

Abstract
Inflammatory critical illness is a syndrome that is characterized by acute inflammation and organ injury, and it is triggered by infections and noninfectious tissue injury, both of which activate innate immune receptors and pathways. Although reports suggest an anti-inflammatory role for the mitogen-activated protein kinase (MAPK) extracellular signal–regulated kinase 5 (ERK5), we previously found that ERK5 mediates proinflammatory responses in primary human cells in response to stimulation of Toll-like receptor 2 (TLR2). We inhibited the kinase activities and reduced the abundances of ERK5 and MEK5, a MAPK kinase directly upstream of ERK5, in primary human vascular endothelial cells and monocytes, and found that ERK5 promoted inflammation induced by a broad range of microbial TLR agonists and by the proinflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor–α (TNF-α). Furthermore, we found that inhibitors of MEK5 or ERK5 reduced the plasma concentrations of proinflammatory cytokines in mice challenged with TLR ligands or heat-killed Staphylococcus aureus, as well as in mice that underwent sterile lung ischemia-reperfusion injury. Finally, we found that inhibition of ERK5 protected endotoxemic mice from death. Together, our studies support a proinflammatory role for ERK5 in primary human endothelial cells and monocytes, and suggest that ERK5 is a potential therapeutic target in diverse disorders that cause inflammatory critical illness.
INTRODUCTION
Acute inflammation and organ injury are caused by diverse conditions, including trauma, hemorrhage, vascular occlusion, transplantation, cardiac arrest, and sepsis. These bodily insults activate the innate immune system, which initiates a well-coordinated series of events that have evolved to contain the microbial threat, eliminate the microbes or damaged tissues, initiate an appropriate adaptive immune response, and instigate tissue repair (1–5). After more extreme injury, mechanisms designed to limit inflammation and to restore immune homeostasis become overwhelmed and dysregulated, leading to uncontrolled systemic inflammation with resultant organ injury, organ failure, and even death (6, 7). Coagulopathy, a disorder characterized by impaired clotting of blood that can cause pathologic microvascular thrombosis, and vascular leak are believed to potentiate inflammatory organ injury through effects on nutrient and oxygen delivery, tissue edema, and through secondary ischemia-reperfusion (IR) injury (2, 8–12). Indeed, multiple organ failure is a leading cause of death of patients hospitalized with sepsis or traumatic injuries (13–16).
Vascular endothelial cells (ECs) and leukocytes mediate critical aspects of the innate immune response to infection and injury (1, 17). Receptors on these cells engage invariant microbial pathogen–associated molecular patterns (PAMPs) and host-derived, danger-associated molecular patterns (DAMPs), and, in conjunction with the complement system, they initiate an acute inflammatory response that is enforced by the immediate release of proinflammatory mediators, such as interleukin-1β (IL-1β) and tumor necrosis factor–α (TNF-α) (18–23). Activated leukocytes and ECs secrete cytokines and chemokines [for example, IL-6, IL-8, CCL2 (also known as MCP-1), and CCL3 (also known as MIP-1α)] and increase the number of adhesion molecules [for example, E-selectin, P-selectin, vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecule 1 (ICAM-1)] at their cell surface to facilitate the localization and subsequent diapedesis of leukocytes, such as neutrophils, to sites of infection and injury (1, 22–25). Furthermore, factors that induce and perpetuate fibrin deposition and thrombosis [for example, plasminogen activator inhibitor–1 (PAI-1)] become increased in abundance, whereas those factors [for example, tissue plasminogen activator (tPA)] that are involved in the breakdown of clots (fibrinolysis) become decreased in abundance, resulting in the initiation and persistence of intravascular thrombosis (22, 23, 26, 27). Increased amounts of PAI-1, which inhibits fibrinolysis, are associated with an increased incidence of organ failure and death in sepsis, pneumonia, and acute lung injury (ALI) (28). Increased PAI-1 activity likely facilitates the persistence of microvascular thrombi and impairs blood flow, which may help to contain invading microbes, as well as promotes vascular permeability and leukocyte trafficking. During acute inflammatory critical illness, excessive inflammation, leukocyte recruitment to organs, vascular leak, and intravascular thrombosis cause organ injury. The failure to shift to a healthy, proresolving phase of inflammation is in part driven by the sustained engagement of innate immune receptors by PAMPs, DAMPs, and host inflammatory mediators (29). Understanding these processes is vitally important in critical care medicine; however, the biochemical origins and components of the initial stages of the innate immune response and their role in the development of acute inflammatory critical illness are still poorly understood.
Central to all innate immune receptor signaling pathways are the transcription factor nuclear factor κB (NF-κB) and the intracellular mitogen-activated protein kinases (MAPKs) (30). NF-κB promotes the expression of genes that encode inflammatory cytokines, whereas the MAPKs primarily act by modulating gene and protein expression and stability and by regulating the integrity of protein complexes within the cell (31, 32). The classical MAPKs p38, c-Jun N-terminal kinase (JNK), extracellular signal–regulated kinase 1 (ERK1), and ERK2 are the most thoroughly studied; however, several other family members exist, including ERK5 [also known as big MAPK (BMK1) or MAPK7], ERK3/4, ERK7/8, and Nemo-like kinase (NLK) (31, 33). The classical MAPKs have been considered as therapeutic targets to treat inflammatory disorders for some time, but several clinical trials, particularly those that used p38α inhibitors, were discontinued as a result of poor safety profiles or low long-term efficacy (30, 34). The other understudied MAPK family members, such as ERK5, may be of interest as therapeutic targets in acute inflammatory critical illness because of their distinct structures and more restricted roles in cells, which may limit the occurrence of potential side effects. We reported that ERK5 mediates the Toll-like receptor 2 (TLR2)–dependent activation of inflammatory responses in human monocytes and ECs, and that it promotes the TLR2-dependent activity of PAI-1 in ECs, indicating that other understudied MAPK family members also function in innate immune signaling pathways (23).
ERK5 and its upstream activator MAP/ERK kinase 5 (MEK5) were independently described in 1995 (35–37). Compared to other MAPK family members, ERK5 is substantially larger and has a distinct C-terminal domain, which contains a proline-rich region, a nuclear localization signal (NLS), and a transcriptional activation domain (TAD) that is regulated by autophosphorylation (fig. S1) (38–42). In resting cells, ERK5 is mainly located in the cytoplasm, but upon phosphorylation of ERK5, the NLS is exposed, and ERK5 translocates to the nucleus where it stimulates the activity of several transcription factors (43–54). In addition, ERK5 has several cytoplasmic substrates and binding partners, such as other kinases, adaptors, scaffolds, and junctional proteins (41, 42, 55–60). ERK5 plays an essential role in embryonic development and invascular integrity, as highlighted in studies of global and EC-specific ERK5 knockout mice (61–65). However, the ablation of ERK5 in other cells, including T cells and neuronal cells, is not lethal, although neuronal deletion affects aspects of neurogenesis and impairs pheromone-mediated behaviors (66–71). ERK5 and MEK5 are also implicated in solid tumors and hematologic malignancies, and inhibitors of ERK5 and MEK5 are being explored as potential cancer therapies (1, 72–77). Specifically, overexpression and constitutive activation of ERK5 and MEK5 have been noted in different tumor cells, and they are reported to correlate with more aggressive cancer behaviors, development of metastases, chemoresistance, and tumor-associated angiogenesis (77–88). In contrast, the role of ERK5 in inflammation is more controversial. Some studies suggest that ERK5 is anti-inflammatory (44, 45, 47, 89–92), whereas other studies, including our own, have reported a proinflammatory role for ERK5 in various cell types (23, 93–99).
Here, we expand on our previously reported observation that ERK5 contributes to proinflammatory signaling downstream of TLR2 (23), testing the hypothesis that ERK5 mediates acute inflammation induced by microbial ligands as well as by endogenous host factors. In experiments with cultured primary human ECs and monocytes, we showed that ERK5 promoted inflammation downstream of diverse inflammatory mediators in vitro, including fibroblast-stimulating ligand 1 (FSL-1) (a ligand of TLR2 and TLR6), lipopolysaccharide (LPS; a TLR4 agonist), IL-1β, and TNF-α. In addition, we found that inhibition of ERK5 activity markedly improved the survival rates of endotoxemic mice and reduced the amounts of circulating inflammatory mediators in mice treated with the TLR1 and TLR2 agonist Pam3Cys, LPS, or heat-killed Staphylococcus aureus (HKSA), as well as in mice that underwent sterile lung IR injury. Together, our studies support a proinflammatory role for ERK5 in primary human ECs and monocytes, and suggest that ERK5 is a potential therapeutic target in diverse disorders that cause inflammatory critical illness.
RESULTS
ERK5 activity promotes the secretion of proinflammatory cytokines by ECs
We previously showed that ERK5 mediates TLR2 signaling in human ECs, human peripheral blood mononuclear cells (PBMCs), and the monocytic cell line THP-1, and that the bacterial lipopeptide TLR1/2 agonist Pam3Cys stimulates the activation of endogenous ERK5 in human umbilical vein endothelial cells (HUVECs) (23). To determine whether ERK5 also promoted EC activation downstream of other exogenous (for example, FSL-1 and LPS) and endogenous (for example, TNF-α and IL-1β) inflammatory mediators, we first analyzed the effects of the ERK5 inhibitors XMD8-92 and XMD17-109 (also known as ERK5-IN-1) on the FSL-1–, LPS-, TNF-α–, and IL-1β–induced production of IL-6 and IL-8 by human lung microvascular endothelial cells (HMVEC-lung cells) and HUVECs (75, 100, 101). We found that both ERK5 inhibitors inhibited the secretion of both IL-6 and IL-8 from HMVEC-lung cells treated with any of the inflammatory agonists (Fig. 1, A to H). We also performed a detailed analysis of the absolute half-maximal inhibitory concentration (IC50) values for the inhibitory effects of XMD8-92 and XMD17-109 on IL-6 and IL-8 secretion by both HUVECs and HMVEC-lung cells (Tables 1 and 2). These results suggest that XMD17-109 has a modestly higher efficacy than XMD8-92 in both HUVECs and HMVEC-lung cells, and that both inhibitors had a higher efficacy in HMVEC-lung cells than in HUVECs. Additionally, the ERK5 inhibitors more potently inhibited the secretion of IL-6 than that of IL-8. None of the inhibitors substantially affected cell viability at the concentrations used in our assays as measured by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay (fig. S2). Furthermore, we found that initiating treatment of HMVEC-lung cells with XMD8-92 anytime from 2 hours before through to 2 hours after the addition of LPS reduced the amounts of IL-6 and IL-8 secreted by the cells (fig. S3, A and B). These data suggests that ERK5 promotes the secretion of IL-6 and IL-8 by HUVECs and HMVEC-lung cells in response to both endogenous and exogenous inflammatory mediators.
Fig. 1. The kinase activities of ERK5 and MEK5 promote the inflammatory activation of ECs in vitro.
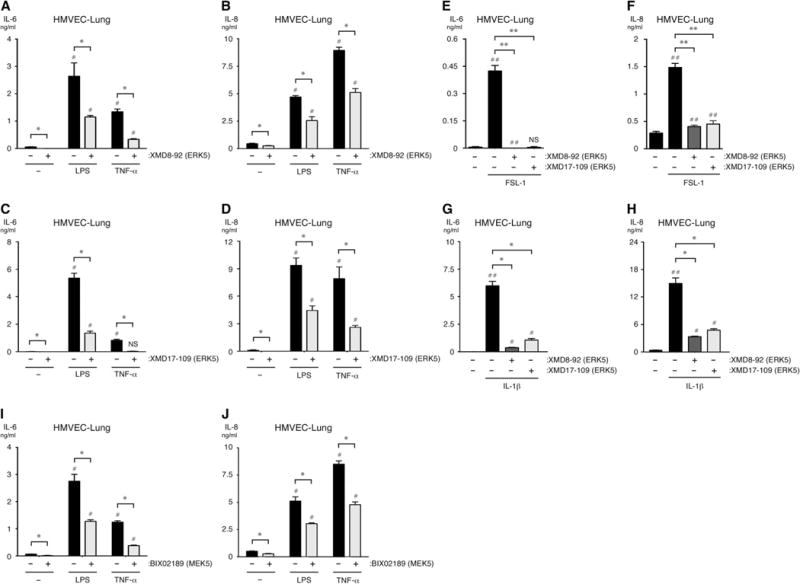
(A to J) HMVEC-lung cells were pretreated for 1 hour with vehicle [dimethyl sulfoxide (DMSO)], 5 μM XMD8-92, 1 μM XMD17-109, or 10 μM BIX02189 before being treated for an additional 6 hours with vehicle (0.9% saline), LPS (10 μg/ml), TNF-α (100 ng/ml), FSL-1 (10 μg/ml), or IL-1β (0.1 ng/ml) while in the continuous presence of DMSO or inhibitor. The concentrations of IL-6 and IL-8 that were secreted by the cells into the culture medium were determined by enzyme-linked immunosorbent assay (ELISA). NS, not significant. *P < 0.05 and **P < 0.01 when comparing between cells treated with inflammatory stimulus in the presence or absence of inhibitor; #P < 0.05 and ##P < 0.01 when comparing between cells treated with vehicle alone and cells treated with inflammatory stimulus in the presence or absence of inhibitor. Data are means ± SD of four sample wells per group and are representative of three independent experiments.
Table 1.
Micromolar IC50 values for XMD8-92 in different ECs.
| Agonist | HUVECs
|
HMVEC-lung cells
|
||
|---|---|---|---|---|
| IL-6 | IL-8 | IL-6 | IL-8 | |
| Pam3Cys | ||||
| 10 μg/ml | 1.17 | 0.91 | 0.70 | 0.93 |
| FSL-1 | ||||
| 10 μg/ml | 3.18 | 4.68 | 1.70 | 1.55 |
| LPS | ||||
| 10 μg/ml | 1.69 | 3.67 | 2.77 | 4.22 |
| 1 μg/ml | 2.95 | 2.91 | 1.92 | 2.47 |
| TNF-α | ||||
| 10 ng/ml | 2.01 | 4.33 | 0.93 | 2.96 |
| IL-1β | ||||
| 0.1 ng/ml | 1.60 | 3.24 | 0.95 | 0.98 |
| Average IC50 | 2.10 | 3.29 | 1.50 | 2.19 |
Table 2.
Micromolar IC50 values for XMD17-109 in different ECs. n.d., no data.
| Agonist | HUVECs
|
HMVEC-lung cells
|
||
|---|---|---|---|---|
| IL-6 | IL-8 | IL-6 | IL-8 | |
| Pam3Cys | ||||
| 10 μg/ml | 0.38 | 0.39 | 0.39 | 0.45 |
| FSL-1 | ||||
| 10 μg/ml | 2.02 | 1.83 | 0.82 | 0.79 |
| LPS | ||||
| 10 μg/ml | 0.80 | 0.74 | 0.66 | 1.17 |
| 1 μg/ml | 1.37 | 1.88 | 0.68 | 0.96 |
| TNF-α | ||||
| 10 ng/ml | 0.61 | 2.63 | 0.43 | 0.59 |
| IL-1β | ||||
| 10 ng/ml | 3.46 | 3.68 | n.d. | n.d. |
| 1 ng/ml | 3.26 | 1.98 | 0.58 | 0.67 |
| 0.1 ng/ml | 0.58 | 0.63 | 0.41 | 0.48 |
| Average IC50 | 1.56 | 1.72 | 0.57 | 0.73 |
MEK5 activity in ECs promotes the secretion of proinflammatory cytokines
The MAPK kinase (MAPKK) MEK5 binds to ERK5 and phosphorylates its activation loop to induce ERK5 activity (fig. S1) (102). Therefore, we tested whether the MEK5 inhibitor BIX02189 could reduce the LPS- and TNF-α–induced secretion of IL-6 and IL-8 by HMVEC-lung cells. Indeed, we found that BIX02189 substantially reduced the amounts of IL-6 and IL-8 secreted by ECs (Fig. 1, I and J). As before, we found that BIX02189 had no substantial effect on cell viability at the concentrations used in our assays (fig. S2C). This result suggests that the MEK5-ERK5 signaling axis promotes an inflammatory response in human ECs.
ERK5 and MEK5 promote the secretion of proinflammatory cytokines by ECs
To further confirm the roles of MEK5 and ERK5 in the inflammatory activation of HMVEC-lung cells, we used RNA interference (RNAi)–mediated protein knockdown to reduce the abundance of either ERK5 or MEK5 in HMVEC-lung cells. The short inhibitory RNAs (siRNAs) specific for MAPK7 (which encodes ERK5) and MAP2K5 (which encodes MEK5) substantially reduced the abundances of ERK5 and MEK5, respectively, but did not affect the abundances of ERK1 and ERK2 or of MEK1 and MEK2, which have kinase domain sequences most similar to those of ERK5 and MEK5, respectively (Fig. 2, A to D) (36, 37). Consistent with the ERK5 inhibitor studies, the specific knockdown of ERK5 reduced the amounts of both IL-6 and IL-8 that were secreted by HMVEC-lung cells treated with either LPS or TNF-α (Fig. 2, E and F), again suggesting a role for ERK5 in inflammatory pathways in ECs. Knockdown of MEK5, however, reduced the amount of IL-8, but not IL-6, that was secreted by HMVEC-lung cells in response to either LPS or TNF-α (Fig. 2, G and H). It is possible that whereas the incomplete knockdown of MEK5 was sufficient to substantially inhibit IL-8 secretion, the same was not true for IL-6 secretion. Alternatively, an off-target effect of the MAP2K5-specific siRNA may have compensated for the effect of MEK5 knockdown on IL-6 secretion. Indeed, we performed an extensive BLAST search to find potential off-target genes for the siRNAs contained in the MAPK7 siRNAs that we used. We found that three of the four siRNAs had 13 of their 19 nucleotides overlap with sequences in MAP2K4 (the gene that encodes MEK4), which is an upstream MAPKK for p38, JNK1, and JNK2. Thus, the off-target knockdown of this kinase could potentially result in a compromised inflammatory response (103). However, we did not observe a substantial effect on MEK4 protein abundance in HMVEC-lung cells transfected with MAPK7-specific siRNA, suggesting that knockdown of MEK4 was not likely responsible for the observed outcomes (Fig. 2, A and B). Together, the results of our RNAi-mediated knockdown studies indicate that the MEK5-ERK5 axis in human ECs promotes the secretion of proinflammatory cytokines in response to TLR4 and TNF receptor (TNFR) signaling.
Fig. 2. ERK5 and MEK5 proteins promote the inflammatory activation of ECs in vitro.
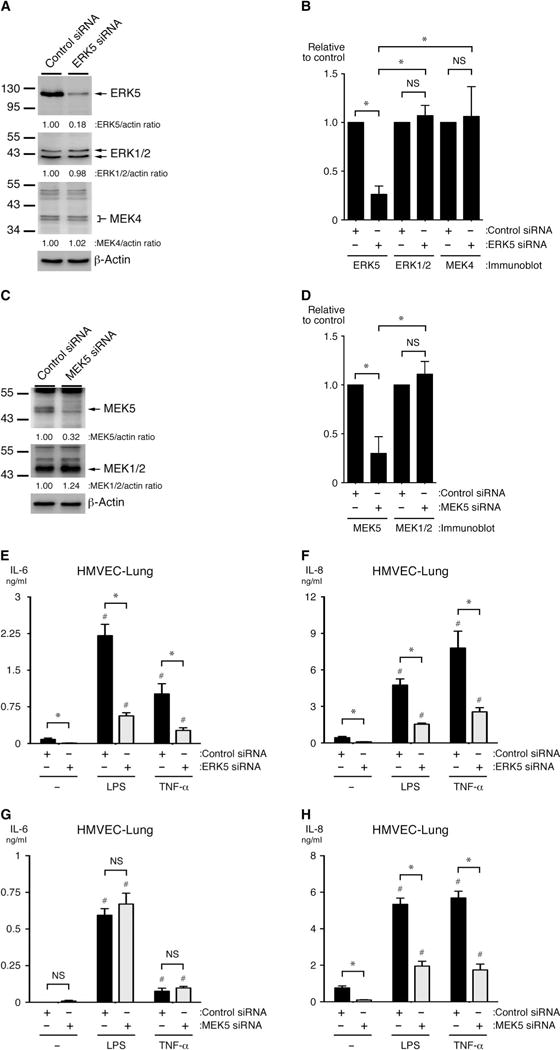
(A to D) HMVEC-lung cells were transfected with (A and B) MAPK7-specific (to knockdown ERK5), (C and D) MAP2K5-specific (to knockdown MEK5), or (A to D) control siRNAs. Seventy-two hours later, cell lysates were analyzed by Western blotting with antibodies specific for the indicated proteins. Western blots were subjected to densitometric analysis. Bar graphs show the ratio of the abundances of (B) ERK5 and (D) MEK5 normalized to that of actin and are expressed relative to those in untreated control cells. *P < 0.05 when comparing relative densities between cells transfected with kinase-specific siRNA and those transfected with control siRNA. Data in (B) and (D) are means ± SD of the specific protein densitometry values from four independent Western blotting experiments. (E to H) HMVEC-lung cells transfected with MAPK7-specific, MAP2K5-specific, or control siRNAs were subsequently treated with vehicle (0.9% saline), LPS (10 μg/ml), or TNF-α (100 ng/ml) for 6 hours. The concentrations of IL-6 and IL-8 secreted by the cells into the culture medium were then determined by ELISA. *P < 0.05 when comparing control siRNA–transfected cells treated with inflammatory stimulus with specific siRNA–transfected cells treated with inflammatory stimulus; #P < 0.05 when comparing cells treated with vehicle alone with siRNA-transfected cells treated with inflammatory stimulus. Data in (E) to (H) are means ± SD of four sample wells per group and are representative of three independent experiments.
ERK5 promotes the adhesion of neutrophils to activated HMVEC-lung cells
To analyze the contribution of ERK5 in ECs to leukocyte adhesion, we used a neutrophil adhesion assay with primary human neutrophils and HMVEC-lung cells (104). After a 3-hour treatment of monolayers of HMVEC-lung cells with FSL-1, LPS, or TNF-α in the presence of either of the ERK5 inhibitors XMD8-92 or XMD17-109, there was a marked decrease in the numbers of neutrophils that adhered to the activated HMVEC-lung cell monolayers compared to those that adhered to monolayers of control cells that were treated with proinflammatory mediators in the absence of ERK5 inhibitors (Fig. 3, A to C). Similar results were obtained from experiments with the MEK5 inhibitor BIX02189, although this inhibitor had a statistically significant effect on adhesion only in experiments in which the monolayers were treated with LPS or TNF-α (fig. S4, A and B). These observations are consistent with our previous study that showed that knockdown of ERK5 in ECs reduces the adherence of neutrophils to HMVEC-lung cells activated with Pam3Cys (23). These data suggest that the ERK5 signaling pathway is necessary for maximal neutrophil adhesion to activated ECs.
Fig. 3. ERK5 promotes the binding of neutrophils to activated ECs.
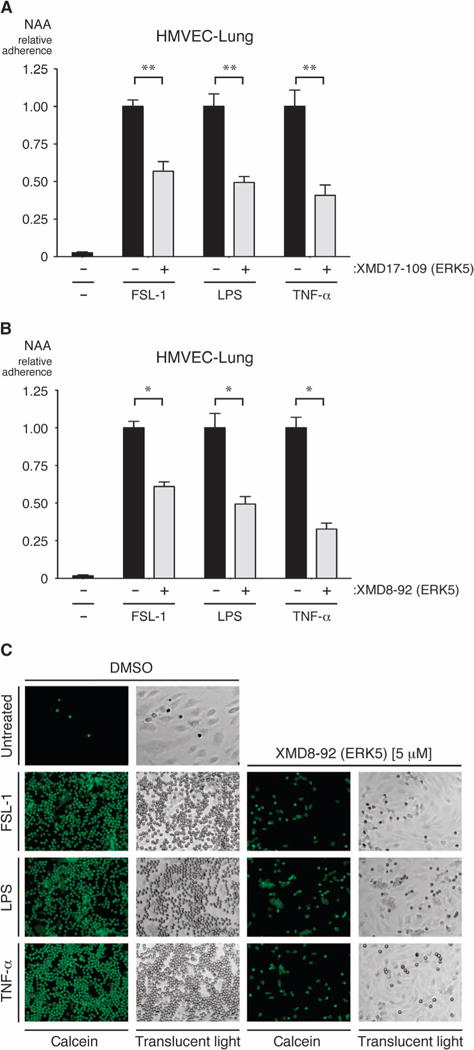
(A and B) Confluent monolayers of HMVEC-lung cells grown in 48-well tissue culture plates were pretreated for 1 hour with vehicle (DMSO), 1 μM XMD17-109 (A), or 5 μM XMD8-92 (B) before being treated with vehicle (0.9% saline), FSL-1 (10 μg/ml), LPS (10 μg/ml), or TNF-α (100 ng/ml) for an additional 3 hours while in the continuous presence of DMSO or inhibitor. Calcein AM–labeled neutrophils were added at 3 × 105 to 6 × 105 cells per well and allowed to adhere for 20 min at 37°C before being washed with phosphate-buffered saline (PBS) to remove nonadherent cells. Pre-and postwashing fluorescence was read in a fluorescent plate reader. The relative numbers of the remaining adherent neutrophils were then calculated. *P < 0.05 and **P < 0.01 when comparing between cells treated with inflammatory stimulus in the absence or presence of inhibitor. Data are means ± SD of four (A) or six (B) sample wells per group and are representative of two independent experiments. NAA, neutrophil adhesion assay. (C) Representative images of calcein-labeled neutrophils bound to HMVEC-lung cells in the presence and absence of XMD8-92. Fluorescence images taken with a fluorescein filter set are shown in the left columns of each group, whereas translucent light microscopy images of the same field of reference are shown in the right-hand columns (under ×10 magnification). Images are representative of two independent experiments.
ERK5 promotes the secretion of proinflammatory cytokines from PBMCs and purified monocytes
We previously found that ERK5 is a critical intermediary in the inflammatory activation of PBMCs in response to the stimulation of TLR1 and TLR2 (23). Here, we assessed the role of ERK5 in inflammatory signaling in human PBMCs and purified monocytes in response to stimulation with FSL-1 or LPS. We found that the amounts of IL-6 and IL-8 that were secreted by either cell population were reduced in the presence of the ERK5 inhibitor XMD8-92 or XMD17-109 (Fig. 4, A to H). We also found that the ERK5 inhibitors reduced the amounts of TNF-α secreted by PBMCs and monocytes in response to LPS, but not FSL-1 (Fig. 4, I to L); however, FSL-1 stimulated the secretion of a negligible amount of TNF-α from both PBMCs and monocytes. Note that because monocytes constitute about 10 to 30% of human PBMCs, these results also suggest that most of the IL-6 and IL-8 secreted by LPS-treated PBMCs is derived from monocytes (Fig. 4, compare C with G and D with H) (105). Together, these data suggest that the MEK5-ERK5 axis promotes the TLR2- and TLR4-mediated secretion of cytokines by PBMCs and monocytes in addition to ECs.
Fig. 4. ERK5 promotes the inflammatory activation of human PBMCs and monocytes in vitro.
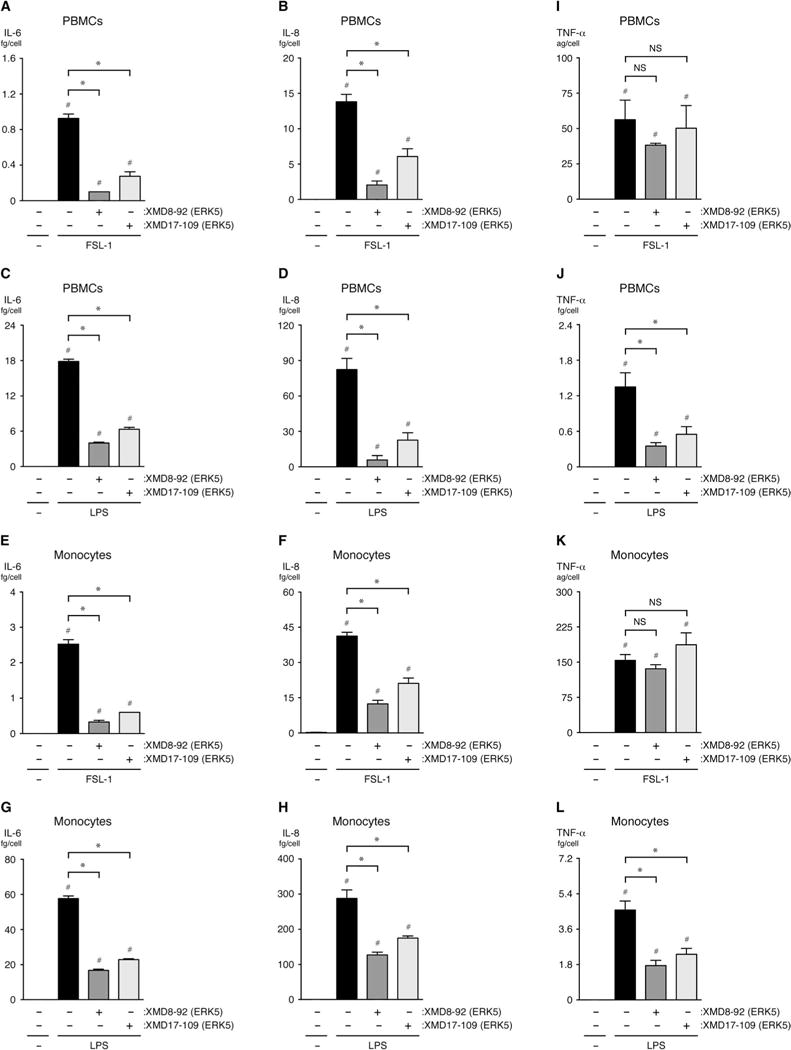
(A to L) PBMCs or monocytes (as indicated) were pretreated for 1 hour with vehicle (DMSO), 5 μM XMD8-92, or 1 μM XMD17-109 before being treated with vehicle (0.9% saline), FSL-1 (10 μg/ml), or LPS (10 μg/ml) for an additional 6 hours while in the continuous presence of DMSO or inhibitor. The concentrations of IL-6, IL-8, and TNF-α secreted by the cells into the culture medium were determined by ELISA. *P < 0.05 when comparing between cells treated with inflammatory stimulus in the presence or absence of inhibitor; #P < 0.05 when comparing between cells treated with vehicle alone and cells treated with inflammatory stimulus in the presence or absence of inhibitor. Data are means ± SD of four sample wells per group and are representative of three independent experiments. ag, attogram; fg, femtogram.
Inhibitors of ERK5 and MEK5 reduce the systemic concentrations of proinflammatory cytokines and PAI-1 activity in mice treated with LPS or Pam3Cys
To investigate the contribution of ERK5 to systemic inflammation and coagulopathy in vivo, we pretreated mice with the ERK5 inhibitor XMD8-92 and then challenged them intravenously with Pam3Cys or LPS. The plasma concentrations of IL-6, CCL2, and CCL3 were determined to assess the role of ERK5 in systemic inflammation, whereas PAI-1 activity was chosen as a measure of coagulation because increased amounts of PAI-1, which inhibits fibrinolysis, are associated with an increased incidence of organ failure and death in sepsis, pneumonia, and ALI (28). Mice were also pretreated with the ERK5 inhibitor XMD17-109 and then challenged intravenously with LPS for comparison. The plasma concentrations of IL-6, CCL2, and CCL3 and the activity of PAI-1 2 hours after challenge with either Pam3Cys or LPS were all substantially reduced in mice treated with ERK5 inhibitors compared to those of control mice treated with vehicle (Fig. 5, A to L). Twenty-four hours after challenge with LPS, mice treated with the ERK5 inhibitor XMD8-92 had substantially decreased plasma PAI-1 activity and reduced concentrations of CCL2, but not IL-6 or CCL3 (fig. S5, A to D). Note that the plasma concentrations of IL-6, CCL2, and CCL3 and the activity of PAI-1 in vehicle- and inhibitor-treated mice were much lower 24 hours after challenge with LPS than they were at 2 hours after challenge.
Fig. 5. ERK5 promotes the secretion of inflammatory mediators and enhances PAI-1 activity after the systemic challenge of mice with Pam3Cys or LPS.
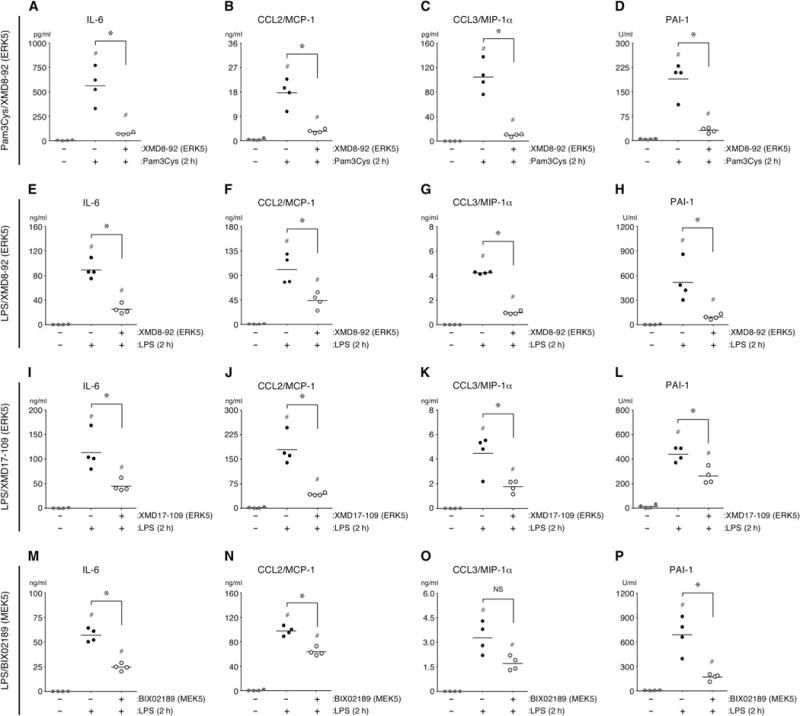
(A to P) Wild-type mice were treated intraperitoneally with XMD8-92 (50 mg/kg), XMD17-109 (50 mg/kg), BIX02189 (25 mg/kg), or vehicle [30% (2-hydroxypropyl)-β-cyclodextrin with or without 5% DMSO] 30 min before they were injected intravenously with Pam3Cys (2.5 mg/kg), LPS (10 mg/kg), or vehicle (0.9% saline). The plasma concentrations of IL-6, CCL2, and CCL3 and the activity of PAI-1 were quantified 2 hours after challenge. *P < 0.05 when comparing Pam3Cys- or LPS-treated mice in the presence or absence of inhibitor; #P < 0.05 when comparing untreated control mice with mice treated with Pam3Cys or LPS in the presence or absence of inhibitor. Data are means ± SD of four mice per group and are representative of two independent experiments.
We next assessed the contribution of MEK5 to LPS-induced inflammation and coagulopathy by pretreating mice with the MEK5 inhibitor BIX02189 before challenging them intravenously with LPS. Two hours after challenge, the activity of PAI-1 and the concentrations of IL-6 and CCL2, but not CCL3, were substantially reduced in BIX02189-treated mice compared to those in vehicle-treated mice (Fig. 5, M to P). These data support a role for the MEK5-ERK5 signaling axis in the induction of acute inflammation and coagulopathy in mice challenged systemically with bacterial PAMPs. To further test this hypothesis, we pretreated mice with the ERK5 inhibitor XMD8-92 and then challenged them intravenously with HKSA, which contain multiple PAMPs. We observed that, as compared with vehicle-treated mice, those mice treated with the ERK5 inhibitor had substantially reduced plasma PAI-1 activity and decreased concentrations of IL-6 and CCL2, but not CCL3, 2 hours after challenge with HKSA (Fig. 6, A to D).
Fig. 6. ERK5 promotes the secretion of inflammatory mediators and enhances PAI-1 activity after the systemic challenge of mice with HKSA.
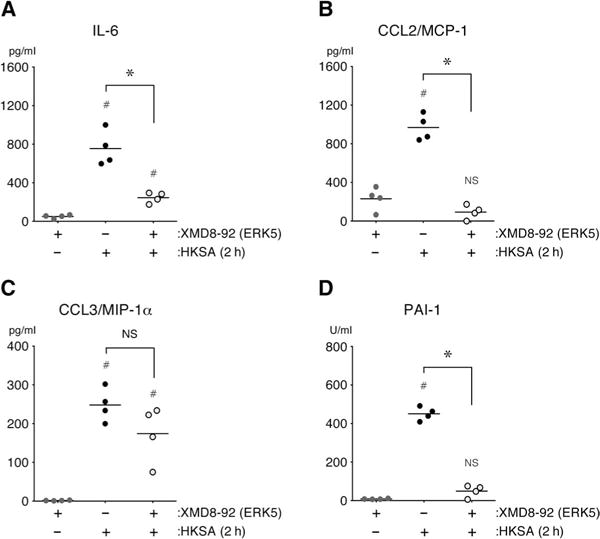
(A to D) Wild-type mice were treated intraperitoneally with XMD8-92 (50 mg/kg) or vehicle [30% (2-hydroxypropyl)-β-cyclodextrin] 30 min before being injected intravenously with HKSA (2.5 × 1010 bacteria/kg) or vehicle (0.9% saline). The plasma concentrations of IL-6, CCL2, and CCL3 and the activity of PAI-1 were quantified 2 hours after the mice were challenged. *P < 0.05 when comparing HKSA-treated mice in the presence or absence of inhibitor; #P < 0.05 when comparing untreated control mice with mice treated with HKSA in the presence or absence of inhibitor. Data are means ± SD of four mice per group and are representative of two independent experiments.
Inhibition of ERK5 activity protects mice from LPS-induced mortality
To test the hypothesis that inhibition of ERK5 activity may improve survival in inflammatory critical illness, we assessed the effects of ERK5 inhibition in an LPS-induced (endotoxemic) mortality model. Mice were treated with the ERK5 inhibitor XMD8-92 30 min before or 1 hour after being challenged with an intraperitoneal injection of LPS [lethal dose (LD)30 to LD50]. In both instances, the inhibition of ERK5 activity conferred the mice with marked protection from LPS-induced lethality, reducing the mortality of mice at 72 hours from 50 to 0% in mice pretreated with the inhibitor and from 29 to 0% in mice treated with the inhibitor after exposure to LPS (Fig. 7, A and B).
Fig. 7. Inhibition of ERK5 protects mice from endotoxemic mortality.
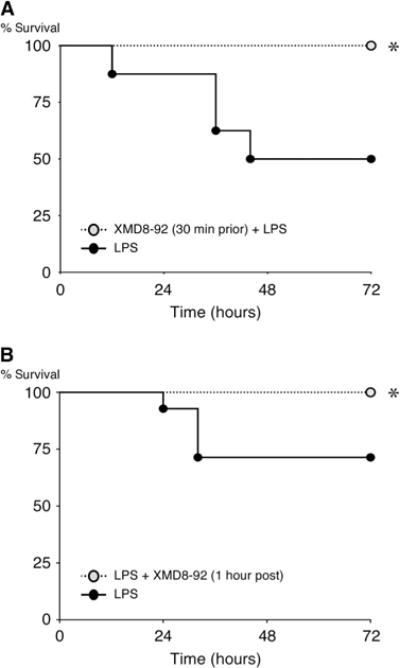
(A and B) Wild-type mice were treated intraperitoneally with XMD8-92 (50 mg/kg) or vehicle [30% (2-hydroxypropyl)-β-cyclodextrin] 30 min before (A) or 1 hour after (B) being injected intraperitoneally with LPS [15 mg/kg (A) or 12.5 mg/kg (B)] or vehicle (0.9% saline). The survival of the mice was monitored for up to 72 hours. Data are from 8 (A) or 14 (B) mice per group from a single experiment and are representative of two independent experiments. The concentration of LPS used was chosen to achieve a 30 to 50% mortality rate (LD30 to LD50) in control mice after 72 hours. *P < 0.05 when comparing control mice with inhibitor-treated mice. Mortality data were analyzed with Kaplan-Meier curves.
ERK5 promotes the production of proinflammatory cytokines during lung IR injury in vivo
Although the underlying stimuli of acute inflammatory critical illness differ considerably, they all lead to variable degrees of IR injury, which results from reduced blood flow as a result of microvascular thrombosis and decreased arterial blood pressure (12, 106). Reduced perfusion of tissues leads to cellular stress and even necrosis, with the release of DAMPs that exacerbate local and systemic inflammation and promote organ injury and failure (5, 107). In experiments with a mouse model of ventilated lung IR injury, we found that IR injury increases the abundance of proinflammatory cytokines and chemokines in the bloodstream and in lung tissue compared to that in mice undergoing a sham surgical procedure (108). To test the hypothesis that ERK5 mediates inflammation after IR injury in the lung, we pretreated mice with the ERK5 inhibitor XMD8-92 just before we temporarily occluded the left pulmonary artery for 30 min, and then we quantified the plasma concentrations of IL-6 and CCL2 1 hour after re-perfusion. Similar to our results from experiments with LPS and Pam3Cys, the plasma concentrations of IL-6 and CCL2 in response to IR injury were substantially less in mice treated with XMD8-92 than they were in vehicle-treated mice (Fig. 8, A and B). These results suggest that in the lung subjected to IR injury, and potentially in other areas of reduced blood flow, ERK5 promotes the production of proinflammatory cytokines and chemokines.
Fig. 8. ERK5 promotes cytokine secretion in a mouse model of lung IR injury.
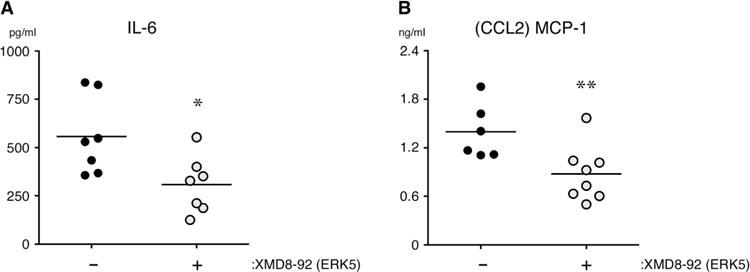
(A and B) Wild-type mice were injected intraperitoneally with XMD8-92 (50 mg/kg) or vehicle [30% (2-hydroxypropyl)-β-cyclodextrin] just before being subjected to ventilated lung IR injury as described in Materials and Methods. The concentrations of IL-6 and CCL2 in the plasma of the indicted mice were quantified 1 hour after reperfusion (ischemia time, 30 min). Each point represents plasma from a single mouse. Data are from 6 to 10 mice per group from a single experiment and are representative of two independent experiments. *P < 0.05 and **P < 0.01 when comparing vehicle-treated mice with inhibitor-treated mice.
DISCUSSION
Here, we expanded on our initial observation that ERK5 promotes inflammation downstream of TLR2 in both ECs and monocytes, and we provided data to suggest that ERK5 promotes acute inflammatory events in vitro and in vivo (23). We found that ERK5 promoted the release of proinflammatory cytokines from primary human monocytes and ECs, as well as the adherence of neutrophils to ECs and the release of PAI-1 from ECs in response to agonists of TLR2 and TLR4, as well as to IL-1β and TNF-α. Our data support roles for ERK5 and MEK5 activity in mediating inflammation and coagulopathy induced by systemic bacterial infection, as evidenced by the reduced concentrations of inflammatory mediators and the decrease in PAI-1 activity in ERK5 inhibitor–treated mice that were subsequently challenged with Pam3Cys, LPS, or HKSA. Indeed, we showed that abating ERK5 activity improved the survival of endotoxemic mice. Supporting a role for ERK5 in inflammation induced by noninfectious injuries is our finding that an ERK5 inhibitor reduced the plasma concentrations of inflammatory mediators in a mouse model of lung IR injury. Together, our results suggest that ERK5 promotes inflammatory pathways downstream of innate immune receptors and, critically, that the MEK5-ERK5 axis could potentially be exploited therapeutically to treat various infectious and noninfectious disorders that cause critical inflammatory illness.
In contrast with our observations, several studies support an anti-inflammatory role for ERK5 (44, 45, 47, 91). There are several differences between our studies and those previously reported. First, we used two different specific inhibitors of ERK5 activity, XMD8-92 and XMD17-109, to assess the role of ERK5 in acute inflammation (75, 100, 101). XMD17-109 is a highly specific inhibitor of ERK5, and the crystal structure of XMD17-109 bound to the kinase domain of ERK5 has been solved (101). Second, we found that the MEK5 inhibitor BIX02189 reduced inflammatory responses in vitro and in endotoxemic mice; however, in some instances, it was less effective than were the ERK5 inhibitors. BIX02189 is a more promiscuous inhibitor than either of the ERK5 inhibitors used in our studies. For example, BIX02189 also potently inhibits the kinases Lck, Src, colony-stimulating factor 1 (CSF-1) receptor, and ribosomal S6 kinase 2/4 (RSK2/4). This may account for its reduced efficacy in limiting inflammatory activation in some of our in vitro and in vivo assays (72). Third, we verified the results from our in vitro experiments using inhibitors with RNAi-mediated knockdown of both ERK5 and MEK5 proteins in primary human ECs, and we analyzed the function of endogenous ERK5 and MEK5 in primary human cells in vitro, bypassing the need for transfection studies. Last, we found that inhibition of ERK5 reduced systemic inflammation in multiple in vivo models of acute inflammation, including toxicity models of bacterial sepsis and a model of sterile acute inflammation. We recognize that competitive inhibitors of the adenosine triphosphate (ATP)–binding sites of kinase domains will undoubtedly have off-target effects, and we have tried to extensively address this possibility in our in vitro studies by using inhibitors of not only ERK5 but also MEK5 and by complementing these studies with siRNA-mediated knockdown of endogenous ERK5 and MEK5 in ECs. We observed highly consistent effects on inflammatory outcomes with each of these methods. Although it is possible that the inhibition of another protein in addition to ERK5 or MEK5 may have contributed to the outcomes that we observed, the sum total of our in vitro data suggests that the effects that we observed in vivo were a result of inhibition of the MEK5-ERK5 signaling pathway. Together, our results suggest that ERK5 promotes acute inflammation in vitro and in vivo.
We speculate that blood flow conditions may determine whether ERK5 serves a proinflammatory or anti-inflammatory role. Under normal physiologic conditions, blood flow is laminar, whereas sepsis and tissue injury or distress can cause decreased and nonlaminar blood flow to occur. Perhaps under laminar blood flow, ERK5 activation protects against vascular inflammation, whereas under nonlaminar blood flow, ERK5 promotes inflammation. Our hypothesis that vascular flow conditions determine whether ERK5 serves a proinflammatory or anti-inflammatory role is supported by our finding that the induction of inflammation by temporarily occluding blood flow to the mouse lung is dependent on ERK5 activity. Further support for this differential role of ERK5 comes from a report that shows that modification of ERK5 with the small ubiquitin-like modifier (SUMO) promotes inflammatory pathways under conditions of disrupted laminar flow, which suggests that posttranslational modifications of ERK5 may switch its function from a protective to a proinflammatory one (90). Therefore, a potential paradigm for the role of ERK5 in inflammation emerges: under normal flow conditions in the vasculature, ERK5 may protect against the inflammatory activation of ECs and therefore maintain endothelial homeostasis and vascular integrity; however, during an infectious or traumatic insult, when blood flow is compromised, ERK5 may promote the inflammatory activation of ECs.
The signaling pathways that lead to the activation of ERK5 downstream of the innate immune receptors are not known. With an appropriate stimulus, MAPKs are activated through a highly conserved sequence of phosphorylation events; that is, MAPKK kinases (MAPKKKs) activate MAPKKs, which in turn activate a specific MAPK (30, 33). The MAPKKKs MAPK/ERK kinase kinase 2 (MEKK2) and MEKK3 are thought to phosphorylate, and thus activate, MEK5, the only kinase known to activate ERK5 (35, 38, 39, 109). Tumor progression locus 2 (Tpl2), another MAPKKK, also activates the MEK5-ERK5 pathway (110). Notably, Tpl2 can promote inflammation by enhancing the formation of active NF-κB heterodimers (111). There is currently no consensus on whether MEKK2, MEKK3, or Tpl2 is involved in the activation of ERK5 downstream of innate immune receptors; indeed, other MAPKKKs, such as transforming growth factor–β (TGF-β)–activated kinase 1 (TAK1) or apoptosis signal– regulating kinase 1 (ASK1), may be involved (30).
Within the nucleus, ERK5 stimulates various transcription factors (43–53, 112–114). In particular, ERK5 stimulates members of the MEF2 family (including MEF2A, MEF2C, and MEF2D) and the AP-1 family (including c-Jun and c-Fos) of transcription factors (50). For example, the ERK5-dependent activation of MEF2C and MEF2D induces expression of the gene encoding c-Jun, and, downstream of Tpl2, ERK5 stimulates the promoter of the same gene concomitantly with JNK and p38 (53, 110, 115). Furthermore, ERK5 may increase the abundance and activation of c-Fos in certain instances (46, 49). These studies all point to a role for ERK5 in the regulation of AP-1, which is of particular interest because the AP-1 family members c-Jun and c-Fos induce the expression of genes whose products mediate inflammation and cell survival (116–118). ERK5 is thought to both stimulate and inhibit NF-κB activity. In this regard, ERK5 may decrease NF-κB activity by increasing the abundance of the transcription factor Krüppel-like factor 2 (KLF2) (119, 120). In ECs, an increase in the amount of KLF2 reduces the abundances of VCAM-1, E-selectin, tissue factor, and PAI-1, but not ICAM-1, as well as potently inhibits vascular endothelial growth factor (VEGF)–mediated vascular permeability (121–123). Furthermore, KLF2 is proposed to have anti-inflammatory properties in healthy ECs under laminar flow, but not in areas of compromised blood flow (124–126). In contrast with these findings, ERK5 promotes NF-κB activity in other cell systems, as well as during the cell cycle (127–129).
In conclusion, we have identified a previously uncharacterized pro-inflammatory role for ERK5 downstream of multiple innate immune receptors that engage exogenous and endogenous inflammatory mediators. Furthermore, we found that ERK5 mediated the increased production of proinflammatory cytokines involved in leukocyte recruitment and activation in mice exposed to microbial factors or tissue ischemia. Placing our findings into the context of the literature, we speculate that blood flow conditions may determine whether ERK5 serves a proinflammatory or anti-inflammatory role. We propose that under steady-state conditions, ERK5 may help to maintain vascular integrity and endothelial homeostasis, perhaps in part through KLF2, but that during sepsis or tissue injury, ERK5 promotes the proinflammatory activation of ECs, which, if sustained, could lead to organ injury and failure. Together, our studies suggest that ERK5, with its distinct structure and limited role in cells, may be a potential therapeutic target in the fight against inflammatory critical illness, a common and often lethal condition in humans.
MATERIALS AND METHODS
Cells
HUVECs (passages 2 to 6 from multiple donors, Lonza) and HMVEC-lung cells (passages 4 to 9 from female and male donors, PromoCell) were incubated at 37°C under humidified 5% CO2. HUVECs and HMVEC-lung cells were cultured in endothelial growth medium–2 (EGM-2) and EGM-2 microvascular medium (Lonza), respectively. Human PBMCs and neutrophils were isolated by gradient centrifugation with Lymphoprep and Polymorphprep (Axis-Shield), respectively, of heparinized whole blood collected by venipuncture from healthy human volunteers. PBMCs were isolated according to the manufacturer’s instructions, whereas neutrophils were isolated as described previously (104). Human CD14+ monocytes were isolated from PBMCs by positive selection with CD14 MicroBeads and LS columns (Miltenyi Biotec) according to the manufacturer’s instructions.
Inhibitor and agonist treatments
All ECs were grown to confluence before being treated with inhibitors or agonists. Unless otherwise noted, cells in all experiments were preincubated with vehicle (DMSO), with the ERK5 inhibitor XMD8-92 (5 μM, Axon Medchem) or XMD17-109 (1 μM, Medchem Express), or with the MEK5 inhibitor BIX02189 (10 μM, Axon Medchem) for 1 hour before treatment and then continuously during treatment with vehicle (0.9% saline) or the inflammatory agonists Pam3Cys (10 μg/ml, EMC Microcollections), FSL-1 (10 μg/ml, EMC Microcollections), LPS (10 μg/ml, List Laboratories), recombinant human TNF-α (100 ng/ml, PeproTech Inc.), or IL-1β (0.1, 1, or 10 ng/ml, R&D Systems) for the times indicated in the figure legends. Preparations of XMD8-92, XMD17-109, BIX02189, Pam3Cys, FSL-1, and TNF-α contained LPS (<0.1 endotoxin unit/ml) based on the limulus amebocyte lysate endotoxin assay (Thermo Scientific).
Transfection of HMVEC-lung cells with siRNAs
MAPK7-specific (ERK5, L-003513-00-0005), MAP2K5-specific (MEK5, L-003966-00-0005), and control (L-005120-01-0005) siRNAON-TARGETplus SMARTpools (Thermo Scientific) were used according to the manufacturer’s suggested protocol for the transfection of HUVECs with the DharmaFECT 4 Transfection Reagent. In some cases, whole-cell lysates were prepared 72 hours after transfection, whereas cell culture media were collected 79 hours after transfection (which included 7 hours of treatment with inhibitor and agonist).
Western blotting analysis
Cells were lysed with radioimmunoprecipitation assay lysis buffer [4 mM sodium dihydrogen phosphate (pH 7.0), 6 mM disodium hydrogen phosphate (pH 7.0), 150 mM sodium chloride, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 2 mM EDTA, 50 mM sodium fluoride, 0.1 mM sodium orthovanadate] containing protease inhibitor cocktail (Sigma-Aldrich). Total proteins were resolved by SDS–polyacrylamide gel electrophoresis and then transferred onto polyvinylidene difluoride (PVDF) membranes (Pall Corp). The membranes were blocked in 3% bovine serum albumin (BSA) for 45 min at room temperature and then incubated with primary antibody solution overnight at 4°C. The PVDF membranes were washed and then incubated with the appropriate horseradish peroxidase–conjugated secondary antibodies (Jackson ImmunoResearch). Western blots were developed with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific), and signals were detected with a Gel Logic 2200 Imaging System (Kodak) run on Carestream imaging software (Carestream Health). Antibodies specific for ERK5 (Cell Signaling), ERK1/2 (Cell Signaling), MEK5 (Millipore), MEK1/2 (R&D Systems), MEK4 (Antibodies Online), and β-actin (Sigma-Aldrich) were used for Western blotting analysis.
ELISAs and MTT assays
The concentrations of cytokines secreted by cells were quantified by analysis of cell culture media with ELISA kits specific for IL-6 (R&D Systems), IL-8 (BD Biosciences), TNF-α (R&D Systems), CCL2/MCP-1 (R&D Systems), and CCL3/MIP-1α (R&D Systems) according to the manufacturers’ instructions. Active PAI-1 was detected with the PAI-1 functional assay ELISA kit (Molecular Innovations). MTT assays (Biotium) were performed in 96-well plates according to the manufacturer’s instructions.
Neutrophil adhesion assay
The neutrophil adhesion assay was performed as described previously (104). Briefly, neutrophils were resuspended in RPMI 1640 medium (2 × 106 cells/ml) and labeled with 3 μM calcein AM (Life Technologies). Immediately before the labeled neutrophils were added, 48-well plates containing monolayers of HMVEC-lung cells were washed three times with RPMI 1640 medium containing 3% BSA. The labeled neutrophils were then added at 3 × 105 to 6 × 105 cells per well and allowed to incubate for 20 min at 37°C in the dark before being washed five times with PBS to remove nonadherent cells. Pre- and post-wash fluorescence were read at an excitation of 485 nm and an emission of 520 nm in a FLUOstar OPTIMA fluorescent plate reader. The relative adherence was then calculated. Neutrophil adhesion was visualized with a Zeiss Axio Imager D1 microscope.
Challenge of mice with saline, Pam3Cys, LPS, or HKSA
All animal study protocols were approved by the University of California San Francisco (UCSF) Animal Care and Use Committee. Eight-week-old C57BL/6J male mice (The Jackson Laboratory) were injected intraperitoneally with XMD8-92 (50 mg/kg) or XMD17-109 (50 mg/kg) dissolved in 30% (2-hydroxypropyl)-β-cyclodextrin or with BIX02189 (25 mg/kg) dissolved in 5% DMSO containing 30% (2-hydroxypropyl)-β-cyclodextrin, or with vehicle alone [30% (2-hydroxypropyl)-β-cyclodextrin with or without 5% DMSO] 30 min before being injected intravenously (“challenged”) with either 0.9% saline, Pam3Cys (2.5 mg/kg), LPS (10 mg/kg), or HKSA (Invivogen) at 2.5 × 1010 bacteria/kg. In each experiment, four mice were used per condition. The doses of Pam3Cys, LPS, and HKSA that were used were established in pilot dose-response studies. Comparisons were made between mice that were treated with the ERK5 or MEK5 inhibitors and control mice that were treated with vehicle alone.
Analysis of LPS-induced mortality in mice
Eight-week-old C57BL/6J male mice were injected intraperitoneally with XMD8-92 (50 mg/kg) dissolved in 30% (2-hydroxypropyl)-β-cyclodextrin or vehicle [30% (2-hydroxypropyl)-β-cyclodextrin] alone 30 min before or 1 hour after the mice were injected intraperitoneally with LPS at 15 and 12.5 mg/kg, respectively. For inhibitor pretreatment experiments, eight mice per group were used, whereas for inhibitor posttreatment experiments, 14 mice per group were used. Mice were monitored by an individual who was blinded to the treatment group at 4 hours, 8 hours, and then every 8 hours up to 72 hours after the mice were challenged with LPS. Mice that became moribund were euthanized and were counted among the mice that died without the need for euthanasia. A moribund state, which is defined as inanition, the lack of response to gentle shaking of the cage, and the inability of the mice to right themselves when placed on their sides are uniform presages for death in endotoxemic mice.
Ventilated lung IR injury induced by left pulmonary artery occlusion
A mouse model of unilateral left pulmonary artery occlusion was used, as described previously (108). Fifteen-week-old C57BL/6J male mice were anesthetized by intraperitoneal administration of tribromoethanol (also known as Avertin), endotracheally intubated, and placed on a ventilator (with tidal volumes of 225 μl and a respiratory rate of 180 breaths/min for a 30-g mouse) and were given buprenorphine intraperitoneally. The mice underwent a left thoracotomy through the interspace between the second and third ribs, after which the left pulmonary artery was identified, and an 8-0 Prolene monofilament suture was passed between the left pulmonary artery and the left bronchus while visualizing under a high-magnification microscope. A slip-knot suture was tied such that the end of the suture was externalized through a narrow bore (27-gauge) needle to the anterior chest wall. Before closure of the thorax, the left lung was inflated to occupy the left thoracic cavity. Local anesthetic (three to four drops of 0.25% bupivacaine) was applied topically before skin closure. The total period of mechanical ventilation was about 20 to 25 min. After the skin was closed, the mice were allowed to recover from anesthesia, mechanical ventilation was discontinued, and the endotracheal tube was removed. After 30 min of ischemia, reperfusion was established by removing the externalized suture. At the end of a 1-hour reperfusion period, blood was collected into a heparinized syringe by cardiac puncture under deep anesthesia and was then centrifuged at 14,000g for 5 min before the plasma was stored at −80°C. For the sham procedure, mice underwent left thoracotomy and all of the other procedures that were described earlier; however, the left pulmonary artery was not isolated and a slip knot was not tied or externalized. Blood from these animals was collected at the same time intervals as were used for the experimental group. Mice that died before the completion of surgery, yielded insufficient blood volumes, or had inadvertent esophageal intubation were excluded from the analysis.
Statistical analysis
Data from in vitro and in vivo experiments were analyzed in GraphPad Prism with Mann-Whitney or Kruskal-Wallis nonparametric-based biostatistics, except for the data from the endotoxemic mortality experiments, which were analyzed with Kaplan-Meier curves. P ≤ 0.05 was considered to be statistically significant for all data, and the data in graphs are presented as means ± SD.
Supplementary Material
Fig. S1. Domain organization of ERK5.
Fig. S2. XMD8-92, XMD17-109, and BIX02189 have no substantial effects on cell viability.
Fig. S3. The ERK5 inhibitor XMD8-92 reduces the amounts of proinflammatory cytokines secreted by HMVEC-lung cells when it is added before or after they are treated with LPS.
Fig. S4. MEK5 promotes the binding of neutrophils to activated ECs.
Fig. S5. ERK5 promotes the secretion of CCL2 and enhances PAI-1 activity in mice 24 hours after they are challenged with LPS.
Acknowledgments
Funding: This work was supported by the UCSF Department of Anesthesia, the National Science Foundation Graduate Research Fellowship Program (#1144247), and the San Francisco Foundation. The Gray laboratory acknowledges the support of the Linde Program in Chemical Biology.
Footnotes
SUPPLEMENTARY MATERIALS
Author contributions: K.W., F.X., A.P., and J.H. designed the research; K.W., F.X., K.F., A.T., S.K., S.S., and A.P. performed the research; J.W. and N.S.G. contributed new reagents and analytic tools; K.W., F.X., K.F., and S.S. analyzed data; and K.W. and J.H. wrote the paper.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 3.Zarbock A, Ley K. Mechanisms and consequences of neutrophil interaction with the endothelium. Am J Pathol. 2008;172:1–7. doi: 10.2353/ajpath.2008.070502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirsiger S, Simmen HP, Werner CML, Wanner GA, Rittirsch D. Danger signals activating the immune response after trauma. Mediators Inflamm. 2012;2012:1–10. doi: 10.1155/2012/315941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaffer U, Wade RG, Gourlay T. Cytokines in the systemic inflammatory response syndrome: A review. HSR Proc Intensive Care Cardiovasc Anesth. 2010;2:161–175. [PMC free article] [PubMed] [Google Scholar]
- 7.Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury. 2007;38:1336–1345. doi: 10.1016/j.injury.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schouten M, Wiersinga WJ, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008;83:536–545. doi: 10.1189/jlb.0607373. [DOI] [PubMed] [Google Scholar]
- 10.Levi M, de Jonge E, van der Poll T. Sepsis and disseminated intravascular coagulation. J Thromb Thrombolysis. 2003;16:43–47. doi: 10.1023/B:THRO.0000014592.27892.11. [DOI] [PubMed] [Google Scholar]
- 11.Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis, thrombosis and organ dysfunction. Thromb Res. 2012;129:290–295. doi: 10.1016/j.thromres.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 12.Kwaan HC. Microvascular thrombosis: A serious and deadly pathologic process in multiple diseases. Semin Thromb Hemost. 2011;37:961–978. doi: 10.1055/s-0031-1297375. [DOI] [PubMed] [Google Scholar]
- 13.Durham RM, Moran JJ, Mazuski JE, Shapiro MJ, Baue AE, Flint LM. Multiple organ failure in trauma patients. J Trauma. 2003;55:608–616. doi: 10.1097/01.TA.0000092378.10660.D1. [DOI] [PubMed] [Google Scholar]
- 14.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 15.Mayr VD, Dünser MW, Greil V, Jochberger S, Luckner G, Ulmer H, Friesenecker BE, Takala J, Hasibeder WR. Causes of death and determinants of outcome in critically ill patients. Crit Care. 2006;10:R154. doi: 10.1186/cc5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakr Y, Lobo SM, Moreno RP, Gerlach H, Ranieri VM, Michalopoulos A, Vincent JL, SOAP Investigators Patterns and early evolution of organ failure in the intensive care unit and their relation to outcome. Crit Care. 2012;16:R222. doi: 10.1186/cc11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hickey MJ, Kubes P. Intravascular immunity: The host–pathogen encounter in blood vessels. Nat Rev Immunol. 2009;9:364–375. doi: 10.1038/nri2532. [DOI] [PubMed] [Google Scholar]
- 18.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 19.Chen GY, Nuñez G. Sterile inflammation: Sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rittirsch D, Redl H, Huber-Lang M. Role of complement in multiorgan failure. Clin Dev Immunol. 2012;2012:962927. doi: 10.1155/2012/962927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 22.Shin HS, Xu F, Bagchi A, Herrup E, Prakash A, Valentine C, Kulkarni H, Wilhelmsen K, Warren S, Hellman J. Bacterial lipoprotein TLR2 agonists broadly modulate endothelial function and coagulation pathways in vitro and in vivo. J Immunol. 2011;186:1119–1130. doi: 10.4049/jimmunol.1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilhelmsen K, Mesa KR, Lucero J, Xu F, Hellman J. ERK5 protein promotes, whereas MEK1 protein differentially regulates, the Toll-like receptor 2 protein-dependent activation of human endothelial cells and monocytes. J Biol Chem. 2012;287:26478–26494. doi: 10.1074/jbc.M112.359489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-κB and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 25.Wilhelmsen K, Mesa KR, Prakash A, Xu F, Hellman J. Activation of endothelial TLR2 by bacterial lipoprotein upregulates proteins specific for the neutrophil response. Innate Immun. 2012;18:602–616. doi: 10.1177/1753425911429336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwaki T, Urano T, Umemura K. PAI-1, progress in understanding the clinical problem and its aetiology. Br J Haematol. 2012;157:291–298. doi: 10.1111/j.1365-2141.2012.09074.x. [DOI] [PubMed] [Google Scholar]
- 27.Gando S. Role of fibrinolysis in sepsis. Semin Thromb Hemost. 2013;39:392–399. doi: 10.1055/s-0033-1334140. [DOI] [PubMed] [Google Scholar]
- 28.Mesters RM, Florke N, Ostermann H, Kienast J. Increase of plasminogen activator inhibitor levels predicts outcome of leukocytopenic patients with sepsis. Thromb Haemost. 1996;75:902–907. [PubMed] [Google Scholar]
- 29.Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40:315–327. doi: 10.1016/j.immuni.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arthur JSC, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 31.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 33.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 34.Cohen P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr Opin Cell Biol. 2009;21:317–324. doi: 10.1016/j.ceb.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 35.Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 36.English JM, Vanderbilt CA, Xu S, Marcus S, Cobb MH. Isolation of MEK5 and differential expression of alternatively spliced forms. J Biol Chem. 1995;270:28897–28902. doi: 10.1074/jbc.270.48.28897. [DOI] [PubMed] [Google Scholar]
- 37.Lee JD, Ulevitch RJ, Han JH. Primary structure of BMK1: A new mammalian map kinase. Biochem Biophys Res Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- 38.Nithianandarajah-Jones GN, Wilm B, Goldring CEP, Müller J, Cross MJ. ERK5: Structure, regulation and function. Cell Signal. 2012;24:2187–2196. doi: 10.1016/j.cellsig.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 39.Mody N, Campbell DG, Morrice N, Peggie M, Cohen P. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5(MKK5) in vitro. Biochem J. 2003;372:567–575. doi: 10.1042/BJ20030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morimoto H, Kondoh K, Nishimoto S, Terasawa K, Nishida E. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J Biol Chem. 2007;282:35449–35456. doi: 10.1074/jbc.M704079200. [DOI] [PubMed] [Google Scholar]
- 41.Hayashi M, Tapping RI, Chao TH, Lo JF, King CC, Yang Y, Lee JD. BMK1 mediates growth factor-induced cell proliferation through direct cellular activation of serum and glucocorticoid-inducible kinase. J Biol Chem. 2001;276:8631–8634. doi: 10.1074/jbc.C000838200. [DOI] [PubMed] [Google Scholar]
- 42.Cameron SJ, Malik S, Akaike M, Lerner-Marmarosh N, Yan C, Lee J-D, Abe J-I, Yang J. Regulation of epidermal growth factor-induced connexin 43 gap junction communication by big mitogen-activated protein kinase 1/ERK5 but not ERK1/2 kinase activation. J Biol Chem. 2003;278:18682–18688. doi: 10.1074/jbc.M213283200. [DOI] [PubMed] [Google Scholar]
- 43.Kim M, Kim S, Lim JH, Lee C, Choi HC, Woo CH. Laminar flow activation of ERK5 protein in vascular endothelium leads to atheroprotective effect via NF-E2-related factor 2 (Nrf2) activation. J Biol Chem. 2012;287:40722–40731. doi: 10.1074/jbc.M112.381509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clark PR, Jensen TJ, Kluger MS, Morelock M, Hanidu A, Qi Z, Tatake RJ, Pober JS. MEK5 is activated by shear stress, activates ERK5 and induces KLF4 to modulate TNF responses in human dermal microvascular endothelial cells. Microcirculation. 2011;18:102–117. doi: 10.1111/j.1549-8719.2010.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohnesorge N, Viemann D, Schmidt N, Czymai T, Spiering D, Schmolke M, Ludwig S, Roth J, Goebeler M, Schmidt M. Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Krüppel-like factor 4 (KLF4) J Biol Chem. 2010;285:26199–26210. doi: 10.1074/jbc.M110.103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sasaki T, Kojima H, Kishimoto R, Ikeda A, Kunimoto H, Nakajima K. Spatiotemporal regulation of c-Fos by ERK5 and the E3 ubiquitin ligase UBR1, and its biological role. Mol Cell. 2006;24:63–75. doi: 10.1016/j.molcel.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 47.Woo C-H, Massett MP, Shishido T, Itoh S, Ding B, McClain C, Che W, Vulapalli SR, Yan C, Abe J-I. ERK5 activation inhibits inflammatory responses via peroxisome proliferator-activated receptor δ (PPAR δ) stimulation. J Biol Chem. 2006;281:32164–32174. doi: 10.1074/jbc.M602369200. [DOI] [PubMed] [Google Scholar]
- 48.Akaike M, Che W, Marmarosh N-L, Ohta S, Osawa M, Ding B, Berk BC, Yan C, Abe J-I. The hinge-helix 1 region of peroxisome proliferator-activated receptor γ1 (PPARγ1) mediates interaction with extracellular signal-regulated kinase 5 and PPARγ1 transcriptional activation: Involvement in flow-induced PPARγ activation in endothelial cells. Mol Cell Biol. 2004;24:8691–8704. doi: 10.1128/MCB.24.19.8691-8704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terasawa K, Okazaki K, Nishida E. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells. 2003;8:263–273. doi: 10.1046/j.1365-2443.2003.00631.x. [DOI] [PubMed] [Google Scholar]
- 50.Kato Y, Zhao M, Morikawa A, Sugiyama T, Chakravortty D, Koide N, Yoshida T, Tapping RI, Yang Y, Yokochi T, Lee JD. Big mitogen-activated kinase regulates multiple members of the MEF2 protein family. J Biol Chem. 2000;275:18534–18540. doi: 10.1074/jbc.M001573200. [DOI] [PubMed] [Google Scholar]
- 51.Yang CC, Ornatsky OI, McDermott JC, Cruz TF, Prody CA. Interaction of myocyte enhancer factor 2 (MEF2) with a mitogen-activated protein kinase, ERK5/BMK1. Nucleic Acids Res. 1998;26:4771–4777. doi: 10.1093/nar/26.20.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.English JM, Pearson G, Baer R, Cobb MH. Identification of substrates and regulators of the mitogen-activated protein kinase ERK5 using chimeric protein kinases. J Biol Chem. 1998;273:3854–3860. doi: 10.1074/jbc.273.7.3854. [DOI] [PubMed] [Google Scholar]
- 53.Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buschbeck M, Ullrich A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J Biol Chem. 2005;280:2659–2667. doi: 10.1074/jbc.M412599200. [DOI] [PubMed] [Google Scholar]
- 55.Iñesta-Vaquera FA, Campbell DG, Arthur JSC, Cuenda A. ERK5 pathway regulates the phosphorylation of tumour suppressor hDlg during mitosis. Biochem Biophys Res Commun. 2010;399:84–90. doi: 10.1016/j.bbrc.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 56.Ranganathan A, Pearson GW, Chrestensen CA, Sturgill TW, Cobb MH. The MAP kinase ERK5 binds to and phosphorylates p90 RSK. Arch Biochem Biophys. 2006;449:8–16. doi: 10.1016/j.abb.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 57.Xu B-E, Stippec S, Lenertz L, Lee B-H, Zhang W, Lee Y-K, Cobb MH. WNK1 activates ERK5 by an MEKK2/3-dependent mechanism. J Biol Chem. 2004;279:7826–7831. doi: 10.1074/jbc.M313465200. [DOI] [PubMed] [Google Scholar]
- 58.Zheng Q, Yin G, Yan C, Cavet M, Berk BC. 14-3-3 β binds to big mitogen-activated protein kinase 1 (BMK1/ERK5) and regulates BMK1 function. J Biol Chem. 2004;279:8787–8791. doi: 10.1074/jbc.M310212200. [DOI] [PubMed] [Google Scholar]
- 59.Sun W, Wei X, Kesavan K, Garrington TP, Fan R, Mei J, Anderson SM, Gelfand EW, Johnson GL. MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol Cell Biol. 2003;23:2298–2308. doi: 10.1128/MCB.23.7.2298-2308.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun W, Kesavan K, Schaefer BC, Garrington TP, Ware M, Johnson NL, Gelfand EW, Johnson GL. MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J Biol Chem. 2001;276:5093–5100. doi: 10.1074/jbc.M003719200. [DOI] [PubMed] [Google Scholar]
- 61.Sohn SJ, Sarvis BK, Cado D, Winoto A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J Biol Chem. 2002;277:43344–43351. doi: 10.1074/jbc.M207573200. [DOI] [PubMed] [Google Scholar]
- 62.Yan L, Carr J, Ashby PR, Murry-Tait V, Thompson C, Arthur JSC. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev Biol. 2003;3:11. doi: 10.1186/1471-213X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Regan CP, Li W, Boucher DM, Spatz S, Su MS, Kuida K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc Natl Acad Sci USA. 2002;99:9248–9253. doi: 10.1073/pnas.142293999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roberts OL, Holmes K, Müller J, Cross DAE, Cross MJ. ERK5 and the regulation of endothelial cell function. Biochem Soc Trans. 2009;37:1254–1259. doi: 10.1042/BST0371254. [DOI] [PubMed] [Google Scholar]
- 66.Pan YW, Zou J, Wang W, Sakagami H, Garelick MG, Abel G, Kuo CT, Storm DR, Xia Z. Inducible and conditional deletion of extracellular signal-regulated kinase 5 disrupts adult hippocampal neurogenesis. J Biol Chem. 2012;287:23306–23317. doi: 10.1074/jbc.M112.344762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pan YW, Kuo CT, Storm DR, Xia Z. Inducible and targeted deletion of the ERK5 MAP kinase in adult neurogenic regions impairs adult neurogenesis in the olfactory bulb and several forms of olfactory behavior. PLOS One. 2013;8:e49622. doi: 10.1371/journal.pone.0049622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ananieva O, Macdonald A, Wang X, McCoy CE, McIlrath J, Tournier C, Arthur JSC. ERK5 regulation in naïve T-cell activation and survival. Eur J Immunol. 2008;38:2534–2547. doi: 10.1002/eji.200737867. [DOI] [PubMed] [Google Scholar]
- 69.Li T, Pan YW, Wang W, Abel G, Zou J, Xu L, Storm DR, Xia Z. Targeted deletion of the ERK5 MAP kinase impairs neuronal differentiation, migration, and survival during adult neurogenesis in the olfactory bulb. PLOS One. 2013;8:e61948. doi: 10.1371/journal.pone.0061948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zou J, Pan YW, Wang Z, Chang SY, Wang W, Wang X, Tournier C, Storm DR, Xia Z. Targeted deletion of ERK5 MAP kinase in the developing nervous system impairs development of GABAergic interneurons in the main olfactory bulb and behavioral discrimination between structurally similar odorants. J Neurosci. 2012;32:4118–4132. doi: 10.1523/JNEUROSCI.6260-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang W, Pan YW, Wietecha T, Zou J, Abel GM, Kuo CT, Xia Z. Extracellular signal-regulated kinase 5 (ERK5) mediates prolactin-stimulated adult neurogenesis in the subventricular zone and olfactory bulb. J Biol Chem. 2013;288:2623–2631. doi: 10.1074/jbc.M112.401091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tatake RJ, O’Neill MM, Kennedy CA, Wayne AL, Jakes S, Wu D, Kugler SZ, Jr, Kashem MA, Kaplita P, Snow RJ. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophys Res Commun. 2008;377:120–125. doi: 10.1016/j.bbrc.2008.09.087. [DOI] [PubMed] [Google Scholar]
- 73.Flaherty PT, Chopra I, Jain P, Monlish D, Cavanaugh J. Structure–activity relationships of benzimidazole-based selective inhibitors of the mitogen activated kinase-5 signaling pathway. Bioorg Med Chem. 2010;18:8054–8060. doi: 10.1016/j.bmc.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 74.Flaherty PT, Chopra I, Jain P, Yi S, Allen E, Cavanaugh J. Identification of benzimidazole-based inhibitors of the mitogen activated kinase-5 signaling pathway. Bioorg Med Chem Lett. 2010;20:2892–2896. doi: 10.1016/j.bmcl.2010.03.033. [DOI] [PubMed] [Google Scholar]
- 75.Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR, III, Gray NS, Lee JD. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010;18:258–267. doi: 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Álvarez-Fernández S, Ortiz-Ruiz MJ, Parrott T, Zaknoen S, Ocio EM, San Miguel J, Burrows FJ, Esparís-Ogando A, Pandiella A. Potent antimyeloma activity of a novel ERK5/CDK inhibitor. Clin Cancer Res. 2013;19:2677–2687. doi: 10.1158/1078-0432.CCR-12-2118. [DOI] [PubMed] [Google Scholar]
- 77.Yang Q, Lee JD. Targeting the BMK1 MAP kinase pathway in cancer therapy. Clin Cancer Res. 2011;17:3527–3532. doi: 10.1158/1078-0432.CCR-10-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Montero JC, Ocaña A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparís-Ogando A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLOS One. 2009;4:e5565. doi: 10.1371/journal.pone.0005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, Leung HY. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene. 2003;22:1381–1389. doi: 10.1038/sj.onc.1206154. [DOI] [PubMed] [Google Scholar]
- 80.Sticht C, Freier K, Knöpfle K, Flechtenmacher C, Pungs S, Hofele C, Hahn M, Joos S, Lichter P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC) Neoplasia. 2008;10:462–470. doi: 10.1593/neo.08164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weldon CB, Scandurro AB, Rolfe KW, Clayton JL, Elliott S, Butler NN, Melnik LI, Alam J, McLachlan JA, Jaffe BM, Beckman BS, Burow ME. Identification of mitogen-activated protein kinase kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery. 2002;132:293–301. doi: 10.1067/msy.2002.125389. [DOI] [PubMed] [Google Scholar]
- 82.Esparís-Ogando A, Díaz-Rodríguez E, Montero JC, Yuste L, Crespo P, Pandiella A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol Cell Biol. 2002;22:270–285. doi: 10.1128/MCB.22.1.270-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nagel S, Burek C, Venturini L, Scherr M, Quentmeier H, Meyer C, Rosenwald A, Drexler HG, MacLeod RAF. Comprehensive analysis of homeobox genes in Hodgkin lymphoma cell lines identifies dysregulated expression of HOXB9 mediated via ERK5 signaling and BMI1. Blood. 2007;109:3015–3023. doi: 10.1182/blood-2006-08-044347. [DOI] [PubMed] [Google Scholar]
- 84.Ramos-Nino ME, Blumen SR, Sabo-Attwood T, Pass H, Carbone M, Testa JR, Altomare DA, Mossman BT. HGF mediates cell proliferation of human mesothelioma cells through a PI3K/MEK5/Fra-1 pathway. Am J Respir Cell Mol Biol. 2008;38:209–217. doi: 10.1165/rcmb.2007-0206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Garaude J, Cherni S, Kaminski S, Delepine E, Chable-Bessia C, Benkirane M, Borges J, Pandiella A, Iñiguez MA, Fresno M, Hipskind RA, Villalba M. ERK5 activates NF-κB in leukemic T cells and is essential for their growth in vivo. J Immunol. 2006;177:7607–7617. doi: 10.4049/jimmunol.177.11.7607. [DOI] [PubMed] [Google Scholar]
- 86.Hayashi M, Fearns C, Eliceiri B, Yang Y, Lee JD. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005;65:7699–7706. doi: 10.1158/0008-5472.CAN-04-4540. [DOI] [PubMed] [Google Scholar]
- 87.McCracken SRC, Ramsay A, Heer R, Mathers ME, Jenkins BL, Edwards J, Robson CN, Marquez R, Cohen P, Leung HY. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene. 2008;27:2978–2988. doi: 10.1038/sj.onc.1210963. [DOI] [PubMed] [Google Scholar]
- 88.Ramsay AK, McCracken SRC, Soofi M, Fleming J, Yu AX, Ahmad I, Morland R, Machesky L, Nixon C, Edwards DR, Nuttall RK, Seywright M, Marquez R, Keller E, Leung HY. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br J Cancer. 2011;104:664–672. doi: 10.1038/sj.bjc.6606062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wu K, Tian S, Zhou H, Wu Y. Statins protect human endothelial cells from TNF-induced inflammation via ERK5 activation. Biochem Pharmacol. 2013;85:1753–1760. doi: 10.1016/j.bcp.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 90.Heo K-S, Chang E, Le N-T, Cushman H, Yeh ETH, Fujiwara K, Abe J-I. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow–induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res. 2013;112:911–923. doi: 10.1161/CIRCRESAHA.111.300179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li L, Tatake RJ, Natarajan K, Taba Y, Garin G, Tai C, Leung E, Surapisitchat J, Yoshizumi M, Yan C, Abe J-I, Berk BC. Fluid shear stress inhibits TNF-mediated JNK activation via MEK5–BMK1 in endothelial cells. Biochem Biophys Res Commun. 2008;370:159–163. doi: 10.1016/j.bbrc.2008.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Le NT, Takei Y, Izawa-Ishizawa Y, Heo KS, Lee H, Smrcka AV, Miller BL, Ko KA, Ture S, Morrell C, Fujiwara K, Akaike M, Abe JI. Identification of activators of ERK5 transcriptional activity by high-throughput screening and the role of endothelial ERK5 in vasoprotective effects induced by statins and antimalarial agents. J Immunol. 2014;193:3803–3815. doi: 10.4049/jimmunol.1400571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mackesy DZ, Goalstone ML. Extracellular signal-regulated kinase-5: Novel mediator of insulin and tumor necrosis factor α-stimulated vascular cell adhesion molecule-1 expression in vascular cells. J Diabetes. 2014;6:595–602. doi: 10.1111/1753-0407.12132. [DOI] [PubMed] [Google Scholar]
- 94.Luerman GC, Nguyen C, Samaroo H, Loos P, Xi H, Hurtado-Lorenzo A, Needle E, Noell GStephen, Galatsis P, Dunlop J, Geoghegan KF, Hirst WD. Phosphoproteomic evaluation of pharmacological inhibition of leucine-rich repeat kinase 2 reveals significant off-target effects of LRRK-2-IN-1. J Neurochem. 2014;128:561–576. doi: 10.1111/jnc.12483. [DOI] [PubMed] [Google Scholar]
- 95.Sun J-L, Xiao C, Lu B, Zhang J, Yuan X-z, Chen W, Yu L-N, Zhang F-J, Chen G, Yan M. CX3CL1/CX3CR1 regulates nerve injury-induced pain hypersensitivity through the ERK5 signaling pathway. J Neurosci Res. 2013;91:545–553. doi: 10.1002/jnr.23168. [DOI] [PubMed] [Google Scholar]
- 96.Kim S, Lim JH, Woo CH. ERK5 inhibition ameliorates pulmonary fibrosis via regulating Smad3 acetylation. Am J Pathol. 2013;183:1758–1768. doi: 10.1016/j.ajpath.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 97.Zhu W, Downey JS, Gu J, Di Padova F, Gram H, Han J. Regulation of TNF expression by multiple mitogen-activated protein kinase pathways. J Immunol. 2000;164:6349–6358. doi: 10.4049/jimmunol.164.12.6349. [DOI] [PubMed] [Google Scholar]
- 98.Hii CS, Anson DS, Costabile M, Mukaro V, Dunning K, Ferrante A. Characterization of the MEK5-ERK5 module in human neutrophils and its relationship to ERK1/ERK2 in the chemotactic response. J Biol Chem. 2004;279:49825–49834. doi: 10.1074/jbc.M406892200. [DOI] [PubMed] [Google Scholar]
- 99.Ahmad R, Shihab PK, Jasem S, Behbehani K. FSL-1 induces MMP-9 production through TLR-2 and NF-κB/AP-1 signaling pathways in monocytic THP-1 cells. Cell Physiol Biochem. 2014;34:929–942. doi: 10.1159/000366310. [DOI] [PubMed] [Google Scholar]
- 100.Elkins JM, Wang J, Deng X, Pattison MJ, Arthur JSC, Erazo T, Gomez N, Lizcano JM, Gray NS, Knapp S. X-ray crystal structure of ERK5 (MAPK7) in complex with a specific inhibitor. J Med Chem. 2013;56:4413–4421. doi: 10.1021/jm4000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deng X, Elkins JM, Zhang J, Yang Q, Erazo T, Gomez N, Choi HG, Wang J, Dzamko N, Lee JD, Sim T, Kim N, Alessi DR, Lizcano JM, Knapp S, Gray NS. Structural determinants for ERK5 (MAPK7) and leucine rich repeat kinase 2 activities of benzo[e]pyrimido-[5,4-b]diazepine-6(11H)-ones. Eur J Med Chem. 2013;70:758–767. doi: 10.1016/j.ejmech.2013.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Glatz G, Gógl G, Alexa A, Reményi A. Structural mechanism for the specific assembly and activation of the extracellular signal regulated kinase 5 (ERK5) module. J Biol Chem. 2013;288:8596–8609. doi: 10.1074/jbc.M113.452235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin A, Minden A, Martinetto H, Claret FX, Lange-Carter C, Mercurio F, Johnson GL, Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 104.Wilhelmsen K, Farrar K, Hellman J. Quantitative in vitro assay to measure neutrophil adhesion to activated primary human microvascular endothelial cells under static conditions. J Vis Exp. 2013:e50677. doi: 10.3791/50677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de Almeida MC, Silva AC, Barral A, Barral Netto M. A simple method for human peripheral blood monocyte isolation. Mem Inst Oswaldo Cruz. 2000;95:221–223. doi: 10.1590/s0074-02762000000200014. [DOI] [PubMed] [Google Scholar]
- 106.Pfeiler S, Massberg S, Engelmann B. Biological basis and pathological relevance of microvascular thrombosis. Thromb Res. 2014;133(Suppl 1):S35–S37. doi: 10.1016/j.thromres.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 107.De Backer D, Orbegozo Cortes D, Donadello K, Vincent J-L. Pathophysiology of micro-circulatory dysfunction and the pathogenesis of septic shock. Virulence. 2014;5:73–79. doi: 10.4161/viru.26482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Prakash A, Mesa KR, Wilhelmsen K, Xu F, Doddo JM, Hellman J. Alveolar macrophages and Toll-like receptor 4 mediate ventilated lung ischemia reperfusion injury in mice. Anesthesiology. 2012;117:822–835. doi: 10.1097/ALN.0b013e31826a4ae3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chao TH, Hayashi M, Tapping RI, Kato Y, Lee JD. MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J Biol Chem. 1999;274:36035–36038. doi: 10.1074/jbc.274.51.36035. [DOI] [PubMed] [Google Scholar]
- 110.Chiariello M, Marinissen MJ, Gutkind JS. Multiple mitogen-activated protein kinase signaling pathways connect the cot oncoprotein to the c-jun promoter and to cellular transformation. Mol Cell Biol. 2000;20:1747–1758. doi: 10.1128/mcb.20.5.1747-1758.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Belich MP, Salmerón A, Johnston LH, Ley SC. TPL-2 kinase regulates the proteolysis of the NF-κB-inhibitory protein NF-κB1 p105. Nature. 1999;397:363–368. doi: 10.1038/16946. [DOI] [PubMed] [Google Scholar]
- 112.English JM, Pearson G, Hockenberry T, Shivakumar L, White MA, Cobb MH. Contribution of the ERK5/MEK5 pathway to Ras/Raf signaling and growth control. J Biol Chem. 1999;274:31588–31592. doi: 10.1074/jbc.274.44.31588. [DOI] [PubMed] [Google Scholar]
- 113.Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999;274:26563–26571. doi: 10.1074/jbc.274.37.26563. [DOI] [PubMed] [Google Scholar]
- 114.Yoon HJ, Jeon SB, Kim IH, Park EJ. Regulation of TLR2 expression by prostaglandins in brain glia. J Immunol. 2008;180:8400–8409. doi: 10.4049/jimmunol.180.12.8400. [DOI] [PubMed] [Google Scholar]
- 115.Han TH, Prywes R. Regulatory role of MEF2D in serum induction of the c-jun promoter. Mol Cell Biol. 1995;15:2907–2915. doi: 10.1128/mcb.15.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 117.Zenz R, Eferl R, Scheinecker C, Redlich K, Smolen J, Schonthaler HB, Kenner L, Tschachler E, Wagner EF. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res Ther. 2008;10:201. doi: 10.1186/ar2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Karin M. Inflammation-activated protein kinases as targets for drug development. Proc Am Thorac Soc. 2005;2:386–390. doi: 10.1513/pats.200504-034SR. discussion 394–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sohn SJ, Li D, Lee LK, Winoto A. Transcriptional regulation of tissue-specific genes by the ERK5 mitogen-activated protein kinase. Mol Cell Biol. 2005;25:8553–8566. doi: 10.1128/MCB.25.19.8553-8566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zahlten J, Steinicke R, Opitz B, Eitel J, N’Guessan PD, Vinzing M, Witzenrath M, Schmeck B, Hammerschmidt S, Suttorp N, Hippenstiel S. TLR2- and nucleotide-binding oligomerization domain 2-dependent Krüppel-like factor 2 expression down-regulates NF-κB–related gene expression. J Immunol. 2010;185:597–604. doi: 10.4049/jimmunol.0901798. [DOI] [PubMed] [Google Scholar]
- 121.SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, Michel TM, Gimbrone MA, Jr, García-Cardeña G, Jain MK. KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA, Jr, Balasubramanian V, García-Cardeña G, Jain MK. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:48–57. doi: 10.1161/01.RES.0000159707.05637.a1. [DOI] [PubMed] [Google Scholar]
- 123.Bhattacharya R, Senbanerjee S, Lin Z, Mir S, Hamik A, Wang P, Mukherjee P, Mukhopadhyay D, Jain MK. Inhibition of vascular permeability factor/vascular endothelial growth factor-mediated angiogenesis by the Kruppel-like factor KLF2. J Biol Chem. 2005;280:28848–28851. doi: 10.1074/jbc.C500200200. [DOI] [PubMed] [Google Scholar]
- 124.Boon RA, Horrevoets AJG. Key transcriptional regulators of the vasoprotective effects of shear stress. Hamostaseologie. 2009;29:39–43. [PubMed] [Google Scholar]
- 125.Kinderlerer AR, Ali F, Johns M, Lidington EA, Leung V, Boyle JJ, Hamdulay SS, Evans PC, Haskard DO, Mason JC. KLF2-dependent, shear stress-induced expression of CD59: A novel cytoprotective mechanism against complement-mediated injury in the vasculature. J Biol Chem. 2008;283:14636–14644. doi: 10.1074/jbc.M800362200. [DOI] [PubMed] [Google Scholar]
- 126.Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA, Jr, García-Cardeña G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pearson G, English JM, White MA, Cobb MH. ERK5 and ERK2 cooperate to regulate NF-κB and cell transformation. J Biol Chem. 2001;276:7927–7931. doi: 10.1074/jbc.M009764200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cude K, Wang Y, Choi HJ, Hsuan SL, Zhang H, Wang CY, Xia Z. Regulation of the G2–M cell cycle progression by the ERK5–NFκB signaling pathway. J Cell Biol. 2007;177:253–264. doi: 10.1083/jcb.200609166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Simões AES, Pereira DM, Gomes SE, Brito H, Carvalho T, French A, Castro RE, Steer CJ, Thibodeau SN, Rodrigues CMP, Borralho PM. Aberrant MEK5/ERK5 signalling contributes to human colon cancer progression via NF-κB activation. Cell Death Dis. 2015;6:e1718. doi: 10.1038/cddis.2015.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Domain organization of ERK5.
Fig. S2. XMD8-92, XMD17-109, and BIX02189 have no substantial effects on cell viability.
Fig. S3. The ERK5 inhibitor XMD8-92 reduces the amounts of proinflammatory cytokines secreted by HMVEC-lung cells when it is added before or after they are treated with LPS.
Fig. S4. MEK5 promotes the binding of neutrophils to activated ECs.
Fig. S5. ERK5 promotes the secretion of CCL2 and enhances PAI-1 activity in mice 24 hours after they are challenged with LPS.