

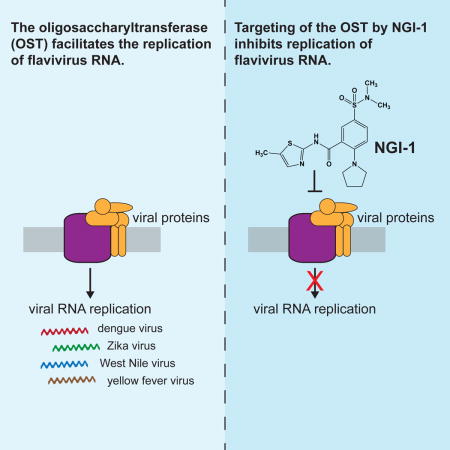
Summary
The mosquito-borne flaviviruses include important human pathogens such as dengue, Zika, West Nile and yellow fever virus, which pose a serious threat for global health. Recent genetic screens identified ER-membrane multiprotein complexes including the oligosaccharyltransferase (OST) complex as critical flavivirus host factors. Here, we show that a chemical modulator of the OST complex termed NGI-1 has promising antiviral activity against flavivirus infections. We demonstrate that NGI-1 blocks viral RNA replication, and that antiviral activity does not depend on inhibition of the N-glycosylation function of the OST. Viral mutants adapted to replicate in cells deficient of the OST complex showed resistance to NGI-1 treatment reinforcing the on-target activity of NGI-1. Lastly, we show that NGI-1 also has strong antiviral activity in primary and disease-relevant cell types. This study provides an example for advancing from the identification of genetic determinants of infection to a host-directed antiviral compound with broad activity against flaviviruses.
Graphical abstract
Introduction
The mosquito-borne flaviviruses comprise a group of important human pathogens including dengue virus (DENV), West Nile virus (WNV), yellow fever virus (YFV), and Zika virus (ZIKV), which all pose a significant threat to global health. Despite the high number of cases, the increase of global spread, the emergence and re-emergence of flaviviral outbreaks, and the risk of severe clinical outcomes, there are currently no approved antiviral therapies against these viruses available.
Traditionally, the development of antivirals is focused on targeting viral proteins by small molecules such as nucleoside analogs or viral protease inhibitors. Alternatively, strategies that inhibit host cellular factors critical for viral infection rather than viral proteins have the potential to be more broad spectrum, more refractory to developing drug resistant mutants and provide a different mode of action that complements direct-antiviral drugs (Kaufmann et al., 2017). Recent genome-wide genetic screens revealed several endoplasmic reticulum (ER)-localized protein complexes to be essential for viral infection (Ma et al., 2015; Marceau et al., 2016; Savidis et al., 2016; Zhang et al., 2016). In particular, deletion of oligosaccharyltransferase (OST) subunits resulted in a >99% reduction of flaviviral infections in cell culture demonstrating its promise as an antiviral target (Marceau et al., 2016). In its cellular function the OST complex catalyzes the N-linked glycosylation of newly synthesized proteins. Mammalian cells have two OST protein isoforms, which are multiprotein complexes composed of a catalytic subunit (encoded by the paralogues STT3A or STT3B) and accessory subunits (Shrimal et al., 2015). Interestingly, we found that DENV RNA replication is dependent on the presence of both OST isoforms, while ZIKV, YFV and WNV exclusively depend on the STT3A OST complex (Marceau et al., 2016).
Here, we used a recently developed, cell-permeable small molecule compound called NGI-1 that targets the OST complex (Lopez-Sambrooks et al., 2016). We show that NGI-1 exhibits pan-flaviviral activity by blocking the viral RNA synthesis. We further demonstrate that NGI-1 specifically targets the OST complex and that its antiviral activity does not depend on the inhibition of the N-glycosylation activity. Lastly, we demonstrate a promising antiviral effect in several disease-relevant cell types for DENV and ZIKV infections.
Results
The oligosaccharyltransferase inhibitor NGI-1 inhibits dengue and Zika virus infection
NGI-1 is an aminobenzamide-sulfonamide compound that targets both OST isoforms and therefore may exhibit antiviral activity against flaviviruses (Figure 1A). To test its inhibitory properties, we infected HEK293 cells with luciferase expressing DENV or ZIKV, treated cells with increasing concentrations of NGI-1 and measured viral replication 48 hours post-infection (hpi) (Figure 1B and 1C). Significant reduction of viral replication was observed at 1 μM and higher. The half maximal effective concentration (EC50) values were 0.85 μM and 2.2 μM for DENV and ZIKV inhibition, respectively. Furthermore, we observed significant reduction of viral particle formation in the supernatant of DENV or ZIKV infected cells (Figure 1D) and a marked effect on the infectivity of the human hepatocyte cell line Huh7 (Figure 1E). To evaluate post-exposure antiviral activity, we treated cells 24 hours after infection and observed a ~80% decrease in DENV infection, which was somewhat lower compared to the immediate treatment (~99% decrease)(Figure S1A). As inhibition of the OST complex may cause cell cycle arrest and reduced proliferation (Lopez-Sambrooks et al., 2016), we determined the effects of increasing concentrations of NGI-1 on HEK293 proliferation. The half maximum cytotoxic concentration (CC50) value in HEK293 cells was 34.9 μM and 33.1 μM using Cell Titer Glo (Figure 1F) and Trypan Blue exclusion assay (Figure S1B), respectively, confirming that the antiviral effects were not caused by reduced proliferation. This resulted in a selectivity index (CC50/EC50) of ~16 and 41 for the treatment of ZIKV and DENV infections, respectively. Additionally, infection with coxsackievirus B3, a picornavirus not known to require the OST complex, did not appear to be affected by NGI-1, even at high concentrations (Figure S1C). By contrast, guanidine hydrochloride (GnHCl), a known picornavirus replication inhibitor, decreased viral infection. This suggests that the effect of NGI-1 is specific to the inhibition of DENV and ZIKV replication, and not due to a general, reduced cellular proliferation.
Figure 1. Targeting the OST complex with a small molecule (NGI-1) inhibits infection with mosquito-borne flaviviruses.
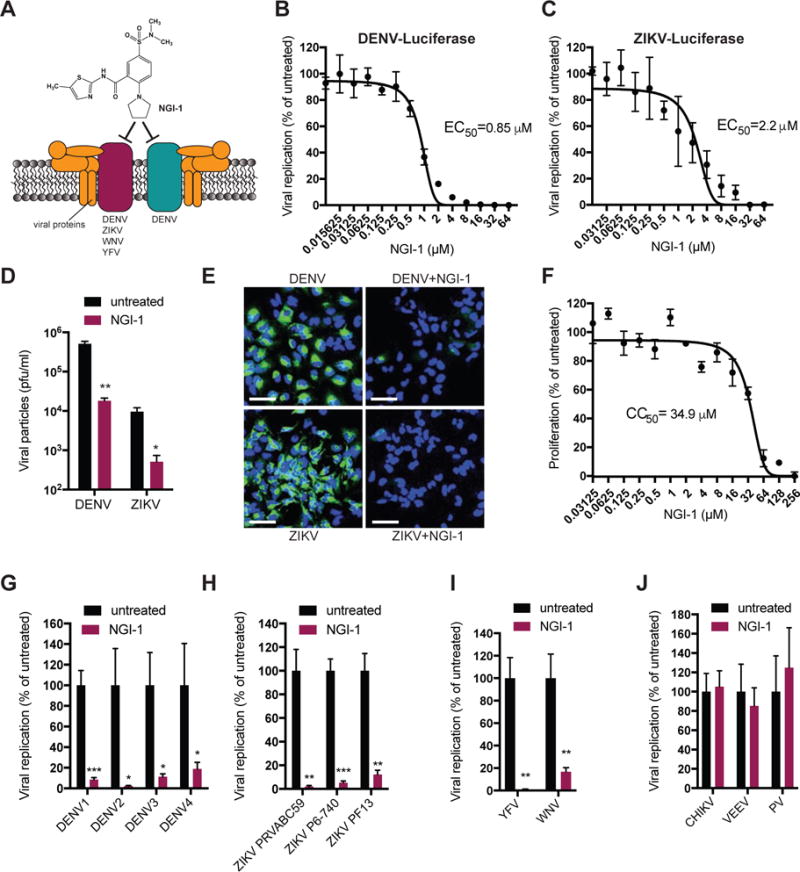
(A) Schematic depiction of the oligosaccharyltransferase complex interacting with flaviviral replication complexes.
(B) Dose-response curve of NGI-1 for DENV2 luciferase infection in HEK293 cells 48hpi.
(C) Dose-response curve of NGI-1 for ZIKV-luciferase infection in HEK293 cells 48hpi.
(D) Plaque-forming assay for DENV2 and ZIKV PRVABC59 harvested from infected HEK293 cells untreated and treated with 8 μM NGI-1 for 48h.
(E) Immunofluorescence of DENV2- or ZIKV PRVABC59-infected Huh7 cells with or without NGI-1 treatment for 48h. Infection was assessed by fluorescence microscopy of stained viral envelope protein (green) and cell nuclei (DAPI). Scale bar is 70 μm.
(F) Proliferation assay for HEK293 cells treated with different NGI-1 concentrations for 48h.
(G–J) Quantitative RT-PCR for RNA of the four different DENV serotypes (DENV1-4), different ZIKV strains, YFV and WNV, and Chikungunya (CHIKV), Venezuelan equine encephalitis (VEEV) and Polio virus (PV), untreated and treated with 8 μM NGI-1 for 48h.
In all figures, experiments were performed 2 or 3 times with 3 biological replicates in each experiment, and one representative is shown, Values are shown as mean ± SD in (B)–(F), and as mean ± SEM in (G)–(J). P-values are defined as ∗ p < 0.05; ∗∗ p < 0.01; *** p < 0.001.
NGI-1 exhibits broad antiviral activity against multiple flaviviruses
Dengue is caused by four DENV serotypes (DENV1-4), which are genetically diverse and differ in virulence and endemic capacity. Multiple serotype often co-circulate thus severely increasing the risk of dengue hemorrhagic fever through cross-reactivity of antibodies against one serotype against a second serotype (OhAinle et al., 2011). Therefore a useful antiviral should be efficacious against all four DENV serotypes. We demonstrated that NGI-1 treatment led to a significant reduction of viral RNA of DENV1-4 isolates (Figure 1G), which recapitulated the genetic requirement of the OST complex for all serotypes (Marceau et al., 2016). Similarly, we showed that NGI-1 had activity against different ZIKV strains from Puerto Rico (PRVABC59), Malaysia (P6–740) and French Polynesia (PF13) in infections of HEK293 cells (Figure 1H) making it a promising drug candidate for both the current and potentially future epidemics.
Moreover, we noted substantial decrease of viral WNV and YFV RNA in NGI-1 treated HEK293 cells (Figure 1I). By contrast, we did not observe a reduction in viral RNA for the infection with the mosquito-borne alphaviruses Chikungunya virus (CHIKV) and Venezuelan equine encephalitis virus (VEEV) as well as the picornavirus poliovirus (Figure 1J). These results highlight the potential of NGI-1 to be a specific, broad-spectrum, pan-flavivirus antiviral compound.
NGI-1 blocks viral RNA replication independent of inhibition of the catalytic activity of the OST complex
We previously pinpointed the role of the OST complex to viral RNA replication during DENV infection (Marceau et al., 2016). To determine which step of the flavivirus life cycle is inhibited by NGI-1, we used a viral replicon assay, which bypasses entry and allows analysis of the effect of NGI-1 on translation and replication of viral RNA. While we did not see a difference in the first 10 hours post viral RNA electroporation, which is reflective of the initial translation of the viral (+)RNA genome, viral RNA replication (apparent at time points after 10 hours) was reduced by NGI-1 treatment (Figure 2A).
Figure 2. NGI-1 blocks viral RNA replication and antiviral effect is independent of inhibiting the N-linked glycosylation activity of the OST complex.

(A) Time course of electroporated DENV replicon expressing Renilla luciferase in untreated or NGI-1 treated HEK293 cells.
(B) Quantitative RT-PCR for ZIKV PRVABC59 RNA in HAP1 cells expressing either WT or catalytically inactive STT3A, untreated or treated with NGI-1.
(C) Immunoblot analysis of DENV prM, E and NS1 proteins from lysates of DENV infected cells under treatment of different NGI-1 concentrations for 48h.
(D) Replication of DENV expressing Renilla luciferase in WT Huh7 cells, isogenic MAGT1 KO cells, and MAGT1 KO cells complemented with wt MAGT1, catalytically dead MAGT1, mCherry vector or empty vector.
In all figures, experiments were performed 2 or 3 times with 3 biological replicates in each experiment, and one representative is shown, Values are shown as mean ± SD in (A) and (D), and as mean ± SEM in (B). P-values are defined as ∗ p < 0.05; ∗∗ p < 0.01; *** p < 0.001; **** p < 0.0001.
We also previously found that DENV RNA replication does not require the catalytic N-glycosylation activity of the OST complexes (Marceau et al., 2016). This opens the possibility of targeting the OST complex without the inhibition of catalytic activity or glycosylation per se, while still blocking viral infection. The uncoupling of the antiviral and the cellular activity could thus lead to the development of a molecule with low cytotoxicity. To explore this hypothesis, we investigated ZIKV, which is only dependent on the presence of STT3A but does not require STT3B for RNA replication (Figure S2A). We observed that ZIKV replication was similarly affected in treated cells containing catalytically inactive STT3A as in WT cells (Figure 2B) suggesting that the antiviral effect of NGI-1 is largely independent from blocking glycosylation of viral or cellular proteins.
Additionally, given the importance of N-glycosylation for prM, E and NS1 viral proteins and the effect of NGI-1 on cellular OST activity, we performed immunoblot analysis for viral glycoproteins under NGI-1 treatment (Figure 2C). For prM, we observed an appearance of a lower band at higher NGI-1 concentrations representing the deglycosylated form of the protein. This may result in a small fraction of unglycosylated/immature virions that may exhibit a reduced infectivity. However, the fraction of the deglycosylated form of prM was less than half based on band intensity and thus unlikely to account for the full antiviral effect of NGI-1 (10–100 fold decrease in viral RNA or particle formation). Moreover, we pinpointed the role of the OST complex in the viral RNA replication phase using a replicon system (Figure 2A) but an additional effect on infectious virion assembly and spread cannot fully be ruled out. DENV E already showed two bands in the untreated condition, which could represent a certain fraction of unglycosylated protein or a degradation product. We did not see an increase of deglycosylated proteins with increasing NGI-1 concentrations. This makes it difficult to conclude whether NGI-1 has a de-glycosylation effect on DENV E protein. Finally, for DENV NS1, we observed an appearing faint band at higher NGI-1 concentrations. Overall, we conclude that at this dose range the antiviral effect is not largely due to an inhibition of glycosylation of viral proteins. While NGI-1 most likely impairs glycosylation of viral proteins to a certain degree, the majority of viral proteins are still properly glycosylated at concentrations where we observed a significant reduction of viral infection.
Lastly, we examined the oxidoreductase catalytic activity of MAGT1, a subunit of the STT3B OST isoform, which is necessary for N-glycosylation of cysteine-proximal acceptor sites in glycoproteins and was recently reported to be critical for DENV infection (Cherepanova et al., 2014; Lin et al., 2017; Schulz et al., 2009). Oxidoreductase activity is mediated by a thioredoxin-like fold with a characteristic CxxC active-site motif (Sevier and Kaiser, 2002). We generated isogenic MAGT1 knockout cells, complemented with wt (CxxC) MAGT1 or a catalytically dead version (SxxS) of MAGT1, and found that both wt and catalytically dead MAGT1 were able to restore DENV replication (Figure 2D, S2B). This indicates that redox activity mediated by the CxxC active site of MAGT1 is not required for DENV infection, contrary to the previous report (Lin et al., 2017). Thus, the mode of action of NGI-1 is not through inhibition of the oxidoreductase activity. The different outcome in the other study may be due to the use of a different active site mutant (AxxA in the Lin et al study versus SxxS in our study) although both mutations completely abolish classical redox activity.
Together, this data shows that the antiviral effect of NGI-1 is independent of the N-glycosylation and oxidoreductase activities of the OST complex, suggesting it may be possible to develop a compound that targets the viral function of the OST complex more selectively and without interfering with the cellular function.
Viral adaptation to STT3A and STT3B knockout cells shows that NGI-1 directly acts on OST complex for its antiviral effect
To further corroborate the OST complex as an antiviral target of NGI-1, we employed a viral evolution strategy. We performed serial passaging of DENV on STT3A- or STT3B-KO cells, and detected four distinct mutations in the isolated and sequenced viral RNA (Figure 3A). We compared the mutations that arose to all complete genome sequences of DENV2 in the NCBI database. Interestingly, we found that the acquired nucleotides are extremely rare (<0.5%, with the exception of T4098C, which is a synonymous mutation) in those genome positions in naturally occurring DENV genomes (Figure S3A). Thus, the likelihood that a circulating virus would already be resistant to the OST inhibitor is low. We then tested whether the isolated adapted viruses are insensitive to NGI-1 treatment, as they have lost the requirement for STT3A and STT3B for viral replication. Strikingly, while we observed a significant reduction in viral RNA for the WT DENV upon NGI-1 treatment, the adapted viruses were completely unaffected (Figure 3B), suggesting that the acquired mutations confer resistance to the drug. We introduced the mutations individually and in combination in a DENV infectious clone expressing luciferase and observed that single or double mutations did not confer the ability to replicate in STT3A- or STT3B-KO cells, that the three shared mutations led to a moderate level of replication, and that all four mutations enabled DENV to replicate more efficiently in KO cells (Figure S3B). Importantly, the adapted DENV reporters were significantly less affected than the WT reporter virus at increasing NGI-1 concentrations (Figure 3C).
Figure 3. DENV adaptation reveals that NGI-1 directly acts on STT3A and STT3B for its antiviral activity.
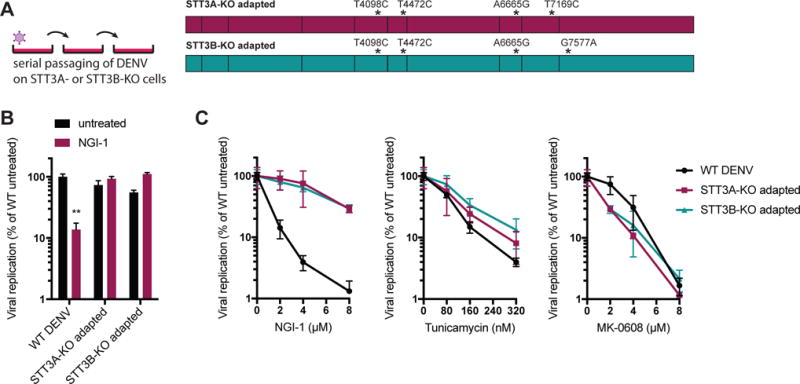
(A) Sequencing of DENV passaged on STT3A- or STT3B-KO cells revealed four distinct mutations in viral genomes. 1 and 3 viruses were independently isolated for STT3B-KO and STT3A-KO adaptations, respectively.
(B) Quantitative RT-PCR for DENV RNA of WT and STT3A- and STT3B-adapted viruses untreated or treated with NGI-1. The experiment was performed 3 times with 3 biological replicates each, and one representative is shown. Values are shown as mean ± SEM and p-value is defined as ∗∗ p < 0.01.
(C) Replication of dengue luciferase reporter virus with WT genomic sequence or containing the four adaptive mutations in HEK293 cells treated with different concentrations of NGI-1, Tunicamycin or MK-0608. The experiment was performed 2 times with 3 biological replicates each, and one representative is shown. Values are shown as mean ± SD.
We also tested whether the acquired mutations confer specific physical independence from the OST complex or a general loss of the requirement for N-linked glycosylation of viral proteins. Treatment with tunicamycin, a global N-glycosylation inhibitor that blocks the transfer of N-acetylglucosamine-1-phosphate to dolichol monophosphate upstream of the OST-mediated glycan transfer to the nascent polypeptide (Heifetz et al., 1979), led to marked reduction of replication of all three viruses (Figure 3C), indicating that adapted mutations and mode of action of NGI-1 are specific to the OST complex but independent of the N-glycosylation. To further exclude that the adaptive mutations facilitate faster replication and/or broader antiviral drug resistance, we treated the viruses with MK-0608, a nucleoside analogue shown to potently inhibit replication of several mosquito-borne flaviviruses including DENV (Chen et al., 2015) and ZIKV (Eyer et al., 2016). We did not observe any differences in antiviral activity of MK-0608 between the WT and the adapted viruses (Figure 3C). Taken together, these results suggest that NGI-1 inhibits the direct utilization of the OST complex for the viral replication machinery, independent of the cellular N-glycosylation function.
Inhibition of the OST complex decreases dengue and Zika virus infection in primary cell types
Human skin is the initial point-of-entry for these mosquito-borne viruses. Upon infection skin immune cells including dendritic cells take up and disseminate the virus (Wu et al., 2000). Additionally, ZIKV has been shown to infect the placenta and the brain of both fetuses and adults (Martines et al., 2016; Mlakar et al., 2016; Tang et al., 2016).
Therefore the efficacy of NGI-1 in various primary and disease-relevant cell types was examined. For DENV, we observed marked reduction of replication in normal human dermis fibroblasts (NHDF), monocyte-derived dendritic cells (MoDC) and Raji DC-SIGN, treated with NGI-1 (Figures 4A–C). Similarly, ZIKV infection was decreased in NHDF, MoDC, JEG-3 placental cells and human neural progenitor cells (hNPC) when treated with NGI-1 (Figures 4D–G). Finally, we also saw a significant reduction of viral infection in immunofluorescence experiments (Figure 4H–I).
Figure 4. NGI-1 has antiviral activity against DENV and ZIKV in disease-relevant and primary cell types.

(A–G) Replication of dengue or Zika luciferase virus (purple) as measured by luciferase activity and cellular proliferation (black) of Normal Human Dermis Fibroblasts (NHDF), monocyte-derived dendritic cells (MoDC), Raji DC-SIGN lymphocytes, JEG-3 placental cells and human Neural Progenitor Cells (hNPC) treated with different concentrations of NGI-1. Note that cells used in proliferation assays were uninfected, and that same proliferation data is used in Fig. 4A and 4D for NHDF, and in Fig. 4B and 4E for MoDC.
(H) Immunofluorescence of ZIKV-infected JEG-3 and hNPC, and DENV-infected NHDF cultured with and without NGI-1. Infection was assessed by fluorescence microscopy of stained viral envelope protein (green) and cell nuclei (DAPI). Scale bar is 70 μm.
(I) Percentage of DENV-infected Raji DC-SIGN (untreated or treated with NGI-1) as measured by flow cytometry.
In all figures, experiments were performed 2 or 3 times with 3 biological replicates in each experiment; one representative is shown. Values are shown as mean ± SD and p-value is defined as ∗∗∗∗ p < 0.0001.
Discussion
Genome-scale genetic knockout screens are a powerful approach to reveal host factors essential for viral replication and provide new candidate targets for antiviral drug development (Puschnik et al., 2017). Previously, other studies identified compounds that showed promising antiviral activity against DENV or ZIKV. For example, repurposing FDA-approved drugs is a valuable strategy to identify new antivirals with acceptable toxicity and a potential fast path to the clinic (Barrows et al., 2016; Xu et al., 2016). Furthermore, natural products can exhibit antiviral activity (Estoppey et al., 2017; Rausch et al., 2017). Recently, the fungal compound cavinafungin was shown to be potent and selectively active against DENV and ZIKV (Estoppey et al., 2017). Interestingly, genome-wide profiling in human cells identified the signal peptidase as a cellular target. Remarkably, this is in congruence with the results of genetic screens for flaviviral host factors (Marceau et al., 2016; Zhang et al., 2016) underscoring the potential of host-directed antiviral drug targets. Similarly, we observed that targeting the OST complex using a small molecule protected cells from flaviviral infection. Several properties of the OST complex and its use in viral replication open the possibility for further improvement of the lead compound: Firstly, the catalytic subunit of the OST complex is duplicated into two paralogues STT3A and STT3B in mammalian cells and each isoform is present in distinct protein complexes. While the STT3A complex is important for the co-translational N-linked glycosylation of the majority of glycoproteins, and the STT3B complex is important for the co-translational or post-translational glycosylation of acceptor sites that have been skipped by the STT3A complex, they largely fulfill redundant functions (Ruiz-Canada et al., 2009). This allows the development of selective STT3A or STT3B inhibitors, which presumably have fewer effects on global cellular glycosylation. Secondly, flaviviruses do not require the catalytic activity of the OST complex for their replication and NGI-1 blocked viral replication of STT3A dependent ZIKV in the setting of catalytically inactive STT3A. This suggests the possibility to modulate viral replication without inhibiting the catalytic function of the OST complex and thus compromising cellular N-glycosylation. Lastly, we pinpointed activity of NGI-1 to the interference of the OST usage of the viral replication machinery by utilizing adapted viruses. Interestingly, both the STT3A- and STT3B adapted virus contained 4 distinct mutations, 3 of which were shared. This highlights both the commonality as well as the divergence in the use of the two OST isoforms. The requirement for 4 escape mutations may further suggest that the barrier of resistance to NGI-1 is quite high compared to the commonly observed single or double escape mutations for direct-acting antivirals. Additionally, escape mutants of direct-acting antivirals usually contain mutations in the enzymatic target domains (protease or RNA polymerase domain). This suggests that there is no expected cross-resistance, which may be favorable for combination therapy.
In conclusion, our study demonstrates the advancement from the identification of a drug target through a genome-scale genetic screen to the development of a host-directed antiviral therapy with defined mechanism of action, broad antiviral activity and a potentially high barrier of resistance.
Experimental Procedures
Reporter virus assay
Cells were plated in 96-well plates and infected with DENV-2 luciferase reporter at a multiplicity of infection (moi) of 0.1 pfu/cell and ZIKV-luciferase from undiluted cell supernatant, respectively, and treated with different concentrations of NGI-1. Cells were lysed 48–96hpi. For luciferase reporter Coxsackievirus B3 cells were pretreated with NGI-1 for 24 hours, then infected under continuous treatment and lysed 24 hours post infection. Luciferase expression was measured using the Renilla Luciferase Assay system (Promega).
Cell proliferation and viability assays
Cells were plated in 96-well plates and treated with different concentrations of NGI-1. After 48h cell growth was measured using CellTiter-Glo assay (Promega) according to the manufacturer’s instructions or Trypan Blue solution.
Plaque-forming units assay
HEK293FT cells were plated in 96-well plates, infected with DENV-2 or ZIKV PRVABC59 at a moi of 0.1 pfu/cell and treated with 8 μM NGI-1. After 48 hours supernatant was harvested and a 10-fold dilutions series was performed. The dilutions were added to Huh7.5.1 cells plated in a 6-well plate and incubated for 2 hours and then aspirated. Cells were overlaid with 2% low melting point agarose/DMEM, grown for 6 days and then fixed using 15% paraformaldehyde. The cells were then stained overnight with crystal violet (0.1% crystal violet in 20% ethanol). Next day, the wells were extensively washed with water then dried and the resulting plaques were counted and plaque-forming units (pfu) per ml were calculated.
Immunofluorescence
Cells were plated in 8-well μ-slides (ibidi GmbH). Huh7 and JEG-3 cells were infected with DENV-2 or ZIKV PRVABC59 at a moi of 0.1 pfu/cell, NHDF and hNPC at a moi of 0.5 pfu/cell. Cells were then treated with 8 μM NGI-1 and after 40–48 hours fixed using 4% paraformaldehyde. Viral envelope protein was stained using Anti-Flavivirus Group Antigen antibody, clone D1-4G2-4-15 (EMD Millipore) at 1:1000 in blocking buffer (PBS containing 1% saponin, 1% Triton X-100, 5% FBS, 0.1% azide) for 1 hour, followed by 2 washes and by incubation with goat anti-mouse-IgG-Alexa-488 (Life technologies) and DAPI (Insitus) for 30min. After 3 washes with PBS cells were visualized using confocal microscopy. For the quantitative analysis of infectivity three random fields of view were acquired for each condition. DAPI and Alexa-488 signals were automatically counted using the “Analyze Particles” feature in ImageJ and the percentage of infected cells was calculated by dividing the number of Alexa-488 positive cells by the number of DAPI positive cells.
Quantitative RT-PCR
Cells were plated in 96-well plates (in triplicates for each condition), infected with a moi of 0.1 pfu/cell and treated with 8 μM NGI-1 for HEK293FT cells and 4 μM NGI-1 for HAP1 cells. RNA was harvested using the Ambion Cells-to-CT kit (ThermoFisher Scientific) at 48 hours post infection for flaviviruses, 24 hours for CHIKV and VEEV, and 16 hours for poliovirus. After RT reaction quantitative PCR was performed, where viral RNA levels were normalized to 18S RNA levels.
Replicon assay
The construction of the dengue replicon plasmid and the production of replicon RNA were previously described (Marceau et al., 2016). 3μg of purified replicon RNA was mixed with 106 HEK293FT cells in electroporation buffer and cells were electroporated using Bio-Rad Gene Pulser Xcell electroporator using the square wave protocol (volts= 120, pulse length= 1.5ms, number of pulses= 10, pulse interval= 1.5s, cuvette= 1mm). Electroporated cells were resuspended in cell culture medium without antibiotics and plated into 96-well plates. 4 μM NGI-1 was added 2 hours after electroporation. Cells were harvested at different timepoints and luciferase expression was measured using Renilla Luciferase Assay system (Promega).
Immunoblot assay
Immunoblot assays were performed as previously described (Marceau et al., 2016). HEK293 cells were infected with DENV (MOI=0.5) and treated with different concentrations of NGI-1 for 52h. As control, cells were treated with 10μg/ml tunicamycin for 4h before harvest. To detect DENV proteins, anti-prM (Genetex GTX128092) at a dilution of 1:1000, anti-E (Genetex GTX127277) at a dilution of 1:2000, and anti-NS1 (Genetex GTX630556) at a dilution of 1:2000 were used. As loading control anti-p84 (Genetex GTX70220) at a dilution of 1:5000 was used.
Flow cytometry
Raji-DC-SIGN cells were infected with DENV-2 at a moi of 0.5 pfu/cell and cultured untreated or treated with 8 μM NGI-1. 60hpi cells were fixed with 4% paraformaldehyde and permeabilized in Perm/Wash buffer (BD Biosciences). Viral antigen was stained using Anti-Flavivirus Group Antigen Antibody, clone D1-4G2-4-15 (EMD Millipore) at 1:800 in Perm/Wash buffer for 30min, followed by 2 washes and by incubation with goat anti-mouse-IgG-Alexa-488 (Life technologies) at 1:500 for 20min. After 3 washes samples were measured using a BD LRSFortessa LX-20 and data was analyzed using FlowJo 9.
Statistical analysis
GraphPad Prism 7 was used to analyze data. Untreated and treated conditions were compared using unpaired t-test. Curve fits were performed by non-linear regression with log(inhibitor) vs. normalized response and variable slope.
Supplementary Material
Acknowledgments
The work was funded in part by National Institutes of Health (NIH) DP2 AI104557 (JEC), NIH U19 AI109662 (JEC), David and Lucile Packard Foundation (JEC), Stanford Graduate Fellowship (ASP), Boehringer Ingelheim Fonds (ASP) and NSF-GFRP (CDM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
ASP, JNC and JEC designed the study. ASP performed viral infection and cell proliferation experiments. CDM generated adapted viruses and knockout cell lines. YSO generated complemented cell lines and viral stocks. KM produced viral stocks. NR synthesized NGI-1 compound. ASP and JEC wrote the manuscript.
References
- Barrows NJ, Campos RK, Powell ST, Prasanth KR, Schott-Lerner G, Soto-Acosta R, Galarza-Munoz G, McGrath EL, Urrabaz-Garza R, Gao J, et al. A Screen of FDA-Approved Drugs for Inhibitors of Zika Virus Infection. Cell host & microbe. 2016;20:259–270. doi: 10.1016/j.chom.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, Yokokawa F, Shi PY. The search for nucleoside/nucleotide analog inhibitors of dengue virus. Antiviral research. 2015;122:12–19. doi: 10.1016/j.antiviral.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Cherepanova NA, Shrimal S, Gilmore R. Oxidoreductase activity is necessary for N-glycosylation of cysteine-proximal acceptor sites in glycoproteins. The Journal of cell biology. 2014;206:525–539. doi: 10.1083/jcb.201404083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estoppey D, Lee CM, Janoschke M, Lee BH, Wan KF, Dong H, Mathys P, Filipuzzi I, Schuhmann T, Riedl R, et al. The Natural Product Cavinafungin Selectively Interferes with Zika and Dengue Virus Replication by Inhibition of the Host Signal Peptidase. Cell reports. 2017;19:451–460. doi: 10.1016/j.celrep.2017.03.071. [DOI] [PubMed] [Google Scholar]
- Eyer L, Nencka R, Huvarova I, Palus M, Joao Alves M, Gould EA, De Clercq E, Ruzek D. Nucleoside Inhibitors of Zika Virus. J Infect Dis. 2016;214:707–711. doi: 10.1093/infdis/jiw226. [DOI] [PubMed] [Google Scholar]
- Heifetz A, Keenan RW, Elbein AD. Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry. 1979;18:2186–2192. doi: 10.1021/bi00578a008. [DOI] [PubMed] [Google Scholar]
- Kaufmann SHE, Dorhoi A, Hotchkiss RS, Bartenschlager R. Host-directed therapies for bacterial and viral infections. Nat Rev Drug Discov. 2017 doi: 10.1038/nrd.2017.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DL, Cherepanova NA, Bozzacco L, MacDonald MR, Gilmore R, Tai AW. Dengue Virus Hijacks a Noncanonical Oxidoreductase Function of a Cellular Oligosaccharyltransferase Complex. mBio. 2017;8 doi: 10.1128/mBio.00939-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sambrooks C, Shrimal S, Khodier C, Flaherty DP, Rinis N, Charest JC, Gao N, Zhao P, Wells L, Lewis TA, et al. Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nature chemical biology. 2016;12:1023–1030. doi: 10.1038/nchembio.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, Abraham S, Choi JG, Shi G, Qi L, et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell reports. 2015;12:673–683. doi: 10.1016/j.celrep.2015.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature. 2016;535:159–163. doi: 10.1038/nature18631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martines RB, Bhatnagar J, Keating MK, Silva-Flannery L, Muehlenbachs A, Gary J, Goldsmith C, Hale G, Ritter J, Rollin D, et al. Notes from the Field: Evidence of Zika Virus Infection in Brain and Placental Tissues from Two Congenitally Infected Newborns and Two Fetal Losses--Brazil, 2015. MMWR Morb Mortal Wkly Rep. 2016;65:159–160. doi: 10.15585/mmwr.mm6506e1. [DOI] [PubMed] [Google Scholar]
- Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodusek V, et al. Zika Virus Associated with Microcephaly. N Engl J Med. 2016;374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- OhAinle M, Balmaseda A, Macalalad AR, Tellez Y, Zody MC, Saborio S, Nunez A, Lennon NJ, Birren BW, Gordon A, et al. Dynamics of dengue disease severity determined by the interplay between viral genetics and serotype-specific immunity. Sci Transl Med. 2011;3:114ra128. doi: 10.1126/scitranslmed.3003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puschnik AS, Majzoub K, Ooi YS, Carette JE. A CRISPR toolbox to study virus-host interactions. Nat Rev Microbiol. 2017 doi: 10.1038/nrmicro.2017.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch K, Hackett BA, Weinbren NL, Reeder SM, Sadovsky Y, Hunter CA, Schultz DC, Coyne CB, Cherry S. Screening Bioactives Reveals Nanchangmycin as a Broad Spectrum Antiviral Active against Zika Virus. Cell reports. 2017;18:804–815. doi: 10.1016/j.celrep.2016.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Canada C, Kelleher DJ, Gilmore R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell. 2009;136:272–283. doi: 10.1016/j.cell.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell reports. 2016;16:232–246. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Schulz BL, Stirnimann CU, Grimshaw JP, Brozzo MS, Fritsch F, Mohorko E, Capitani G, Glockshuber R, Grutter MG, Aebi M. Oxidoreductase activity of oligosaccharyltransferase subunits Ost3p and Ost6p defines site-specific glycosylation efficiency. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11061–11066. doi: 10.1073/pnas.0812515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 2002;3:836–847. doi: 10.1038/nrm954. [DOI] [PubMed] [Google Scholar]
- Shrimal S, Cherepanova NA, Gilmore R. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin Cell Dev Biol. 2015;41:71–78. doi: 10.1016/j.semcdb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell stem cell. 2016;18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SJ, Grouard-Vogel G, Sun W, Mascola JR, Brachtel E, Putvatana R, Louder MK, Filgueira L, Marovich MA, Wong HK, et al. Human skin Langerhans cells are targets of dengue virus infection. Nature medicine. 2000;6:816–820. doi: 10.1038/77553. [DOI] [PubMed] [Google Scholar]
- Xu M, Lee EM, Wen Z, Cheng Y, Huang WK, Qian X, Tcw J, Kouznetsova J, Ogden SC, Hammack C, et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nature medicine. 2016;22:1101–1107. doi: 10.1038/nm.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature. 2016;535:164–168. doi: 10.1038/nature18625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.