

Abstract
Monoclonal antibodies targeting the immune checkpoint Programmed Death-1 (aPD-1 mAbs) have demonstrated impressive benefits for the treatment of some cancers; yet, these drugs are not always effective and we still have a limited understanding of the mechanisms that contribute to their efficacy or lack thereof. Here we employed in vivo imaging to uncover the fate and activity of aPD-1 mAbs in real-time and at subcellular resolution in mice. We show that aPD-1 mAbs effectively bind PD-1+ tumor-infiltrating CD8+ T cells at early time-points after administration. However, this engagement is transient, as aPD-1 mAbs are captured within minutes from the T cell surface by PD-1− tumor-associated macrophages. We further show that macrophage accrual of aPD-1 mAbs depends both on the drug’s Fc domain glycan and on Fcγ-receptors (FcγRs) expressed by host myeloid cells, and extend these findings to the human setting. Finally, we demonstrate that in vivo blockade of FcγRs prior to aPD-1 mAb administration substantially prolongs aPD-1 mAb binding to tumor-infiltrating CD8+ T cells and enhances immunotherapy-induced tumor regression in mice. These investigations yield new insight into aPD-1 target engagement in vivo and identify specific Fc : FcγR interactions that can be modulated to improve checkpoint blockade therapy.
Introduction
Immune checkpoint blockade is a recent development in cancer therapy that has shown remarkable results in certain cancers and patient groups (1–3). Currently approved immune checkpoint blockers are monoclonocal antibodies (mAbs) that target the programmed cell death-1 (PD-1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) pathways, while agents targeting other pathways are in clinical development (e.g. OX40, Tim-3, LAG-3) (4). Checkpoint inhibitors are used to reactivate exhausted tumor-specific T cells and reinstate cancer immuno-surveillance (5, 6). Indeed, some cancer tissues limit anti-tumor immunity by upregulating immunosuppressive factors such as PD-1 ligand (PD-L1) that binds to PD-1 on tumor-specific CD8+ T cells (7). Drugs targeting the PD-1/PD-L1 immune checkpoint axis can block immunosuppressive signals and enable T cell-mediated elimination of cancer cells (8). However, immune checkpoint blockade is not always effective and we lack a complete understanding of the mechanisms that contribute to efficacy and resistance (9).
At present, experimental and clinical evidence suggest that a pre-existing tumor infiltrate of CD8+ T cells is one of the most favorable prognostic indicators of checkpoint inhibitor response (10). Also, patients with the highest degree of neoantigen burden (high mutational load) in their cancers may have increased tumor infiltration by T cells and more robust responses to checkpoint blockade (11,12). Histology and sequencing methodologies have been used to define metrics of cytotoxic T cell infiltration in tumors (13), with a focus on the identification of tumor neoantigens and the resultant antigen-specific T cell expansion following immunotherapy (11). These studies have provided insight into the mechanism of aPD-1 mAb-induced antitumor T cell activation and spurred efforts focused on identifying new strategies that foster T cell recruitment to tumors (14–16).
Much less is known about checkpoint inhibitors’ in vivo pharmacokinetics and interactions with host components in the tumor bed. Studying these parameters is likely essential to identifying resistance mechanisms and developing improved therapeutic options. Here, we used intravital imaging to follow fluorescently-labeled aPD-1 mAbs in real-time and at subcellular resolution. Because tumor microenvironments are home to diverse host cell types, and immune checkpoint blockers are unlikely to solely act on T cells, we focused on aPD-1 mAb interactions with various host components by simultaneously assessing aPD-1 mAbs, tumor cells, CD8 T cells, and myeloid cells/macrophages. We investigated tumor-infiltrating CD8+ T cells since they express PD-1 and are the expected targets of aPD-1 mAbs. We also investigated myeloid cells because they are frequently found in the stroma of growing tumors (17) and emerging evidence indicates they can affect virtually all therapeutic modalities, including immunotherapy (18). Our results confirm existing knowledge on PD-1 inhibition but also uncover important new findings with therapeutic implications to further improve immunotherapy.
RESULTS
Global aPD-1 mAb biodistribution
We initially sought to track the temporal distribution of aPD-1 mAbs in vivo at the organ level. We thus covalently labeled aPD-1 mAb (clone 29F.1A12) with an Alexa-Fluor 647 dye (AF647-aPD-1) using NHS chemistry. For maximal brightness without dye quenching, we optimized the labeling conditions to achieve ~4 fluorochrome molecules per antibody. For in vitro studies, the EL-4 mouse lymphoma cell line was used as a T cell model because of its stable PD-1 expression and its broad adaptation to in vitro culture (19). Using this cell line, we confirmed that fluorescent labeling of aPD-1 did not interfere with the drug’s binding specificity (Fig. 1A). Additionally, AF647-aPD-1 retained therapeutic activity in the ovalbumin-expressing MC38 tumor model, which is responsive to single-agent aPD-1 therapy (Fig. 1B) (20). Collectively, these data indicate that AF647-aPD-1 retains PD-1 tropism and anti-tumor activities.
Figure 1. Anti-PD-1 mAb labeling facilitates tracking of tissue biodistribution.
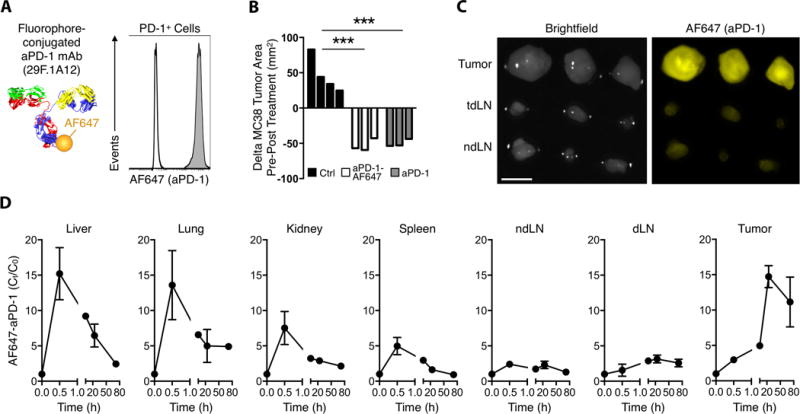
(A) Conjugation of Alexa Fluor 647 via NHS ester linkage to rat anti-mouse PD-1 29F.1A12 clone was confirmed by flow cytometry to not alter the binding tropism to PD-1+ EL4 cells (grey). Rat IgG2a isotype control (white). (B) MC38 tumors were equally responsive to single-dose AF647-aPD-1 and unlabeled aPD-1, while tumor areas increased 72 h after control IgG2a treatment. (C) Fluorescence reflectance imaging of 3 tumors compared to draining (dLN) and contralateral (cLN) lymph nodes 24 hours after AF647-aPD-1 treatment (AF647: λex = 620–650 nm, λem = 680–710 nm). Scale bars represent 5 mm. (D) Quantified AF647-aPD-1 in each tissue demonstrating tumor accumulation. Values represent SEM and n = 3 unless otherwise noted. ***P < 0.001; unpaired, two-tailed t-test.
We next examined the in vivo biodistribution of AF647-aPD-1 in the wild-type MC38 tumor model, which also responds to aPD-1 treatment, but less efficiently than to the ovalbumin-expressing MC38 counterpart in which aPD-1 treatment results in uniform tumor rejection. Mouse cohorts were sacrificed at times ranging from 0.5 h to 72 h post-treatment, and organs were removed for fluorescence measurements (Fig. 1C). AF647-aPD-1 signal was primarily retained within tumors over time (Fig. 1D and fig. S1). We observed a large spike of AF647-aPD-1 in the liver, lungs, kidney, and spleen at 0.5 hours post-injection, followed by a subsequent decrease over time that coincided with increases in AF647-aPD-1 signal in the tumor (Fig. 1D). Collectively, these data indicate that the drug started to accumulate in tumors within minutes after injection but that maximal aPD-1 accumulation in the tumor was achieved after 24 hours.
Cellular kinetics and dynamics of aPD-1
After observing that aPD-1 mAbs collect within the tumor microenvironment shortly after administration, we further aimed to study whether the drugs locally bind their intended target T cells. To this end, we used intravital microscopy in dorsal skin-fold chambers, which enabled us to examine the distribution and tropism of AF647-aPD-1 at subcellular resolution within the tumor stroma and longitudinally following drug administration (Fig. 2A). The experimental system allowed simultaneous tracking of four components: aPD-1 mAbs (labeled with AF647); MC38 tumor cells (labeled with H2B-mApple); T cells (labeled with GFP or YFP); and tumor-associated macrophages (labeled with PacificBlue-dextran nanoparticles). We used two different reporter mouse models to visualize T cells: i) DPE-GFP mice (21) in which all GFP-expressing cells are CD90+ (fig. S2A), and ii) interferon gamma reporter (GREAT) mice (22), which were useful because tumor-infiltrating YFP+ cells in these mice were almost exclusively CD8+ T cells (fig. S2B). The fluorescent PacificBlue-dextran nanoparticle has been validated for intratumoral macrophage identification (23) and we confirmed its specificity for macrophages (F4/80+ cells) in the tumor stroma (fig. S2C). Finally, we verified that single agent aPD-1 treatment was able to control MC38-H2B-mApple tumor growth in the window chamber system (Fig. 2B).
Figure 2. In vivo temporal aPD-1 mAb pharmacokinetics reveals drug accumulation in TAMs.
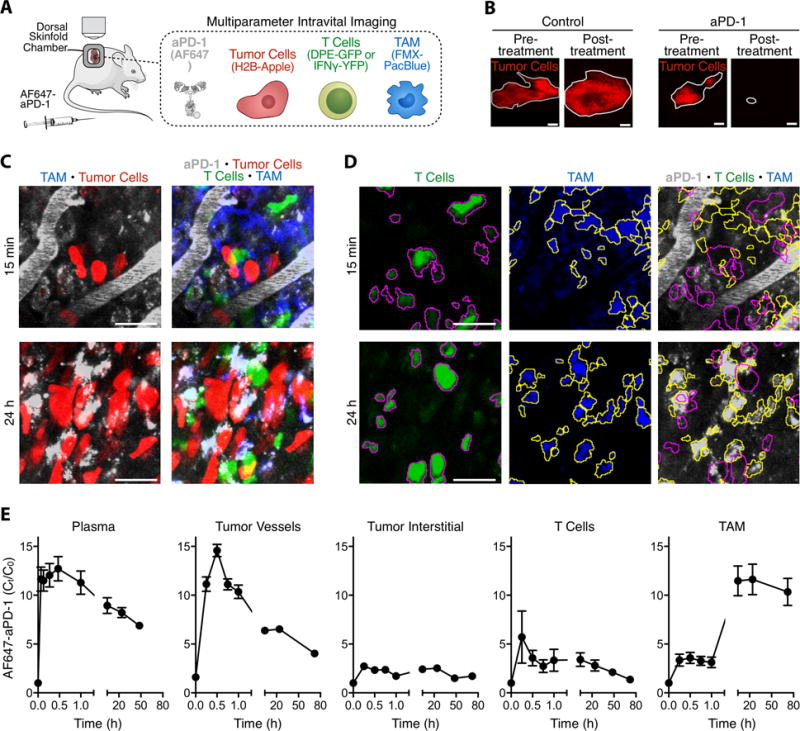
(A) Diagram depicting intravital imaging setup with labeled aPD-1, MC38 tumor cells, T cells, and tumor associated macrophages (TAMs). (B) Treatment with single-dose aPD-1 mAb can achieve significant remission in the MC38/H2B-mApple tumor model. Tumors are outlined in grey; scale bars represent 2 mm. (C) Z-projections of an MC38/H2B-mApple tumor in a DPE-GFP mouse injected i.v. with AF647-aPD-1 after 15 minutes (top) or 24 hours (bottom). (D) Micrographs of T cells (magenta outline) identified as GFP+ cells and TAMs (yellow outline) identified by Pacific Blue signal. Outlines are overlaid on micrographs of the corresponding AF647-aPD-1 channel; scale bars represent 30 μm. (E) IVM biodistribution studies indicate early aPD-1 binding to T-cells and long-term accumulation in TAMs. Data are representative of 5 independently-treated DPE-GFP mice and normalized to autofluorescent signal.
Upon administration of AF647-aPD-1, we found that the antibody rapidly perfused tumor vessels and gradually disseminated out of the vasculature and into the tumor interstitium (Fig. 2C and movie S1). AF647-aPD-1 was observed on GFP-labeled T cells as early as 5 minutes after injection, and these were essentially the first cells in the tumor microenvironment to be detectably labeled by the drug. AF647-aPD-1 binding to tumor-infiltrating T cells was initially peri-cellular, but within minutes formed puncta on the cell surface (fig. S3). These rearrangements occurred without apparent decreased T cell motility (fig. S4, A-C). Later time points revealed that tumor-associated macrophages, which were stationary in the tumor microenvironment (fig. S4D), had collected most of the AF647-aPD-1; T cells were not associated with AF647-aPD-1 at these time points (Fig. 2, D and E). T cells were present in the tumor microenvironment at all times examined, removing the possibility that the drug biodistribution was an artifact of T cell loss post-therapy. Also, tracing AF647-aPD-1 across all time-points failed to show binding to tumor cells, precluding the possibility that aPD-1 mAbs had direct effects on cancer cells (fig. S5).
Quantification of the tumor microenvironment images taken at 15 minutes or 24 h after drug administration showed a clear pattern of AF647 signal loss on T cells over time, and a concomitant AF647 signal increase on macrophages (Fig. 3A). While DPE-GFP labels CD90+ lymphocytes (fig. S2A), the GREAT mouse model allowed us to focus specifically on IFNγ-expressing cells (movie S2), which are almost exclusively CD8+ T cells in the tumor microenvironment (fig. S2B). Intravital imaging of these cells confirmed surface binding of AF647-aPD-1 within minutes after drug administration (fig. S3). Longitudinal studies further showed that T cells eventually lose AF647-aPD-1 mAbs, which are physically transferred to, and retained by, neighboring macrophages (Fig. 3B). Consistent with our observations in DPE-GFP mice, we found in IFNγ reporter mice that aPD-1 exchange from T cells to macrophages greatly limited the overall duration of drug binding to their intended target cells (Fig. 3C).
Figure 3. In vivo imaging reveals aPD-1 mAb transfer from CD8+ T cells to TAMs.
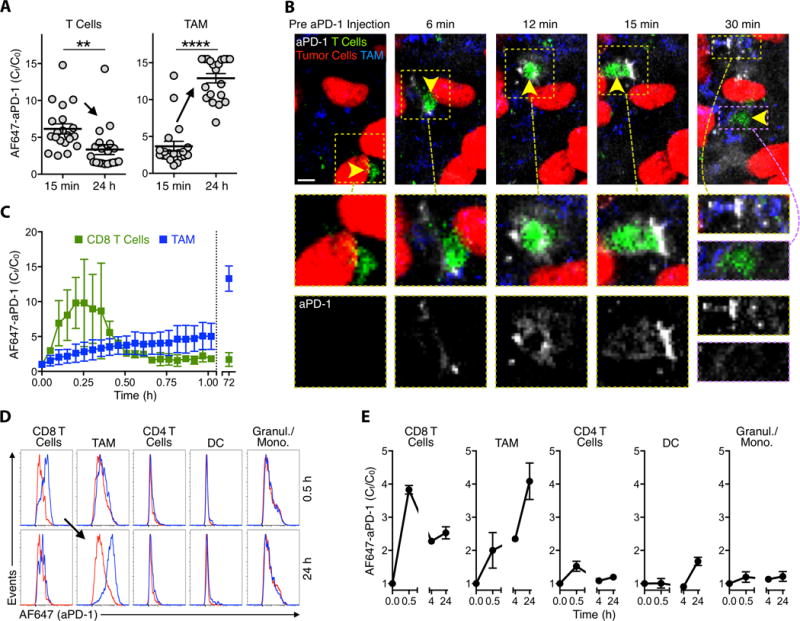
(A) Quantified AF647 signal on T cells and TAMs from a representative DPE-GFP mouse demonstrates collection of AF647-aPD-1 in T cells at 15 minutes and in TAMs at 24 hours following injection of therapy. (B) Early time-course IVM images acquired in an IFNγ-YFP reporter mouse with an MC38-H2B-mApple tumor. Yellow arrows indicate site of aPD-1 mAb binding to an IFNγ+ CD8 T-cell at 6-15 minutes and macrophage internalization at times >21 min. (C) Quantification of aPD-1-mAb on IFNγ-expressing CD8+ lymphocytes and TAMs reveals a narrow window of target binding. (D) Flow cytometry histograms pre-gated for 7-AAD−/CD45+ show AF647-aPD-1 signal (x-axis, logarithmic scale) on immune cell populations at 0.5 hr and 24 hours post administration (blue). Cell populations from untreated control animals (red) were used as reference. (E) aPD-1 mAb binds to CD8+ lymphocytes early but accumulates in TAMs at later time points. Scale bars represent 30 μm. **P < 0.01; ****P < 0.0001; unpaired, two-tailed t-test.
To address whether checkpoint blockade agent uptake by macrophages could be independently validated using a bulk tissue measurement, we performed flow cytometry of tissues excised from tumor-bearing animals at 0.5 and 24 h after AF647-aPD-1 administration (Fig. 3D). We evaluated several relevant cell populations for aPD-1 binding and confirmed our intravital microscopy observations: aPD-1 mAbs were bound mostly to CD8+ T cells at 0.5 h, but to macrophages at 24 h (Fig. 3, D and E). Tumor macrophages were positive for rat IgG2a (fig. S6), confirming that the aPD-1 mAb, and not just the fluorophore, was transferred in vivo. Other cell types investigated, including CD4+ T cells, CD45+ CD11b+ F4/80− cells (which can include granulocytes and monocytes), and dendritic cells did not display significant binding of aPD-1 at any time-point tested (Fig. 3, D and E).
Analysis of B16 melanoma and KP1.9 lung adenocarcinoma tumor models also demonstrated that most AF647-aPD-1 mAbs accumulate within tumor-associated macrophages at 24 h, similar to findings obtained with MC38 colon adenocarcinoma (fig. S7A). The majority of intratumoral macrophages were bound by AF647-aPD-1 in all three tumor models (fig. S7B). Linear regression analysis of combined data further indicated that anti-PD-1 mAb uptake by macrophages is independent of macrophage number and can also occur when tumor-associated macrophage numbers are relatively low (fig. S7C).
AF647-aPD-1 transfer is mediated by macrophage FcγRs
To explore the removal of aPD-1 mAbs on T cells by macrophages, we first asked whether the latter may also express PD-1 on their surface. However, ex vivo flow cytometry analysis indicated that tumor-associated macrophages, in contrast to tumor-infiltrating CD8+ T cells, were PD-1− (Fig. 4A). PD-1 was also absent in NK and B cells in these tumors (fig. S8). We then designed an in vitro co-culture system combining bone marrow derived macrophages and T cells that constitutively express PD-1 to create a controlled system to study the mechanism of drug collection by macrophages (Fig. 4B). T cells were pre-incubated with AF647-aPD-1 to emulate the antibody initially bound to T cells we observed in vivo and co-cultured with macrophages. Using this experimental setting, AF647-aPD-1 mAbs effectively relocated from T cells to macrophages within several minutes, as detected by the formation of drug puncta within macrophages (Fig. 4, B and C; movie S3). The transfer could not be attributed solely to phagocytosis of cell debris because it occurred even in the presence of the phagocytosis inhibitor dynasore (Fig. 4B). No macrophage uptake was observed when using an AF647-labeled isotype control IgG antibody, which did not bind T cells (fig. S9).
Figure 4. aPD-1 mAb transfer to macrophages is mediated by FcγRs.
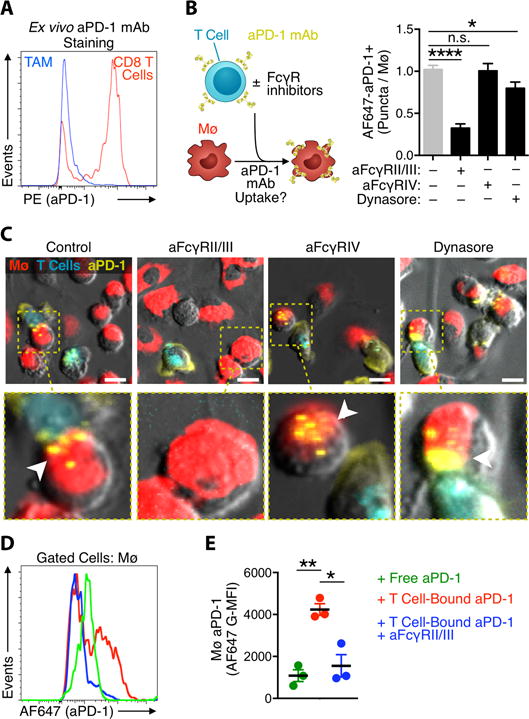
(A) Ex vivo flow cytometry histograms of MC38 tumors stained with PE-aPD-1 show CD8+ T cells but not TAMs express cell surface PD-1. (B) Co-culture of bone marrow-derived macrophages (Mø) and AF647-aPD-1 coated EL4 lymphocytes was used to quantify the AF647-aPD-1 puncta in macrophages pre-blocked with FcγR inhibitors or the phagocytosis inhibitor, dynasore. *P < 0.05; ****P < 0.0001; one-way ANOVA. (C) Representative images of macrophages labeled with PKH-red and observed by time-lapse imaging for aPD-1 mAb (yellow) transfer from neighboring lymphocytes (cerulean) after 30 min. FcγRII/III inhibition blocks antibody transfer (highlighted); Scale bars represent 10 μm. (D-E) Flow cytometry was used to estimate AF647-aPD-1 transfer to F4/80+ BMDMs in co-culture assays when the mAb was added directly (green), bound to EL4 cells (red) or bound to EL4 cells in the presence of FcγRII/III neutralizing antibody (blue). Results in D are a representative histogram of AF647 signal in macrophages and E is G-MFI value of conditions presented in D. Data are from 3 independent experiments. *P < 0.05; ** P < 0.01; two-way ANOVA with Tukey’s multiple comparisons test. Values represent SEM for three separate experiments.
Since tumor macrophages did not capture AF647-aPD-1 in substantial quantities early after administration, did not express PD-1 on their cell surface, and withdrew drug bound to the T cell surface, we reasoned that AF647-aPD-1 mAbs must accumulate in macrophages through a non-antigen specific mechanism. The aPD-1 clone 29F.1A12 is a rat IgG2a isotype that is used to mimic the biological properties of human IgG4. Both rat IgG2a and human IgG4 have been demonstrated to bind inhibitory FcγRs (mouse FcγRIIb and human FcγRIIB, respectively) (24, 25). Importantly, adding FcγRIIb/III blocking antibodies to the in vitro co-culture system diminished AF647-aPD-1 transfer from T cells to macrophages (Fig. 4, B and C). The effect was specific because blocking FcγRIV, an Fc receptor that does not bind rat IgG2a, failed to inhibit AF647-aPD-1 transfer (Fig. 4, B and C).
To substantiate these findings, we developed a flow cytometry-based antibody transfer assay: macrophages were incubated with T cells previously labeled with AF647-aPD-1 mAbs for 30 min and analyzed by flow cytometry. AF647-aPD-1 signal was detected on macrophages in this experimental setting (Fig. 4D); however, aPD-1 transfer could be neutralized by adding a blocking antibody to FcγRIIb/III to the co-culture system (Fig. 4, D and E). AF647-aPD-1 mAbs added directly to the culture medium in the absence of T cells was not efficiently taken up by macrophages, further suggesting that T cells are the major source of aPD-1 for macrophages (Fig. 4, D and E). We also tested whether aPD-1 loss on the T cell surface might be due to receptor internalization independent of macrophage uptake. T cells were exposed to AF647-aPD-1 for 1 h at 37℃, treated with an acid solution to remove cell surface antibodies, and analyzed by flow cytometry to assess remaining (internalized) fluorescent signal (fig. S10A). Acid stripping strongly reduced AF647-aPD-1 detection, indicating that antibody internalization is likely not the primary contributor to aPD-1 loss on T cells (fig. S10B).
Macrophages co-cultured with AF647-aPD-1-coated T cells were positive not only for AF647, but also for rat IgG2a, as assessed by ELISA (fig. S11A). The eventual decline in macrophage rat IgG2a was not accompanied by release of IgG2a into the supernatant, suggesting that acquired antibody is eventually degraded by the macrophage (fig. S11B). Collectively, these data indicate that aPD-1 mAb removal from the T cell surface receptor by macrophages is a pharmacologic end-point elicited by FcγR interactions with T cell-bound antibody complexes.
To assess whether the PD-1 receptor is transferred during aPD-1 shaving, PD-1+ T cells were exposed to unlabeled aPD-1, co-cultured with macrophages (to enable aPD-1 capture), and re-probed for cell surface PD-1 expression with a fluorescent aPD-1 mAb. Transfer of unlabeled aPD-1 together with PD-1 would prevent re-probing with fluorescent aPD-1. Instead, T cells from which aPD-1 had been captured were efficiently re-probed, indicating that aPD-1 removal frees up PD-1 molecules, which become available to fluorescent aPD-1 mAb (fig. S12A). Control experiments confirmed that the increased re-probing signal was not contributed by new PD-1 molecules on the surface of T cells (fig. S12B). Taken together, these data indicate that PD-1 remains on the T cell membrane after aPD-1 capture.
Because Fc : FcγR binding interactions of many IgG subclasses depend upon mAb Fc glycosylation (26), we profiled the glycan structures from the murine aPD-1 mAb (29F.1A12) and further extended our analysis to the human aPD-1 mAb nivolumab. Both antibodies were treated with PNGase F, a glycosidase that cleaves N-linked glycan, to remove glycan from each antibody and the digested products were analyzed by HPLC (Fig. 5A). We found that murine and human aPD-1 mAbs share the same predominant glycoform that lacks terminal galactose residues (G0F), and is fucosylated on the penultimate N-acetylglucosamine (GlcNac, fig. S13). Both mouse and human aPD-1 contained substantial fractions of terminally galactosylated glycoforms, indicating a high degree of Fc glycan heterogeneity in these aPD-1 mAb preparations.
Figure 5. Nivolumab shares similar glycan patterns to mouse aPD-1 and is transferred to macrophages via FcγRs.
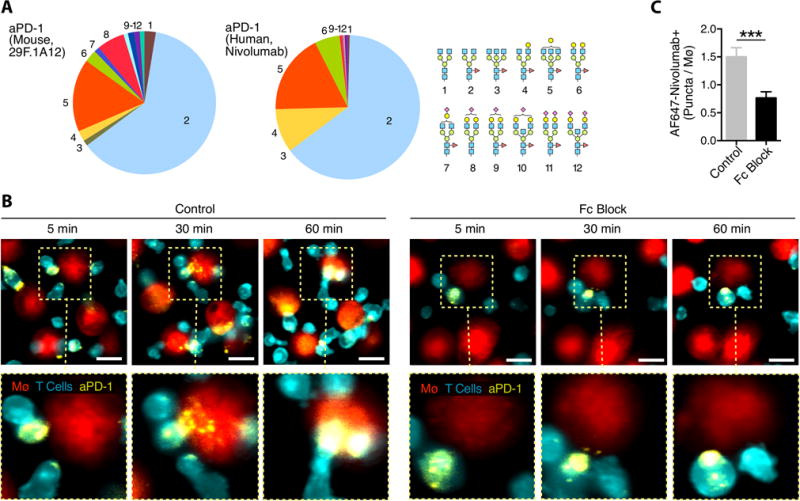
(A) HPLC analysis of the glycan patterns found on the mouse aPD-1 mAb and nivolumab shows the G0F isoform to be predominant, but glycosylation is not uniform. (B) AF647-labeled nivolumab (yellow) was used to stain the surface of aCD3 stimulated PKH-green labeled human CD8+ T cells (cerulean) co-incubated with PKH-red labeled peripheral blood mononuclear cell-derived macrophages in the presence or absence of Fc Block. Scale bars represent 20 μm. (C) Quantification of AF647+ puncta per macrophage confirms that nivolumab is transferred via FcγRs. Values represent SEM for 4 separate experiments.
The similarity in glycan pattern between mouse and human aPD-1 mAbs led us to hypothesize that FcγR-mediated antibody transfer is relevant to human aPD-1 interactions. We fluorescently labeled the anti-human PD-1 mAb nivolumab with AF647, and adapted the in vitro co-culture system (Fig. 4B) to primary human cells. We differentiated human macrophages from blood monocytes using macrophage colony-stimulating factor (M-CSF), and PD-1-expressing CD8+ T cells were generated by isolating primary human CD8+ T cells and stimulating them with plate-bound aCD3 mAbs for 3 days (27). Co-culture of AF647-nivolumab-labeled human CD8+ T cells with macrophages resulted in mAb transfer from CD8+ T cells to macrophages (movie S4), and the transfer was inhibited by blocking FcγRs using aFcγRIIB/III (Fig. 5B). There was no evidence of CD8+ T cell membrane components in the macrophages, consistent with the antibody alone being removed from the surface PD-1 receptor (fig. S14). Furthermore, blocking FcγRs decreased AF647-nivolumab puncta inside of macrophages, implying that FcγRs also regulate nivolumab uptake in human cells (Fig. 5C).
Improving immunotherapy
In an attempt to minimize aPD-1 : FcγR interactions, we used PNGase F to remove the glycan from murine aPD-1 mAbs and confirmed cleavage of glycan by LCA lectin blot (fig. S15A). The PNGase F-treated aPD-1 was labeled with AF647 and flow cytometry confirmed that the presence of glycan was not required for aPD-1 tropism as PD-1+ T cells were still efficiently labeled with PNGase F-treated AF647-aPD-1 (fig. S15B). However, live cell imaging methods demonstrated that glycan removal significantly diminished antibody transfer from T cells to macrophages (Fig. 6A) and these results were confirmed by flow cytometry (Fig. 6, B and C).
Figure 6. Disrupting Fc binding affects macrophage uptake of aPD-1 and improves treatment efficacy.
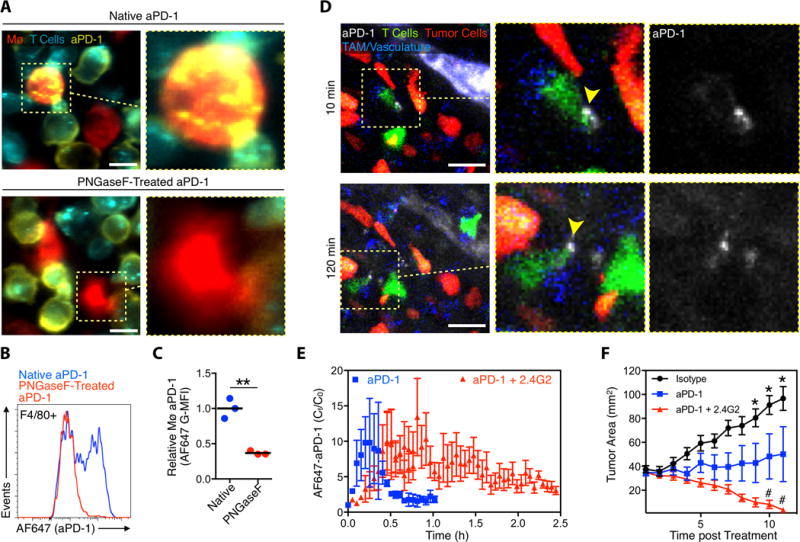
(A) PKH-green labeled EL4 cells (cerulean) stained with native AF647-aPD-1 or deglycosylated AF647-aPD-1 co-cultured with PKH-red labeled bone marrow-derived macrophages (Mø). Images are representative of 3 separate experiments. Scale bars represent 10 μm. (B) FACS plots of mouse Mø (gated on F4/80+) co-cultured with PD-1+ EL4 cells labeled with either AF647-aPD-1 mAb (blue) or deglycosylated AF647-aPD-1 mAb (red). (C) aPD-1-mAb deglycosylation substantially reduces the transfer from EL4 cells to Mø (n = 3). **P < 0.01; Unpaired 2-tailed t test. (D) Intravital images 10 and 120 minutes after AF647-aPD-1 drug extravasation in a representative IFNγ-YFP reporter mouse MC38-H2B-mApple tumor pre-treated with the FcγRII/III blocking antibody (2.4G2). (E) Quantified AF647-aPD-1 from three 2.4G2-treated mice shows prolonged aPD-1 binding to tumor T cells, relative to mice treated with AF647 aPD-1 alone (data repeated from Fig. 3C for comparison). (F) MC38 tumor growth curves of mice (n ≥ 5) treated with isotype control (black), aPD-1 (blue), and aPD-1 plus 2.4G2 (red). *P < 0.05 One-Way ANOVA with Tukey’s multiple comparison test.
We then aimed to uncover the in vivo activity of aPD-1 mAbs when aPD-1 : FcγR interactions were therapeutically diminished. To this end, we tracked aPD-1, CD8+ T cells, macrophages and tumor cells in mice in which we inhibited FcγRs by infusing FcγRIIb/III blocking Abs (2.4G2 clone) before delivering AF647-aPD-1 mAbs (Fig. 6D). Remarkably, administration of the FcγR blocking agent substantially prolonged the occupancy time of AF647-aPD-1 mAbs on CD8+ T cells in the tumor bed (Fig. 6E). Furthermore, whereas the response of MC38 tumors to aPD-1 therapy typically varies among subjects (fig. S16), blocking FcγR interactions completely eliminated the fraction of non-responders observed, with complete tumor rejection in all subjects that received the combination treatment (Fig. 6F). These data provide evidence that mAb : FcγR interactions abbreviate aPD-1 mAb occupancy time on tumor-infiltrating CD8+ T cells and limit therapy response; conversely, aPD-1 mAb therapy can be improved by blocking FcγR interactions (fig. S17).
DISCUSSION
Many cancer patients do not respond to immune checkpoint blockade therapy and we lack a complete understanding of the mechanisms that contribute to treatment efficacy and resistance. Here, by using time-lapse intravital microscopy, we uncovered in real-time how the immune checkpoint blocker aPD-1 mAb distributes in tumors and physically interacts with tumor microenvironment components. This approach enabled us to detect aPD-1 mAb association with cytotoxic T cells infiltrating tumors in vivo. Furthermore, by following the drug’s pharmacokinetics over time, we found the drug to be rapidly removed from PD-1+ CD8+ T cells and transferred to neighboring PD-1− tumor-associated macrophages. The transfer of aPD-1 mAbs from T cells to macrophages was unexpected because macrophages do not directly take up aPD-1 mAbs in culture. We further identify that aPD-1 uptake by macrophages depends both on the Fc domain of the antibody and on FcγRs expressed by macrophages. Interactions between the drugs and macrophages are likely important, because blocking Fc : FcγR binding inhibited aPD-1 transfer from CD8+ T cells to macrophages in vivo and enhanced aPD-1 therapeutic efficacy.
Although clinical aPD-1 have an extensive circulation half-life (~26 days), our observations suggest that the time of target engagement in the local tumor environment may be much shorter. This engagement time is shortened at least in part by FcγRII/III, which mediate aPD-1 (IgG2a) mAb uptake from T cells to macrophages. Accordingly, previous work has shown that IgG2a isotypes preferentially bind FcγRIIb/III and that aPD-1 therapy is more effective in FcγRIIb knockout mice (25). The human aPD-1 drugs nivolumab and pembrolizumab were designed as human IgG4 antibody isotypes that are not known to fix complement or trigger ADCC (27). However, IgG4 can bind FcγRI and FcγRIIb, and these interactions are can have profound clinical consequences (28, 29). Awareness of the FcγR binding profile of a mAb offers an opportunity to improve upon existing monoclonal therapies, exemplified by obinutuzumab, a de-fucosylated IgG1 bio-similar of rituximab designed to bind FcγRIIIA and enhance ADCC against CD20+ cells in chronic lymphocytic leukemia (30). Human germ-line variants of FcγRs that display altered Fc binding tropism have been identified and are an important focus in the effort to understand responses to mAb therapies that rely on FcγRs for therapeutic function, like cetuximab (31), rituximab (32), trastuzumab (33), and other mAb therapies (34).
We suggest that nivolumab and other IgG4-based mAbs are not exceptions to the rules of FcγR binding and therefore Fc interactions should be considered in pharmacologic models. This is particularly important since there is growing interest in immune checkpoint molecules as diagnostic tools to identify e.g. PD-1/PD-L1+ tumors. Prior efforts to image PD-1 expression using PET radio-ligands have focused on gross tissue distribution and lack the resolution to identify cellular tropisms in vivo. Natarajan et. al. used hamster anti-mouse PD-1 (35), but it is not fully understood how hamster mAbs interact with mouse FcγRs in this context. A secondary aPD-1 PET imaging study reported the use of the RMP1-14 rat IgG2a anti-mouse PD-1 clone to cross-correlate with ex vivo PD-1 staining (36), but drug withdrawal by macrophages could complicate the relationship between PET signal and PD-1 expression. Future pre-clinical diagnostic efforts to image PD-1 expression should consider imaging agents that avoid FcγR interactions; however, antibodies meant to mimic nivolumab and pembrolizumab should accurately represent the Fc status of the human IgG4 antibodies.
Our data also suggest that FcγR-mediated aPD-1 removal does not involve transfer of membrane components, or trogocytosis, which has been described for other mAbs including rituximab (37, 38). However, aPD-1 uptake by macrophages is favored when the mAb is bound to PD-1 on T cells (in contrast, unbound aPD-1 is not taken up by macrophages in culture), which aligns with previous findings that IgGs bind FcγRs more efficiently when they form immune complexes (39, 40). From our understanding of Fc interactions with aPD-1, Fc-engineered IgG variants that abrogate FcγR binding and mAb effector functions (41), or combination with therapies that inhibit FcγR binding in vivo, may enhance the effects of treatment.
The data presented here indicate that myeloid cells are immune components that interface with aPD-1 cancer immunotherapy. Recently identified correlates of aPD-1 response in tumors suggest that alterations in macrophage gene signatures are associated with non-responsiveness to aPD-1 (42). Substantial preclinical studies and recruitment for clinical trials are underway with macrophage targeting therapeutics, such as CSF-1R inhibitors (43). It is conceivable that therapies designed to target tumor macrophages, when in combination with aPD-1, may derive additional benefit by increasing immune checkpoint blockade drug delivery to CD8+ T cells, thereby enhancing activity of immunotherapy.
MATERIALS AND METHODS
Study Design
The objective of this research was to understand the binding tropism of aPD-1 mAb therapy in tumors. Flow cytometry, ex vivo microscopy, and intravital microscopy techniques were used for longitudinal investigation of checkpoint blockade pharmacokinetics. The expectation was that the fluorescently-tagged drug would allow tumor infiltrating lymphocytes to be tracked during pharmacodynamic response, however accumulation of drug in tumor associated macrophages prompted us to hypothesize that the Fc region of these monoclonal antibodies can also affect the drug biodistribution. All in vitro and in vivo studies were performed using C57BL/6J mice, and human primary cells were collected from healthy volunteers for ex vivo studies. Sample sizes were decided using data from preliminary caliper measurements of tumor growth (standard deviation = 25% with a 99% difference between treatment and control groups, α = 0.05, β = 0.2). Data were analyzed by Grubbs’ test for statistical outliers, which were pre-defined using an alpha value of 0.01. Data are representative of at least 3 independent experiments as indicated in the figure legends. Treatment cohorts were assigned to ensure that tumors were size-matched at the start of the intervention. Caliper measurements were acquired by researchers blinded to the intervention and group allocation. Microscopy imaging was performed unblinded using predetermined data collection methods that allowed multiple regions to be studied for each sample, permitting thousands of cells to be analyzed.
Animal Models
Animal research was performed in accordance with the Institutional Animal Care and Use Committees at MGH. DPE-GFP mice (21) were generously provided by Ulrich von Andrian (Dept. of Microbiology and Immunobiology, Harvard Medical School), IFN-γ reporter with endogenous polyA tail (GREAT) mice (22) were kindly provided by Andrew Luster (Division of Rheumatology, Allergy and Immunology, Massachusetts General Hospital). Tumor growth was monitored by caliper measurement and the area (A) of these predominantly two-dimensional tumors was calculated using the formula A = length * width. Tumor implantation was performed by intradermal injection of tumor cells (2 × 106 MC38/MC38-H2B-mApple, 5 × 105 B16-F1-Ova, and 5 × 105 KP1.9). MC38 cells were gifted by Dr. Mark Smyth (QIMR Berghofer, Brisbane, Australia), B16 cells were from ATCC, and KP1.9 cells were a gift from Dr. Alfred Zippelius (University Hospital Basel, Switzerland). Experiments were generally started when tumors became vascularized, which was after 8 days. For aPD-1 and AF647-aPD-1 treatments, mice were given 200 μg i.p. of the 29F.1A12 aPD-1 clone. For in vivo Fc blocking experiments, mice were infused i.p. with 200 μg of monoclonal antibody specific to mouse Fc gamma receptors II and III (clone 2.4G2, Bioxcell) daily for 5 days. Control mice received 200 μg rat IgG2a isotype control (clone 2A3, Bioxcell).
In vivo microscopy
Intravital microscopy was performed in dorsal skin-fold window chambers installed on DPE-GFP or GREAT mice inoculated with MC38-H2B-mApple tumors. Mouse macrophages and/or vasculature were labeled with Pacific Blue ferumoxytol and dextran, respectively. AF647-aPD-1 (200 μg) was delivered i.v. and its tumor distribution was observed using an Olympus FluoView FV1000MPE confocal imaging system (Olympus America), as described previously (44). Pacific Blue, GFP/YFP, mApple, and AF647 were imaged sequentially using 405, 473, 559, and 635 nm lasers and BA430-455, BA490-540, BA575-620, BA575-675 emission filters with DM473, SDM560, and SDM 640 beam splitters, all sourced from Olympus America. Time lapse images were acquired continually over the first hour following AF647-aPD-1 injection, after which the mice were allowed to recover before subsequent imaging.
Statistical Analysis
Data points were compiled in Microsoft Excel and statistical analyses were performed using GraphPad Prism 6. Alpha levels of 0.05 were used to define statistical significance, and error bars represent SEM unless otherwise noted.
Supplementary Material
Summary.
Tumor-associated macrophages limit anti-PD-1 effects by removing the antibody from CD8+ T cells.
Acknowledgments
We thank Drs. Andrew Luster and Uli von Andrian for mice and reagents, Dr. Thorsten Mempel, Camilla Engblom, and Christina Pfirschke for helpful discussions and Greg Wojtkiewicz and Benoit Tricot for technical assistance. Funding: This work was supported in part by the Samana Cay MGH Research Scholar Fund; grants from the NIH (R01AI084880, R01CA164448, R21CA190344, U54-CA151884, 5P50CA086355, DP2AR068272-01 and HL084312), DoD (PC140318); and the David H. Koch–Prostate Cancer Foundation Award in Nanotherapeutics, G.J.F. was supported by P50CA101942. S.P.A., M.C. and M.A.M. were supported by T32 CA79443 and C.S.G. was supported by F31 CA196035.
Footnotes
Author contributions: S.P.A., C.S.G., R.W. and M.J.P. developed the concept; S.P.A, R.W. and M.J.P. developed intravital imaging studies; S.P.A., C.S.G. and M.J.P. designed the experiments; S.P.A, C.S.G., R.K, M.C., K.Y, M.K., M.A.M., and J.C. performed experiments; S.P.A., C.S.G, R.W., M.J.P wrote the paper; all authors analyzed the results and edited the manuscript.
Competing interests: G.J.F. has patents and potential royalties on the PD-1 pathway.
Data and materials availability: All cell lines were obtained through material transfer agreements or are commercially available. Requests for collaboration involving materials used in this research will be fulfilled provided that a written agreement is executed in advance between Massachusetts General Hospital and the requesting parties.
REFERENCES AND NOTES
- 1.Leach DR, Krummel MF, Allison JP. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 2.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 5.Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting - Annual Review of Immunology, 22(1):329-360. Annu Rev Immunol. 2004 doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 6.Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 7.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, Lee JS, Hellmann MD, Hamid O, Goldman JW, Soria JC, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L. KEYNOTE-001 Investigators, Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 10.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 13.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin YJ, Wojtkiewicz G, Iwamoto Y, Mino-Kenudson M, Huynh TG, Hynes RO, Freeman GJ, Kroemer G, Zitvogel L, Weissleder R, Pittet MJ. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity. 2016;44:343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minn AJ, Wherry EJ. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell. 2016;165:272–275. doi: 10.1016/j.cell.2016.03.031. [DOI] [PubMed] [Google Scholar]
- 16.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 19.Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 Regulates PD-1 Expression upon T Cell Activation. The Journal of Immunology. 2008;181:4832–4839. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, Boles KS, Andrews DM, Smyth MJ, Colonna M. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J Exp Med. 2008;205:2965–2973. doi: 10.1084/jem.20081752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, von Andrian UH. Regulatory T Cells Reversibly Suppress Cytotoxic T Cell Function Independent of Effector Differentiation. Immunity. 2006;25:129–141. doi: 10.1016/j.immuni.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 22.Reinhardt RL, Liang HE, Bao K, Price AE, Mohrs M, Kelly BL, Locksley RM. A Novel Model for IFNg-Mediated Autoinflammatory Syndromes. The Journal of Immunology. 2015;194:2358–2368. doi: 10.4049/jimmunol.1401992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater. 2014;13:125–138. doi: 10.1038/nmat3780. [DOI] [PubMed] [Google Scholar]
- 24.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 25.Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell. 2015;28:285–295. doi: 10.1016/j.ccell.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 26.Feige MJ, Nath S, Catharino SR, Weinfurtner D. Structure of the Murine Unglycosylated IgG1 Fc Fragment. J Mol Bio. 2009;391:599–608. doi: 10.1016/j.jmb.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, Garcia C, Wu Y, Kuhne M, Srinivasan M, Singh S, Wong S, Garner N, Leblanc H, Bunch RT, Blanset D, Selby MJ, Korman AJ. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014;2:846–856. doi: 10.1158/2326-6066.CIR-14-0040. [DOI] [PubMed] [Google Scholar]
- 28.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 29.Hussain K, Hargreaves CE, Roghanian A, Oldham RJ, Chan HTC, Mockridge CI, Chowdhury F, Frendeus B, Harper KS, Strefford JC, Cragg MS, Glennie MJ, Williams AP, French RR. Upregulation of Fc RIIb on monocytes is necessary to promote the superagonist activity of TGN1412. Blood. 2015;125:102–110. doi: 10.1182/blood-2014-08-593061. [DOI] [PubMed] [Google Scholar]
- 30.Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, Chagorova T, de la Serna J, Dilhuydy MS, Illmer T, Opat S, Owen CJ, Samoylova O, Kreuzer KA, Stilgenbauer S, Döhner H, Langerak AW, Ritgen M, Kneba M, Asikanius E, Humphrey K, Wenger M, Hallek M. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370:1101–1110. doi: 10.1056/NEJMoa1313984. [DOI] [PubMed] [Google Scholar]
- 31.Sclafani F, Gonzalez de Castro D, Cunningham D, Wilson S Hulkki, Peckitt C, Capdevila J, Glimelius B, Keränen S Roselló, Wotherspoon A, Brown G, Tait D, Begum R, Thomas J, Oates J, Chau I. FcγRIIa and FcγRIIIa polymorphisms and cetuximab benefit in the microscopic disease. Clin Cancer Res. 2014;20:4511–4519. doi: 10.1158/1078-0432.CCR-14-0674. [DOI] [PubMed] [Google Scholar]
- 32.Manches O, Lui G, Chaperot L, Gressin R, Molens JP, Jacob MC, Sotto JJ, Leroux D, Bensa JC, Plumas J. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood. 2003;101:949–954. doi: 10.1182/blood-2002-02-0469. [DOI] [PubMed] [Google Scholar]
- 33.Arnould L, Gelly M, Penault-Llorca F, Benoit L. British Journal of Cancer - Abstract of article: Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? British Journal of Cancer. 2006;94:259–267. doi: 10.1038/sj.bjc.6602930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 35.Natarajan A, Mayer AT, Xu L, Reeves RE, Gano J, Gambhir SS. Novel Radiotracer for ImmunoPET Imaging of PD-1 Checkpoint Expression on Tumor Infiltrating Lymphocytes. Bioconjug Chem. 2015;26:2062–2069. doi: 10.1021/acs.bioconjchem.5b00318. [DOI] [PubMed] [Google Scholar]
- 36.Hettich M, Braun F, Bartholomä MD, Schirmbeck R, Niedermann G. High-Resolution PET Imaging with Therapeutic Antibody-based PD-1/PD-L1 Checkpoint Tracers. Theranostics. 2016;6:1629–1640. doi: 10.7150/thno.15253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beum PV, Kennedy AD, Williams ME, Lindorfer MA, Taylor RP. The shaving reaction: rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia cells by THP-1 monocytes. J Immunol. 2006;176:2600–2609. doi: 10.4049/jimmunol.176.4.2600. [DOI] [PubMed] [Google Scholar]
- 38.Taylor RP, Lindorfer MA. Fcγ-receptor-mediated trogocytosis impacts mAb-based therapies: historical precedence and recent developments. Blood. 2015;125:762–766. doi: 10.1182/blood-2014-10-569244. [DOI] [PubMed] [Google Scholar]
- 39.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M. Specificity and affinity of human Fc receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 40.Lux A, Yu X, Scanlan CN, Nimmerjahn F. Impact of Immune Complex Size and Glycosylation on IgG Binding to Human Fc Rs. The Journal of Immunology. 2013;190:4315–4323. doi: 10.4049/jimmunol.1200501. [DOI] [PubMed] [Google Scholar]
- 41.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29:457–466. doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
- 42.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A, Lo RS. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Macrophage IL-10 Blocks CD8(+) T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell. 2014;26:623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thurber GM, Yang KS, Reiner T, Kohler RH, Sorger P, Mitchison T, Weissleder R. Single-cell and subcellular pharmacokinetic imaging allows insight into drug action in vivo. Nat Commun. 2013;4:1504. doi: 10.1038/ncomms2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guile GR, Rudd PM, Wing DR, Prime SB, Dwek RA. A rapid high-resolution high-performance liquid chromatographic method for separating glycan mixtures and analyzing oligosaccharide profiles. Anal Biochem. 1996;240:210–226. doi: 10.1006/abio.1996.0351. [DOI] [PubMed] [Google Scholar]
- 46.Kurlander RJ, Hall J. Comparison of intravenous gamma globulin and a monoclonal anti-Fc receptor antibody as inhibitors of immune clearance in vivo in mice. J Clin Invest. 1986;77:2010–2018. doi: 10.1172/JCI112530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang KS, Kohler RH, Landon M, Giedt R, Weissleder R. Single cell resolution in vivo imaging of DNA damage following PARP inhibition. Sci Rep. 2015;5:10129. doi: 10.1038/srep10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beum PV, Peek EM, Lindorfer MA, Beurskens FJ, Engelberts PJ, Parren PWHI, van de Winkel JGJ, Taylor RP. Loss of CD20 and bound CD20 antibody from opsonized B cells occurs more rapidly because of trogocytosis mediated by Fc receptor-expressing effector cells than direct internalization by the B cells. The Journal of Immunology. 2011;187:3438–3447. doi: 10.4049/jimmunol.1101189. [DOI] [PubMed] [Google Scholar]
- 49.Alieva M, Ritsma L, Giedt RJ, Weissleder R. Imaging windows for long-term intravital imaging: General overview and technical insights. IntraVital. 2014;3(2) doi: 10.4161/intv.29917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mempel TR, Henrickson SE, von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 51.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23:41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.