

Abstract
Introduction
Rett syndrome (RTT) is a pervasive neurodevelopmental disorder that presents with deficits in brain functioning leading to language and learning regression, characteristic hand stereotypies and developmental delay. Different mutations in the gene implicated in RTT—methyl-CpG-binding protein 2 (MECP2) establishes RTT as a disorder with divergent symptomatology ranging from individuals with severe to milder phenotypes. A reliable and single multidimensional questionnaire is needed that can embrace all symptoms, and the relationships between them, and can map clinically meaningful data to symptomatology across the lifespan in patients with RTT. As part of the HealthTracker-based Tailored Rett Intervention and Assessment Longitudinal (TRIAL) database, the Rett Evaluation of Symptoms and Treatments (REST) Questionnaire will be able to marry with the physiological aspects of the disease obtained using wearable sensor technology, along with genetic and psychosocial data to stratify patients. Taken together, the web-based TRIAL database will empower clinicians and researchers with the confidence to delineate between different aspects of disorder symptomatology to streamline care pathways for individuals or for those patients entering clinical trials. This protocol describes the anticipated development of the REST questionnaire and the TRIAL database which links with the outcomes of the wearable sensor technology, and will serve as a barometer for longitudinal patient monitoring in patients with RTT.
Methods and analysis
The US Food and Drug Administration Guidance for Patient-Reported Outcome Measures will be used as a template to inform the methodology of the study. It will follow an iterative framework that will include item/concept identification, item/concept elicitation in parent/carer-mediated focus groups, expert clinician feedback, web-based presentation of questionnaires, initial scale development, instrument refinement and instrument validation.
Ethics and dissemination
The study has received favourable opinion from the National Health Service (NHS) Research Ethics Committee (REC): NHS Research Ethics Committee (REC)—London, Bromley Research Ethics Committee (reference: 15/LO/1772).
Keywords: biomarkers, health trackerTM, questionnaire development and validation, rett syndrome, tailored rett intervention and assessment longitudinal (TRIAL) database, wearable sensor technology
Strengths and limitations of the study.
The new Rett Evaluation of Symptoms and Treatments (REST) questionnaire will capture clinically meaningful change of symptomatology in individuals with Rett syndrome across the lifespan.
The HealthTrackerTM-based Tailored Rett Intervention and Assessment Longitudinal (TRIAL) database will link the behavioural data with the physiological aspects of the disease and has the potential to be used globally, allowing for quicker development of decision-support analytics and personalised care.
The use of HealthTracker, a multimodal eHealth web-based monitoring platform, will make the TRIAL database as user friendly as possible and allows it to be tailored to the individual participant.
The TRIAL database will enable the streamlining of treatment and expedite triaging of care by signposting patients to correct specialists sooner to enable timely intervention.
Participation might be time consuming for families.
Introduction
Rett syndrome (RTT) can trace its genesis to a clinical waiting room in Vienna (1965)1 where Dr Rett first observed the clinical signs and symptoms of RTT. Symptoms usually appear between 6 and 18 months after birth2 and clinically RTT presents with impairments in brain functioning leading to language and learning regression, hand stereotypies (hand washing/wringing) and developmental stagnation. RTT is predominantly found in young females with an incidence of about 1:10000 live births.3 There are geographical variations4 with one Australian study indicating a prevalence of about 1:90005. The prevalence is probably underscored by the high clinical variability of the disease and hence the frequency could be underestimated. Loss of function in the methyl-CpG-binding protein 2 (MECP2) gene is responsible for the disorder in the vast majority of cases,6 with rarer cases being attributed to mutations in CDKL5 and FOXG1 gene7 8 leading to atypical or variant RTT. More recently, mutations in other less known candidate genes have emerged such as those encoding ankyrin repeat proteins and neuronal acetylcholine receptor subunits.9 MeCP2 is a highly conserved nuclear protein abundant in the mammalian brain10 and notably the disorder is reversible in mice models of RTT.11
MeCP2 acts as a critical epigenetic modulator in the mammalian brain, controlling overlapping mechanisms such as DNA methylation and post-translational mechanisms.12 Through differential post-translational modification at serine 164,13 MeCP2 may help in limiting transcriptional noise14 of other genes. For example, mutations in the gene switch-insensitive 3 family member A (SIN3A), a MECP2 interactor and transcriptional repressor—crucial for cortical integrity, causes intellectual disability and autism spectrum disorder (ASD)15 and the MECP2R306C mutation prevents MeCP2 from interacting with the NCoR/histone deacetylase 3 (HDAC3) complex resulting in impediments in social and cognitive functioning in animal models.16 MeCP2 has complex genome level modalities, and the general opinion is that loss of the transcriptional repressor function of MECP2 impacts other genes crucial for postnatal neuronal development and has led others to suggest that this leads to a suboptimal brain.17 This seems to be the significant driver for the classical RTT clinical phenotype. Genes in neuronal development tend to be long (100 kb or larger)18 and as the transcriptional repression function of MECP2 is biased towards longer genes,19 it is likely that impairments of long genes associated with neuronal development dictates the functional and developmental versatility of the MeCP2 protein seen in RTT. This has a knock-on effect on the homoeostasis of excitatory and inhibitory pathways20 21 in RTT brains leading to the clinical versatility that is commonly observed.
MECP2 being an X-linked gene has an impact on the phenotype of patients with RTT and on the clinical severity. The X chromosome inactivation can cause uneven expression of wild type and mutant alleles resulting in skewed patterns of RTT phenotype severity22 23 and the degree of DNA methylation-dependent long gene repression.19 The range of functional ability in patients with RTT is, therefore, broad and depending on the type of genetic mutation ranges from patients with severe functional impairments to those with milder symptoms24; hence assessment and care pathways must be individually tailored to each affected person.
Although there have been considerable advances in understanding the genetics and into the genetic testing of RTT, the diagnosis of RTT is based on the 2010 revised consensus clinical criteria3 (see table 1 in Ref. 3) and recommends that all individuals with RTT should be first be assessed according to the revised clinical criteria followed by a thorough genetic test for MECP2. Given that about 3%–5% of RTT individuals who fulfil the diagnostic clinical criteria do not have MECP2 mutations, and this is even higher for atypical RTT cases,25 more recently clinical predictors that can facilitate a clinician’s decision making to order genetic testing for RTT have been provided.26 This showed that the likelihood of a having a positive MECP2 test was greatest in patients with partial or complete attenuation of hand skills. Impairments in gait and hand stereotypies were also strong predictors. Of interest was that loss of speech did not discriminate whether an individual was MECP2+ or MECP2-.
Table 1.
Measures to be administered during stage 2 (Validation) and stage 3 (Wearable Sensor Technology) of the study
| Administered to: | |||||||
| Measure | Key information | Individual with RTT | Individual with ASD | Healthy subjects | Parent/carer of child with RTT/*ASD | Parent/carer/partner of adult with RTT/ASD* | Clinician/researcher |
| Rett Natural History study32 | More than 1000 participants with RTT providing information on important aspects of disorder symptomatology | X | |||||
| RSBQ38 | Provides an accurate measure of the behavioural features of RTT | X | X | ||||
| RSSS42 | Provides information on the overall clinical severity and severity across individual parameters:
|
X | |||||
| REST questionnaire | A multidimensional questionnaire that can capture clinically meaningful data across the lifespan in individuals with RTT and improve treatment pathways | X | X | X | |||
| Wearable sensor technology | Captures real-time biometric physiological data (heart rate variability, skin conductance, blood volume pressure, perspiration and temperature) | X | X | X | |||
| Anticipated administration time (minutes) | 30 | 30 | 30 | ~60 | ~60 | ~60 | |
*Participants in the ASD cohort will be asked to complete only the relevant questions in the questionnaire battery that would be applicable and relevant to them
ASD, autism spectrum disorder; REST, Rett Evaluation of Symptoms and Treatments Questionnaire; RSBQ, Rett Syndrome Behavioural Questionnaire; RSSS, Rett Syndrome Severity Score; RTT, Rett syndrome.
Pre-existing measures in RTT
As far as we are aware, no complete instrument has been developed for individuals with RTT that can capture longitudinal pharmacological, behavioural, genetic and psychosocial information, as well as an ability to correlate this with the physiological aspects of the disease. Previous datasets/instruments have been inconsistent and provide limited information on the behavioural and physiological facets of the disease. While some might provide information on the genetic diagnoses of individuals with RTT, there is a lack of consistency. First, RettBase collected mainly molecular genetic data from the Australian cohort of patients with RTT.27 Some other instruments have included both genetic and clinical data, although the clinical data were limited. InterRETT, an Australian Rett syndrome database, was based on data collection by distributing a questionnaire to families.28 The Italian Rett Database and Biobank consisted of 357 patients and had 20 structured and seven descriptive clinical items along with 17 structured genetic items.29 The British Isles Rett Syndrome Survey, included 275 British Rett patients and had 271 structured and 94 descriptive clinical items, and six structured genetic items.30 An American survey collected data on the natural history of the disease that allowed researchers and physicians to access comprehensive patient data on more than 1000 individuals with RTT.31 32 These datasets were preserved and integrated into the Rett Networked Database30 and offers an amalgamated data repository for researchers to access anonymised patient information. Elsewhere, the Japanese RTT database includes the clinical data from 102 females with a median age of 11 years old.33
Capture of disease severity and sensitivity to change throughout the lifespan in patients are important elements that need to be considered when developing clinically meaningful outcome measures. The Unified Parkinson’s Disease Rating Scale is a good example of an outcome measure that is effective and can capture disease severity and clinically meaningful change of symptoms of Parkinson’s disease.34 With rare diseases, the Sanfilippo Behaviour Rating Scale, a 68-item questionnaire developed using 44 families, is also effective and can map the behavioural phenotype of children with Sanfilippo syndrome to disease progression and/or results from treatment across the lifespan.35 In RTT, the current outcome measures are inadequate in their ability to capture disease severity across the lifespan, although others have made significant headway in this area. The 37-item motor–behavioural assessment (MBA) incorporates historical items with items from direct clinician evaluations and has been used to describe clinical severity in RTT,36 37 while the Rett Syndrome Behavioural Questionnaire (RSBQ), a validated checklist, was designed to differentiate individuals with RTT compared with those with severe intellectual disability.38 Other measures tested in RTT include the Anxiety Depression and Mood Scale (ADAMS),39 the clinician based International Scoring System40 41 to evaluate the disease severity, Vineland Adaptive Behaviour Scale,42 the 13-item Rett Clinical Severity Scale (RCSS)37 43 and its modified version.42 Others have developed RTT specific anchors such as for the Clinical Global Impression Severity Scale based on scores from the RCSS for improved outcome measures in clinical trials.44 Quality of Life measures such as the Child Health Questionnaire-P50 have also been used in RTT45 including a recent phase II open-label clinical trial using glatiramer acetate.46 Some of these measures such as the MBA, RSBQ, ADAMS and RCSS have been implemented into clinical trials to evaluate the effect of insulin-like growth factor (IGF-1)47 or sarizotan48 in individuals with RTT or to develop a novel scoring tool (Rett Severity Score (RSS)) to assess the impact of IGF-1 treatment in RTT.41 Other scales, such as the Mullen Scales for Early Learning used in other rare disorders,49 have also been adapted for use in RTT.47 These measures are not without their faults. Some have suggested that the MBA can be difficult to use with some items that describe disease regression having not been validated.24 This is important given that in some patients with RTT, disease regression has been described as transient or often goes unrecognised.50 Others such as the RSBQ although are suitable to measure some aspects of behaviour such as mood and anxiety51 might not be able to capture the salient features of behaviour as an outcome measure in a clinical trial in patients with RTT. Furthermore, there is differing reliability of anxiety scales in RTT, with ADAMS especially its Social Avoidance subscale having the best psychometric properties in comparison to the RSBQ.52 While no outcome measure will be perfect, these studies have paved the way for more sensitive outcome measures to be developed such as the validated 15-item Gross Motor Scale for individuals with RTT.53
Autonomic function in RTT
Large cross-sectional studies investigating the genotype–phenotype relationships have revealed divergence in the phenotype seen in individuals with RTT.54 55 These were the first studies of sufficient sample size that bestowed important information on the genotype and phenotype relationships in RTT, and have been elegantly summarised elsewhere.24 Some mutations or variants dictate a more severe phenotype when it comes to motor abilities24 54 55 and cardiorespiratory phenotypes.24 56 Moreover, at present it is unknown whether autonomic dysfunction is governed by any specific mutation in RTT.24 56 Assessing the autonomic dysfunction in individuals in RTT is therefore a pressing clinical concern.
Autonomic dysfunction is a pivotal factor that requires consideration when managing patients with rare disorders such as RTT. From our clinical experience when managing patients in the Centre for Interventional Paediatric Psychopharmacology and Rare Diseases,57 autonomic dysfunction is often found in patients who do not respond to treatment and those with significant functional disability. Autonomic dysfunction co-occurs in the context of emotional and behavioural dysregulation and recently using wearable sensor technology, we have shown that Emotional, Behavioural and Autonomic Dysregulation (EBAD) is a crucial factor that needs to be considered when managing patients with RTT.58 59 Although autonomic dysfunction has been investigated in individuals with RTT,60–63 the progression of autonomic dysfunction and the developmental trajectory of EBAD has never been researched. Moreover, the components of EBAD in a questionnaire that can map across other symptomatology longitudinally in individuals with RTT has not previously been shown.
Aim
The objective of this study is to develop and validate a comprehensive multisystem questionnaire (Rett Evaluation of Symptoms and Treatments (REST)) that can profile the symptomatology of patients with RTT and is sensitive to change across the lifespan allowing better understanding of patient needs. In parallel, information collected using wearable sensor technology58 59 will be linked to data obtained from the REST questionnaire, genetic data and information about available psychosocial support from the patient and their family, to form a comprehensive Tailored Rett Intervention and Assessment Longitudinal (TRIAL) database. The TRIAL database will streamline treatment approaches to expedite triaging of care by signposting patients to correct specialists earlier than is currently happening (figure 1). Specifically, the functionality of the multimodal HealthTracker platform will be exploited so that anonymised data from the TRIAL database can be used to develop a parent/carer alert system to signal when it may be useful to request unscheduled clinician appointments. Using this functionality, the TRIAL database will also be able to stratify patients to inform adaptive clinical trial design, by allowing pre-existing datasets to be used so that rare disease trials can be done in a more cost-effective manner.
Figure 1.
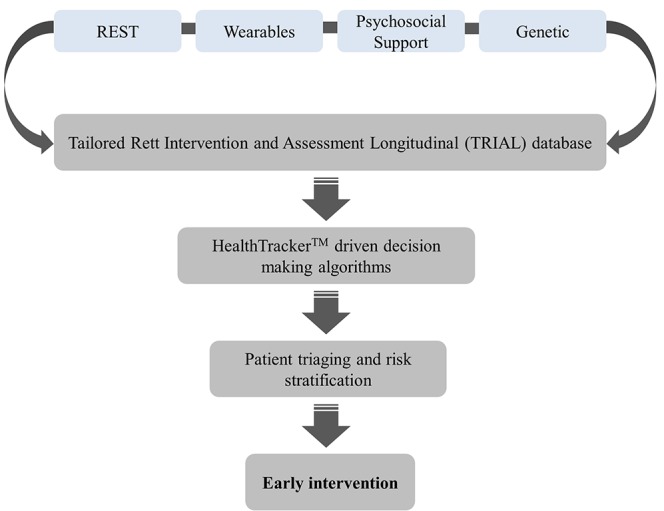
Flow diagram illustrating the sequence of steps showing how data obtained from the TRIAL database can be used to provide timely intervention to streamline treatment outcomes in patients with RTT syndrome. REST, Rett Evaluation of Symptoms and Treatments; RTT, Rett syndrome.
Methods and analyses
The title of this questionnaire was based on the feedback of the focus groups involving parents and carers of children with RTT from the parent-based charities such as Reverse Rett UK and clinician feedback. It will incorporate elements from previous scales40 52 and standardised RTT questionnaires—data from the Natural History Study,3 RSBQ38 and the modified version of the RSS Scale (RSSS).42 It is anticipated that the questionnaire will not take more than 30 min to complete.
The US Food and Drug Administration Guidance for Patient-reported Outcome Measures (PROM)64 will be used as a template to guide the methodology in the study. It was described in Santosh et al 57 65 and will follow an iterative framework that will involve item/concept identification, item/concept elicitation in parent/carer mediated focus groups, clinician feedback, web-based presentation of questionnaires, initial scale development, instrument refinement and instrument validation.
Stage 1: qualitative development of the REST questionnaire
Concept identification
For this initial phase, a systematic literature review will be conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses66 to identify signs and symptoms that are deemed to be problematic in RTT. A draft version will be reviewed by expert clinicians who have substantial experience in RTT and ASD. Common themes will be identified and draft version of the questionnaire will be prepared based on their feedback.
Concept elicitation
This stage will involve parents/carers of individuals aged between 6 and 40 years with RTT. A series of focus groups anticipated to last about 1.5 hours will be conducted as part of the concept elicitation stage. These focus groups will include parents/carers of individuals with RTT from the parent-based charities, such as Reverse Rett UK, and clinicians who see patients with RTT. The groups will follow a semistructured format using open-ended questions to allow participants to discuss their experiences and views. Some of the focus groups will be on item generation while others may centre on reviewing draft versions of the questionnaire identifying pertinent themes. Focus groups will be audio recorded and each group will include approximately 4–6 parents/carers of children with RTT. Up to two researchers may be present for the focus groups, which will be led by a consultant child and adolescent psychiatrist/specialist. All participants will also be asked to complete a demographic questionnaire.
Web-based presentation of questionnaires on the HealthTracker platform
HealthTracker, a web-based health monitoring platform,67 has been successfully trialled in multicentric EU FP7 studies68 69 and in a questionnaire development and validation study.70 Parents and carers will be shown how the REST questionnaire might appear on the HealthTracker platform, how the response options to the questionnaire could be presented and whether a choice of single or multiple-choice questions would be appropriate. The various views of the focus groups will be used to choose the most optimal web-based visualisation of the questionnaire.
Tool review
As far as the authors are aware, no questionnaire exists that not only is RTT focused but can capture a broad range of problematic themes, in particular, the developmental trajectory of EBAD. Nor do these existing questionnaires/scales attempt to marry this with the physiological measurements from wearable sensor technology. At this stage, a further literature review will be conducted to identify any themes that may have been missed during the focus groups and whether any further areas of RTT symptomatology that were not highlighted in the focus groups needs to be addressed. In addition, parents/carers from Reverse Rett UK will be consulted and any feedback incorporated into the tool review stage.
Following the focus groups, study participants will be sent a copy of the draft version of the questionnaire (via e-mail or post). Once this part has been completed, a draft operating beta version of the questionnaire will be finalised.
Stage 2: validation of the REST questionnaire
This stage of the study will involve parents/carers of individuals aged between 6 and 40 years with RTT. Questionnaires to assess the longitudinal trajectory of symptomatology in rare diseases have proven to be difficult to validate.71 72 To broach this conundrum, it is important to focus on the symptoms and not just the clinical diagnosis, and be able to evaluate the symptom level across other comparable patient groups. At the neuronal level, the brains of RTT and ASD patients share many core features.73 Both RTT and ASD exhibit behaviours that might overlap that is, there are deficits in social behaviour and speech and in both cases individuals may share common stereotypical behaviours.74 Due to these similarities and based on consultation with clinicians with expertise in ASD, as a comparator group, this stage of the study will also include parents/carers/partners of individuals aged between 6 and 40 years with ASD with significant intellectual disability. It will also involve clinicians who see patients with RTT and ASD who will test the clinician version of the questionnaire. Participants (parents/carers and clinicians) will be recruited to complete the respective versions of the REST questionnaire as well as other standardised questionnaires—namely the RSSS and the RSBQ (table 1). The RSSS37 42 43 75 and the RSBQ38 51 have previously been used in studies in patients with RTT. Pertinent information will also be taken from the RTT Natural History Study.31 32 It is anticipated that 50 participants in the RTT cohort and 50 in the ASD with significant intellectual disability cohort will complete the questionnaire battery. Although there is significant symptom overlap in patients with RTT and ASD, participants in the ASD cohort will be asked to complete only the relevant questions in the questionnaire battery that would be applicable and relevant to them.
The questionnaire battery will be presented to study participants in HealthTracker, a multimodal web-based portal for remote online completion using developmentally appropriate interfaces. Participants will be given a unique ID number and log-in information and will be asked to complete the questionnaires independently. The research team will be able to support participants with questionnaire completion should they need it. Where applicable, participants will also be able to complete paper versions of the questionnaires if they request them. Participant medical records will be accessed only by members of the study team to validate the questionnaire against details of diagnoses obtained from patient case notes as well as against the Development and Well-Being Assessment (DAWBA)76 and treatment/medication status if they are available in case notes. Patient records will also be used to gain genetic information on the specific mutation and diagnosis. Consent will be obtained to access medical notes.
All participants will be asked to complete the questionnaire battery, at baseline, again after 1 week and then between 4 and 6 months after first completion to assess questionnaire stability.
Stage 3: wearable sensor technology
The use of wearable sensor technology to improve treatment outcomes has gathered momentum in recent years77 and is currently being used to develop new outcome measures in patients with complex neurodisability such as amyotrophic lateral sclerosis.78 Using wearable sensor technology as a PROM is not without its challenges. In RTT, wearable technology has been used to explore respiratory and cardiac function in observational studies79 80 and in two recent clinical trials,46 47 however, inherently captured biometric data can be noisy especially from quasiperiodic oscillations from cardiac rhythms. Wrist worn devices might be particularly susceptible to this type of noise. To mitigate these issues, we have applied the methods described previously81 82 to analyse heart rate variability and electrodermal activity as metrics when evaluating wrist sensor biometric data and autonomic function in a 15-year-old girl with RTT. We were able to demonstrate a recalibration of the autonomic equilibrium from pretreatment to post-treatment using buspirone (30 mg/day),58 and subsequent improvement in EBAD in this girl. Quasiperiodic oscillations cannot be easily quantified using conventional methods. To manage this, phase-rectified signal averaging83 may be used in conjunction with spectral factorisation and applied to the beat-to-beat interval data, which is particularly prone to extraneous noise. This methodology coupled with EDA assessment will provide more sensitive methods to capture changes in autonomic physiology in patients with RTT. In the context of this study, the outcomes of the wearable sensor technology will marry into the outcomes of the newly developed questionnaire (REST), with psychosocial and genetic data to create the TRIAL database. The technology will be evaluated in individuals with RTT, ASD and healthy controls.
Sample size
Justification for Sample Size
Owing to the small sample population of individuals with rare and complex genetic disorders, formal modelling to obtain sample size estimates will not be readily applicable.
Stage 1: Questionnaire Development Stage
It is anticipated that the total number of participants for the questionnaire development stage of the study will be between 10 and 20 (including participants and clinicians). In our experience, focus groups involving families with children with rare diseases are well versed with the problems associated with the condition in question. Often, the themes that need to be addressed get saturated after a couple of focus groups, leading to us getting the basic structure of the items needed to be tested in stage 2.
Stage 2: Questionnaire Validation Stage
The total number of participants for the questionnaire validation stage of the study will be 150 participants (n=100 RTT cohort and n=50 ASD cohort). The number in the ASD cohort will be split so that 25 parents/carers will either have a male or female diagnosed with ASD.
Stage 3: Wearable Sensor Technologies Stage
The total number of participants for the wearable technology stage of the study is expected to be 100 participants (n=50 RTT cohort and n=50 (25 male and 25 female) ASD cohort). This part of the study will also include a matched healthy control group.
Stage 4: Longitudinal Monitoring in Patients with RTT
Longitudinal data capture on a 3-monthly basis from 80 to 100 parents/carers of individuals with RTT will be undertaken using the REST questionnaire over a 12–18 month period. Ethics submission for stage 4 of the study will be done after stages 1–3 have been completed.
Recruitment
Information sheets (and age appropriate information sheets where relevant) will be provided for all participants, in addition to consent forms. Information sheets will emphasise that participant involvement in the research is voluntary and they have the right to withdraw from the research at any time, without giving a reason. In addition, participants will be advised that participating or withdrawing from the research will have no impact on their usual care that they are currently receiving or will receive in the future. A minimum of 24 hours will be given between providing study information and recruitment of participants into the study.
Stage 1: Questionnaire Development Stage Recruitment
Parents/carers of individuals with RTT and clinicians who work with individuals with RTT will be recruited. Due to the group-based nature of the focus groups, parents/carers or clinicians who have not provided consent will not be able to partake in focus groups and will be excluded. The focus groups comprise parents/carers of individuals with RTT and clinicians working with patients with RTT. Depending on the nature of the focus groups about 4–6 participants will take part in each focus group.
Questionnaire Development Stage: Inclusion Criteria
Parents/carers/partners/relatives of individuals aged between 6 and 40 years with RTT.
Clinicians who work within healthcare settings in South London and Maudsley (SLaM) National Health Service (NHS) Foundation Trust that see children and/or adults with RTT and associated developmental conditions.
Without any exclusion for concurrent stable medication.
Questionnaire Development Stage: Exclusion Criteria
Parents/carers whom do not have a reasonable level of English. This is because a reasonable level of English will be required to engage in the focus groups.
Stage 2: questionnaire validation stage recruitment
For this stage of the study, parents/carers of individuals with RTT and those with ASD will be recruited via clinician/researcher invite. Study participants will be under the care of a service within SLaM NHS Foundation Trust. Where relevant, parents/carers/partners/relatives of individuals with RTT and ASD will be asked to provide details of their clinician at the time of consent so that they can also be contacted by the research team and invited to take part.
Questionnaire Validation Stage: Inclusion Criteria
Parents/carers of individuals aged between 6 and 40 years with RTT or ASD.
Questionnaire Validation Stage: Exclusion Criteria
If parents/carers of individuals aged between 6 and 40 years with RTT or ASD are not able to (or expected to not be able to) complete questionnaires they will be excluded from the study.
Parents/carers who do not have a reasonable level of English will be excluded from the validation stage of the study. This is because a reasonable level will be required to complete questionnaires, which will only be available in English at the validation stage. A research assistant my assist the parent/carer in completion.
Stage 3: wearable sensor technologies stage recruitment
Individuals aged between 6 and 40 years with RTT and ASD and parents/carers/partners/relatives of individuals with RTT and ASD will be recruited via clinician/researcher invite. Information sheets (and age appropriate information sheets where relevant) will be provided for all participants, in addition to consent forms and where applicable assent forms. Healthy controls will be recruited via clinician/researcher invite using widely used and appropriate advertising channels.
Wearable Sensor Technologies Stage: Inclusion Criteria
RTT
Females aged 6–40 years with confirmed diagnosis of RTT (via clinician/researcher invite)
Parents/carers/partners/relatives of individuals aged 6–40 years with RTT
ASD
Males and females aged 6–40 years with ASD (via clinician/researcher invite)
Parents/carers/partners/relatives of individuals aged 6–40 years with ASD
Healthy controls
Males and females aged 6–40 years considered to be healthy for their age (via clinician/researcher invite)
Are capable of understanding and complying with the requirements of the protocol
Wearable Sensor Technologies Stage: Exclusion Criteria
RTT
Individuals aged 6–40 years with RTT who are not able to (or expected to not be able to) wear the wearable sensor technology will be excluded from the study
Parents/carers/partners/relatives of individuals aged 6–40 years with RTT who do not have a reasonable level of English
ASD
Individuals (aged 6–40 years with ASD who are not able to (or expected to not be able to) wear the sensor technology will be excluded from the study.
Parents/carers/partners/relatives of individuals aged 6–40 years with ASD who do not have a reasonable level of English
Healthy controls
Individuals who are not able to (or expected to not be able to) wear the sensor technology will be excluded from the study.
Individuals who do not have a reasonable level of English
Analyses plan
Questionnaire development
Data obtained from the focus groups will be recorded securely and transcribed accurately, paying close attention to the identified themes and issues. The analysis will be performed as described previously.65 In brief, the focus group data will be organised into clinically meaningful themes using thematic and content analysis. Following this, to manage the qualitative data generated from the focus groups, NVivo software (version 11) will be used and the data analysis will be guided by the framework for thematic analysis.84
Questionnaire validation
The quantitative data will be analysed using the latest version of the SPSS statistical package (version 24).
Internal consistency
Internal consistency of the measures will be reported using Cronbach’s alpha. Alpha coefficients ≥0.85 will be indicative of reasonable evidence of internal reliability.85 Where applicable, ‘alpha if deleted analyses’ will be performed to see if omitting any item(s) from the (subthemes of the) questionnaire would strengthen the measure.
Test–retest reliability
Intraclass correlation (ICC) will be used to assess test–retest reliability on subscale and total scores as described.86 Given the exploratory nature of this study, weighted Cohen’s kappa values will also be determined to assess test–retest reliability at the item level. The ICC will also be performed after 4 to 6 months after initial completion of the questionnaire to assess the long-term stability of the new questionnaire.
Validity
Validity (discriminative power) of the new questionnaire will be assessed using Receiver Operating Characteristic (ROC) analyses as described in Santosh et al.57 65 As there are no gold standard questionnaires for patients with RTT, where applicable the ROC analyses will also be performed on the scores on the RTT Natural History Study, the RSBQ and the RSSS. Where necessary and if data are available, analysis of variance (ANOVA; general linear model) will be performed with grouping variable DAWBA diagnoses (coded in 1 for positive and coded 0 for negative diagnosis) so that the differences in REST scoring can be assessed.
Factor analysis
Studies involving small sample sizes have often been plagued by the inappropriate use of exploratory factor analysis (EFA) or principal component analysis (PCA) to identify clinically meaningful factor items.87 Many recommendations have been put forward regarding sample sizes but there does not seem to be an overall consensus.88 Some have suggested improbable sample sizes that would not be feasible for studies of rare and complex genetic diseases.89 In these instances, methods to reveal the multidimensional aspects of factor structure are not as straightforward. Recently, the regularised exploratory factor analysis (REFA) was introduced90 that is recommended over EFA and PCA, when samples sizes are less than 50. Despite this new approach, it is unclear whether the REFA would be applicable for a multidimensional questionnaire in a condition with many variables. In the context of the new questionnaire, the robustness of the REST will be evaluated using tools applicable for smaller samples sizes90 and those used in exploratory studies as described recently91.
Gender differences
In stage 3 (Wearable Sensor Technologies Stage), if the data meet the requirements for parametric testing, the general linear model (ANOVA) covaried for gender will be applied to the RTT and ASD cohorts.
Study dates
The study is expected to complete by January 2019.
Dissemination
The goal of this study is to develop and validate a new RTT questionnaire. Data from the REST questionnaire will be linked with the data from the wearable sensor technology as well as psychosocial and genetic information to construct the TRIAL database, which will improve the overall healthcare delivery for individuals with RTT. Using the functionality of the HealthTracker platform, the TRIAL database will provide all the necessary information to clinicians and researchers about different aspects of the disease and serve as a barometer for improving treatment pathways in individuals. This will allow algorithms to be developed alerting parent/carers to request unscheduled clinician appointments when symptoms deviate significantly from one another thereby streamlining the patient care pathway. As the HealthTracker-based TRIAL database is web based, with appropriate funding, it has the potential to be used globally, allowing for quicker development of decision-support analytics and personalised care.
Rare disorders such as RTT have a limited patient population and it is therefore crucial for patients to be stratified using phenotype and biomarkers (such as those obtained through wearable sensor monitoring). Adaptive clinical trial design using Bayesian methodology has been suggested to augment the statistical power and decrease the number of patients required for a rare disease trial.72 In this view, the TRIAL database will serve for the recruitment of patients into clinical trials, as baseline information would already be available so the clinical trial can be conducted with fewer patients and in a more cost-effective manner.
Results stemming from this study will be disclosed unreservedly and the findings published in scientific journals and will also be presented in meetings and conferences for professionals, patients as well as carers and families. Reverse Rett UK will lead on wider dissemination of the applied research findings to engage policy-makers, key professional groups and service managers and parents/carers of children with RTT.
Supplementary Material
Acknowledgments
We are indebted to Reverse Rett UK for their helpful comments and suggestions on the study design.
Footnotes
Contributors: JS drafted, wrote and revised the manuscript and wrote the documentation required for ethical approval of the study. KL provided important intellectual review of the manuscript and reviewed the documentation required for ethical approval of the study. FF reviewed the statistical components and reviewed the manuscript. PS secured funding and conceived the study and revised the manuscript critically for important intellectual content.
Funding: A part of this research study was funded by Reverse Rett UK (Ref No: PCCTABR) and HealthTracker for significantly subsidising the costs for this study.
Competing interests: PS is the coinventor of the HealthTracker and is the chief executive officer and shareholder in HealthTracker. FF is a data analyst and KL is a project manager employed by HealthTracker. JS is on the professional advisory board for Reverse Rett UK and acts as a scientific advisor.
Ethics approval: NHS Research Ethics Committee (REC)—London, Bromley Research Ethics Committee (reference: 15/LO/1772).
Provenance and peer review: Not commissioned; externally peer reviewed.
Correction notice: This paper has been amended since it was published Online First. Owing to a scripting error, some of the publisher names in the references were replaced with 'BMJ Publishing Group'. This only affected the full text version, not the PDF. We have since corrected theseerrors and the correct publishers have been inserted into the references.
References
- 1. Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood]. Wien Med Wochenschr 1966;116:723–6. [PubMed] [Google Scholar]
- 2. Zoghbi HY. Rett Syndrome and the Ongoing Legacy of Close Clinical Observation. Cell 2016;167:293–7. 10.1016/j.cell.2016.09.039 [DOI] [PubMed] [Google Scholar]
- 3. Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 2010;68:944–50. 10.1002/ana.22124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vorsanova S, Iourov I, Yurov Y. Neurological, genetic and epigenetic features of Rett syndrome. Journal of Pediatric Neurology 2004;02:179–90. 10.1055/s-0035-1557218 [DOI] [Google Scholar]
- 5. Fehr S, Bebbington A, Nassar N, et al. Trends in the diagnosis of Rett syndrome in Australia. Pediatr Res 2011;70:313–9. 10.1203/PDR.0b013e3182242461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bienvenu T, Carrié A, de Roux N, et al. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum Mol Genet 2000;9:1377–84. 10.1093/hmg/9.9.1377 [DOI] [PubMed] [Google Scholar]
- 7. Evans JC, Archer HL, Colley JP, et al. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur J Hum Genet 2005;13:1113–20. 10.1038/sj.ejhg.5201451 [DOI] [PubMed] [Google Scholar]
- 8. Philippe C, Amsallem D, Francannet C, et al. Phenotypic variability in Rett syndrome associated with FOXG1 mutations in females. J Med Genet 2010;47:59–65. 10.1136/jmg.2009.067355 [DOI] [PubMed] [Google Scholar]
- 9. Lucariello M, Vidal E, Vidal S, et al. Whole exome sequencing of Rett syndrome-like patients reveals the mutational diversity of the clinical phenotype. Hum Genet 2016;135:1343–54. 10.1007/s00439-016-1721-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lewis JD, Meehan RR, Henzel WJ, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992;69:905–14. 10.1016/0092-8674(92)90610-O [DOI] [PubMed] [Google Scholar]
- 11. Guy J, Gan J, Selfridge J, et al. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007;315:1143–7. 10.1126/science.1138389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katz DM, Bird A, Coenraads M, et al. Rett Syndrome: crossing the threshold to clinical translation. Trends Neurosci 2016;39:100–13. 10.1016/j.tins.2015.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stefanelli G, Gandaglia A, Costa M, et al. Brain phosphorylation of MeCP2 at serine 164 is developmentally regulated and globally alters its chromatin association. Sci Rep 2016;6:28295 10.1038/srep28295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Skene PJ, Illingworth RS, Webb S, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell 2010;37:457–68. 10.1016/j.molcel.2010.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Witteveen JS, Willemsen MH, Dombroski TC, et al. Haploinsufficiency of MeCP2-interacting transcriptional co-repressor SIN3A causes mild intellectual disability by affecting the development of cortical integrity. Nat Genet 2016. 2016;48:877–87. 10.1038/ng.3619 [DOI] [PubMed] [Google Scholar]
- 16. Nott A, Cheng J, Gao F, et al. Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat Neurosci 2016;19:1497–505. 10.1038/nn.4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shah RR, Bird AP. MeCP2 mutations: progress towards understanding and treating Rett syndrome. Genome Med 2017;9:17 10.1186/s13073-017-0411-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zylka MJ, Simon JM, Philpot BD. Gene length matters in neurons. Neuron 2015;86:353–5. 10.1016/j.neuron.2015.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gabel HW, Kinde B, Stroud H, et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015;522:89–93. 10.1038/nature14319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meng X, Wang W, Lu H, et al. Manipulations of MeCP2 in glutamatergic neurons highlight their contributions to Rett and other neurological disorders. Elife 2016;5 pii: :e14199 10.7554/eLife.14199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ure K, Lu H, Wang W, et al. Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of Rett Syndrome. Elife 2016;21 pii: :e14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gibson JH, Williamson SL, Arbuckle S, et al. X chromosome inactivation patterns in brain in Rett syndrome: implications for the disease phenotype. Brain Dev 2005;27:266–70. 10.1016/j.braindev.2004.07.002 [DOI] [PubMed] [Google Scholar]
- 23. Xinhua Bao, Shengling Jiang, Fuying Song, et al. X chromosome inactivation in Rett Syndrome and its correlations with MECP2 mutations and phenotype. J Child Neurol 2008;23:22–5. 10.1177/0883073807307077 [DOI] [PubMed] [Google Scholar]
- 24. Leonard H, Cobb S, Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol 2017;13:37–51. 10.1038/nrneurol.2016.186 [DOI] [PubMed] [Google Scholar]
- 25. Dolce A, Ben-Zeev B, Naidu S, et al. Rett syndrome and epilepsy: an update for child neurologists. Pediatr Neurol 2013;48:337–45. 10.1016/j.pediatrneurol.2012.11.001 [DOI] [PubMed] [Google Scholar]
- 26. Knight VM, Horn PS, Gilbert DL, et al. The Clinical Predictors That Facilitate a Clinician's Decision to Order Genetic Testing for Rett Syndrome. Pediatr Neurol 2016;63:66–70. 10.1016/j.pediatrneurol.2016.06.016 [DOI] [PubMed] [Google Scholar]
- 27. Christodoulou J, Grimm A, Maher T, et al. RettBASE: The IRSA MECP2 variation database-a new mutation database in evolution. Hum Mutat 2003;21:466–72. 10.1002/humu.10194 [DOI] [PubMed] [Google Scholar]
- 28. Louise S, Fyfe S, Bebbington A, et al. InterRett, a model for international data collection in a rare genetic disorder. Res Autism Spectr Disord 2009;3:639–59. 10.1016/j.rasd.2008.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sampieri K, Meloni I, Scala E, et al. Italian Rett database and biobank. Hum Mutat 2007;28:329–35. 10.1002/humu.20453 [DOI] [PubMed] [Google Scholar]
- 30. Grillo E, Villard L, Clarke A, et al. Rett networked database: an integrated clinical and genetic network of Rett syndrome databases. Hum Mutat 2012;33:1031–6. 10.1002/humu.22072 [DOI] [PubMed] [Google Scholar]
- 31. Percy AK, Lane JB, Childers J, et al. Rett syndrome: North American database. J Child Neurol 2007;22:1338–41. 10.1177/0883073807308715 [DOI] [PubMed] [Google Scholar]
- 32. Percy AK, Neul JL, Glaze DG, et al. Rett syndrome diagnostic criteria: lessons from the Natural History Study. Ann Neurol 2010;68:951–5. 10.1002/ana.22154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Itoh M, Saikusa T, Tanioka T, et al. A study of Methyl-CpG Bing Protein 2 mutations and locomotion ability; Consideration from Japanese Rett Syndrome Database. Rett Syndrome – RTT50.1. Wiener Medizinische Wochenschrift 2016:11–12. [Google Scholar]
- 34. Lang AE, Eberly S, Goetz CG, et al. Movement disorder society unified Parkinson disease rating scale experiences in daily living: longitudinal changes and correlation with other assessments. Mov Disord 2013;28:1980–6. 10.1002/mds.25671 [DOI] [PubMed] [Google Scholar]
- 35. Shapiro EG, Nestrasil I, Ahmed A, et al. Quantifying behaviors of children with Sanfilippo syndrome: the Sanfilippo Behavior Rating Scale. Mol Genet Metab 2015;114:594–8. 10.1016/j.ymgme.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. FitzGerald PM, Jankovic J, Percy AK. Rett syndrome and associated movement disorders. Mov Disord 1990;5:195–202. 10.1002/mds.870050303 [DOI] [PubMed] [Google Scholar]
- 37. Tarquinio DC, Motil KJ, Hou W, et al. Growth failure and outcome in Rett syndrome: specific growth references. Neurology 2012;79:1653–61. 10.1212/WNL.0b013e31826e9a70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mount RH, Charman T, Hastings RP, et al. The Rett Syndrome Behaviour Questionnaire (RSBQ): refining the behavioural phenotype of Rett syndrome. J Child Psychol Psychiatry 2002;43:1099–110. 10.1111/1469-7610.00236 [DOI] [PubMed] [Google Scholar]
- 39. Esbensen AJ, Rojahn J, Aman MG, et al. Reliability and validity of an assessment instrument for anxiety, depression, and mood among individuals with mental retardation. J Autism Dev Disord 2003;33:617–29. 10.1023/B:JADD.0000005999.27178.55 [DOI] [PubMed] [Google Scholar]
- 40. Kerr AM, Nomura Y, Armstrong D, et al. Guidelines for reporting clinical features in cases with MECP2 mutations. Brain Dev 2001;23:208–11. 10.1016/S0387-7604(01)00193-0 [DOI] [PubMed] [Google Scholar]
- 41. Pini G, Congiu L, Benincasa A, et al. Illness severity, social and cognitive ability, and EEG analysis of ten patients with Rett syndrome treated with mecaserminome (Recombinant Human IGF-1). Autism Res Treat 2016;2016:1–9. 10.1155/2016/5073078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaufmann WE, Tierney E, Rohde CA, et al. Social impairments in Rett syndrome: characteristics and relationship with clinical severity. J Intellect Disabil Res 2012;56:233–47. 10.1111/j.1365-2788.2011.01404.x [DOI] [PubMed] [Google Scholar]
- 43. Tarquinio DC, Hou W, Berg A, et al. Longitudinal course of epilepsy in Rett syndrome and related disorders. Brain 2017;140:306–18. 10.1093/brain/aww302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neul JL, Glaze DG, Percy AK, et al. Improving treatment trial outcomes for Rett syndrome: the development of Rett-specific anchors for the Clinical Global Impression Scale. J Child Neurol 2015;30:1743–8. 10.1177/0883073815579707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lane JB, Lee HS, Smith LW, et al. Clinical severity and quality of life in children and adolescents with Rett syndrome. Neurology 2011;77:1812–8. 10.1212/WNL.0b013e3182377dd2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Djukic A, Holtzer R, Shinnar S, et al. Pharmacologic Treatment of Rett Syndrome With Glatiramer Acetate. Pediatr Neurol 2016;61:51–7. 10.1016/j.pediatrneurol.2016.05.010 [DOI] [PubMed] [Google Scholar]
- 47. Khwaja OS, Ho E, Barnes KV, et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci U S A 2014;111:4596–601. 10.1073/pnas.1311141111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. https://www.clinicaltrials.gov/ct2/show/NCT02790034?term=newron&rank=2 (accessed 25 February 2017).
- 49. Kolevzon A, Bush L, Wang AT, et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol Autism 2014;5:54 10.1186/2040-2392-5-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Naidu S, Johnston MV. Neurodevelopmental disorders: clinical criteria for Rett syndrome. Nat Rev Neurol 2011;7:312–4. 10.1038/nrneurol.2011.64 [DOI] [PubMed] [Google Scholar]
- 51. Robertson L, Hall SE, Jacoby P, et al. The association between behavior and genotype in Rett syndrome using the Australian Rett Syndrome Database. Am J Med Genet B Neuropsychiatr Genet 2006;141B:177–83. 10.1002/ajmg.b.30270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barnes KV, Coughlin FR, O'Leary HM, et al. Anxiety-like behavior in Rett syndrome: characteristics and assessment by anxiety scales. J Neurodev Disord 2015;7:30 10.1186/s11689-015-9127-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Downs J, Stahlhut M, Wong K, et al. Validating the Rett Syndrome Gross Motor Scale. PLoS One 2016;11:e0147555 10.1371/journal.pone.0147555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bebbington A, Anderson A, Ravine D, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology 2008;70:868–75. 10.1212/01.wnl.0000304752.50773.ec [DOI] [PubMed] [Google Scholar]
- 55. Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008;70(16):1313–21. 10.1212/01.wnl.0000291011.54508.aa [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Halbach N, Smeets EE, Julu P, et al. Neurophysiology versus clinical genetics in Rett syndrome: A multicenter study. Am J Med Genet A 2016;170:2301–9. 10.1002/ajmg.a.37812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Santosh PJ, Bell L, Fiori F, et al. Pediatric antipsychotic use and outcomes monitoring. J Child Adolesc Psychopharmacol 2016. (Sep 8). 10.1089/cap.2015.0247 [DOI] [PubMed] [Google Scholar]
- 58. Singh J, Santosh P. Psychopharmacology of neurodevelopmental disorders in children Child and Adolescent Psychiatry: Asian Perspectives. Edition 1: Springer, 2017. pp. :325–62. [Google Scholar]
- 59. Santosh PJ, Bell L, Lievesley K, et al. Paradoxical physiological responses to propranolol in a Rett syndrome patient: a case report. BMC Pediatr 2016;16:194 10.1186/s12887-016-0734-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Julu PO, Kerr AM, Hansen S, et al. Functional evidence of brain stem immaturity in Rett syndrome. Eur Child Adolesc Psychiatry 1997;6 Suppl 1(Suppl 1):47–54. [PubMed] [Google Scholar]
- 61. Julu PO, Kerr AM, Apartopoulos F, al AFet, et al. Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch Dis Child 2001;85:29–37. 10.1136/adc.85.1.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Julu PO, Witt Engerström i . Assessment of the maturity-related brainstem functions reveals the heterogeneous phenotypes and facilitates clinical management of Rett syndrome. Brain Dev 2005;27 Suppl 1(Suppl. 1):S43–S53. 10.1016/j.braindev.2005.02.012 [DOI] [PubMed] [Google Scholar]
- 63. Julu PO, Engerström IW, Hansen S, et al. Cardiorespiratory challenges in Rett's syndrome. Lancet 2008;371:1981–3. 10.1016/S0140-6736(08)60849-1 [DOI] [PubMed] [Google Scholar]
- 64. US Department of Health and Human Services Food and Drug Administration. Guidance for industry: patient-reported outcome measures: use in medical product development to support labelling claims. Secondary Guidance for industry: patient-reported outcome measures: use in medical product development to support labelling claims. 2009. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf.
- 65. Santosh P, Tarver J, Gibbons F, et al. Protocol for the development and validation of a questionnaire to assess concerning behaviours and mental health in individuals with autism spectrum disorders: the Assessment of Concerning Behaviour (ACB) scale. BMJ Open 2016;6:e010693 10.1136/bmjopen-2015-010693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liberati A, et al. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of studies that evaluate health care interventions: explanation and elaboration. Ann Intern Med 2009;151:W 10.7326/0003-4819-151-4-200908180-00136 [DOI] [PubMed] [Google Scholar]
- 67.https://www.healthtracker.co.uk
- 68. Santosh P. STOP study aims to monitor suicidality. EU Research 2014:36–9. [Google Scholar]
- 69. http://cordis.europa.eu/project/rcn/110200_en.html (accessed 25 February 2017)
- 70. Santosh P, Gringras P, Baird G, et al. Development and psychometric properties of the parent version of the Profile of Neuropsychiatric Symptoms (PONS) in children and adolescents. BMC Pediatrics 2015;15:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lilford RJ, Thornton JG, Braunholtz D. Clinical trials and rare diseases: a way out of a conundrum. BMJ 1995;311:1621 5. 10.1371/journal.pone.0120981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hampson LV, Whitehead J, Eleftheriou D, et al. Elicitation of expert prior opinion: application to the MYPAN trial in childhood polyarteritis nodosa. PLoS One 2015;10:e0120981 10.1371/journal.pone.0120981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Theoharides TC, Athanassiou M, Panagiotidou S, et al. Dysregulated brain immunity and neurotrophin signaling in Rett syndrome and autism spectrum disorders. J Neuroimmunol 2015;279:33–8. 10.1016/j.jneuroim.2014.12.003 [DOI] [PubMed] [Google Scholar]
- 74. Neul JL. The relationship of Rett syndrome and MECP2 disorders to autism. Dialogues Clin Neurosci 2012;14:253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Carter JC, Lanham DC, Pham D, et al. Selective cerebral volume reduction in Rett syndrome: a multiple-approach MR imaging study. AJNR Am J Neuroradiol 2008;29:436–41. 10.3174/ajnr.A0857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Goodman R, Ford T, Richards H, et al. The Development and Well-Being Assessment: description and initial validation of an integrated assessment of child and adolescent psychopathology. J Child Psychol Psychiatry 2000;41:645–55. 10.1111/j.1469-7610.2000.tb02345.x [DOI] [PubMed] [Google Scholar]
- 77. http://www.proteus.com/ (accessed 25 February 2017)
- 78. https://clinicaltrials.gov/ct2/show/NCT02447952 (accessed 25 February 2017)
- 79. Weese-Mayer DE, Lieske SP, Boothby CM, et al. Autonomic nervous system dysregulation: breathing and heart rate perturbation during wakefulness in young girls with Rett syndrome. Pediatr Res 2006;60:443–9. 10.1203/01.pdr.0000238302.84552.d0 [DOI] [PubMed] [Google Scholar]
- 80. Weese-Mayer DE, Lieske SP, Boothby CM, et al. Autonomic dysregulation in young girls with Rett Syndrome during nighttime in-home recordings. Pediatr Pulmonol 2008;43:1045–60. 10.1002/ppul.20866 [DOI] [PubMed] [Google Scholar]
- 81. Tarvainen MP, Niskanen JP, Lipponen JA, et al. Kubios HRV--heart rate variability analysis software. Comput Methods Programs Biomed 2014;113:210–20. 10.1016/j.cmpb.2013.07.024 [DOI] [PubMed] [Google Scholar]
- 82. Benedek M, Kaernbach C. A continuous measure of phasic electrodermal activity. J Neurosci Methods 2010;190:80–91. 10.1016/j.jneumeth.2010.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bauer A, Kantelhardt JW, Bunde A, et al. Phase-rectified signal averaging detects quasi-periodicities in non-stationary data. Physica A 2006;364:423–34. 10.1016/j.physa.2005.08.080 [DOI] [Google Scholar]
- 84. Braun V, Clarke V. Using thematic analysis in psychology. Qual Res Psychol 2006;3:77–101. 10.1191/1478088706qp063oa [DOI] [Google Scholar]
- 85. Portney LG, Watkins MP. Foundations of clinical research: Applications to practice. 3rd ed Upper Saddle River, N.J: Pearson/Prentice Hall, 2009. [Google Scholar]
- 86. Weir JP. Quantifying test-retest reliability using the intraclass correlation coefficient and the SEM. J Strength Cond Res 2005;19:231–40. 10.1519/00124278-200502000-00038 [DOI] [PubMed] [Google Scholar]
- 87. MacCallum RC, Austin JT. Applications of structural equation modeling in psychological research. Annu Rev Psychol 2000;51:201–26. 10.1146/annurev.psych.51.1.201 [DOI] [PubMed] [Google Scholar]
- 88. de Winter JC, Dodou D, Wieringa PA. Exploratory Factor Analysis With Small Sample Sizes. Multivariate Behav Res 2009;44:147–81. [DOI] [PubMed] [Google Scholar]
- 89. Rouquette A, Falissard B. Sample size requirements for the internal validation of psychiatric scales. Int J Methods Psychiatr Res 2011;20:235–49. 10.1002/mpr.352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jung S, Lee S, Soonmook K. Exploratory factor analysis for small samples. Behav Res Methods 2011;43:701–9. 10.3758/s13428-011-0077-9 [DOI] [PubMed] [Google Scholar]
- 91. Vindras P, Desmurget M, Baraduc P. When one size does not fit all: a simple statistical method to deal with across-individual variations of effects. PLoS One 2012;7:e39059 10.1371/journal.pone.0039059 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.