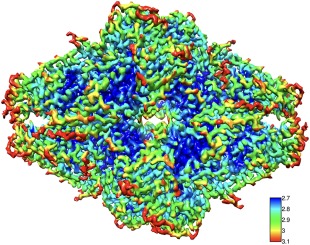
Local resolution (program blocres) of a reconstruction of ß‐galactosidase (detailed in Fig. 1). The local kernel (box) edge was 40 pixels (25 Å) and FSC0.3 estimates were used
Masking
Real space masks are typically used to remove background noise in images and reconstructions. They can be used during alignment (in programs borient and brefine), applied to reconstructions and used in FSC calculations (program bresolve). For alignment and FSC calculation, the nature of the mask is critical in obtaining valid results. The ideal mask would have zero background in real space, and zero amplitudes beyond a low‐pass limit in frequency space. However, to satisfy both conditions is impossible, because any signal cannot be both real and frequency space limited (uncertainty principle or Gabor limit44). The best solution is to generate a real space mask with soft edges, but then to low‐pass filter it to a lower resolution than expected in the data (Supporting Information Fig. S10). This will produce some ripples in the background in real space, but still suppress most of the noise, while avoiding any high frequency terms that may enter into projection‐matching or FSC calculations.
Filtering
The final reconstruction typically shows suppressed higher frequencies, rendering the visual appearance somewhat fuzzy and making modeling more challenging. The amplitudes can be modified in several ways using the program bampweigh. If a reference map is available from X‐ray crystallography, the reconstruction amplitudes can be scaled to the radial power spectrum of the reference. Alternatively, the amplitudes can be scaled so that their radial averages are equivalent to the carbon electron scattering cross section (the cross section is taken from Peng et al.45). The latter has been applied to a map of ß‐galactosidase (shown in Fig. 5, Supporting Information Fig. S11), reconstructed from a publicly available data set (https://www.ebi.ac.uk/pdbe/emdb/empiar/entry/10013/).22
Phase 4: Interpreting Maps
The interpretation of a 3D map comprises identifying and annotating parts, dividing it into regions with meaning, and building models on an atomic or coarse‐grained scale. The tools in Bsoft allow the user to segment maps, create masks, and handle various types of models. These cover a broad field and only a few examples are given here.
Segmentation
The program bflood uses a very common segmentation method, the watershed by flooding (see Supporting Information Fig. S12 for an example). The algorithm starts with a definition of the segments as those regions connected at the initial threshold that the user chooses. This starting threshold can be selected in bshow by finding a gray scale level where the regions of interest are separated. The algorithm then progressively enlarges these regions by lowering the threshold until they touch or until the minimum threshold is reached. The regions/segments are then encoded in a multi‐level mask that can be used to extract parts of the map. In bshow the “Model” tool can be used to mark specific regions and extract those as a combined mask.
An example is the segmentation of the herpes simplex type 1 capsid46, 47 (Fig. 6). The watershed approach was used to generate multi‐level masks denoting the different subunits of the capsid. These were then manipulated to produce masks that correspond to each type of subunit. The masks in turn were used to extract maps from the original capsid reconstruction, each with only one type of component. Another example is the segmentation of the bacteriophage φ6 map to isolate the different components (see Supporting Information Fig. S12).
Figure 6.

Segmentation of the herpes simplex virus type 1 capsid, showing the major capsid protein, VP5, in blue (hexons in lighter blue and pentons in darker blue), the triplexes (each composed of VP19C and two VP23) in green, and VP26 in orange. The inset shows the E‐hexon up close
Quantification of density
The density in a reconstruction is not directly quantifiable. Therefore, the user needs to define the foreground, the background, and the particle area of interest. In Bshow, the Selection tool can be used to calculate the average density in a defined region, and do it for the three types of regions. A better way is to generate a mask with multiple levels: 1 = particle density of interest, 2 = reference density, 3 = background density, 0 = ignore. For instance, the stoichiometry of a protein that binds to a virus capsid can be determined by composing a mask with 1 at the protein, 2 at the capsid shell, 3 at the background, and 0 where everything else should be ignored (such as nucleic acid inside the capsid). The individual levels are derived from the segmentation of the reconstruction (see previous section). The mask is then used in the program, bdens, to calculate the averages in the different regions (see Supporting Information Fig. S12). The fractional contribution of the density‐of‐interest is then:
| (5) |
where d i, d f, and d b refer to the levels 1–3 in the mask, respectively. The program bspr can use the mask to calculate the fractional contribution in each individual particle—typically applied to icosahedral particles where a single reconstruction carries significant information.
Modeling
Bsoft offers modeling tools addressing from full atomic representations to coarse components. While these have been used mostly in the context of tomography, they are also useful for single particle maps. One example is the rigid body fit of the protrusion domain of the herpes simplex virus type 1 capsid major protein into the maps along the maturation pathway (see below for more details).48
Illustrative Case Studies
Bsoft has been used in many studies with diverse goals. Here are examples of studies aimed at specific biological questions.
Single, Well‐Defined Structures
In some cases the specimen is a purified biomolecule with a structure that shows little variance within the expected achievable resolution. The approach is then straightforward and Bsoft has been used to produce several reconstructions of this nature. In Sen et al.,49 we used a synthetic reference map (Supporting Information Fig. S5) with some of the features of the bacteriophage φ6 procapsid based on previous studies.50 We further improved the maps to locate minor components of the procapsid.51 For the antifeeding prophage particle from Seratia entomophila, I started with a reference composed of a cylinder with a sphere at the end to simulate the baseplate.52
Often a previous map of the same or similar specimen exists, and this can be used as starting reference filtered to a suitably low resolution. Examples of these are the Kaposi's sarcoma‐associate herpes virus capsid,53 and the capsid and the primary enveloped virion of herpes simplex virus type 1.54, 55 The initial map for the ß‐galatosidase reconstruction shown in Figures 5 and Supporting Information Figure S11 was obtained from the EMDB (EMDataBank.org: EMD‐5995) and low‐pass filtered to 60 Å.
A different approach is to start with random orientations and allow the alignment processing to sort out coherence. Sanz‐García et al.34 showed that this is difficult with an asymmetric specimen. If the specimen has high symmetry, this can work, as found with icosahedral particles.56 In a similar approach, an analysis of the variance in one‐particle reconstructions with high symmetry can lead to suitable initial references.57 Cardone et al.58 used this in reconstructing a small icosahedral particle formed by the Rous sarcoma virus capsid protein.
Dynamic Structures
If the specimen shows significant variation in conformation or composition, some form of classification is required. The Bsoft approach is to generate multiple 3D starting references and run each as a separate alignment of all the particles (MPA: multiple particle analysis59). The resultant parameter files are then compared (program bpartmulti) and the particles assigned to the reference classes based on a figure‐of‐merit (such as the correlation coefficient). The starting reference maps can be produced in any conceivable way. A simple way is to divide the micrographs into subsets and process them separately to yield several maps. Another approach is to use the bootstrap method60 to calculate a set of maps from particles aligned to a single reference (in Bsoft, each such map is generated using the –bootstrap options in bpartsel and breconstruct).
As an example, we studied the maturation of the herpes simplex virus type 1 procapsid, imaging the capsids at various time intervals. Each of the resultant micrographs contain capsids in several different stages of maturation.46 We started by aligning the particles to a series of reference maps, morphed between previous procapsid and mature capsid maps. The key was to limit the cross‐correlation to those resolution shells that show the biggest difference between the procapsid and mature capsid. After several rounds of refining and reclassifying the particles, we produced 17 maps illustrating the transformations during maturation. At the time part of the major capsid protein, its protrusion domain, was solved by X‐ray crystallography.61 We did a rigid body fit into the series of maps, showing how the protrusion domain moves during hexon consolidation48 (Supporting Information Fig. S13).
The bacteriophage φ6 capsid undergoes large‐scale conformational changes during genome packaging. We obtained micrographs with different conformations, allowing us to classify the capsid into the four states adopted during expansion.62
Compositional Analysis
It is often important to identify the components of particles, or understand their stoichiometry. For the bacteriophage φ6 capsid, we had mutants in the minor procapsid components, allowing us to generate particles of varying composition (see Supporting Information Fig. S12 for a segmentation of an early map). We reconstructed maps with and without the minor components, locating them using difference maps, and calculated copy numbers.49, 51 In a similar study on clathrin cages, we showed through classification and difference mapping the binding sites of Hsc70, the chaperone responsible for dissociating the cages.63
Masked Alignment
The encapsulin particles from Myxococcus xanthus contain large amounts of iron aggregate, giving a strong, irregular signal in cryo‐electron micrographs.64 The initial reconstructions were generated using random orientations but imposing icosahedral symmetry and masking the centers of the particles. These were later used in refining the structure from recombinant protein without iron clusters.
Helical Processing
Bsoft offers tools for the interpretation of helical diffraction patterns in bshow (see Ref. 65 and 66 for examples). However, the SPA approach has become the method of choice for helical reconstruction, as first implemented as IHRSR (iterative helical real‐space reconstruction).67 We analyzed filaments of the HIV protein REV, obtaining maps to subnanometer resolution.65 Additional tools are available for analyzing filaments in terms of width (program bfilwidth) and calculating helical cross sections (program bhelcross). We used these to show that α‐synuclein amyloid fibrils have two protofibrils.68
Conclusion
I will continue developing Bsoft as a cross‐platform software package for image processing and interpretation for 3D electron microscopy, with better integration in processing environments such as Scipion69 and EMIRA.70 The main challenges are dealing with large amounts of data and efficient processing through automation while ensuring valid results. One key issue that needs to be further improved is particle picking: It essentially is a segmentation problem complicated by the high variability in micrograph contents. However, even with most of the current automated picking algorithms, the successes are highly encouraging for routinely solving biomolecular structures. With sufficient optimization and automation, the time between data acquisition and final reconstruction can be shortened to hours and days rather than weeks and months.
Supporting information
Supporting Information
Acknowledgments
I wish to thank all the contributors to Bsoft, in particular Drs. David Belnap, Giovanni Cardone, and Juha Huiskonen. This work was supported by the Intramural Research Program of the National Institute for Arthritis, Musculoskeletal and Skin Diseases, NIH. The author has no conflicts of interest to declare.
Impact Statement: Bsoft offers a rich and powerful environment for image processing in general, and specifically for calculating three‐dimensional maps of biomolecules from electron microscope images. With increasing use of electron microscopy to solve biomolecular structures to high resolution, it is important to validate the results. This paper focusses on approaches to produce valid structures using Bsoft, as well as automation to handle the large amount of data generated for high resolution work.
Footnotes
An essay on the use of the term “gold standard” 21. Claassen JA. 2005. [‘gold standard’, not ‘golden standard’]. Ned Tijdschr Geneeskd. 149(52):2937.
References
- 1. Kuhlbrandt W (2014) Biochemistry. The resolution revolution. Science 343:1443–1444. [DOI] [PubMed] [Google Scholar]
- 2. Ruska E (1987) Nobel lecture. The development of the electron microscope and of electron microscopy. Biosci Rep 7:607–629. [DOI] [PubMed] [Google Scholar]
- 3. Crewe AV, Eggenberger DN, Wall J, Welter LM (1968) Electron gun using a field emission source. Rev Sci Instrum 39:576–583. [Google Scholar]
- 4. Bisogni MG, Campbell M, Conti M, Delogu P, Fantacci ME, Heijne EHM, Maestro P, Magistrati G, Marzulli VM, Meddeler G, Mikulec B, Pernigotti E, Rosso V, Schwarz C, Snoeys W, Stumbo S, Watt J (1998) Performance of a 4096‐pixel photon counting chip. Proc SPIE 3445:298–304. [Google Scholar]
- 5. Faruqi AR, Henderson R (2007) Electronic detectors for electron microscopy. Curr Opin Struct Biol 17:549–555. [DOI] [PubMed] [Google Scholar]
- 6. De Rosier DJ, Klug A (1968) Reconstruction of three dimensional structures from electron micrographs. Nature 217:130–134. [DOI] [PubMed] [Google Scholar]
- 7. Thon F (1966) Notizen: Zur defokussierungsabhängigkeit des phasenkontrastes bei der elektronenmikroskopischen abbildung. Zeitschrift für Naturforschung A 476. [Google Scholar]
- 8. Unwin PNT, Henderson R (1975) Molecular structure determination by electron microscopy of unstained crystalline specimens. J Mol Biol 94:425–440. [DOI] [PubMed] [Google Scholar]
- 9. Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y (2013) Electron counting and beam‐induced motion correction enable near‐atomic‐resolution single‐particle cryo‐em. Nat Methods 10:584–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bai XC, Fernandez IS, McMullan G, Scheres SH (2013) Ribosome structures to near‐atomic resolution from thirty thousand cryo‐em particles. Elife 2:e00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heymann JB (2001) Bsoft: image and molecular processing in electron microscopy. J Struct Biol 133:156–169. [DOI] [PubMed] [Google Scholar]
- 12. Heymann JB, Belnap DM (2007) Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol 157:3–18. [DOI] [PubMed] [Google Scholar]
- 13. Heymann JB, Cardone G, Winkler DC, Steven AC (2008) Computational resources for cryo‐electron tomography in bsoft. J Struct Biol 161:232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henderson R (2013) Avoiding the pitfalls of single particle cryo‐electron microscopy: Einstein from noise. Proc Natl Acad Sci USA 110:18037–18041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Heel M (2013) Finding trimeric hiv‐1 envelope glycoproteins in random noise. Proc Natl Acad Sci USA 110:E4175–E4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subramaniam S (2013) Structure of trimeric hiv‐1 envelope glycoproteins. Proc Natl Acad Sci 110:E4172–E4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, Jiang W, Ludtke SJ, Medalia O, Penczek PA, Rosenthal PB, Rossmann MG, Schmid MF, Schroder GF, Steven AC, Stokes DL, Westbrook JD, Wriggers W, Yang H, Young J, Berman HM, Chiu W, Kleywegt GJ, Lawson CL (2012) Outcome of the first electron microscopy validation task force meeting. Structure 20:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frank J, Verschoor A, Boublik M (1981) Computer averaging of electron micrographs of 40s ribosomal subunits. Science 214:1353–1355. [DOI] [PubMed] [Google Scholar]
- 19. Saxton WO, Baumeister W (1982) The correlation averaging of a regularly arranged bacterial cell envelope protein. J Microsc 127:127–138. [DOI] [PubMed] [Google Scholar]
- 20. Grigorieff N (2000) Resolution measurement in structures derived from single particles. Acta Cryst D56:1270–1277. [DOI] [PubMed] [Google Scholar]
- 21. Scheres SH, Chen S (2012) Prevention of overfitting in cryo‐em structure determination. Nat Methods 9:853–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bartesaghi A, Matthies D, Banerjee S, Merk A, Subramaniam S (2014) Structure of beta‐galactosidase at 3.2‐a resolution obtained by cryo‐electron microscopy. Proc Natl Acad Sci USA 111:11709–11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heymann JB (2015) Validation of 3d em reconstructions: the phantom in the noise. AIMS Biophys 2:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosenthal PB, Henderson R (2003) Optimal determination of particle orientation, absolute hand, and contrast loss in single‐particle electron cryomicroscopy. J Mol Biol 333:721–745. [DOI] [PubMed] [Google Scholar]
- 25. Brunger AT (1992) Free r value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355:472–475. [DOI] [PubMed] [Google Scholar]
- 26. Shaikh TR, Trujillo R, LeBarron JS, Baxter WT, Frank J (2008) Particle‐verification for single‐particle, reference‐based reconstruction using multivariate data analysis and classification. J Struct Biol 164:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shaikh TR, Hegerl R, Frank J (2003) An approach to examining model dependence in em reconstructions using cross‐validation. J Struct Biol 142:301–310. [DOI] [PubMed] [Google Scholar]
- 28. Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, Henderson R (2013) High‐resolution noise substitution to measure overfitting and validate resolution in 3d structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Belnap DM, Olson NH, Baker TS (1997) A method for establishing the handedness of biological macromolecules. J Struct Biol 120:44–51. [DOI] [PubMed] [Google Scholar]
- 30. Vargas J, Oton J, Marabini R, Carazo JM, Sorzano CO (2016) Particle alignment reliability in single particle electron cryomicroscopy: a general approach. Sci Rep 6:21626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grant T, Grigorieff N (2015) Measuring the optimal exposure for single particle cryo‐em using a 2.6 a reconstruction of rotavirus vp6. Elife 4:e06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fernandez J‐J, Sanjurjo J, Carazo J‐M (1997) A spectral estimation approach to contrast transfer function detection in electron microscopy. Ultramicroscopy 68:267. [Google Scholar]
- 33. Zhu J, Penczek PA, Schroder R, Frank J (1997) Three‐dimensional reconstruction with contrast transfer function correction from energy‐filtered cryoelectron micrographs: procedure and application to the 70s escherichia coli ribosome. J Struct Biol 118:197–219. [DOI] [PubMed] [Google Scholar]
- 34. Sanz‐García E, Stewart AB, Belnap DM (2010) The random‐model method enables ab initio 3d reconstruction of asymmetric particles and determination of particle symmetry. J Struct Biol 171:216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker TS, Cheng RH (1996) A model‐based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J Struct Biol 116:120–130. [DOI] [PubMed] [Google Scholar]
- 36. Lanzavecchia S, Bellon PL (1995) A bevy of novel interpolating kernels for the shannon reconstruction of high‐bandpass images. J Vis Commun Image Represent 6:122–131. [Google Scholar]
- 37. Lanzavecchia S, Bellon PL, Radermacher M (1999) Fast and accurate three‐dimensional reconstruction from projections with random orientations via radon transforms. J Struct Biol 128:152–164. [DOI] [PubMed] [Google Scholar]
- 38. Cardone G, Heymann JB, Steven AC (2013) One number does not fit all: mapping local variations in resolution in cryo‐em reconstructions. J Struct Biol 184:226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Louder RK, He Y, López‐Blanco JR, Fang J, Chacón P, Nogales E (2016) Structure of promoter‐bound tfiid and model of human pre‐initiation complex assembly. Nature 531:604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hashem Y, des Georges A, Fu J, Buss SN, Jossinet F, Jobe A, Zhang Q, Liao HY, Grassucci RA, Bajaj C, Westhof E, Madison‐Antenucci S, Frank J (2013) High‐resolution cryo‐electron microscopy structure of the trypanosoma brucei ribosome. Nature 494:385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang K, Ren Z, Raushel FM, Zhang J (2016) Structures of the carbon‐phosphorus lyase complex reveal the binding mode of the nbd‐like phnk. Structure. 24:37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang W, MacKinnon R (2017) Cryo‐em structure of the open human ether‐a‐go‐go‐related k+ channel herg. Cell 169:422–430 e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. He Y, Yan C, Fang J, Inouye C, Tjian R, Ivanov I, Nogales E (2016) Near‐atomic resolution visualization of human transcription promoter opening. Nature 533:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gabor D (1946) Theory of communication. J IEE 93:429–457. [Google Scholar]
- 45. Peng L‐M, Ren G, Dudarev SL, Whelan MJ (1996) Robust parameterization of elastic and absorptive electron atomic scattering factors. Acta Cryst A52:257–276. [Google Scholar]
- 46. Heymann JB, Cheng N, Newcomb WW, Trus BL, Brown JC, Steven AC (2003) Dynamics of herpes simplex virus capsid maturation visualized by time‐lapse cryo‐electron microscopy. Nat Struct Biol 10:334–341. [DOI] [PubMed] [Google Scholar]
- 47. Steven AC, Heymann JB, Cheng N, Trus BL, Conway JF (2005) Virus maturation: dynamics and mechanism of a stabilizing structural transition that leads to infectivity. Curr Opin Struct Biol 15:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heymann JB, Trus BL, Steven AC (2005) Dynamics of the protrusion domain of herpes simplex virus capsid from time‐resolved cryo‐em and molecular modelling. Microsc Microanal 11:1068–1069. [Google Scholar]
- 49. Sen A, Heymann JB, Cheng N, Qiao J, Mindich L, Steven AC (2008) Initial location of the rna‐dependent rna polymerase in the bacteriophage phi6 procapsid determined by cryo‐electron microscopy. J Biol Chem 283:12227–12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huiskonen JT, de Haas F, Bubeck D, Bamford DH, Fuller SD, Butcher SJ (2006) Structure of the bacteriophage phi6 nucleocapsid suggests a mechanism for sequential rna packaging. Structure 14:1039–1048. [DOI] [PubMed] [Google Scholar]
- 51. Nemecek D, Qiao J, Mindich L, Steven AC, Heymann JB (2012) Packaging accessory protein p7 and polymerase p2 have mutually occluding binding sites inside the bacteriophage φ6 procapsid. J Virol 86:11616–11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heymann JB, Bartho JD, Rybakova D, Venugopal HP, Winkler DC, Sen A, Hurst MR, Mitra AK (2013) Three‐dimensional structure of the toxin‐delivery particle antifeeding prophage of serratia entomophila. J Biol Chem 288:25276–25284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Trus BL, Heymann JB, Nealon K, Cheng N, Newcomb WW, Brown JC, Kedes DH, Steven AC (2001) Capsid structure of kaposi's sarcoma‐associated herpesvirus, a gammaherpesvirus, compared to those of an alphaherpesvirus, herpes simplex virus type 1, and a betaherpesvirus, cytomegalovirus. J Virol 75:2879–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trus BL, Newcomb WW, Cheng N, Cardone G, Marekov L, Homa FL, Brown JC, Steven AC (2007) Allosteric signaling and a nuclear exit strategy: binding of ul25/ul17 heterodimers to DNA‐filled hsv‐1 capsids. Mol Cell 26:479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Newcomb WW, Fontana J, Winkler DC, Cheng N, Heymann JB, Steven AC (2017) The primary enveloped virion of herpes simplex virus 1: its role in nuclear egress. mBio 6:e01525–01515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yan X, Dryden KA, Tang J, Baker TS (2007) Ab initio random model method facilitates 3d reconstruction of icosahedral particles. J Struct Biol 157:211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cantele F, Lanzavecchia S, Bellon PL (2004) Viva (from virus variance), a library to reconstruct icosahedral viruses based on the variance of structural models. Comput Methods Programs Biomed 76:155–162. [DOI] [PubMed] [Google Scholar]
- 58. Cardone G, Purdy JG, Cheng N, Craven RC, Steven AC (2009) Visualization of a missing link in retrovirus capsid assembly. Nature 457:694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Heymann JB, Conway JF, Steven AC (2004) Molecular dynamics of protein complexes from four‐dimensional cryo‐electron microscopy. J Struct Biol 147:291–301. [DOI] [PubMed] [Google Scholar]
- 60. Penczek PA, Frank J, Spahn CM (2006) A method of focused classification, based on the bootstrap 3d variance analysis, and its application to ef‐g‐dependent translocation. J Struct Biol 154:184–194. [DOI] [PubMed] [Google Scholar]
- 61. Bowman BR, Baker ML, Rixon FJ, Chiu W, Quiocho FA (2003) Structure of the herpesvirus major capsid protein. EMBO J 22:757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nemecek D, Cheng N, Qiao J, Mindich L, Steven AC, Heymann JB (2011) Stepwise expansion of the bacteriophage φ6 procapsid: possible packaging intermediates. J Mol Biol 414:260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Heymann JB, Iwasaki K, Yim YI, Cheng N, Belnap DM, Greene LE, Eisenberg E, Steven AC (2005) Visualization of the binding of hsc70 atpase to clathrin baskets: implications for an uncoating mechanism. J Biol Chem 280:7156–7161. [DOI] [PubMed] [Google Scholar]
- 64. McHugh CA, Fontana J, Nemecek D, Cheng N, Aksyuk AA, Heymann JB, Winkler DC, Lam AS, Wall JS, Steven AC, Hoiczyk E (2014) A virus capsid‐like nanocompartment that stores iron and protects bacteria from oxidative stress. EMBO J 33:1896–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. DiMattia MA, Watts NR, Cheng N, Huang R, Heymann JB, Grimes JM, Wingfield PT, Stuart DI, Steven AC (2016) The structure of hiv‐1 rev filaments suggests a bilateral model for rev‐rre assembly. Structure 24:1068–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jaballah SA, Bailey GD, Desfosses A, Hyun J, Mitra AK, Kingston RL (2017) In vitro assembly of the rous sarcoma virus capsid protein into hexamer tubes at physiological temperature. Sci Rep 7:2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Egelman EH (2000) A robust algorithm for the reconstruction of helical filaments using single‐particle methods. Ultramicroscopy 85:225–234. [DOI] [PubMed] [Google Scholar]
- 68. Dearborn AD, Wall JS, Cheng N, Heymann JB, Kajava AV, Varkey J, Langen R, Steven AC (2016) Alpha‐synuclein amyloid fibrils with two entwined, asymmetrically associated protofibrils. J Biol Chem 291:2310–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. de la Rosa‐Trevin JM, Quintana A, Del Cano L, Zaldivar A, Foche I, Gutierrez J, Gomez‐Blanco J, Burguet‐Castell J, Cuenca‐Alba J, Abrishami V, Vargas J, Oton J, Sharov G, Vilas JL, Navas J, Conesa P, Kazemi M, Marabini R, Sorzano CO, Carazo JM (2016) Scipion: a software framework toward integration, reproducibility and validation in 3d electron microscopy. J Struct Biol 195:93–99. [DOI] [PubMed] [Google Scholar]
- 70. Radermacher M (2013) (A new environment for modular image reconstruction and data analysis. Microsc Microanal 19:762–763. [Google Scholar]
- 71. Claassen JA (2005) [‘gold standard’, not ‘golden standard’]. Ned Tijdschr Geneeskd 149:2937. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information