

Abstract
This paper describes the current update on macromolecular model validation services that are provided at the MolProbity website, emphasizing changes and additions since the previous review in 2010. There have been many infrastructure improvements, including rewrite of previous Java utilities to now use existing or newly written Python utilities in the open‐source CCTBX portion of the Phenix software system. This improves long‐term maintainability and enhances the thorough integration of MolProbity‐style validation within Phenix. There is now a complete MolProbity mirror site at http://molprobity.manchester.ac.uk. GitHub serves our open‐source code, reference datasets, and the resulting multi‐dimensional distributions that define most validation criteria. Coordinate output after Asn/Gln/His “flip” correction is now more idealized, since the post‐refinement step has apparently often been skipped in the past. Two distinct sets of heavy‐atom‐to‐hydrogen distances and accompanying van der Waals radii have been researched and improved in accuracy, one for the electron‐cloud‐center positions suitable for X‐ray crystallography and one for nuclear positions. New validations include messages at input about problem‐causing format irregularities, updates of Ramachandran and rotamer criteria from the million quality‐filtered residues in a new reference dataset, the CaBLAM Cα‐CO virtual‐angle analysis of backbone and secondary structure for cryoEM or low‐resolution X‐ray, and flagging of the very rare cis‐nonProline and twisted peptides which have recently been greatly overused. Due to wide application of MolProbity validation and corrections by the research community, in Phenix, and at the worldwide Protein Data Bank, newly deposited structures have continued to improve greatly as measured by MolProbity's unique all‐atom clashscore.
Keywords: all‐atom contact analysis, electron‐cloud hydrogen position, Asn/Gln/His flip, CCTBX, Top8000, CaBLAM, cis non‐proline
Introduction
MolProbity is a widely used system of model validation for protein and nucleic acid structures, accessed at http://molprobity.biochem.duke.edu. It builds upon the work of earlier systems such as ProCheck,1 WhatIf,2 and Oops,3 which introduced the use of validation by Ramachandran‐plot and sidechain rotamer criteria. It complements systems for validating data4, 5 and model‐to‐data match such as Rfree6 or real‐space residual.7 MolProbity has some features specifically tailored for X‐ray crystallography, and is also suitable, and used, for cryoEM, neutron, NMR, and computational models. MolProbity's unique feature of all‐atom contact analysis (including hydrogens) was described in 1999,8, 9 followed by its complementary rotamer, Ramachandran, and Cβ deviation criteria,10, 11 and the initial MolProbity web service.12 Validation of RNA backbone, interfaces, and NMR ensembles, a large speedup for Reduce, and an entirely new web interface were described in 2007.13 Implementation of many MolProbity validations inside the Phenix crystallography package, an emphasis on helping users correct specific types of outliers, and the resulting improvements in clashscore and Asn/Gln/His flips were reported in 2010.14
We here describe the many additions and updates to MolProbity since 2010, along with background on its underlying principles and a tour of the components in its typical workflow. Notable new developments include adoption of MolProbity criteria by the wwPDB (worldwide Protein Data Bank15) and continued improvement of our unique scores seen for new mid‐resolution depositions to the PDB worldwide. Major infrastructure developments include translation of the former Java utilities to use the Python CCTBX utilities in Phenix, a MolProbity mirror site at the University of Manchester, GitHub version control and distribution, new Top8000 and RNA11 datasets, stricter protocols for Met and ring‐plane methyls, a “OneDotEach” streamlined but non‐pairwise contact analysis, an improved method of generating output coordinates for Asn/Gln/His flip corrections, and the update and use of electron‐cloud hydrogen positions for X‐ray crystallography. New validation measures (described in context of the website workflow) include additional file interpretation, conversions and cleaning at upload, expanded and more nuanced Ramachandran and rotamer criteria from the Top8000 dataset, use of a conformation‐dependent library of backbone geometry for validation if it was used in refinement, better handling for ensemble structures, CaBLAM Cα and carbonyl virtual‐backbone analyses for low resolution, and the flagging of twisted and cis non‐proline peptides.
Improving the worldwide database
The primary aim of structure validation, as we see it, is not just to identify mistakes, but rather to help fix them. We calibrate our success at that goal by annually tracking the “clashscore” (all‐atom steric clash overlaps ≥0.4Å per thousand atoms) for new worldwide PDB depositions in the resolution range 1.8‐2.2Å. All‐atom clashes are an especially sensitive and powerful indicator of local fitting problems, and still are provided only by MolProbity. Before the advent of MolProbity in 2002, clashscores were constant, but since then they have steadily improved, now by about a factor of 3, as shown in Figure 1(A). The change seems to be leveling off, as it must, since it cannot go below zero. In fact, this measure is sometimes interpreted too stringently: the goal is few clashes, as in the best reference data, not zero clashes.16, 17
Figure 1.
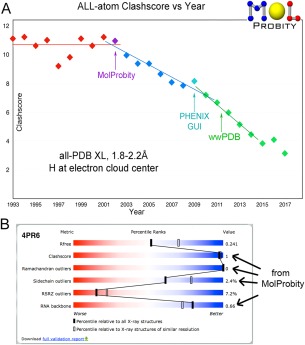
A, Time course showing strong improvement of MolProbity clashscores for the mid‐resolution half of deposits to the wwPDB from 1993 to mid‐2017. B, The validation “slider” and percentile system on the wwPDB web sites, which includes four criteria from MolProbity, illustrated for the 4pr6 HDV ribozyme at 2.3Å resolution.65
The wwPDB has adopted four MolProbity criteria (clashscore, Ramachandran, rotamer, and RNA backbone) along with other validation types, at deposition, as PDF reports for reviewers, and as visual “sliders” showing relative percentile scores on each structure summary page at the RCSB, PDBe, and PDBj websites [Fig. 1(B)]. They are now used for X‐ray crystallography18, 19 and for nuclear magnetic resonance,20 and may also be adopted for cryo electron microscopy. Complete MolProbity validation and its ongoing updates are integrated into the Phenix structural biology software suite, in the graphical user interface as well as for automated procedures and command‐line use.21, 22, 23
Results: Infrastructure Changes since 2010
CCTBX and python
The full range of MolProbity validation has now been incorporated into the Phenix crystallography suite21 to provide frequent user feedback, and specific MolProbity criteria are also used directly within the automated Phenix workflows. To accomplish this, MolProbity's mid‐level Java utilities have been re‐implemented in Python and use the same open‐source CCTBX (Computational Crystallography Toolbox) codebase24 that underlies Phenix. For Phenix users, MolProbity validation is accessible through the Phenix GUI, with real‐time links to outlier locations for fixup in Coot, or through the command‐line as phenix.molprobity. The individual components of MolProbity, including Reduce, Probe, Clashscore, Ramalyze, Rotalyze, Omegalyze, CaBLAM, Cbetadev, and Suitename are also separately available through the Phenix command‐line. The cctbx_project portion of Phenix is open source and freely available at https://github.com/cctbx/cctbx_project.
The specific validation functions on the MolProbity website call the same Python utilities and other CCTBX functions, so there is now only a single codebase for this part of the validation. Building MolProbity on the CCTBX project assures that validation on the MolProbity webserver and within Phenix stay synchronized. It also takes advantage of the robust support that the CCTBX project offers for access to the diffraction data and for evolving file types like mmCIF and other new code requirements.
Manchester mirror
Since 2016 there has been an identical MolProbity mirror website at the University of Manchester, UK (http://molprobity.manchester.ac.uk), with a link from the main site at Duke. It is hosted by Simon Lovell, who worked on early development of the validation criteria.9, 10, 11 This provides redundancy for rare downtimes and a closer site for users in the UK or Europe.
GitHub
GitHub now serves MolProbity both for version control and for distribution. The MolProbity source code and its key dependencies are all open source and freely available from the Richardson Lab GitHub repositories at https://github.com/rlabduke. MolProbity, Reduce, Probe, Suitename, and KiNG (as javadev) are each available as their own repositories. The reference_data repository contains the Top8000 dataset versions and the Ramachandran, rotamer, and CaBLAM distributions derived from it. This availability is used for Phenix nightly builds, and it allows users to install their own local MolProbity server, if they have limited internet access, confidentiality concerns, or a need to script many MolProbity runs using the command‐line tools; CCTBX is archived on GitHub, also open source. The GitHub pull request interface has also allowed community members outside the lab to submit suggested code improvements, which we have implemented.
Top8000 and RNA11 datasets
Many MolProbity validations rely on statistical expectations for macromolecular structure. These expectations are drawn from data of high‐quality residues in high‐resolution protein and nucleic acid structures. As the number of depositions in the PDB has grown, we have increased both the size and the accuracy of our reference datasets to better capture the depth and diversity of real molecular structure.
The latest iterations of our reference datasets are the Top8000 for proteins and RNA11 for RNA (see Methods for details of their construction). The “standard” Top8000 used for Ramachandran and CaBLAM is filtered at the 70% homology level and contains 7957 protein chains, up by an order of magnitude from the previous Top500.11 RNA11 contains 311 RNA structures (including proteins if present, to allow study of the interactions), up from 171 in the previous RNA05 dataset used in defining the community‐consensus RNA suite backbone conformers,25 with classification by content and function.
Chain‐level filtering is an important first step in preparing a high‐quality dataset. However, even high‐resolution structures almost always contain regions of local disorder. To guard against inclusion of the resulting local modeling errors, residue‐level filtering is a necessity. In preparation of Ramachandran and CaBLAM contours, we excluded residues having any mainchain atom with a B‐factor > 30. This simple filter worked quite well at excluding poor conformations and producing quite clean, reproducible contours. The enforced deposition of structure‐factor data along with each new entry in the worldwide PDB lets us use local electron‐density criteria in addition to B‐factors, for a more accurate and complete residue‐level filter (see Methods for specific details). All future validations and revisions will take advantage of this improved residue‐level filtering, applied across the atom types appropriate for backbone or for sidechain criteria.
Methyl orientations
Rotation of all methyl groups was originally written into Reduce but was deprecated almost immediately, partly because of the computational expense but mainly because steric clashes of methyl H atoms were nearly always caused by misplaced parent C atoms, so that curing them by methyl rotation was not the correct approach. Most methyls rotate no more than about 15° off stagger, but terminal Met methyls (with a longer S‐C bond) can rotate as much as 40–50°, so for many years we allowed rotation of Met methyls. However, in a survey of sub‐1Å resolution crystal structures we discovered that even for those structures such freedom produced the wrong answer more often than the right one, so we now keep Met methyls staggered also.
Methyl groups attached to planar aromatic rings (on groups such as hemes, thymines, or modified bases) are an interestingly different case, since the joining of a planar sp2 to a tetrahedral sp3 atom produces a flatter energy profile for rotation. Instead of a 3‐fold stagger, there is a preference for one of the two conformations with one H perpendicular to the ring plane and the other two 30° from it, confirmed by a survey of very high‐resolution H difference peaks. Figure 2 shows an example methyl on a heme at 0.88Å, illustrating clear positive difference peaks (blue) for all three H atoms in one of those preferred orientations, the incorrect positioning of our previous default (white bonds, off by 30°), and the resulting small, incorrect hydrogen clash (hotpink spikes). The new system solves such problems by trying out the two preferred orientations and choosing the better of the two (green lines) by all‐atom contact criteria, using a procedure analogous to the determination of Asn/Gln/His flips but not requiring an extended interaction network.
Figure 2.
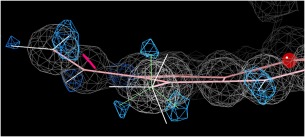
Improved positioning of methyl hydrogens attached to planar rings. White bond vectors show the old, incorrect default and green lines the new result, which uses one of the two preferred orientations and matches the H difference peaks at +2.8σ (blue). From the 1gwe Micrococcus lysodeikticus catalase at 0.88Å resolution.67
All‐atom contacts and OneDotEach
The standard MolProbity all‐atom contact representation,8 both for scoring and for visualization, consists of paired patches of dots (or overlap spikes) on the surface of each atom of the contacting pair. All hydrogen atoms are included. The intuition here is that atoms can be treated as interacting at the effective surfaces of their electron clouds (approximated by van der Waals radii if nuclear positions are being used). Thus contact is characterized by surface‐to‐surface interaction, as distinct from overlap volume or from pairwise center‐to‐center distance. That surface interaction is attractive until overlap, then repulsive unless defined to be a hydrogen bond. Since surface‐to‐surface approach defines interaction, a third intervening atom occludes pairwise interaction of the original two. Scalings are tuned by overall score match with observed occurrence in very well built experimental crystallographic models.
The many‐dots Probe protocol starts by placing at each atom position a sphere of surface dots approximately uniformly distributed (16 dots per Å2 by default). The scoring algorithm uses nested loops measuring unoccluded line‐of‐sight distances ≤0.5Å (by default) between each dot on a source atom and all dots of neighboring, not‐covalently‐bonded target atoms. The result is better than pairwise‐from‐centers algorithms at giving an intuitive understanding of packing inside and between molecules, and is more powerful for diagnosing and guiding corrections of fitting errors in the model.
However, this calculation both produces verbose output and is inherently slow, suitable for one‐time evaluations but not usable in a fast computational inner loop. As a first step in correcting that problem, Probe now includes an optional calculation called OneDotEach. It solves the verbosity and helps the speed problem without losing the non‐pairwise aspect, but it does not yet incorporate the smooth derivatives necessary for dynamic calculations such as refinement.
OneDotEach starts from the simple center‐to‐center vector between each non‐covalently‐bonded pair of atoms tested (those closer than van der Waals contact plus 0.5Å, by default). It then tests whether that vector passes within any other nearby atom's van der Waals radius of its center: this is our definition of a third atom occluding a contact. If so, the potential contact pair is rejected. Of course, in compensation there will be an all‐atom contact between the occluding atom and one of the original pair. The logic was simplified by the realization that, except in a direly bad‐geometry model, the only type of third atom that can occlude a pair within 0.5Å of each other is a hydrogen covalently bonded to one of the pair atoms. The output of OneDotEach is a single dot for each atom in an accepted pair, at the position where the interatomic vector intersects its surface. The main application of OneDotEach so far has been in calculating the distributions of nearest‐neighbor distances used in defining new van der Waals radii for the H parameter update (see below).
Better‐idealized output coordinates from NQH flips
In order to perform all‐atom contact analysis, MolProbity uses the C++ program Reduce to add and optimize hydrogen atoms, including analysis of each complete H‐bond network, rotation of OH, SH, NH3 (but not methyls), and consideration of first‐layer waters.9 As part of that process, Reduce optionally performs automated “flips” on Asn, Gln, and His residues to correct a common error where one of these sidechains (near‐symmetric in electron density) is modeled with its terminal χ angle 180° from the correct position. Such an error usually produces a pattern of clashes and missing H‐bonds that can be recognized automatically. When Reduce recognizes such a pattern, it will recommend a “flip” of that sidechain. The automated assignments are very reliable, but 3D “flipkin” kinemages show views with animation to compare the pattern of contacts between alternatives, allowing the user to see the evidence behind each potential flip and make an informed personal decision if they wish.
The NQH flip was previously performed by simply renaming the terminal N and O atoms of Asn and Gln or the C and N ring atoms of His, without changing any of the modeled coordinates. This method has the important advantage of preserving the fit‐to‐density of those atoms perfectly, and of showing the two competing sets of all‐atom contacts most correctly. Therefore the scores, flip decisions, and flipkins are still produced this way. However, the bond geometry around the His ring and between the C‐O and C‐N bonds is not quite symmetrical. Performing the flip by renaming atoms generated aberrant bond lengths and angles around the renamed atoms, by up to 6σ (0.1Å and 5° for Gln). This geometry would be easily corrected by another round of refinement after performing flips, but enough users have deposited structures without refining again that the PDB now contains a statistically significant population of aberrant bonds resulting from Reduce. To protect the integrity of the database, we developed a new method for producing the output coordinates from NQH flips.
The goal of the new method is to closely maintain the optimized fit of the relevant sidechain atoms in their electron density (and thus also their contacts), but without distorting covalent geometry. The intuitive, simple 180° rotation is not acceptable, because asymmetry of the sidechain head (the His ring or the terminal C/N/O of Asn and Gln) changes atom positions significantly. We chose a three‐step docking procedure. First, the sidechain head is rotated 180°, as intuition dictates. Second, the head group is hinged back into the plane of the head group in the original model, compensating for cases in which the head group is not in plane with its stem [Fig. 3(A,B)]. Third and finally, the whole sidechain is three‐point docked as a rigid body [Fig. 3(B,C)]. The Cα position is held constant and the two terminal H‐bonding atoms in the sidechain are docked as closely as possible onto their switched‐identity original positions.
Figure 3.
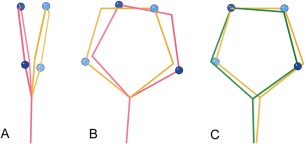
The new NQH flip output protocol starts with a simple 180° rotation, which does not give exactly superimposed atoms even for ideal geometry. That offset, and also more severe distortions, can be nearly corrected by two additional moves. A, The head groups of sidechains are often not in plane with their stems, resulting in a large shift of the terminal atoms when the sidechain head is rotated 180° (pink) from the original position (gold). A hinging motion brings the new head position back into the plane of the original. B, The rotated and hinged sidechain (pink) is still not well aligned to the original (gold) within that plane. C, A three‐point rigid dock motion, keeping the same Cα position, results in a final docked sidechain (green) with atoms nearly on top of the original ones (gold), but without added geometric distortion.
This new flip method still affects sidechain geometry, but much less so. It changes the bond angles between the mainchain and the sidechain at the Cα, but usually by less than one σ. No bond lengths are affected. Users should bear in mind that the output coordinates will now not precisely match what is seen in the flipkins. The new output coordinates correct the database pollution, but should serve as a reminder that all local structure corrections can generate geometry problems where they rejoin the rest of the model, and refinement after correction is always necessary to settle these details.
Re‐definition of H‐atom parameter sets
Explicit hydrogen atoms have increasingly become an important part of both experimental and computational methods for structural biology. In contrast to the accurately determined geometrical parameters for heavier atoms, the various bond‐length and van der Waals parameter sets in current use for hydrogen were derived many decades ago from limited data. Their specific values differ by as much as 20% between libraries, including within our now‐integrated system of MolProbity and Phenix. That is a big discrepancy for an effective bond length, and it can fairly often mean the difference between an acceptable steric contact and a serious steric clash of the van der Waals spheres (>0.4Å overlap counts as serious in MolProbity). Around that threshold of 0.4Å, we aim to optimize the balance between diagnosing serious conformational errors and raising false alarms. Packing analysis and validation both depend on the total system of hydrogen x‐H distances, vdW radii, and the 0.4Å threshold defined for clashes. Although there may not be a single right answer at all resolution levels, several factors convinced us that our previous system was slightly too strict – primarily because even for the best structures, clashscore bottomed out at about 5 clashes per 1000 atoms rather than at zero.
For the above reasons, we set out to provide the scientific basis for two updated sets of H parameters, one specific for the electron‐cloud‐center positions suitable for x‐ray crystallography and one for the nuclear positions used in most other methods, and as accurate as currently feasible across bonded‐atom types and geometries within each set. Many information sources were utilized, including a search of the older literature, spherical‐patch fitting to quantum‐calculated (QM) electron‐density contours, small‐molecule neutron and x‐ray coordinates from the Cambridge Structural Database (CSD26), H difference peaks in <1Å protein crystal structures from the Protein Data Bank and, most decisive of all, a combination of H atom coordinates and electron‐density difference peaks in small‐molecule x‐ray structures from the Crystallography Open Database (COD27). New H van der Waals radii were tuned for use with the new H positions, by pairwise nearest‐neighbor atom‐atom distance distributions in the Top8000 quality‐filtered dataset of protein chains. The extensive methodological details are described in the Supplementary Information. Along the way, several interesting categories of H atom contacts were clarified. Very short carboxyl O‐O pair H‐bonds form the short side of a cleanly bimodal distribution and are unequivocally real, even outside the transition states where they are usually studied;28, 29 however, they require a narrowly specific relative geometry.30 Shortened CH…O interactions are fairly common in β sheet, but for good reference data they show distances within the tail of the overall distribution and no preference for biologically functional sites.
Historically, Reduce used nuclear hydrogen positions because it was originally developed for packing calculations rather than for crystallography, and because the best‐documented set of van der Waals radii was tuned to those positions.31, 32 However, for the major current uses that is the wrong choice, because the electron cloud is what diffracts X‐rays and its outer region is where the atoms actually interact – repelling when separate electron clouds overlap.33 Phenix, appropriately for X‐ray refinement, has placed hydrogens at the shorter distances of the electron‐cloud centroids. The Phenix x‐H distance values were adopted from ShelX34 by way of the CCP4 monomer library,35 but the original procedures were not explicitly documented and there was no value for S‐H. Therefore, the work described here has combined evidence from a variety of sources in order to define optimal contemporary sets of electron‐cloud and of nuclear x‐H bond lengths and corresponding van der Waals parameters. We also re‐examined the nuclear positions for H and D, finding no significant difference, and we carefully proofread large libraries to correct the inevitable few typographical errors. A number of software modifications were made, both for accomplishing the underlying research and for implementing its results.
Heavier atoms such as carbon have electron‐cloud center and nuclear position essentially coincident, but for hydrogens it has long been known that they differ quite significantly.36, 37, 38 The single electron of a hydrogen atom must provide its share of electron occupancy in the covalent bond to its parent heavier atom, which systematically contracts the electron cloud inward from the nuclear position. In addition, the H electron cloud can be shifted sidewise by steric bumps or H‐bonding with surrounding atoms.39 As an example of both these effects, the Hɛ1 hydrogen on Trp 37 of PDB 1yk4 at 0.69Å resolution in Figure 4 was placed by Reduce at the nuclear position and planar to the ring. However, the clear Fo‐Fc difference peak (blue) for Hɛ1 is shifted both inward toward the ring N and also upward toward a line to the H‐bond acceptor, a carboxyl oxygen. This work aims to correctly reflect the systematic shifts along the covalent bond direction, but does not attempt to model second‐order shifts due to local environment. The following sections summarize the database and literature results and the overall decisions, organized by parent‐atom element type.
Figure 4.
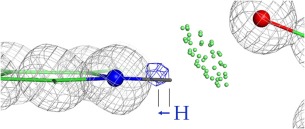
Shift of a high‐resolution H difference peak at 3.2σ (blue contours) toward its parent atom from the nuclear position. Trp Hɛ1 of the 1yk4 Pyrococcus abyssi rubredoxin at 0.69Å resolution.68
C‐H distances
CH nonpolar x‐H distances. There are plentiful data, and all methods agree closer than ±0.02Å for each type of geometry. For nuclear‐position tetrahedral CH, the previous distance value was 1.1Å for all subtypes, as implemented in Reduce and MolProbity. The newly estimated values are:
1.099 ±0.04Å (72 examples), 1.088 ±0.03Å (101 examples), and 1.084 ±0.05 Å (114 examples) for separate CH1, CH2, CH3 from CSD neutron crystallography, with an average of 1.089 ±0.04Å and trending down across the three subtypes
From electron diffraction, we located a tetrahedral CH distance only for methane (which we consider CH3 type), of 1.086 ±0.0024Å,40, 41 and 1.087Å for deuteromethane.
NMR sees a longer effective CαH distance of 1.117 ±0.007Å, presumed to be because dipolar couplings are sensitive to bond‐angle dynamics.42
For nuclear planar CH, the previous MolProbity value was the same as tetrahedral, at 1.1Å. The new estimate is:
1.077 ±0.02Å (68 examples) from CSD neutron data
For electron‐cloud‐center tetrahedral CH, the prior ShelX/Phenix values were 0.98, 0.97, 0.96 Å across CH1, CH2, CH3. The newly estimated values are:
0.958, 0.961, 0.955 ±0.01Å for CH1,2,3 from QM sphere‐fit, averaging 0.956Å without any monotonic trend
0.96, 1.01, 0.94 ±0.1Å for CH1,2,3 from the high‐resolution PDB survey of H difference peaks, averaging 0.97 Å (77 examples) without a trend
0.95 ±0.03, 0.96 ±0.04, 0.967 ±0.04Å for CH1,2,3 from adjusted COD values, averaging 0.96 ±0.036Å (119 examples) with an upward rather than downward trend
For electron‐cloud planar CH, the previous value in ShelX/Phenix was 0.93Å. Newly estimated values are:
0.93 ±0.01Å from the QM sphere‐fit procedure
0.96 ±0.1Å from hi‐resolution PDB H difference peaks
0.942 ±0.03Å (217 examples) from COD data, with adjusted H difference peaks where needed
For tetrahedral CH, the measured trend across CH1–2‐3 is not consistent between atom types or between methods (see Fig. 5 left). Therefore, for the time being, we are adopting single distance values for tetrahedral CH of 1.09Å nuclear and keeping the central ShelX/Phenix value of 0.97Å electron‐cloud although it may be a bit high, since the accuracy of our new data does not justify a 0.01Å change.
Figure 5.
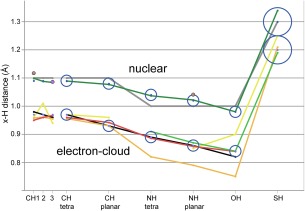
Parent‐atom‐to‐hydrogen (x‐H) distances. Previous values are in gray for MolProbity nuclear and in black for ShelX/Phenix electron‐cloud center. New data sources are in dark green for CSD nuclear, lighter green for CSD X‐ray, gold for QM sphere‐fit, yellow for PDB H peaks, and red for COD adjusted (our most reliable e‐cloud values). Individual datapoints are in brown for NMR and in purple for electron diffraction. Our final adopted values are plotted as circles with an ESD radius, 0.05Å for SH and 0.02Å for all other atom‐pair types.
Going forward, we have adopted 1.08Å for nuclear planar CH. For electron‐cloud planar CH, the COD adjusted average and the direction of most adjustments suggest that the ShelX value of 0.93Å is slightly low, but we judge the current evidence insufficient to justify a change.
N‐H distances
Different methods agree less closely for polar x‐H distances, and the freer rotation of OH and NH3 gives fewer examples with clear H difference peaks, degrading statistics. QM sphere‐fit produces low values for polar x‐H, because the QM calculations were done in vacuo, where the electron draws in even closer along the bond. In confirmation of this effect, a difference of 0.04 to 0.06Å longer was seen in our PDB H‐peak study for hydrogen‐bonded NH or OH versus the rare case of apolar surroundings. QM calculations were also done with a suitably H‐bonded water, but the local sphere‐fit could not be performed because electron density was continuous along the H‐bond direction (see Methods section of Supplementary Material). However, the sphere‐fit values help confirm other methods by showing a very close match in their pattern of change (gold line in Fig. 5).
For nuclear tetrahedral NH, the prior MolProbity value was 1.0Å. In our macromolecular data, tetrahedral NH groups are all NH3. The new estimate is:
1.037 ±0.03Å (165 examples) from CSD neutron coordinates
For nuclear planar NH, the prior MolProbity value was 1.0Å. In our macromolecular data, NH1 and NH2 are planar. The new estimates are:
1.022 ±0.03Å (67 examples) from CSD neutron coordinates
1.041 ±0.006Å from NMR
For electron‐cloud tetrahedral NH, the ShelX/Phenix value was 0.89Å. The new estimates are:
0.82 ±0.01Å from QM sphere‐fit
0.91Å from CSD X‐ray coordinates
0.886 ±0.03Å (154 examples) from adjusted COD H difference peaks
For electron‐cloud planar NH, the ShelX/Phenix value was 0.86Å. The new estimates are:
0.79 ±0.01Å from QM sphere‐fit
0.87Å from CSD X‐ray coordinates
0.85 ±0.1Å from PDB H peaks
0.857 ±0.04Å (57 examples) from adjusted COD H difference peaks
We have adopted new values of 1.04Å for nuclear tetrahedral NH and 1.02Å for nuclear planar NH, and are keeping the ShelX electron‐cloud NH values of 0.89Å tetrahedral and 0.86Å planar.
O‐H distances
OH groups are tetrahedral in macromolecular crystal data: on Ser, Thr, or Tyr sidechains, sugar rings, or waters (virtual H, in MolProbity). Although common, OHs are inherently mobile, and clear H difference peaks are only seldom observed.
For nuclear tetrahedral OH the prior MolProbity value was 1.0Å. The only new estimate is:
0.98 ±0.03Å (51 examples) from CSD neutron coordinates
Electron‐cloud tetrahedral OH distances vary considerably, and are a case where we adopt a value different from the prior ShelX/Phenix value of 0.82Å. New estimates are:
0.75 ±0.015Å from QM sphere‐fit is, again, low
0.90 ±0.1Å (31 examples) from PDB H difference peaks is quite high relative to other trends, and is downweighted because of the large standard deviation
0.84Å (39 examples) from CSD X‐ray
0.839 ±0.03Å (58 examples) from adjusted COD H peaks. The ShelX value at 0.82Å was considerably lower, so we compared raw versus adjusted COD distributions (Supporting Information Fig. S3). The raw data showed a clear bias toward assigning the expected ShelX value: over half of the 76 original datapoints were in a narrow spike at exactly 0.82Å. 58 examples showed clear H difference‐density peaks, and when H coordinates were adjusted to match those peaks if necessary, then the distribution broadened and shifted upward to a mean value of 0.839 ±0.03Å.
0.85 ±0.05Å (68 examples) for water OH, from unadjusted COD; these had no artifact at 0.82Å
We have therefore adopted 0.98Å as the nuclear OH distance and 0.84Å as our best estimate of the true electron‐cloud OH distance.
S‐H distances
Most difficult of all are the SH distances, which have no entry from ShelX, were inadvertently set to a nuclear distance (1.34Å) in Phenix and to a CH distance (0.96Å) in CNS (producing spurious values at or near those), and have few reliable examples in any of the experimental datasets. All are tetrahedral. The prior MolProbity value for nuclear tetrahedral SH was 1.3Å. The new estimate is:
1.34Å (only 5 examples) from CSD neutron coordinates, including the most precise historical measurement of 1.338 ±0.002Å43
The prior electron‐cloud tetrahedral SH distance was missing in ShelX and incorrectly set to a nuclear 1.34Å in Phenix, so those values are irrelevant. The new estimates are:
1.21 ±0.01Å from QM sphere‐fit
1.19 ±0.03Å (24 examples) from CSD X‐ray coordinates (there were no further useful SH examples from the COD
1.25 ±0.1Å (17 examples) from PDB H difference peaks
Compromising across this rather approximate overall data, we have rounded off our SH values to 1.3Å nuclear and 1.2Å electron‐cloud. The consolation is that since free SH groups (not disulfide bonded or metal liganded) are relatively rare, refinement or validation will not be much affected by this particular uncertainty.
Van der Waals radii
It is the combination of x‐H positioning with van der Waals radii of H and other atoms that determines the atom‐atom contacts, both favorable and unfavorable. This requires that the newly determined nuclear and electron‐cloud‐center x‐H “bond lengths” be complemented by new effective radii, optimized for each set. This task was done by analyzing distance distributions of H‐to‐H and H‐to‐heavier atom nearest‐neighbor distance in high‐quality reference data, and tweaking the individual radii so that all pairwise distributions peak close to contact distance (zero “min gap”, shown in Supporting Information Fig. S4). We used the Top8000 reference dataset quality‐filtered at both chain level and residue level (see Methods section in Supplementary Material). We started with CH2‐CH2 methylene distances, as they are the commonest type of cleanly positioned H atoms, and worked outward from there. Our previous MolProbity parameters peaked nicely near zero for H‐to‐heavier atom distributions, but peaked somewhat below zero (overlapped) for H‐to‐H distributions. The two new parameter sets now behave well for both types of cases (Supporting Information Fig. S5). The nuclear radii have not changed, but the new radii for H atoms are larger than before by 0.05 Å.
Overall H parameter results
Table 1 lists the new parameter values for x‐H distances and Table 2 for van der Waals radii, including both electron‐cloud and nuclear cases. The new parameters are implemented consistently both on the MolProbity website and within the integrated Phenix‐MolProbity system. H atom positioning is now accurate to about 0.02Å in most cases, where previously a number of values were inappropriate choices by about 0.2Å.
Table 1.
x‐H Distances Used in Reduce (Å)
| x‐H atoms | e‐cloud | nuclear | example |
|---|---|---|---|
| tetra CH1 | 0.97 | 1.09 | Val Hb |
| tetra CH2 | 0.97 | 1.09 | Glu Hb2,3 |
| tetra CH3 | 0.97 | 1.09 | Ala Hb1,2,3 |
| planar CH | 0.93 | 1.08 | Phe Hd1 |
| tetra NH3 | 0.89 | 1.04 | Lys Hz1,2,3 |
| planar NH | 0.86 | 1.02 | peptide H |
| tetra OH | 0.84 | 0.98 | Ser Hg |
| tetra SH | 1.20 | 1.30 | Cys Hg |
Nuclear x‐H distances are from parent atom to H nucleus; “e‐cloud” x‐H distances are from parent atom to the H electron‐cloud center.
Table 2.
Atomic Radii Used in Probe (Å)
| atom type | e‐cloud | nuclear |
|---|---|---|
| H | 1.22 | 1.17 |
| H aromatic | 1.05 | 1.00 |
| H polar | 1.05 | 1.00 |
| C | 1.70 | 1.70 |
| C aromatic | 1.75 | 1.75 |
| N | 1.55 | 1.55 |
| O | 1.40 | 1.40 |
| P | 1.80 | 1.80 |
| S | 1.80 | 1.80 |
| Se | 1.90 | 1.90 |
The “e‐cloud” radii are used when x‐H distances are specified to the electron‐cloud center, and “nuclear” radii are used when H positions are at the nucleus.
Carbonyl carbons are given the smaller “C” radius.
We have tested the new system for its practical uses in two ways. An analysis of Asn/Gln/His “flips”, for a set of high‐resolution cases where electron density clearly defines the right answer, showed that few decisions differed, but where they did differ the new system was more often correct. More definitive is the comparison shown in Figure 6, where clashscores for high‐quality structures previously bottomed out significantly above zero, while in the new system those clashscores are overall somewhat lower, but most importantly they now satisfactorily asymptote to zero.
Figure 6.
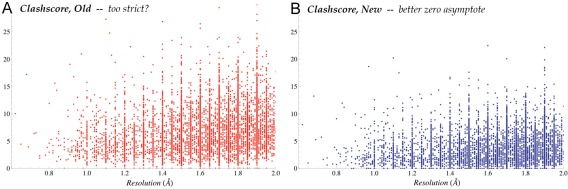
Clashscore vs resolution, for the Top8000 high‐quality reference dataset (see above). A, Clashscore for each structure by the previous MolProbity system (red), where few datapoints are at or just above zero. B, Clashscores for the same structures in the present MolProbity system (blue), where the scores do asymptote satisfactorily to zero.
MolProbity Workflow and New Validations
Upload: interpretation, conversions, and cleaning
The first step in analyzing a structure on the MolProbity webservice is either to upload a coordinate file from your computer or to fetch one from the PDB or other database. About 85% of MolProbity use is by upload (presumably structural biologists analyzing their own models during the process of structure solution) and 15% by fetch (presumably biological, biomedical, bioinformatic, educational, and other end‐users).
Input coordinate files are parsed to provide user feedback about their interpreted contents, such as number of chains, alternate conformations, presence and type of hydrogens, etc. The user should check that the displayed interpretation seems correct, since it is easy to confuse O vs 0 or l vs 1 in a PDB code and fetch the wrong file. Some format problems now generate error messages, such as old PDB format (pre‐v3.0, now rare) which is converted, mispaired MODEL and ENDMDL records, or duplicate atoms (usually a result of missing or inconsistent chain or segid fields), which will fail in CCTBX. Files submitted in mmCIF format are now automatically converted to PDB format on upload or fetch, with the hybrid36 system used for files too large for standard PDB format. Hybrid36 uses 2‐character chain‐ids and numbers atoms normally through atom 99999, then uses a combination of letters and numerals starting with A0000; the equivalent operation for residue number starts after 9999. The hybrid36 format is supported by Phenix and by all MolProbity validations. Due to heightened security issues, we also now check for content that seems to be executable code or not to be an interpretable coordinate file.
Hydrogen addition and NQH flips
On the main page there are edit options, such as dropping extra chain copies, and file‐choice options, such as choosing to keep the H atoms of your own input file rather than have Reduce optimize them. However, the first validation step is nearly always H addition, absolutely necessary to take advantage of the all‐atom contact analysis that is MolProbity's most unique and powerful feature. Roughly half the atoms in a given macromolecule are hydrogens, and the vast majority of interatomic contacts in a macromolecule are between H atoms. However, due to their single, weakly scattering electron, hydrogens are rarely visible except in difference density at ultra‐high resolution. As a result, hydrogens have seldom been modeled historically, and MolProbity must add and optimize H atoms for most structures before full validation can be performed. It uses Reduce for that task. The default is now to place those hydrogens at the electron‐cloud center positions suitable for x‐ray crystallography (see above), but the user can choose to use nuclear positions. In either case, they are optimized across complete H‐bond networks, including optimization of Asn/Gln/His (NQH) “flips” unless turned off by the user. The NQH flips are very reliable and the easiest type of automatic structure correction, so we recommend their use each time your structure has changed substantially. However, they are not perfect, and if you are already near completion and have decided to reject some flips, then by all means turn the process off. Note that you must download the resulting file to take advantage of the improved flips, and that their coordinates will now be better idealized than in previous versions of MolProbity (see above).
Better treatment of ensembles
We have improved the functionality of MolProbity for multi‐model ensemble PDB files. NMR structures are often deposited as ensembles of models, with potentially over a hundred models within a single PDB file. Additionally, ensemble crystallography is becoming more common, as a way of expressing either the modeling uncertainty or the likely conformational heterogeneity within a crystal. Previously, if a user uploaded an ensemble file to MolProbity only a limited set of analysis options were available, primarily an option to generate a multi‐model multi‐criterion kinemage, and a multi‐model Ramachandran plot PDF file. In order to obtain the results of the entire MolProbity validation suite for an ensemble, users were forced to manually select each model and run the analysis on each model one at a time.
In MolProbity 4.4, we have updated the ensemble analysis so that users can run our entire suite of analysis options on the complete ensemble file. Summary charts for each model are presented under separate tabs within the results page, allowing users to easily click between different models and compare their validation results. Additionally, the analysis options have been updated to include all of the improvements documented in this article. In order to maintain a reasonable runtime, we limit the ensemble analysis to the first 80 models, which covers the majority of ensembles currently deposited in the PDB and still provides a good sense of quality for a larger ensemble.
Finally, note that biological‐unit files from the PDB are expressed as multi‐model files, but they have a very different logic, since the “models” are actually separate instances of the same chain within a molecule, rather than alternative models for the same chain in the same place. MolProbity deals with these biological‐unit multi‐model files by attempting to remap the chains in the different models into a single model, giving each chain a new unique chain ID. This code has been updated to use 2‐character chain IDs, which allows analysis of larger biological‐unit files.
Primary validation run and summary
After adding hydrogens, the next step is to run “Analyze all‐atom contacts and geometry”. In that setup, MolProbity turns on appropriate options dynamically depending on the contents, size, and resolution of the structure being analyzed, but you can of course change them. Help explanations are linked from each option to help you decide.
The report of results starts with a summary table coded in stoplight colors, plus full details in graphics and in chart form. The following sections describe each individual validation type, in the order they appear on the summary.
All‐atom contact analysis
The first line in the summary table reports the all‐atom clashscore (number of bad atom‐atom overlaps ≥ 0.4Å per thousand atoms). Clashes are the single most powerful diagnostic for many kinds of local fitting problems. For instance, the backward His in Figure 7(A) has the wrong protonation and N placement as well as clashes, and can be corrected automatically as in panel B. The clashing “water” in Figure 7(C) is really a positive ion, as shown in panel D. If you are the structural biologist, start fixups on the largest clashes first when you return to model rebuilding (you can sort any column in the html chart by severity). Clashes are directional as well as local, so that, in context of the electron density, it is usually possible to figure out the underlying problem. For a severe clash, some group is usually turned around backward or misidentified; just pushing the two atoms apart is almost never a good answer. If you are an end‐user, zoom in on the part that interests you most. If that region is clear of outliers, then the details there are probably quite reliable even if the overall scores are poor.
Figure 7.
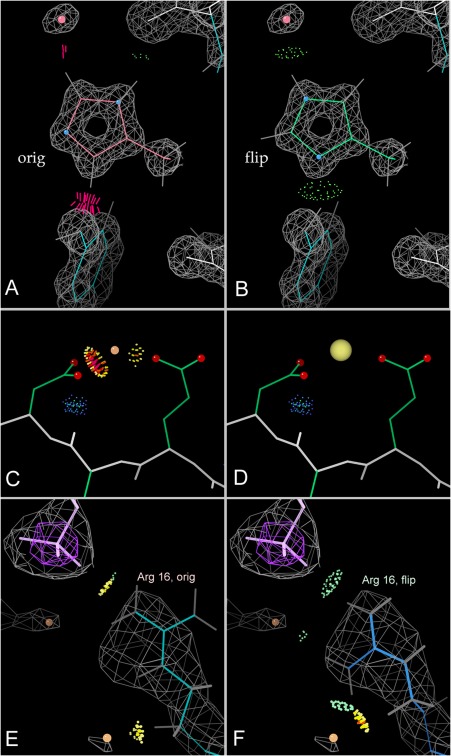
All‐atom contact analysis. A,B, Histidine “flip” from clashing to good H‐bonds; 1bkr His42 at 1.1Å.69 C,D, A peak originally fit as water, with clashes to nearby carboxyl oxygens, rebuilt as a sodium ion before deposition as 1xk8 at 2.7Å.70 E,F, An Arg guanidinium next to an RNA phosphate but making no H‐bonds, then as flipped over to a better position; 1s72 Arg 16 of ribosomal protein L3 at 2.4Å resolution.71
All‐atom contact analysis also evaluates presence or absence of H‐bonds and favorable van der Waals contacts. Those add further useful information, intuitively understandable in the graphics, such as the improbably missing H‐bonds between an Arg sidechain and the adjacent RNA phosphate shown in Figure 7(E) and corrected in Figure 7(F). Although not considered in standard refinement or model building, the absence of H‐bonding in such an arrangement is actually a strong signal of probable misfitting.
Sidechain rotamers
The new Top8000 dataset also allowed us to revisit and improve our empirical distributions for rotamer validation.44 Nearly a million (983,574) non‐Gly, non‐Ala residues passed the new electron‐density and other quality filters to be included in the final reference dataset from which the individual multi‐dimensional distributions are made. This increased size and accuracy allowed us to define 3 validation regions for rotamers as has long been done for Ramachandran‐plot criteria, with 98% of observed reference rotamers in the favored region, 0.3% in the outlier region, and the remainder falling in the allowed region between those cutoffs. Overall outlier frequency in general data has remained about the same, although some rotamer centers have shifted slightly or even divided, and additional rotamer peaks have reached above the outlier level for the long, multi‐dihedral sidechains. See the rotamer paper44 for methods, a complete list of rotamer names, χ values and reference‐data frequencies, and an explanation of why we can call this MolProbity's “ultimate” rotamer library.
Ramachandran backbone criteria
In Ramachandran analysis, for each residue the backbone ϕ and ψ dihedrals are calculated and compared against outer contours for the expected distribution of those angles. Those outer contours match very closely for most amino acid types (general case), but differ strongly for the other 5 types, since they have idiosyncratic configurations near the Cα (see18 supplement). The Top8000 dataset allowed us to add unique distributions for the cis‐proline and the branched Cβ (Ile/Val) cases and to update our existing Gly, Pro, prePro, and general cases (Fig. 8). The cutoffs for Favored vs Allowed vs Outlier are unchanged, with 98% of observed reference datapoints in the favored region and 0.5% (1 in 2000) in the outlier region. The only changes from older scores commonly seen in new Ramachandran validation results are for Ile/Val residues that were acceptable by the old but not the new specific criterion, or general‐case residues newly acceptable which lie along the more continuous region that runs down the positive side of ϕ.
Figure 8.
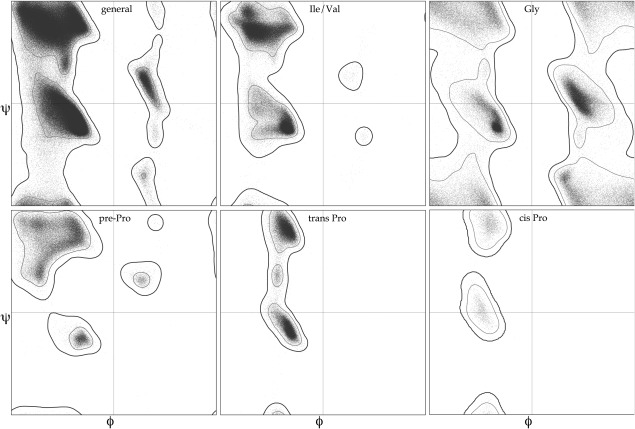
The six Ramachandran plots currently used for backbone ϕ,ψ validation by MolProbity, Phenix, and the wwPDB: general case, Ile/Val, Gly, pre‐Pro, trans Pro, and cis Pro. Based on a million quality‐filtered residues in the Top8000 dataset.
MolProbity score
At the frequent request of users, we developed the MolProbity score as a combined single indicator of model quality.13 It uses a weighted function of clashes, Ramachandran favored, and rotamer outliers, scaled and normalized so that its value approximates the resolution at which that score would be average. For MolProbity score and for clashscore, the summary table includes a percentile relative to structures near the same resolution. MolProbity scores and percentiles give a quick, rough guide for end‐users to compare entries at different resolutions for their molecule of interest, and structural biologists should aim to improve their model if scores are below average.
Covalent geometry
MolProbity performs covalent bond geometry analysis for both mainchain and sidechain atoms in protein and nucleic acids, now using the Phenix geometry libraries. Bond lengths or angles more than 4σ from the expected value are considered outliers, and are flagged in both chart and graphical forms. Another validation unique to MolProbity is the Cβ deviation,11 a combination of covalent angles and chirality around the Cα that flags geometry problems there much more effectively than simply analyzing individual variables. Large Cβ deviations usually mean that either the sidechain or the backbone has been misfit at that residue.
By default, the expected values for bond geometry are drawn from a single‐value library derived from Engh and Huber.45 However, single values, even with standard deviations, do not fully capture covalent bond geometry in the complex environment of macromolecules, since bond geometry is dependent on local backbone conformation. For this, a Conformation‐Dependent Library (CDL) has been developed46, 47 and implemented in Phenix48 for protein refinement. The CDL relates the expected covalent bond geometry to local backbone Ramachandran conformation. Because the expected bond geometry values in the CDL differ from those in the single‐value library (especially for the N‐Cα‐C τ angle), MolProbity validation now uses the CDL values for structures refined with the CDL, as detected from the REMARK 3 information of a submitted file. Similarly, for RNA, geometry targets are dependent on ribose pucker.
Cis or twisted non‐trans peptides
The peptide bond that joins adjacent amino‐acid residues in a protein has partial double‐bond character and therefore assumes a trans, or more rarely a cis, configuration. The cis configuration is significantly more common preceding a proline and results in a unique Ramachandran distribution for cis‐proline. To maintain this special relationship, we associate peptide bonds with their following residue. About 5% of prolines are cis, while only about 0.03% of all non‐proline residues are genuinely cis.
Recently, we were alerted to a surprising and improbable increase in the number of cis non‐proline peptide bonds being modeled,49 as shown in the plot (updated) of Figure 9(A). These are due to model‐building without consideration of prior probabilities, but also in part due to the lack of validation that flagged cis‐nonPro peptides, in MolProbity or other systems. We have therefore implemented a new validation and visual markup for non‐trans peptides. Matching the PDB definition, we define a cis peptide as one with an ω angle between −30° and +30°, and a trans peptide as one with an ω angle > +150° or < −150°. We add an additional definition of “twisted peptides” for ω angles that are more than 30° from planar. Justifiable twisted peptides are even rarer than non‐proline cis,50 and twisted peptides should virtually always be considered modeling errors.
Figure 9.
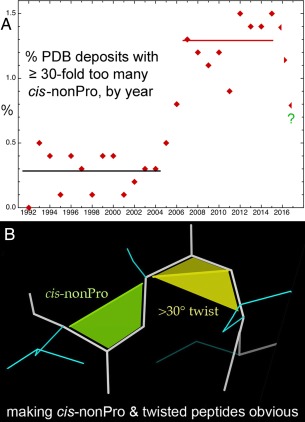
Cis‐nonProline and twisted peptides. A, Time course for percent of PDB deposits each year with ≥ 30‐fold too many cis‐nonProline peptides, in 3 phases: first low, then high for 10 years and, after recognition, now abruptly decreasing. B, MolProbity graphics markup for cis‐nonPro (lime green) and for twisted peptides (>30°, in yellow), with the twist line emphasized.
MolProbity reports on non‐trans peptides by providing counts of cis prolines, cis non‐prolines, and twisted peptides. Counts of cis non‐prolines or twisted peptides that constitute a suspiciously high percentage of the structure are flagged with yellow or red in the summary statistics chart. In the multi‐criterion chart that reports on each residue individually, each non‐trans residue is marked with its category (cis Pro, cis nonPro, twisted Pro, twisted nonPro) and the measured value of its omega peptide dihedral. In the multi‐criterion kinemage, each non‐trans peptide is marked with a surface that fills in the trapezoidal shape between the backbone trace and the Cα trace of a cis peptide [Fig. 9(B)]. These trapezoids are offset slightly from the model for ease of reading and are color‐coded by severity. Cis prolines are marked in gentle sea green, as they are relatively common and expected. Cis non‐prolines are marked in a more aggressive lime green, as they are likely to be errors. The vanishingly rare twisted peptides are marked in a warning yellow. Additionally, the interior angle between the two triangles of the trapezoidal shape indicates the severity of the twist.
Genuine cis‐nonPro peptides, like most very rare conformations, are nearly always found at functional sites, because evolution does not conserve unfavorable conformations unless they are biologically useful. Validation, and subsequent correction, of the incorrect ones is valuable because it improves the signal‐to‐noise for identifying the important, genuine cases. Following the discussions, papers, newsletters [e.g. Williams and Richardson (2015)23], and screaming markups in MolProbity and in Coot,51 it seems from the last few points in Figure 9(A) that this epidemic of overuse is now on the way to being cured.
CaBLAM for lower resolutions
We have introduced another new validation targeted at low‐resolution crystal structures, the Cα Based Low‐resolution Annotation Method, or CaBLAM.52 Low‐resolution structures pose two particular challenges to validation. First, they tend to contain many modeling errors, making it difficult to choose where to start. Second, sensitive validations, which work well in high‐resolution structures, may be confounded by the compound errors common in low‐resolution models. Ramachandran validation is a particular case in point ‐ Ramachandran validation is sensitive to small dihedral changes and is a powerful tool at high resolution. However, very large distortions of the Ramachandran dihedrals, especially multiple distortions in series, can mislead Ramachandran analysis.
CaBLAM is a validation of protein backbone that is more robust in the 2.5–4.0Å regime than Ramachandran analysis. CaBLAM takes advantage of the phenomenon that the overall Cα trace is relatively well represented in low‐resolution electron density and is relatively well modeled by humans and programs even when other details of the model are not. CaBLAM therefore uses Cα geometry to determine the intended structure of a region. It then checks the details of the model against this assessment of intended structure using contours derived from the Top8000, and declares outliers where there is a mismatch, as for most of the black datapoints in Figure 10(A,B).
Figure 10.
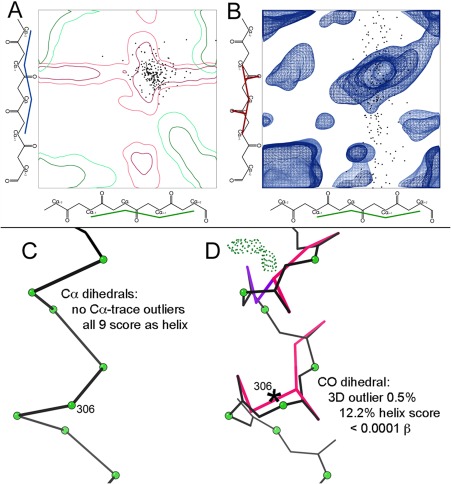
CaBLAM outlier and secondary‐structure diagnosis for 2o01, a large membrane protein at 3.4Å resolution.72 Datapoints (black) for “disguised” helix residues plotted on A, the α − α (μin‐μout) projection and B, the α‐CO (μin‐ν) projection of the 3D CaBLAM plot contoured for general‐case reference data. These points are nearly all inside the red 2‐D contours for helix diagnosis (which are distinct from the green β contours), but about half are shown to be misfit outliers in the 3‐D space, along the CO dihedral axis. C, Details for the distorted model of a particular α‐helix. All nine residues have legal Cα dihedrals which score as helix with good probability, in spite of D, 5 out of 8 COs pointed in the wrong direction (hotpink and purple markup) and only one α‐helical H‐bond.
The measures CaBLAM uses are, for each residue, two Cα pseudo dihedrals we call μin and μout and a dihedral that relates carbonyl oxygen orientation across the residue we call ν. Taken together, μin and μout indicate the intended structure. α helix, 310 helix, and β strands can be identified from μin and μout. The ν dihedral is sensitive to incorrect orientations of the peptide plane, which constitute a variety of common modeling errors at low resolution, especially misplacement of the carbonyl oxygen due to ambiguous or truncated sidechain density.53 Together these measures both identify modeling errors and suggest probable secondary structure elements, as shown on a local region in Figure 10(C,D). In contrast, Ramachandran and DSSP analyses are unhelpful and even misleading in cases such as this.
CaBLAM is most useful for validating secondary structure elements in low‐resolution models. In a high‐quality model, it provides little information beyond Ramachandran validation. Because loop structure is highly varied, CaBLAM's training set cannot recognize all valid loop conformations, and it tends to generate false positives in loops. Nevertheless, CaBLAM provides uniquely useful validation in a resolution regime where other methods struggle. For these reasons, MolProbity runs CaBLAM by default only for structures at 2.5Å or worse from either X‐ray or cryoEM.
RNA pucker and backbone conformers
In addition to the all‐atom contact and covalent geometry criteria applicable to nucleic acids as well as proteins, MolProbity provides conformational criteria tailored for RNA. C3'‐endo vs C2′‐endo ribose pucker cannot be seen directly at typical RNA resolutions, but it can be deduced from features which are observable and reliably modeled: the spherical phosphate density and the direction of the glycosidic bond that connects the base blob to the ribose blob.25, 54 The “Pperp” criterion (see Methods) is easily approximated visually while fitting, as how far the 3' P is out from the plane of the previous base,55 and it has enabled pucker‐specific torsion and geometry targets during Phenix refinement. A pucker outlier by this criterion is nearly always wrong.
In a community consensus study,25 42 clearly valid and several “wannabe” RNA backbone conformers were defined and named. This process was aided by the “suite” parsing of RNA backbone from sugar to sugar rather than the phosphate‐to‐phosphate nucleotide, which relates adjacent bases and within which the dihedrals show higher correlation.56 These backbone conformers, with a few updates, (see Methods) are a validation criterion in MolProbity. They are reported in the chart by name and “suiteness”: 1.0 at the 7‐dihedral cluster mean and 0 at the edge, as calculated by the Suitename program.25 They cover most but not all valid conformations, so each outlier should be examined but may be valid, especially if it is in an extended arrangement.
Multi‐criterion chart and graphics
The multi‐criterion kinemage produced by MolProbity or phenix.kinemage is the most powerful feedback we provide for a detailed exploration of a structure and its challenges. The visual markup employed in the kinemage encodes not just the location of outliers, but also their severity. Larger steric overlaps result in larger clash spikes, the sphere that marks a Cβ deviation has a radius equal to the deviation distance, and the “fans” that mark bond angle outliers extend between the modeled angle and the ideal, as in Figure 11(A). Additionally, the visual density of outlier markup is key to identifying problem regions in a model. We strongly recommend studying the multi‐criterion kinemage as linked either in the Phenix GUI or on‐line in MolProbity. If Java is impossible in your browser, download the kinemage and view it on your computer, using the KiNG program57 (available standalone on GitHub or packaged within Phenix both in the GUI and as a command‐line tool) and Java. We are investigating the addition of MolProbity markup in other software, for alternative future on‐line viewing (see Methods/Software).
Figure 11.
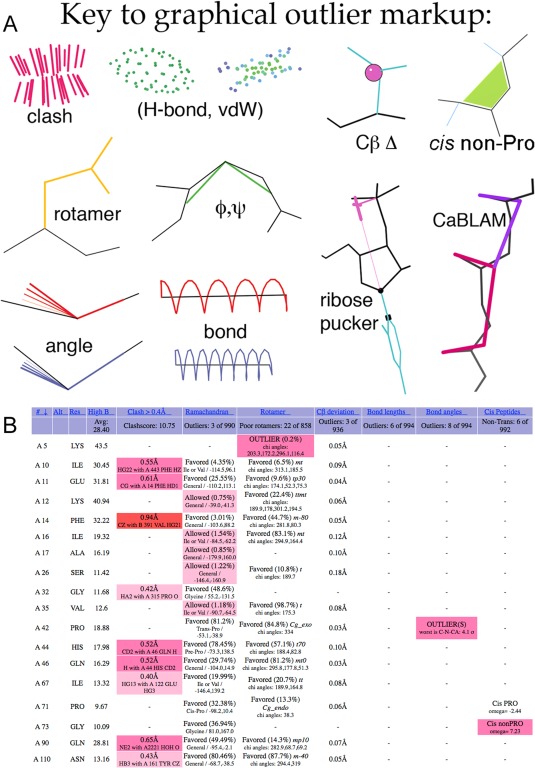
A, Key to MolProbity graphics markup for contacts and validation outliers. CaBLAM and non‐trans peptide markups are new. B, An example of the new three‐color system in the sortable html chart, and of the new non‐trans peptide reports in the right‐hand column, for 1qw9.73 Hotpink cells flag validation outliers, as before; pale pink cells are allowed but disfavored, and red cells are for extreme outliers. The single outlier in the rightmost column is Gly 73 cis‐nonPro; it is one of the rare valid ones, with excellent electron density and at the active site. Overall, however, this structure has more validation issues than usual at 1.2Å resolution. [Note that for large structures such as this, the chart default is to show only residues with an outlier.].
The multi‐criterion chart (an html page) gives detailed, sortable information on every residue, or only on each residue with an outlier if that option was specified. It now uses an enhanced coloring scheme to reproduce some of that visual intuition for outlier severity and problem regions in chart form [Fig. 11(B)]. Previously, any table cell representing an outlier would be colored hot pink, and all other cells would be uncolored. Now there are three colors ‐ light pink, hot pink, and bright red. These colors were selected to be distinguishable in gray‐scale as well.
Light pink is used for less‐favored conformations and minor outliers. During our CASP8 assessment,58 we found that it was often useful to overlook small clashes (with overlaps of 0.4–0.5Å) in favor of larger problems in particularly challenging predicted models. The same proved true for experimental models solved at low resolution. Small clashes are now colored light pink in the chart [Fig. 1(B)], where previously they had been hot pink. Residues in the Ramachandran “Allowed” region were previously only identified by text in the multi‐criterion chart. Now these less favored, but non‐outlier conformations are marked with light pink, as are the new “Allowed” rotamer conformations. Similarly, the new CaBLAM validation uses light pink for residues in its “Disfavored” region. Residues colored light pink in the chart should be considered worthy of attention, but are not necessarily outliers.
Bright red indicates particularly severe outliers. Clashes with an overlap of >0.9Å, bond length and angle outliers of >10σ, Cβ deviations >0.7Å, twisted peptide bonds >45° from planar, and Cα geometry outliers identified by CaBLAM are marked in the chart with bright red [Fig. 11(B)]. Ramachandran and rotamer validations do not currently produce a “severe outlier” designation suitable for marking in this way. The general outliers marked with hot pink are sometimes justified as valid by strong density, hydrogen bonding, structural homology, or other factors. The cutoffs for the severe outliers are set such that any outlier marked with bright red is almost impossible to justify.
Understanding Local Quality and Making Local Corrections
For a really large clash or other outlier colored red in the chart, something is sure to be wrong. However, perhaps it is not literally a steric clash but instead an unmodeled alternate conformation, a misnamed atom, or a bond too long to be recognized as covalent. All outliers are worth looking at, because there are few false alarms, but a fraction of cases such as poor rotamers, or CaBLAM outliers at low resolution will be shown valid by good electron density and some interaction holding them in an unfavorable conformation. Those valid outliers are likely to be functionally significant, because an unfavorable conformation is seldom conserved unless it is biologically useful. Remember that outliers are defined statistically by a low but finite occurrence in the well‐ordered parts of high‐resolution, quality‐filtered reference data. The expectation for a newly solved structure, therefore, is to approach or equal the same low percentage of outliers. Except in a small structure at high resolution, zero outliers usually means overfitting the data. A recent overview from a CCP4 Study Weekend presentation17 gives our best current guidance on how to tackle rebuilding the different types of validation outliers, with examples, and advice on when to stop.
Discussion
For 25 years now, macromolecular structure validation has provided a gatekeeping function at deposition and publication. More recently, its importance is recognized as a way for end‐users of those structures to evaluate reliability both overall and in local detail, and especially for structural biologists to improve the accuracy of their models throughout the process of structure solution. MolProbity continues to enhance its capability at performing all those functions. That enhancement process requires ongoing infrastructure improvement and also the development of new or extended validation criteria that can be optimally helpful for molecular systems at lower resolutions and for the characteristics of new structural biology techniques. New criteria typically come from solidly researched empirical or theoretical recognition of further regularities and relationships in macromolecular structure, and then formulating them in ways that can reliably improve the practice and the results of structural biology. We do such research ourselves, and also seek to apply it from the results of others.
This field still, somewhat surprisingly, needs a good many more ultra‐high‐resolution, careful X‐ray and neutron crystal structures done for the standard small components or pieces of protein, RNA, DNA, and carbohydrates, in order to truly define the geometrical parameters for their hydrogen atoms. From our side, we will aim in future work to research better handling of waters. All‐atom contact analysis is capable of classifying “water” peaks into ions, unmodeled alternate conformations, parts of unmodeled large ligands, or actual waters.59 After such a process, we then need to rescale scoring of their H‐bonds and clashes based on partial occupancy, electron density, and mobility.
In general, MolProbity validates macromolecular models based on their coordinates and does not do model‐to‐data validation. Asn/Gln/His flips are an especially good example, since the electron density is no help in making the choice except at very high resolution, while the combination of all‐atom clashes and H‐bonds does an excellent job. However, we use the evaluation of electron density as a central part of quality‐filtering the reference data from which we develop our detailed validation criteria.
Thorough integration of the complete MolProbity validation system into the Phenix crystallography and cryoEM software package has been a major factor in empowering development of the recent advances described in this paper, and has provided greatly improved accessibility and effectiveness of validation and correction for a large, important community of users. The separate MolProbity website now also uses the Phenix CCTBX utilities, enabling consistent and well‐tested addition of new functions for the website's wider community of users: other crystallography and cryoEM people, NMR and computational structural biologists, the deposition system at the worldwide PDB, biological and biomedical end‐users of structures, and teachers and students.
Methods
Website service
MolProbity is a research, software, and service project that deals with large volumes of complex data. The service component performs comprehensive validation on individual macromolecular structures, where it pays special attention to the local anomalies which are usually errors but sometimes valid and biologically important. After producing effective, user‐friendly software that creates user demand, the over‐riding requirement for service is near‐24/7 uptime of the website. That both requires long‐term management and equipment upgrades to maintain capacity, run speed, and security, and also requires constant short‐term attention: automatic monitoring where feasible, attention to user alerts, and frequent checks of the server state for possible hardware failures, persistent attack trials, or hung jobs. Our team, especially the system manager, almost always notice and fix problems within a few hours, nearly all of which is set up so it can be done remotely.
The website also produces error messages, with an email bug report option. We respond promptly to problems and queries, fix actual bugs as soon as feasible, and prioritize requests for new features if they are of fairly broad use and we have staff that can provide them. File format problems are the most common cause of bug reports, although those have decreased since we now diagnose the most frequent ones at the input stage (either fetch or upload).
Software
Our programs where speed or complexity is an issue are in C or C++. As described in the Infrastructure section, our connecting scripts and utilities formerly in Java have been rewritten in Python, as are new ones. The website is primarily controlled by PHP, especially for its dynamic responses to input, user choice, and intermediate results. Our online 3D interactive molecular display is done by the Java KiNG program.12, 57 Its structure modeling and corrections, docking construction, image and movie creation, and of course interactive display can still be done offline by us and others, but Java is nearly dead for the important online display in MolProbity. We have established collaborative initial systems to read and display 3D MolProbity validation markup on the molecule in ChimeraX60 with Tom Goddard and Tristan Croll, and in the RCSB PDB's new Javascript NGL viewer61 with Alex Rose. We will continue to enhance those capabilities and spread them to as many different viewers as feasible.
As described in Infrastructure, we use Git and GitHub as both our software version control for development and as our open‐source distribution system; it also provides a second backup. As Phenix developers, our components participate in their unit and regression test system. Most importantly, we ourselves both use in our own research and also set up deliberate tests of the detailed results from our software and look critically for unanticipated problems.
Reference datasets
The research component of MolProbity selects and quality filters high‐resolution, non‐redundant structures to obtain our basic chain‐level reference datasets. For each distinct use, those chains are quality filtered at the residue level, eliminating most errors by an optimal compromise between total number of residues and reliability of each one of them. For this reference data, our policy accepts a moderate rate of false negatives (i.e., correct residues not included) in order to get a low rate of false positives, provided that the rejection criteria are not logically connected to the criterion we will be testing and evaluating. In order to ensure satisfying the low false‐positive rate, we examine several dozen individual examples near the proposed thresholds to check for distortions, unconvincing electron density, or other circumstances which could allow acceptance of a problematic local model.
Conformational and validation parameters for each residue of the reference data are stored in, and queried from, a database. Earlier work used MySQL,62 and the recent rotamer work44 set up a Mongo database http://github.com/mongodb/mongo. Results from such structural‐bioinformatics queries are converted into multi‐dimensional data distributions smoothed and contoured by iterative kernel‐density methods.11 The validation measures eventually resulting from this process are characterized in stringency by the percent of outliers in the residue‐filtered, high‐resolution data; currently this varies from 0.03% for cis non‐proline peptides to 0.3% for rotamers to 1% for CaBLAM outliers.
The two primary reference datasets used in the work described here are the Top8000 for proteins and RNA11 for RNA. To construct the Top8000, we considered all protein chains solved by x‐ray crystallography and released by the PDB as of March 25, 2011. To be eligible for inclusion in the dataset, a chain had to have been solved at a resolution better than 2.0Å and to be of good structural quality. General structural quality was enforced by requiring chains to have a MolProbity score (a combination of clash, rotamer, and Ramachandran measures13) better than 2.0, and overall geometric quality was enforced by requiring chains to have ≤5% of residues with Cβ deviations, ≤5% of residues with bond angle outliers, and ≤5% of residues with bond length outliers. Chains that passed these filters were grouped according to PDB homology clusters, separately at 90%, 70%, and 50% sequence‐identity levels, and separately with and without requiring deposited diffraction data. In each homology cluster, the highest quality chain was selected for inclusion in the Top8000, where quality was determined by the average of a chain's resolution and MolProbity score. The “standard” Top8000 used for Ramachandran and CaBLAM is filtered at the 70% homology level and contains 7957 protein chains. In preparation of Ramachandran and CaBLAM contours, we excluded residues having any mainchain atom with a B‐factor > 30. This simple filter provides a proxy for local model fit to the electron density, and it indeed produced quite clean, reproducible contours.
For RNA, high‐resolution data is still relatively sparse, so criteria must be more forgiving, but more hand selection is feasible. For the RNA11 reference dataset, all X‐ray structures as of 11/11/11 containing at least a 3‐nucleotide RNA chain at ≤3.0Å resolution were hand‐selected for homology, in order to allow for inclusion of more than one complex or condition of the same sequence showing a significantly different RNA conformation (such as 5S rRNA alone versus in the ribosome). That process produced a chain‐level set of 311 structures, up from the RNA05 dataset of 171.25
The enforced deposition of structure‐factor data along with each new entry in the worldwide Protein Data Bank (wwPDB15) provides an opportunity for a more accurate and complete residue‐level filter. To this end, we revised the Top8000 in 2015 to require each chain to have deposited structure factors in addition to the other criteria. We then developed a new residue‐level filter combining local real‐space correlation coefficient, 2mFo‐DFc σ value at each atom, and B‐factor.44 Optimization produced threshold criteria using all 3 terms: real‐space correlation coefficient ≥0.7, 2mFo‐DFc σ ≥ 1.1, and atomic B‐factor <40, for all relevant atoms in the residue. Future validations and revisions will take advantage of this improved residue‐level filtering.
Electron‐cloud hydrogen positions
This study involved multiple methods and the work of over half the authors over several years. The method description is therefore very lengthy and is found in the Supplement.
CaBLAM
CaBLAM was developed to be a robust validation even in models where many atoms are placed incorrectly.53 As a result it seeks to use a minimal set of atoms ‐ Cαs and COs ‐ to define its four parameters. The main parameters are Cα pseudo dihedrals μin (defined as Cαi‐2‐Cαi‐1‐Cαi‐Cαi+1 for residue i) and μout (defined as Cαi‐1‐Cαi‐Cαi+1‐Cαi+2 for residue i), as shown in Figure 12. The third dihedral ν is defined using Cαi‐1, Cαi, Cαi+1, Oi‐1, and Oi for residue i. Additional pseudoatom points are constructed: Xi‐1 on the Cαi‐1‐Cαi line at the point closest to Oi‐1, and Xi on the Cαi‐Cαi+1 line at the point closest to Oi. The ν dihedral is then defined as Oi‐1‐Xi‐1‐Xi‐Oi (pink in Fig. 12). Finally, the Cα virtual angle for residue i is defined as Cαi‐1‐Cαi‐Cαi+1.
Figure 12.
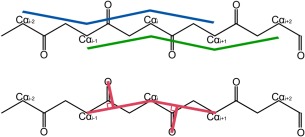
Virtual backbone dihedral angles in CaBLAM: μin (blue) and μout (green) defined by four successive Cα atoms, and ν (pink) to relate the direction between two successive carbonyl oxygens.
These four parameters were calculated for each protein residue in the Top8000. Residues for which any of the atoms used in the calculation were missing or had B‐factor >30 were excluded. Thus only residues from fully modeled and confidently modeled regions were included in the final training set.
Three‐dimensional contours were generated using two different combinations of these parameters: contours in the μin‐μout‐ν parameter space define expected protein backbone behavior, and are used to identify the majority of CaBLAM outliers; contours in the μin‐μout‐Cα virtual angle parameter space define expected Cα trace behavior and are used to identify severe Cα trace modeling errors. As in Ramachandran evaluation, proline residues populate a restricted portion of the CaBLAM parameter spaces and glycine residues populate a less restricted portion than the general case. Therefore, separate contours are defined for the proline, glycine, and general cases of residue type.
A further set of two‐dimensional contours were generated to define the observed behavior of secondary structure in the μin‐μout parameter space. Residues in the training set were identified as α helix, 310 helix, or β strand based on hydrogen bonding pattern. A residue i was identified as α helix if residues i‐1, i, and i + 1 all participated in n to n + 4 hydrogen bonding or if residues i‐1, i, and i + 1 all participated in n‐4 to n hydrogen bonding. These paired, one‐sided definitions of helix allowed the correct identification of helix residues near the ends of helices. A residue i was identified as 310 if residues i‐2, i‐1, and i all participated in n to n + 3 hydrogen bonding and residues i, i + 1, and i + 2 all participated in n‐3 to n hydrogen bonding. A residue i was identified as β strand if it was on a middle strand of a β sheet, and a full cycle of β‐pattern hydrogen bonding (parallel, antiparallel, or a combination of the two) both preceded and followed that residue. This definition of β structure is somewhat restrictive, but was necessary to generate clean contours. This provided a mapping between Cα geometry and hydrogen bonding patterns.
Setting cutoff values for the contours was done heuristically by manual inspection of structures with known modeling errors. For the μin‐μout‐ν CaBLAM space, the outlier cutoff was set at the bottom 1%, and a second “Disfavored” cutoff roughly analogous to the Ramachandran “Allowed” cutoff was set at the bottom 5%. The top 95% are considered favored conformations. For the Cα‐only μin‐μout‐Cα virtual angle space, a single outlier cutoff was set at the bottom 0.5%. This space did not receive an “Allowed” analogue cutoff, as it is intended primarily as a sanity check on Cα trace interpretation. The lower bounds for α and 310 helix contours were set at 0.1%, and for β strand at 0.01%. Additionally, for a residue to be identified as secondary structure, that residue's preceding and following residues are also required to score well on that secondary structure's contours. All these definitions strive to achieve a balance between a generous interpretation of structure appropriate to addressing low‐resolution structures and a requirement for structure continuity that prevents false identifications.
When CaBLAM validation is run on a structure, the four CaBLAM parameters ‐ μin, μout, ν, and Cα virtual angle ‐ are calculated for each protein residue. Much as Ramachandran validation is not possible for the first or last residue in a chain, CaBLAM assessment is not possible for the first two or last two residues because not all atoms necessary to calculate μin and μout are present. For each residue with a complete parameterization, its parameters are compared to the μin‐μout‐ν contours to identify CaBLAM outliers. Each residue is also compared to the μin‐μout‐Cα virtual angle contours to identify Cα geometry outliers. Cα geometry outliers are usually excluded from the following secondary structure identification step on grounds that secondary structure identification is dependent on a reliable Cα trace. Residues are then compared to the μin‐μout contours for secondary structure. Individual residues identified as matching the appropriate μin‐μout geometry are assembled into secondary structure elements ‐ helices and strands ‐ based on adjacency to other residues of like geometry. Residues that are part of these assembled secondary structure elements are reported as probable secondary structure, with their % contour as a score; residues with isolated secondary‐structure‐like geometry are not reported.
CaBLAM validation results are available in several forms. On the Phenix commandline, phenix.cablam produces colon‐delimited text output by default. On the MolProbity website, this same information is available in the structure summary and residue‐level tables. CaBLAM also provides visual markup for outliers in kinemage format. CaBLAM outliers and disfavored residues are marked with lines that follow the ν dihedral of that residues, colored hotpink for outliers and purple for disfavored [see Fig. 9(A)]. Cα geometry outliers are also marked, using red lines that follow the Cα trace along the Cα virtual angle of the residue.
CaBLAM is a unique tool for validating protein backbone, and especially secondary structure, at low resolution or in otherwise difficult models. Because CaBLAM's outlier cutoff is unusually punishing for a validation ‐ excluding a full 1% of observed structure ‐ a significant number of residues are expected to present as outliers even in good structures. As a result, CaBLAM performs best on regular regions of structure and tends to offer false positives in loop regions due to their high irregularity. Ramachandran evaluation is therefore still recommended as the backbone validation of choice for high‐resolution structures. Nevertheless, CaBLAM extends the reach of protein backbone validation into a resolution range at which Ramachandran evaluation is not reliable.
RNA pucker and backbone
Ribose pucker outliers (C3'‐endo when they should be C2′‐endo or vice versa) are checked by the “Pperp” versus δ dihedral criterion from Richardson et al.25 A perpendicular is dropped from the 3′ P atom to the vector direction of the glycosidic bond; its length is >2.9Å for C3′ pucker and ≤2.9Å for C2′. Backbone suite (sugar‐to‐sugar) conformers are defined, both originally and here, by 7‐dimensional suite‐torsion datapoint clustering, shown and analyzed in the Mage63 or KiNG57 display programs. A named conformer requires ≥5 examples with B‐factor <60, or ≥3 if one is at <2.0Å resolution.25 Their validation for new structures is calculated by the Suitename program.
In present work with RNA11, we made use of the ERRASER program,64, 65, 66 which uses MolProbity, Phenix, and Rosetta to perform and evaluate exhaustive backbone‐conformer search, for problematic dinucleotides. It does not use the information of our defined suite conformers, but almost always reproduces them in its output. The most frequent apparently valid RNA backbone outliers we see are for bulged or junction suites with widely separated bases, which have high variability and therefore few examples per conformer. To test whether such a case with good electron density might be a valid new suite conformer, we ran ERRASER on it and related structures to see if a consistent consensus conformer was found. For instance, Figure 13 shows a spread‐out junction at 2.9Å in the 3gx5 riboswitch, previously an unrecognized and thus outlier (!!) conformation, but with completely unambiguous electron‐density support. ERRASER left it as is, and corrected several less well‐fit examples in related structures to match. This conformer has now been named 3h and added to the suite conformer list.
Figure 13.
A new RNA backbone suite conformer (named 3h), its recognition as valid aided by ERRASER calculations for this and related structures. This example forms an extended helix junction in the 3gx5 SAM riboswitch at 2.4Å resolution.74 2mFo‐DFc electron density at 1.2σ (gray) and 3σ (purple).
Supplementary material
MolProbity4_Supplement.pdf describes in detail the methods used in the revision of hydrogen distance parameters and lists all of the structures used.
Supporting information
Supporting Information
Acknowledgments
We thank Laura Murray for compiling RNA05 and starting RNA11, Ralf Grosse‐Kunstleve for the hybrid36 format and its converters, Richard Gildea for scripts in the Olex2 viewer, Dorothee Leibschner for revisiting SH difference peaks in the PDB, and Nat Echols and Billy Poon for the MolProbity GUI in Phenix.
References
- 1. Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) ProCheck: a program to check the stereochemical quality of protein structures. J Appl Cryst 26:283–291. [Google Scholar]
- 2. Hooft RWW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381:272–272. [DOI] [PubMed] [Google Scholar]
- 3. Kleywegt GJ, Jones TA (1996) Efficient rebuilding of protein structures. Acta Cryst 52:829–832. [DOI] [PubMed] [Google Scholar]
- 4. Yeates TO (1997) Detecting and overcoming crystal twinning. Methods Enzymol 276:344–358. [PubMed] [Google Scholar]
- 5. Zwart PH, Grosse‐Kunstleve RW, Adams PW (2005) Xtriage and Fest: automatic assessment of X‐ray data and substructure structure factor estimation. CCP4 Newsletter, Winter, Contribution 7.
- 6. Brünger AT (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355:472–475. [DOI] [PubMed] [Google Scholar]
- 7. Kleywegt GJ, Harris MR, Zou J‐Y, Taylor TC, Wahlby A, Jones TA (2004) The Uppsala electron‐density server. Acta Cryst 60:2240–2249. [DOI] [PubMed] [Google Scholar]
- 8. Word JM, Lovell SC, Labean TH, Taylor HC, Zalis ME, Presley BK, Richardson JS, Richardson DC (1999) Visualizing and quantifying molecular goodness‐of‐fit: small‐probe contact dots with explicit hydrogen atoms. J Mol Biol 285:1711–1733. [DOI] [PubMed] [Google Scholar]
- 9. Word JM, Lovell SC, Richardson JS, Richardson DC (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of side‐chain amide orientation. J Mol Biol 285:1735–1747. [DOI] [PubMed] [Google Scholar]
- 10. Lovell SC, Word JM, Richardson JS, Richardson DC (2000) The penultimate rotamer library. Proteins 40:389–408. [PubMed] [Google Scholar]
- 11. Lovell SC, Davis IW, Arendall WB, III , Bakker PIWD, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins 50:437–450. [DOI] [PubMed] [Google Scholar]
- 12. Davis IW, Murray LW, Richardson JS, Richardson DC (2004) MolProbity: structure validation and all‐atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 32:W615–W619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davis IW, Leaver‐Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen VB, Arendall WBIII, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Cryst 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burley SK, Berman HM, Kleywegt GJ, Markley JL, Nakamura H, Velankar S (2017) Protein Data Bank (PDB): The single global macromolecular structure archive. Methods Mol Biol 1607:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richardson JS, Prisant MG, Richardson DC (2013) Crystallographic model validation: from diagnosis to healing. Curr Opin Struct Biol 23:707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richardson JS, Williams CJ, Hintze BJ, Chen VB, Prisant MG, Videau LL, Richardson DC (2017) Model validation – Local diagnosis, correction, and when to quit. Acta Cryst D in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Read RJ, Adams PD, Arendall WB, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lütteke T, Otwinowski Z, Perrakis A, Richardson JS, Sheffler WH, Smith JL, Tickle IJ, Vriend G, Zwart PH (2011) A new generation of crystallographic validation tools for the Protein Data Bank. Structure 19:1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gore S, Velankar S, Kleywegt GJ (2012) Implementing an X‐ray validation pipeline for the Protein Data Bank. Acta Cryst 68:478–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Montelione GT, Nilges M, Bax A, Güntert P, Herrmann T, Richardson JS, Schwieters CD, Vranken WF, Vuister GW, Wishart DS, Berman HM, Kleywegt GJ, Markley JL (2013) Recommendations of the wwPDB NMR validation task force. Structure 21:1563–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) Phenix: a comprehensive Python‐based system for macromolecular structure solution. Acta Cryst 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Echols N, Grosse‐Kunstleve RW, Afonine PV, Bunkoczi G, Chen VB, Headd JJ, McCoy AJ, Moriarty NW, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Adams PD (2012) Graphical tools for macromolecular crystallography in Phenix. J Appl Cryst 45:581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Williams CJ, Richardson JS (2015) Avoiding excess cis peptides at low resolution or high B. Comp Cryst Newsletter 6:2–6. [Google Scholar]
- 24. Grosse‐Kunstleve RW, Sauter NK, Moriarty NW, Adams PD (2002) The computational toolbox: crystallographic algorithms in a reusable software framework. J Appl Cryst 35:126–136. [Google Scholar]
- 25. Richardson JS, Schneider B, Murray LW, Kapral GJ, Immormino RM, Headd JJ, Richardson DC, Ham D, Hershkovits E, Williams LD, Keating KS, Pyle AM, Micallef D, Westbrook J, Berman HM (2008) RNA Backbone: Consensus all‐angle conformers and modular string nomenclature. RNA 14:465–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Allen FH (2002) The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Cryst 58:380–388. [DOI] [PubMed] [Google Scholar]
- 27. Grazulis S, Chateigner D, Downs RT, Yokochi AT, Quiros M, Lutterotti L, Manakova E, Butkus J, Moeck P, Le Bail A (2009) Crystallography Open Database – an open‐access collection of crystal structures. J Appl Cryst 42:726–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cleland WW, Frey PA, Gerlt JA (1998) The low barrier hydrogen bond in enzymatic catalysis. J Biol Chem 273:25529–‐25532. [DOI] [PubMed] [Google Scholar]
- 29. Ishikita H, Saito K (2013) Proton transfer reactions and hydrogen‐bond networks in protein environments. J Royal Soc Interface 11:20130518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Richardson JS, Prisant MG, Williams CJ, Deis LN, Videau LL, Richardson DC (2017) Fitting Tips #13: O‐pairs: The love‐hate relationship of carboxyl oxygens. Comp Cryst Newsletter 8:2–5. [Google Scholar]
- 31. Bondi A (1964) van der Waals volumes and radii. J Phys Chem 68:441–451. [Google Scholar]
- 32. Gavezzotti A (1983) The calculation of molecular volumes and the use of volume analysis in the investigation of structured media and of solid‐state organic reactivity. J Am Chem Soc 105:5220–5225. [Google Scholar]
- 33. Bader RFW (1985) Atoms in molecules. Acc Chem Res 18:9–15. [Google Scholar]
- 34. Sheldrick GM (2008) A short history of ShelX. Acta Cryst 64:112–122. [DOI] [PubMed] [Google Scholar]
- 35. Vagin AA, Steiner RS, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN (2004) REFMAC5 dictionary: organisation of prior chemical knowledge and guidelines for its use. Acta Cryst 60:2284–2229. [DOI] [PubMed] [Google Scholar]
- 36. Stewart RF, Davidson ER, Simpson WT (1965) Coherent X‐ray scattering for the hydrogen atom in the hydrogen molecule. J Chem Phys 42:3175–3318. [Google Scholar]
- 37. Williams DE, Starr TL (1977) Calculation of the crystal structures of hydrocarbons by molecular packing analysis. Comput Chem 1:173–177. [Google Scholar]
- 38. Allen FH (1986) A systematic pairwise comparison of geometric parameters obtained by X‐ray and neutron diffraction. Acta Cryst 42:515–522. [Google Scholar]
- 39. Deis LN, Verma V, Videau LL, Prisant MG, Moriarty NW, Headd JJ, Chen VB, Adams PD, Snoeyink J, Richardson JS, Richardson DC (2013) Phenix/MolProbity hydrogen parameter update. Comput Cryst Newsletter 4:9–10. [Google Scholar]
- 40. Bartell LS, Kuchitsu K, deNeui RJ (1960) Equilibrium bond lengths in methane and deuteromethane as determined by electron diffraction and spectroscopic methods. J Chem Phys 33:1254–1255. [Google Scholar]
- 41. Bartell LS, Kuchitsu K (1978) Representations of molecular force fields. V. On the equilibrium structure of methane. J Chem Phys 68:1213–1215. [Google Scholar]
- 42. Ottiger M, Bax A (1998) Determination of relative N‐HN, N‐C', Cα‐C', and Cα‐Hα effective bond lengths in a protein by NMR in a dilute liquid crystalline phase. J Am Chem Soc 120:12334–12341. [Google Scholar]
- 43. Takusagawa F, Koetzle TF, Kou WWH, Parthasarathy R (1981) Structure of N‐acetyl‐L‐cysteine: X‐ray (T=295 K) and neutron (T=16 K) diffraction studies. Acta Cryst 37:1591–1596. [Google Scholar]
- 44. Hintze BJ, Lewis SM, Richardson JS, Richardson DC (2016) Molprobity's ultimate rotamer‐library distributions for model validation. Proteins 84:1177–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Engh RA, Huber R, Structure quality and target parameters. Chapter 18.3 In: Rossmann MG, Himmel D, Arnold E, Eds (2006) IUCr's International Tables of Crystallography, Volume F: Crystallography of Biological Macromolecules. Kluwer Academic Press, Dortrecht. [Google Scholar]
- 46. Berkholz DS, Shapovalov MV, Dunbrack RL, Jr , Karplus PA (2009) Conformation dependence of backbone geometry in proteins. Structure 17:1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tronrud DE, Berkholz DS, Karplus PA (2010) Using a conformation‐dependent stereochemical library improves crystallographic refinement of proteins. Acta Cryst 66:834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moriarty NW, Tronrud DE, Adams PD, Karplus PA (2014) Conformation‐dependent backbone geometry restraints set a new standard for protein crystallographic refinement. febs J 281:4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Croll TI (2015) The rate of cis–trans conformation errors is increasing in low‐resolution crystal structures. Acta Cryst 71:706–709. [DOI] [PubMed] [Google Scholar]
- 50. Berkholz DS, Driggers CM, Shapovalov MV, Dunbrack RL, Karplus PA (2012) Nonplanar peptide bonds in proteins are common and conserved but not biased toward active sites. Proc Natl Acad Sci USA 109:449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Williams CJ, Hintze BJ, Richardson DC, Richardson JS (2013) CaBLAM identification and scoring of disguised secondary structure at low resolution. Comput Cryst Newsletter 4:33–35. [Google Scholar]
- 53. Williams CJ (2016) Using C‐alpha geometry to describe protein secondary structure and motifs. Doctoral Dissertation, Duke University 248. pages. [Google Scholar]
- 54. Jain S, Richardson DC, Richardson JS, Computational methods for RNA structure validation and improvement. Chapter 7 In: Woodson S, Allain F, Eds (2015) Structures of large RNA molecules and their complexes. Elsevier, Oxford UK, Methods Enzymol series, vol 558:181–212. [DOI] [PubMed] [Google Scholar]
- 55. Jain S, Kapral G, Richardson D, Richardson J (2014) Fitting Tips #7: Getting the pucker right in RNA structures. Comput Cryst Newsletter 5:4–7. [Google Scholar]
- 56. Murray LW, Arendall WBIII, Richardson DC, Richardson JS (2003) RNA backbone is rotameric. Proc Natl Acad Sci USA 100:13904–13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen VB, Davis IW, Richardson DC (2009) KiNG (Kinemage, Next Generation): A versatile interactive molecular and scientific visualization program. Protein Sci 18:2403–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keedy DA, Williams CJ, Headd JJ, Arendall WBIII, Chen VB, Kapral GJ, Gillespie RA, Block JN, Zemla A, Richardson DC, Richardson JS (2009) The other 90% of the protein: Assessment beyond the Cαs for CASP8 template‐based and high‐accuracy models. Proteins 77:29–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Headd JJ, Richardson JS (2013) Fitting Tips #5: What's with water? Comput Crystallogr Newsletter 4:2–5. [Google Scholar]
- 60. Goddard TD, Huang CC, Meng EC, Petterson EF, Couch GS, Morris JH, Ferrin TE (2017) UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 26:10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rose AS, Hildebrand PW (2015) NGL viewer: A web application for molecular visualization. Nucleic Acids Res 43:W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. MySQL AB (2006) MySQL administrator's guide and language reference. 2nd Ed., MySQL Press, Indianapolis. [Google Scholar]
- 63. Richardson DC, Richardson JS, MAGE, PROBE, and Kinemages. Chapter 25.2.8 In: Rossmann MG, Arnold E, Eds (2001) IUCr's International Tables of Crystallography, Volume F: Crystallography of Biological Macromolecules. Kluwer Academic Press, Dortrecht. [Google Scholar]
- 64. Chou FC, Sripakdeevong P, Dibrov SM, Hermann T, Das R (2013) Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Methods 10:74–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Adams PD, Baker D, Brunger AT, Das R, DiMaio F, Read RJ, Richardson DC, Richardson JS, Terwilliger TC (2013) Advances, interactions, and future developments in the CNS, Phenix, and Rosetta structural biology software systems. Ann Rev Biophys 42:265–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kapral GJ, Jain S, Noeske J, Doudna JA, Richardson DC, Richardson JS (2014) New tools provide a second look at HDV ribozyme structure, dynamics, and cleavage. Nucleic Acids Res 42:12833–12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Murshudov GN, Grebenko AI, Brannigan JA, Antson AA, Barynin VV, Dodson GG, Dauter Z, Wilson KS, Melik‐Adamyan WR (2002) The structures of Micrococcus lysodeikticus catalase, its ferryl intermediate (Compound II) and Nadph complex. Acta Cryst D58:1972–1982. [DOI] [PubMed] [Google Scholar]
- 68. Bonisch H, Schmidt CL, Bianco P, Ladenstein R (2005) Ultra‐high resolution study on Pyrococcus abyssi rubredoxin. I. 0.69 Å x‐ray structure of mutant W4L/R5S. Acta Cryst 61:990–1004. [DOI] [PubMed] [Google Scholar]
- 69. Banuelos S, Saraste M, Carugo KD (1998) Structural comparisons of calponin homology domains: implications for actin binding. Structure 6:1419–1431. [DOI] [PubMed] [Google Scholar]
- 70. Tempel W, Chen L, Liu Z‐J, Lee D, Shah A, Dailey TA, Mayer MR, Arendall WB III, Rose JP, Dailey HA, Richardson JS, Richardson DC, Wang B‐C (2004) Divalent cation tolerant protein CUTA from Homo sapiens O60888. unpublished.
- 71. Klein DJ, Moore PB, Steitz TA (2004) The roles of ribosomal proteins in the structure, assembly, and evolution of the large ribosomal subunit. J Mol Biol 340:141–177. [DOI] [PubMed] [Google Scholar]
- 72. Amunts A, Drory O, Nelson N (2007) The structure of a plant photosystem I supercomplex at 3.4Å resolution. Nature 447:58–63. [DOI] [PubMed] [Google Scholar]
- 73. Hövel K, Shallom D, Niefind K, Belakhov V, Shoham G, Baasov T, Shoham Y, Schomburg D (2003) Crystal structure and snapshots along the reaction pathway of a family 51 alpha‐L‐arabinofuranosidase. embo J 22:4922–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Montange RK, Mondragon E, van Tyne D, Garst AD, Ceres P, Batey RT (2010) Discrimination between closely related cellular metabolites by the SAM‐I riboswitch. J Mol Biol 396:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information