

Abstract
Inflammation arising from central and/or peripheral sources contributes to the pathogenesis of multiple neurodegenerative diseases including Parkinson's disease (PD). Emerging data suggest that differential activation of glia could lead to the pathogenesis and progression of PD. Here, we sought to determine the relationship between lipopolysaccharide (LPS) treatment, loss of dopaminergic neurons and differential activation of glia. Using a model of repeated injections with LPS (1 mg/kg, i.p. for 4 days), we found that LPS induced a 34% loss of dopamine neurons in the substantia nigra 19 days after initiation of treatment, but no further cell loss was observed at 36 days. LPS induced a strong pro-inflammatory response with increased mRNA expression of pro-inflammatory markers, including tumor necrosis factor-α (4.8-fold), inducible nitric oxide synthase (2.0-fold), interleukin-1 beta (8.9-fold), interleukin-6 (10.7-fold), and robust glial activation were observed at 1 day after final dose of LPS. These pro-inflammatory genes were then reduced at 19 days after treatment, when there was a rise in the anti-inflammatory genes Ym1 (1.8-fold) and arginase-1 (2.6-fold). Additionally, 36 days after the last LPS injection there was a significant increase in interleukin-10 (2.1-fold) expression. The qPCR data results were supported by protein data, including cytokine measurements, western blotting, and immunofluorescence in brain microglia. Taken together, these data demonstrate that progressive neurodegeneration in the substantia nigra following LPS is likely arrested by microglia shifting to an anti-inflammatory phenotype. Thus, strategies to promote resolution of neuroinflammation may be a promising avenue to slow the progressive loss of dopamine neurons in PD.
1. Introduction
Parkinson's disease (PD) is an age-related progressive neurodegenerative disease primarily characterized by a constellation of motor symptoms such as bradykinesia, rigidity, postural instability and tremors. However, non-motor symptoms including gastrointestinal dysfunction, autonomic dysfunction and cognitive deficits are increasingly being accepted as part of the clinical manifestation of PD (Sveinbjornsdottir, 2016). Pathologically, PD is characterized by the loss of dopamine neurons in the substantia nigra pars compacta (SNpc), the presence of Lewy bodies containing synuclein and the presence of activated glial cells, including microglia and astrocytes, accompanied by evidence of inflammation (Halliday and Stevens, 2011). Although the precise mechanisms leading to neurodegeneration are not known, increasing evidence points to a pivotal role for neuroinflammation in the initiation and progression of PD pathology (Collins et al., 2012; Wang et al., 2015).
Recent studies have established that both central and peripheral inflammation occurs in PD (Su and Federoff, 2014) and that inflammatory signals promote a cytotoxic environment in the nigrostriatal pathway, which has a large microglial population (Barnum and Tansey, 2010; Ferrari et al., 2006). Experimental studies demonstrate that bacterial lipopolysaccharide (LPS) can induce and/or accelerate dopaminergic neurodegeneration in mice and rats (Dutta et al., 2008). Both single and repeated dose LPS models have been used to study neurodegeneration of dopaminergic cells, in which LPS administration produces a prolonged state of neuroinflammation resulting in loss of dopamine neuron in days, weeks or months depending on the paradigm employed (Bodea et al., 2014; Cunningham et al., 2009; Liu and Bing, 2011; Qin et al., 2007). Frank-Cannon and co-workers showed that parkin null mice treated with LPS exhibited loss of TH-immunoreactive cells by three months of age, whereas untreated parkin null mice did not lose neurons until 6 months of age (Frank-Cannon et al., 2008). Collectively, a central role for inflammation in the initiation and progression of PD is becoming more evident, but the immunological landscape is complex and involves a variety of pro- and anti-inflammatory molecules resulting from interactions between differentially activated microglia and astrocytes (Cherry et al., 2014; Liddelow et al., 2017; Moehle and West, 2015).
The pro- and anti-inflammatory nomenclature regarding microglial states was originally developed according to the two major T cell activation states, Th1 and Th2. Based on this, macrophages and microglia can become activated towards more of a pro- or anti-inflammatory phenotype, oversimplified as M1 or M2, respectively (Cherry et al., 2014; Ransohoff, 2016a; Tang and Le, 2016). Upon stimulation with different cytokines, microglia can undergo classical (M1) or alternative (M2) activation to become pro-inflammatory or anti-inflammatory/resolving, respectively. However, emerging evidence demonstrates that microglia exist as a heterogeneous population in vivo with activation states existing along a continuum defined by the expression of specific genes that play an important role in either promoting inflammation and cytotoxic factor production, attenuating the inflammatory response, removing apoptotic cells or repairing tissue damage (Mills, 2012). The involvement of different types of microglial activation states within distinctive stages of PD is primarily based on established reports of increases in respective cytokines and morphological alteration of microglia (Brodacki et al., 2008; Mogi et al., 1996). Additional evidence supports a predominantly pro-inflammatory (M1) phenotype and neurodegenerative disease including Alzheimer's disease (AD) (Cribbs et al., 2012; Tang and Le, 2015; Varnum and Ikezu, 2012) and PD (Sanchez-Guajardo et al., 2013). However, there is limited information on the relative timecourse of these activation states in experimental animal models and their role in the initiation and progression of neurodegeneration.
In this study, we sought to characterize the timecourse of M1 and M2 inflammatory response in the mouse nigrostriatal pathway following repeated LPS treatment and determine its relationship with dopamine neuron loss. The data demonstrate that M1 activation is associated with dopamine neuron loss and that cessation of the progressive loss of dopamine neurons is accompanied by a switch to an M2 phenotype. Collectively, these data suggest that unresolved inflammation can drive dopamine neuron loss and that targeting resolution of inflammation may lead to cessation of the progressive dopamine neuron loss, providing a potential means to slow the progression of neuron loss observed in PD.
2. Materials and Methods
2.1 Animal care and LPS treatment
Twelve-week-old C57BL/6J male mice from Jackson Labs were randomly assigned and injected intraperitoneally (i.p.) with 100 μL LPS (from Salmonella abortus equi S-form, Enzo Life Sciences) at a dose of 1 μg/g or 100 μL phosphate-buffered saline (PBS; Life Technologies) as vehicle control for 4 consecutive days. This protocol was essentially based on that of Bodea and co-workers (Bodea et al., 2014). Animals were sacrificed at 1 day, 2 weeks and 4 weeks after the last dose of LPS, corresponding to days 5, 19, and 36 of treatment. Animal handling and experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the animal care committee of Rutgers-Robert Wood Johnson Medical School.
2.2 Tissue preparation
Animals were euthanized with CO2 or cervical dislocation and brains were removed and dissected on ice. The left striatum was isolated and frozen in liquid nitrogen, while the remaining forebrain from the right hemisphere and the hindbrain were drop fixed in 4% paraformaldehyde. Fixed tissues were maintained in paraformaldehyde at 4°C for 7 days and then were transferred to 30% sucrose containing 0.1% sodium azide prepared in PBS. For tissue sectioning, coronal sections were cut at 40 μm thickness on a freezing sliding microtome and stored in a cryoprotectant solution containing 25% ethylene glycol and 25% sucrose in PBS at -20°C (Hossain et al., 2015).
For western blots, frozen striatum was homogenized in 500 μL HEPES buffer (pH 7.4) plus protease inhibitor cocktail (Sigma, catalog #P8340) and centrifuged for 5 min at 2,000 rpm at 4°C to pellet cell debris and nuclei. An 100 μl aliquot of the supernatant was diluted with an equal amount of 0.2 N perchloric acid (PCA) containing 6% EDTA.2Na and 5% sodium metabisulfite for high pressure liquid chromatography (HPLC). An additional 100 μl aliquot of the supernatant was used for cytokine measurement of interleukin 1 beta (IL-1β), interleukin-6 (IL-6), interleukin-10 (IL-10), tumor necrosis factor alpha (TNF-α), and interferon gamma (IFN-γ) using a multiplex ELISA detection kit (Bio-Rad) according to manufacturer's protocols. The remainder of the supernatant was centrifuged for additional 45 min at 14,000 rpm at 4°C for western blot analysis.
2.3 Western immunoblotting
Following reconstitution with HEPES buffer and protein concentration quantification, 20 μg of protein/lane was separated on 4-12% Bis-Tris Mini gels (Schuh et al., 2009). Protein bands were then transferred to a PVDF membrane and blocked in 7.5% milk. Membranes were incubated overnight at 4°C with primary antibodies (tyrosine hydroxylase (TH, 1:2000, Millipore, catalog #AB152), dopamine transporter (DAT, 1:750, Millipore, catalog #MAB369), vesicular monoamine transporter 2 (VMAT2, 1:1000, (Cliburn et al., 2016)), glial fibrillary acidic protein (GFAP, 1:2000, Abcam, catalog #AB10062), cluster of differentiation molecule 11B (CD11b, 1:1000, Abcam, catalog #AB52478), TNF-α (1:750, Abcam, catalog #AB9348), arginase-1 (ARG1, 1:500, BD Biosciences, catalog #610708), inducible nitric oxide synthase (iNOS, 1:500, Santa Cruz, catalog #SC-650), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:5000; Santa Cruz. Cat #FL-335), followed by 1 hr incubation with species-appropriate secondary antibody (Bio-Rad, 1:1000-1:5000). Specific antibody bound bands were developed with Pierce super signal kit Dura West and visualized with an Alpha Innotech Fluorchem imaging system.
2.4 Immunohistochemistry and immunofluorescence
Free-floating brain sections were washed with PBS and then steamed in 0.1 M citrate buffer (pH 6.0) for 5 min for antigen retrieval. Sections were then incubated in background buster (INNOVEX Biosciences, catalog #NB306), followed by overnight incubation with primary antibody [TH (1:1000), DAT (1:750), GFAP (Abcam, 1:2000, catalog #AB4674), ARG1 (1:500), iNOS (Abgent, 1:200, catalog #ALS16734), or ionized calcium-binding adapter molecule 1 (IBA1, Wako Diagnostics, 1:850, catalog #019-19741)] in 1% bovine serum albumin (BSA) and 0.3% Triton X-100. Following rinsing, sections were incubated with Alexa Fluor secondary antibody (Thermo-Fisher, 1:1000-1:2000) for 1 h at room temperature. Sections were rinsed and mounted on slides and cover slipped with ProLong Gold reagent with or without DAPI (Life Technologies). TH and DAT were imaged on a fluorescent microscope (Zeiss AxioObserver D1, Carl Zeiss) equipped with AxioVision Software. For glial cell activation and cytokine evaluation, IBA1 (1:1000), GFAP (1:1000), iNos (1:500), and/or ARG1 (1:500) stains were conducted. Confocal z-stack images were acquired from a Biovision spinning disk microscope comprised of an upright Zeiss AxioImager Z1 microscope equipped with a Yokogawa CSUX1-5000 spinning disc. Images were collected under identical parameters and reconstructed using Fiji software (Schindelin et al., 2012).
For immunofluorescent staining of sialoadhesin (CD169, 1:200, Bio Rad, MCA884) and Iba1 (1:1,000), postmortem brain specimens from saline and LPS-treated C57BL/6J mice (male, 8-10 weeks old) were immersion fixed in 10% formalin for 7 days, cryoprotected in a 30% sucrose solution with 0.1% sodium azide, and stored at 4°C prior to sectioning. Free-floating sections (i.e., every 8-12th section beginning in the cortex through the midbrain) were rinsed in PBS (pH 7.4, 5 min × 6) and pre-blocked for 1 h at room temperature in a 10% BSA working stock, 1% Triton X, and PBS. After rinsing, sections then were placed in a primary antibody cocktail of rat CD169 and rabbit IBA1 diluted in PBS and 10% BSA working stock for 24 h at 4°C. The next day, sections were rinsed and incubated in a secondary antibody cocktail (1:200) of goat anti-rabbit Alexa Fluor 488 (Abcam, ab150157), goat anti-rabbit Alexa Fluor 594 (Thermo Fisher, A-11037), PBS, and 10% BSA working stock (1 h, RT). After a final rinse, sections were mounted on slides, dried, coverslipped using Fluoroshield with DAPI (Sigma, E6057), and stored at 4°C. Omission of the primary antibody cocktail was used as a negative control. Photomicrographs of the immunofluorescent staining were taken on a Leica DMi8 confocal microscope at 40× (N.A. 1.15 oil).
2.5 Unbiased stereology
For neuronal quantification, slides were double stained for TH (1:1000) and microtubule-associated protein 2 (MAP2, 1:1000, Sigma, catalog #M1406). The substantia nigra pars compacta (SN) was defined based on TH+ cells, as outlined previously (Alam et al., 2017; Baquet et al., 2009; Bradner et al., 2013). A counting frame of 50 μm × 50 μm with framing space of 200 μm and a height of 10 μm was chosen. Only the cells that came into focus within the counting frame height were counted. The coefficient error for all the animals counted was equal or below 0.1 and a minimum of 100 markers were counted within 40-60 framing sites for each animal. Unbiased stereology was performed using a Leica DM2500 (Leica Microsystems, Chicago, IL), and Stereologer computer assisted stereology system (Stereology Resource Center, St. Petersburg, FL). The SN was delineated using previously described criteria (West et al., 1991) at low magnification and every sixth section throughout the SN was sampled at higher magnification (63×) with the optical fractionator probe.
2.6 High performance liquid chromatography (HPLC)
Catecholamine levels were determined in the striatum using HPLC with electrochemical detection (ECD) as previously described (Richardson and Miller, 2004; Yochum et al., 2014). Briefly, the homogenates in PCA solution described above were centrifuged at 15,000 × g for 20 min at 4°C, the supernatant removed, and filtered through a 0.22 μm filter by centrifugation at 15,000 × g for 20 min at 4°C. The supernatants were analyzed for levels of dopamine (DA), 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 5-hydroxyindoleacetic acid (5-HIAA), and serotonin (5-HT) using HPLC-ECD and expressed as ng catecholamine/mg protein (Sheleg et al., 2013). Protein concentrations were determined by BCA protein assay kit from centrifuged pellets according to manufacturer's instructions (Thermo Scientific). Quantification was made by reference to calibration curves made with individual monoamine standards.
2.7 Quantitative Real-time Polymerase Chain Reaction
RNA was isolated from the striatum by homogenizing in TRIZOL reagent and purified by DNase digestion and column separation using QIAGEN RNeasy mini columns (Quanta) as described previously (Fortin et al., 2013). Reverse transcription was then performed using the iScript cDNA synthesis kit (Bio-Rad). PCR reactions were performed using PerfeCTa SYBER green (Quanta BioSciences) according to manufacturer's protocols. Genes of interest were normalized to Gapdh expression. Primer sequences are provided in Supplemental Table 1.
2.8 Stride length assessment
Stride lengths were measured by a method adapted from D'Hooge et al. (D'Hooge et al., 1999). Briefly, mice were studied longitudinally for variations in stride length at 5, 19, and 36 days after LPS treatment. The apparatus was an open field track (6-cm wide, 50-cm long with borders of 7-cm height) illuminated by a 60W light bulb arranged to lead into a cardboard box containing their home cage. Mice were habituated to the experimenter and to the track one day prior to starting the experiments by performing the runs twice and mice were placed in a new cage at commencement. Mouse fore and hind paws were moistened with commercially available ink before being released to walk across a lit runway containing a strip of paper (4.5-cm wide, 40-cm long). Stride lengths were measured with a ruler for hind and forepaws simultaneously, and the three longest stride lengths (analogous to maximal velocity) were averaged from each run. Prints at the beginning and end of the run were excluded due to changes in velocity as well as obvious decelerations or stops.
3. Statistics
Data are presented as mean ± SEM. Statistical analysis was performed using Prism (Graphpad, Software v6). To compare independent variables (i.e., LPS and time), we used two-way analysis of variance (ANOVA) with a Bonferroni correction. When significant main effects were indicated, Students t test was used to determine differences between control and treated animals. Statistical significance was considered at p value < 0.05.
4. Results
4.1 Progressive dopamine neuron loss follows LPS exposure
The nigrostriatal dopamine system has shown susceptibility to systemic inflammatory insults, and specifically to LPS. Following 4 daily doses of LPS (1 mg/kg), there was no significant loss of TH+ and MAP2+ neurons on experimental day 5, but a significant reduction of 36% and 32%, respectively, was observed on experimental day 19 (Figure 1). No significant additional loss of neurons was observed on day 36 when there remained a similar significant reduction of TH+ and MAP2+ cells (41% and 31% respectively compared with controls). These results indicate that LPS exposure induced progressive dopamine neuronal loss starting after day 5 through day 19, after which there was no additional significant cell loss.
Figure 1.
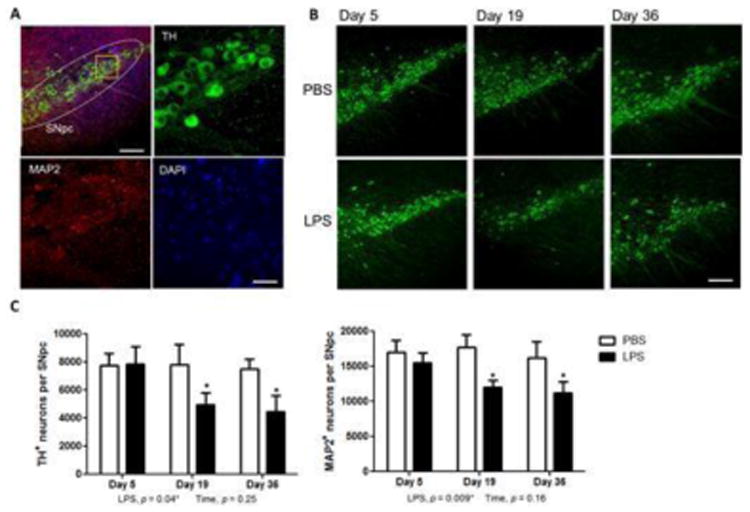
Time course of dopamine neuron loss in the substantia nigra in LPS-treated mice. (A) Combined immunofluorescent staining with TH (green), MAP2 (red) and DAPI (blue) at low magnification and at high magnification showing cell-specific staining. (B) Reduced staining of TH-positive neurons was determined on days 5, 19 and 36 after LPS treatment compared to PBS. (C) Quantification of TH+ and MAP2+ neurons in the SNpc of LPS mice show significant loss of cells on day 19, with no significant increase in cell loss on day 36. Bar: 50 μm (A) and 500 μm (B). Data represent mean ± SEM for 5 mice/group, *p < 0.05 for treatment effects.
4.2 LPS Administration Decreases Dopaminergic Terminal Markers in the Striatum
Following repeated administration of LPS for four days, we assessed the dopamine terminal markers, TH, DAT and VMAT2, and monitored the amount of dopamine and catecholamine levels in the striatum. LPS treatment led to a dramatic and progressive decline in TH and DAT staining in the striatum, with the decline initially developing at day 5 and progressing through day 19 (Figure 2). Changes in DAT levels were less severe than TH, but showed evident reduction by 19 and 36 days (Figure 2).
Figure 2.
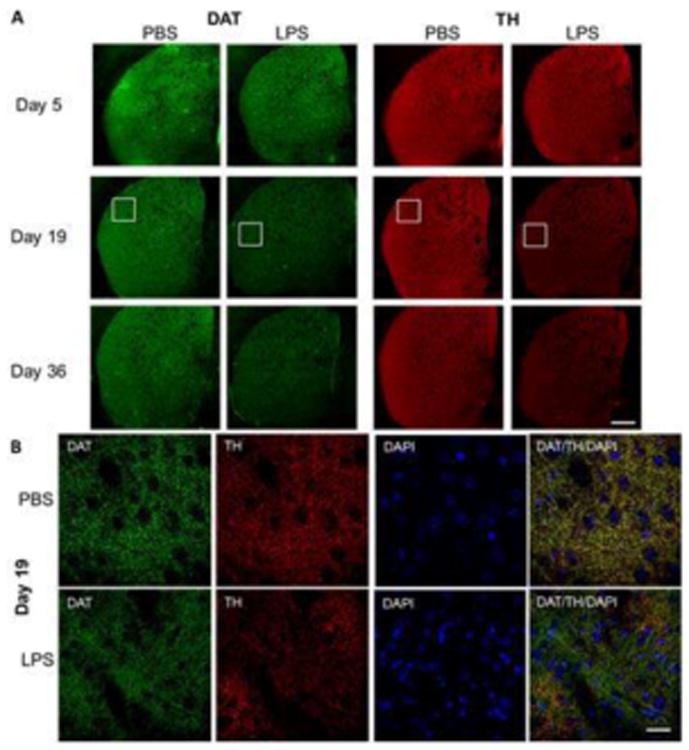
Representative immunofluorescent staining of TH and DAT in striatum of mice exposed to LPS. (A) Brain sections stained for TH (red), DAT (green), and DAPI (blue) is shown at 5, 19 and 36 days since initiation of LPS challenge. (B) Higher magnification pictures for days 5 and 19. Bar = 500 μm (A) and 100 μ (B).
Quantification of this effect in the striatum by western blotting revealed a modest decline in TH, DAT, and VMAT2 on experimental day 5 compared to controls (Figure 3A). However, LPS triggered a dramatic decline in dopamine terminal markers by day 19 which persisted to 36 days. The protein levels of DAT and VMAT2 were reduced by 81% and 57% on experimental day 19 (Figure 3B) and 52% and 46% on day 36, respectively, when compared with saline controls (Figure 3C). In addition, TH levels of LPS-treated mice were reduced by 62% compared to PBS-treated mice on experimental day 19 (Figure 3B), and by 34% on day 36 (Figure 3C). Further reduction in striatal dopaminergic markers beyond 19 days were not observed on day 36, with a moderate recovery of dopaminergic protein marker levels. Together, these results clearly demonstrate the loss of dopaminergic terminals after LPS exposure.
Figure 3.
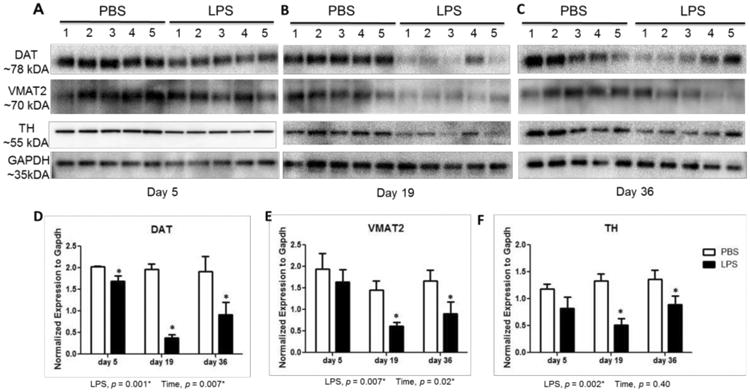
Loss of terminal dopaminergic markers in the striatum of LPS-treated mice. Protein levels in striatum for TH, DAT, and VMAT2 on experimental day 5 (A), day 19 (B), and day 36 (C) shows the loss of dopaminergic markers by day 19. (D) Western blots for DAT (D), VMAT2 (E) and TH (F) were quantified by densitometry and normalized to GAPDH levels. Data represent mean ± SEM for 5 mice/group, *p < 0.05 for treatment effect.
4.3 LPS Decreases Striatal Dopamine Levels
Along with the assessment of relevant protein levels to dynamic changes in dopaminergic neurotransmission, we quantified the levels of striatal dopamine and its metabolites. HPLC analysis of catecholamines in LPS-exposed mice showed deleterious reduction in levels of DA (73%), DOPAC (55%), and HVA (75%) on day 19 compared to PBS (Table 2). DOPAC levels were the first to decrease significantly, as seen by a 50% change at day 5 of LPS exposure. The deficits in striatal dopamine levels rebounded by day 36 after LPS treatment, suggesting a compensatory response. The levels of 5-HT and 5-HIAA were lower than control on day 19, but this did not reach statistical significance (Table 1).
Table 2.
Stride length in control and LPS treated mice.
| Front right | Front left | Rear right | Rear left | |
|---|---|---|---|---|
| Day 5 | ||||
| PBS | 6.92 ± 0.14 | 6.96 ± 0.10 | 7.04 ± 0.16 | 6.85 ± 0.22 |
| LPS | 6.74 ± 0.15 | 6.87 ± 0.18 | 6.84 ± 0.12 | 6.88 ± 0.18 |
| Day 19 | ||||
| PBS | 7.28 ± 0.19 | 7.24 ± 0.21 | 7.12 ± 0.12 | 7.07 ± 0.13 |
| LPS | 6.70 ± 0.10* | 6.68 ± 0.11* | 6.61 ± 0.14* | 6.62 ± 0.22 |
| Day 36 | ||||
| PBS | 7.67 ± 0.14 | 7.51 ± 0.16 | 7.52 ± 0.21 | 7.46 ± 0.11 |
| LPS | 7.01 ± 0.21* | 7.04 ± 0.11* | 6.99 ± 0.25* | 7.03 ± 0.28 |
Average velocity (average of 3 strides) was determined longitudinally in PBS and LPS-treated mice. Data represent mean ± SEM for 5 mice/group.
p < 0.05 for effect of LPS.
Table 1.
Striatal dopamine and metabolite levels in control and LPS treated mice.
| DA | DOPAC | HVA | 5-HIAA | 5-HT | |
|---|---|---|---|---|---|
| Day 5 | |||||
| PBS | 100.0 ± 25.9 | 100.0 ± 18.7 | 100.0 ± 30.4 | 100.0 ± 26.2 | 100.0 ± 26.8 |
| LPS | 66.5 ± 13.6 | 50.5 ± 7.7* | 68.9 ± 0.12 | 90.6 ± 4.0 | 81.6 ± 17.1 |
| Day 19 | |||||
| PBS | 100.0 ± 25.7 | 100.0 ± 23.1 | 100.0 ± 17.7 | 100.0 ± 19.2 | 100.0 ± 20.3 |
| LPS | 27.1 ± 16.7* | 45.0 ± 8.5* | 25.1 ± 0.14* | 67.7 ± 10.2 | 79.0 ± 14.7 |
| Day 36 | |||||
| PBS | 100.0 ± 14.9 | 100.0 ± 5.3 | 100.0 ± 16.5 | 100.0 ± 5.8 | 100.0 ± 10.0 |
| LPS | 61.8 ± 16.2* | 82.2 ± 12.9* | 79.8 ± 0.25* | 97.4 ± 7.8 | 85.1 ± 14.5 |
Monoamine levels were measured using HPLC-ECD. Specific loss of dopamine (DA) and metabolites DOPAC and homovanillic acid (HVA) is shown by 19 days, with some recovery at 36 days. No significant changes were observed for striatal serotonin (5-HT) or its metabolite, 5-hydroxy-indole acetic acid (5-HIAA). Data expressed as mean percent of control ± SEM for 4-5 mice/group.
p < 0.05 for effect of LPS. All statistical analysis was performed on raw data.
4.4 Reduction of Stride Length Accompanies Loss of Striatal Dopamine and Dopamine Neurons
Measurement of stride length has been established as a sensitive measure of dopamine loss in PD models (Tillerson et al., 2002). Here, stride lengths were not significantly affected by LPS on Day 5. However, LPS treatment resulted in significant decreases of stride length on Day 19, which remained significantly decreased on Day 36 (Table 2).
4.5 Glial Cells are Activated in the Striatum of LPS-treated Mice
To examine the glial activation, we labeled microglia and astrocytes using IBA1 and GFAP, respectively (Figure 4A). On day 5, there was increased staining of microglia and astrocyte cells, which peaked at 19 days. In addition, IBA1 stained microglia in LPS-exposed brains displayed increased staining intensity along with morphology indicative of activation, including hypertrophied amoeboid cell soma (Figure 4B). This staining appears to relate to endogenous microglia, since we observed no co-localization of IBA1 staining with CD169, a marker of infiltrating macrophages ((Gu et al., 2016); Supplemental Figure 1), consistent with that observed by Chen and co-workers (Chen et al., 2012).
Figure 4.
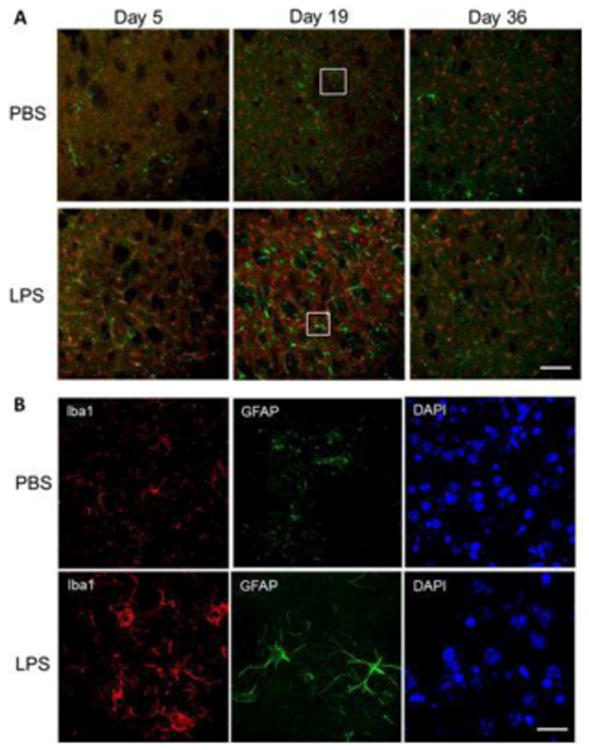
Activated microglia and astrocytes in the striatum of mice treated with LPS. (A) Immunofluorescence staining for microglia marker ionized calcium-binding adapter molecule 1 (Iba1, red), astrocyte marker glial fibrillary acidic protein (GFAP, green), and DAPI (blue) in striatum on experimental days 5, 19 and 36 after treatment with PBS or LPS. Microglial Iba1 levels were highly increased on day 5 and continued to day 36. (B) Higher magnification at 19 days demonstrates the difference in microglia phenotype and activation in LPS-treated mice. Bar: 500 μm (A) and 50 μm (B).
To validate the IBA1 and GFAP immunohistochemical results following LPS treatment, we performed western immunoblotting on striatal extracts and qPCR for mRNA expression of glial cell markers. On experimental day 5, CD11b and GFAP protein increased in LPS-treated mice compared to controls (Figure 5A,B). The CD11b response remained significantly increased (2.1-fold) compared to controls through day 19 but returned to control levels by day 36, whereas GFAP levels were sustained throughout all timepoints.
Figure 5.
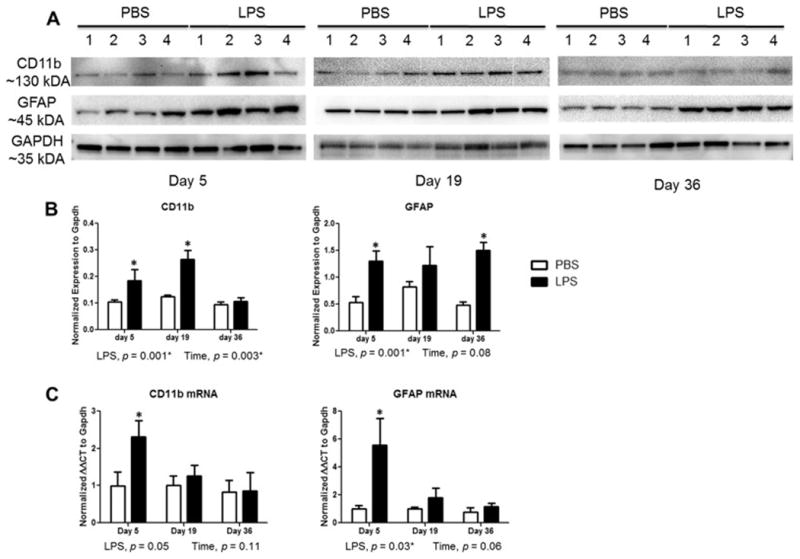
Sustained microglia and astrocyte activation in striatum of mice after systemic challenge with LPS. (A) Western blot analysis shows an increase in microglia marker CD11b and astrocyte marker GFAP following LPS administration. (B) Western data were quantified by densitometry and normalized to GAPDH levels. (C) qPCR analysis of for Cd11b and Gfap in striatum of mice. Data represent mean ± SEM of 4-5 mice/group, *p < 0.05 for treatment effects.
The mRNA data support the protein expression findings, with a significant 5.5-fold increase in Gfap expression in LPS-treated mice compared with controls on experimental day 5, but this was not significantly different than control on day 19 or 36 (Figure 5C). Significantly increased mRNA expression of allograft inflammatory factor (Aif1), also known as IBA1, following LPS treatment was more apparent at earlier time points on days 5 and 19 than day 36 compared to control animals, similar to that observed with the immunofluorescence staining (data not shown). Expression of the microglial marker Cd11b was significantly increased on day 5 following LPS administration compared to controls (Figure 5C), this did not quite reach statistical significance. Gene expression of the microglial phagocytic marker Cd68 was significantly increased by LPS treatment with an increase of 2.1-fold on day 19 (data not shown). Together, we observed increased gene transcription and protein expression of microglia and astrocyte markers after initial exposure of LPS, and found that the levels of these glial activation markers are altered after longer periods of time following LPS.
4.6 Increased Pro-inflammatory Cytokine Production in Striatum Early in Degeneration
To characterize the M1 and M2 phenotypes, we analyzed the cytokine profile in the striatum of controls and LPS-treated mice using multiplexed ELISA. Levels of TNF-α and IL-Iβ were elevated on days 5 and 19 up to 6-fold higher than controls (Figure 6A). Similarly, levels of IL-6 were increased by 1.8-fold on day 5; however, expression returned to control levels by day 19. Monitoring levels of IFN-γ, we found a significant increase (2.6-fold) over controls only on day 19 (Figure 6B). On the other hand, higher levels of the anti-inflammatory protein IL-10 were over 2-fold higher than controls on day 36 (Figure 6B).
Figure 6.
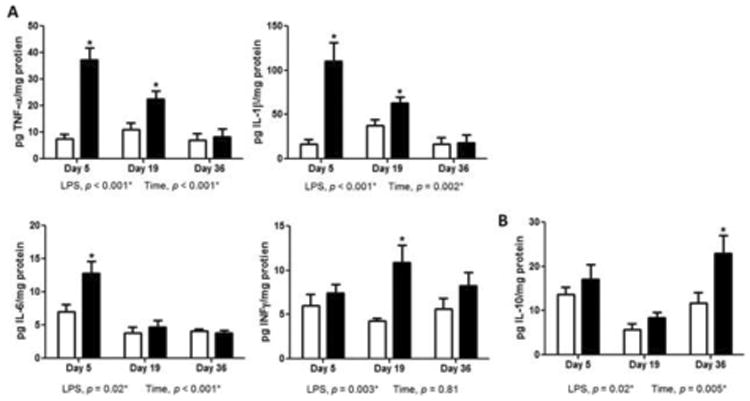
Increased production of proinflammatory cytokines in striatum of mice after introduction to LPS. (A) ELISA measurement of TNF-α, IL-Iβ, and IL-6 and INF-γ protein in striatum homogenate on experimental days 5, 19, and 36. LPS produced pronounced elevation of proinflammatory cytokines on day 5. (B) ELISA measurement of IL-10 protein in striatum during experimental course with PBS or LPS showed elevation on experimental days 36. Data represent mean ± SEM of 4-5 mice/group, *p < 0.05 for treatment effects.
4.7 Cytokine mRNA Profile Shifts from Pro- to Anti-inflammatory Starting at 19 days after LPS Exposure
We next examined a panel of phenotype-specific glial genes by qPCR that reflect the overall inflammatory response in the striatum at the different timepoints following LPS. In general, the pro-inflammatory markers were expressed the highest at day 5, followed by a mixed expression of both pro-and anti-inflammatory markers at day 19, and finally ending with a predominantly anti-inflammatory profile at day 36. On experimental day 5, we found significant elevations of mRNA expressions for iNos by 2-fold, Tnf-α by 4.8-fold, Il-Iβ by 8.9-fold, and Il-6 by 10.7-fold in the striatum of LPS-treated mice (Figure 7A,B). Monocyte chemotactic protein 1 (Mcp1) increased at day 5 after the first LPS exposure, but this did not quite reach statistical significance (Figure 7B). Following 19 days LPS treatment, Ifn-γ was significantly increased (1.9-fold) compared with controls (Figure 7B). Expression profile of M2-like genes began to rise on day 19 as the level of arginase-1 increased and reached to 2.7-fold higher on day 36 when compared to control (Figure 7C). We also found a 2.1-fold elevation of IL-10 and 2.5-fold elevation of mannose receptor, C type 1 (Mrc1) at 36 days after LPS treatment. The mRNA expression levels of other prototypical M2 markers such as resistin-like molecule alpha1 (Fizz1) and chitinase 3-like 3 (Ym1) showed some increases at post exposure day 36 but were not significantly different from control (Figure 7D). In addition, a significant (2.5 fold) elevation of transforming growth factor beta (Tgf-β) by LPS administration was observed on day 5, indicating the presence of mixed inflammatory profiles even at the earliest timepoint.
Figure 7.
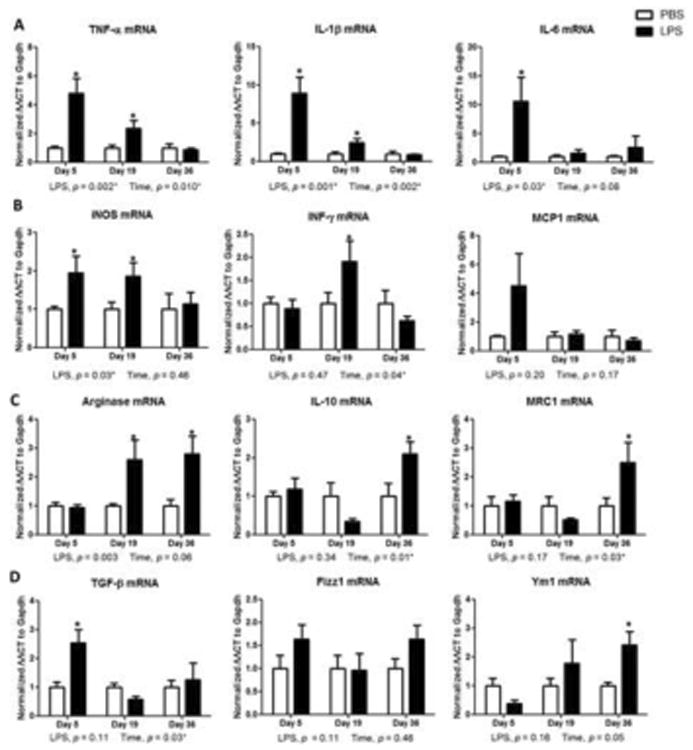
Expression profile of inflammatory genes in striatum of mice. (A) Quantitative PCR for cytokines Tnfα, Il1β, and Il6, and (B) iNos, Inf-γ, and Mcp1 in striatum of mice during course of experiment. LPS induced a strong induction of M1 cytokines at day 5 treatment, and returned to basal levels around day 19. (C) Arginase-1, Il-10, and Mrc1 mRNA expression by qPCR and (D) Tgfβ, Fizz1, and Ym1 in striatum of mice during course of experiment. LPS induced a rise in arginase-1 starting at day 19, and a robust induction of M2 cytokines at day 36 LPS treatment. Data represent mean ± SEM of 4-5 mice/group, *p < 0.05 for treatment effects.
4.8 Protein Data Support Transition from Pro- to Anti-inflammatory Environment
To confirm the mRNA results, we examined protein levels of prototypical microglial markers using western blotting techniques. TNF-α and iNOS levels were significantly increased by 1.8 to 2.3-fold over days 5-19 following LPS treatment (Figure 8A, B, D). Levels were not significantly different from control 36 days after LPS treatment. In contrast, expression of the anti-inflammatory marker arginase-1 increased by 2.2-fold over controls on day 36 (Figure 8C, D).
Figure 8.
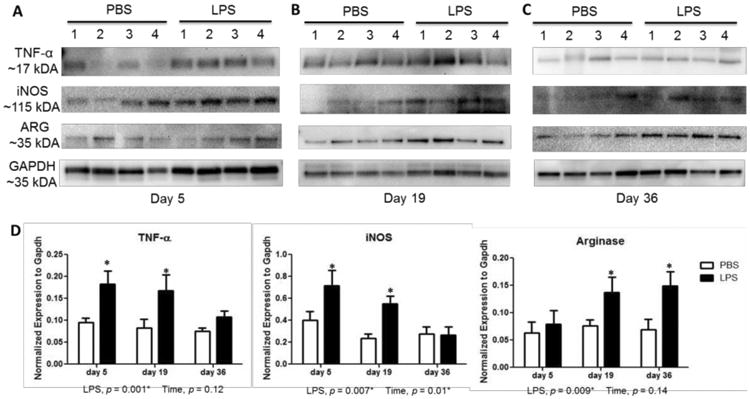
Inflammation phenotype-specific proteins in striatum of mice after systemic challenge with LPS. Western blot analysis shows an increase TNF-α and iNOS at 5 days (A) and 19 days (B) following LPS administration. Induction of Arginase-1 (ARG) protein levels at 19 days (B) and 36 days (C) after LPS treatment. (D) Westerns were quantified by densitometry and normalized to GAPDH levels. Data represent mean ± SEM of 4 mice/group, *p < 0.05 for treatment effects.
Immunoreactivity of the pro-inflammatory marker iNOS and anti-inflammatory marker arginase-1 in glial cells further demonstrated these results. We found increased staining for iNOS in IBA1+ microglia in the SNpc on day 5 (Figure 9A), but these protein levels returned to control levels on day 36. On the other hand, arginase-1 protein levels in IBA1+ microglia (Figure 9B) were no different than control at day 5, but were higher on day 36. Overall, the phenotypic conversion of microglia, evident by the predominant change from iNOS to arginase-1 expression profile, followed a similar course to the cessation of dopamine neuron loss.
Figure 9.
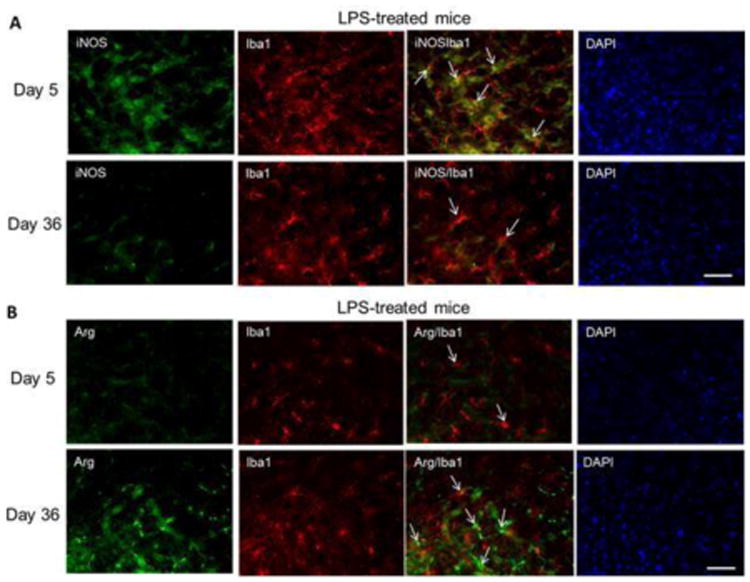
Expression of iNOS in IBA1+ cells 19 days after LPS treatment shifts to higher arginase-1 protein levels at 36 days. (A) Microglia were visualized by immunofluorescence staining for IBA1 (red) in the striatum at day 5 and day 36. Representative images for iNOS (green) and DAPI (blue) on experimental days 5 and 36 after treatment with LPS. (B) Immunofluorescence staining for arginase-1 (green) and DAPI (blue) in striatum on experimental days 5 and 36 after treatment with LPS. Bar: 50 μm.
5. Discussion
Central and peripheral inflammation are known to contribute to the pathophysiology of PD. However, the role of M1 and M2 immune activation in PD is not well established. Here, we used a model of repeated LPS administration to produce a sustained neuroinflammatory response that elicited nigrostriatal dopaminergic lesions and triggered deficits in locomotor behavior. In addition, progressive dopaminergic cell loss occurred while the microglial inflammatory phenotype was predominantly pro-inflammatory or M1, as characterized by the induction of iNOS and the production of M1-indicating cytokines such as TNF-α. There was a significant reduction in TH+ neurons in the SNpc on experimental day 19, which was validated with a significant decline in total MAP2+ cells. Starting on day 19, an anti-inflammatory or M2 phenotype started to appear and was predominant on day 36, as characterized by induction of arginase-1 and the elevation of resolving cytokines such as IL-10. This phenotypic switch coincided with the cessation of dopamine neuron death, as there was no statistically significant increase in cell loss on day 36 when compared to day 19. These results indicate that the resolution of inflammation occurring between day 19 and day 36 following LPS exposure may play an important role in the cessation of progressive dopamine neuron loss in this model.
Evidence for microglial dysfunction is present in PD patients, including higher incidence of dystrophic microglia (de-ramification, shortened cytoplasmic processes, cytoplasmic fragmentation, amoeboid soma morphology) and higher levels of pro-inflammatory mediators such as TNFα (Halliday and Stevens, 2011). In our model, LPS challenge produced a robust M1 microglial response as early as 1 day following the last injection and along with persistently increased astrocyte activation. The microglial markers CD11b and Aif1 were highly elevated at both experimental days 5 and 19 in the striatum. In addition to microglial activation, the astrocytic marker GFAP was consistently elevated at all timepoints. Several studies have examined the inflammatory response using this repeated LPS paradigm (Bodea et al., 2014; Chen et al., 2012); however, this is the first study to examine inflammatory markers and dopamine neuron loss past day 19 in this particular model. Our findings exemplify the need to examine later time points and use full time course studies when exploring neuroinflammation-related questions, including microglial activation phenotypes.
Along with the significant loss of dopaminergic neurons in the SNpc, levels of striatal dopamine and its metabolites were significantly decreased at 19 days, but showed some improvement at 36 days. This rebound in dopamine levels, in contrast with no change in neuronal counts in the SNpc, is likely because of compensatory mechanisms like decreased dopamine metabolism, novo terminal sprouting, and TH upregulation, which have been observed in other toxicant-induced models among rodents and primates (McCormack et al., 2002; Mounayar et al., 2007; Rappold et al., 2011; Song and Haber, 2000). Decreases in striatal dopamine and behavior deficits have been inconsistent among animals exposed to LPS despite the consistent observation of neuroinflammation and dopamine neuron loss (Hoogland et al., 2015; Liu and Bing, 2011). Intra-nigral injection of LPS in rats caused selective dopaminergic neuronal damage (Castano et al., 1998; Hsieh et al., 2002). In contrast, intra-striatal (Choi et al., 2009; Hunter et al., 2009; Hunter et al., 2007), intra-pallidal (Zhang et al., 2005) and systemic administration has been reported to produce limited changes in dopamine content (Bodea et al., 2014). However, additive effects of injection with 750 μg/kg LPS followed 4 months later by injection of 15 mg/kg MPTP significantly reduced stride length in mice (Byler et al., 2009). Overall, loss of dopamine neurons accompanied by reduction in striatal dopamine and a change in gait as a consequence of LPS treatment appear to be a consequence of dose, duration, areas of primary damage, or possibly the vulnerability of specific subsets of dopaminergic neurons responsible for striatal dopamine levels maintenance to pro-inflammatory cytokine challenge (German et al., 1996; Liu and Bing, 2011).
The reduction in striatal dopamine and its metabolites appeared to be relatively specific, as no significant effects were observed for serotonin and its metabolite 5HIAA. Because we did not count neurons and a full inflammation profile was not determined in areas other than the nigrostriatal pathway, we cannot exclude that other subtypes of neurons or neurotransmitters were affected by LPS administration. We did observe increased IBA1+ microglial cells in the frontal cortex in LPS-treated mice (Supplemental Fig. 2), which is similar to recently reported data (Noh et al., 2014), suggesting these regions may also be affected. However, it has also been reported that the systemic LPS treatment preferentially targets the dopaminergic nigrostriatal system (Dutta et al., 2008; Liu and Bing, 2011), and our data are consistent with these reports. A potential explanation for this phenomenon is that microglial cells, although ubiquitously present in the brain, are more densely populated in the hippocampus, olfactory telencephalon, basal ganglia, and substantia nigra compared to other regions (Lawson et al., 1990; Mittelbronn et al., 2001). Similarly, dopamine neurons of the SNpc are postulated to be more susceptible to inflammatory insult, most likely as a consequence of the increased presence of inflammatory cells and increased reactive oxygen species (Di Giovanni et al., 2012; Perry et al., 2007).
The existence of distinct microglial phenotypes (i.e. M1 or M2) and their specific role in neurodegeneration is a hotly debated topic; however, there is considerable evidence that microglia can adopt either a predominantly pro- or anti-inflammatory phenotype (Cherry et al., 2014; Joers et al., 2016; Ransohoff, 2016b; Tang and Le, 2016). Microglia are generally considered pro-inflammatory (M1) if they produce higher levels of the pro-inflammatory factors iNOS, TNFα, IL-1β, and IL-12. On the other hand, increased levels of arginase-1, CD206/Mrc1, Fizz1, Ym1, and IL-10 indicate that the microglia are more of an anti-inflammatory phenotype (M2). Although still poorly understood and somewhat controversial (Ransohoff, 2016a), there are several mechanisms proposed for this phenotypic switch, including prominent roles for IL-4/IL13 and IL-10 (Orihuela et al., 2016). Various studies have investigated the role of M1 microglia and neuroinflammation in the different animal models of PD (Joers et al., 2016), but only recently has research focused on M2 microglial activation dynamics in this disease. Chronic MPTP administration has been shown to progressively reduce the expression of the M2 marker MRC1, which suggests that down-regulation of M2 microglial activation could be a significant contributor to the progression of neuron loss observed in this model (Pisanu et al., 2014). In our study with LPS treatment, indicators of a M2 response became apparent at day 19 and were predominant at day 36, a time at which cessation of LPS-induced dopamine neuron was observed. This culminated with a predominant expression of ARG1 in IBA+ cells, suggesting that these cells exhibited more of an M2a state, likely working through the IL-4/IL-13 pathway (Orihuela et al., 2016). Together, these data suggest the intriguing possibility that the phenotypic switch from more of a M1 to a M2 phenotype may be responsible for the cessation of cell loss. Based on our data and that of others, it could be hypothesized that PD may share features with AD (Mandrekar-Colucci and Landreth, 2010; Theriault et al., 2015) and experimental autoimmune encephalitis (EAE) (Jadidi-Niaragh and Mirshafiey, 2011; Ponomarev et al., 2007), in which an ineffectual downregulation of an acute pro-inflammatory response or a failed anti-inflammatory response appear to be significant contributors to the initiation and progression of neurodegeneration.
Targeting the multiple microglial activation states for therapeutic intervention and neuroprotection is a relatively new line of thinking in the race to stop the progression of PD (Cherry et al., 2014; Tang and Le, 2015; Wang et al., 2015). This concept is complicated by the imprecise translation of characterized activation states in microglia (Hellwig et al., 2013; Mosher and Wyss-Coray, 2014). Indeed, microglial phenotypes in human neurodegenerative diseases have been difficult to characterize because of limitations in specific markers and other confounding factors such as medication usage, co-morbidities and the constantly changing dynamics of glial cells as a function of age and disease (Joers et al., 2016; Moehle and West, 2015; Soreq et al., 2017). Further, abnormal functioning of microglia in the aged or diseased brain may already have impaired restorative M2 activity (Conde and Streit, 2006; Sawada et al., 2008), leading to neurodegeneration (Luo et al., 2010). Recent research has primarily focused on therapeutic approaches based around anti-inflammatory molecules such as NADPH oxidase (i.e. minocycline), COX2 and 5-LO inhibitors that disrupt pro-inflammatory activation (Choi et al., 2011; Kang et al., 2013; Klegeris and McGeer, 2002; Manev et al., 2001). However, there have been mixed results in pre-clinical and clinical studies with minocycline (2008; Diguet et al., 2004; Du et al., 2001; Yang et al., 2003) that suggest it might actually worsen neurodegeneration.
Although there has been less focus on increasing resolution of inflammation, peroxisome proliferator-activated receptor gamma (PPARγ) activation can induce polarization of anti-inflammatory microglia, and shows protection in the MPTP-probenecid model (Pisanu et al., 2014) and reduced AD pathology severity (Mandrekar-Colucci et al., 2012; Yamanaka et al., 2012). However, caution is raised by those data because although sustained IL-1β can reduce amyloid burden, it exacerbates tau pathology in the triple transgenic mouse model of AD (Ghosh et al., 2013). Based on our data and those of Pisanu and co-workers (Pisanu et al., 2014), we believe those interventions targeted to enhance the M2 microglial activation state may mitigate toxicant-induced dopamine neuron loss. Similarly, the repeated LPS injection paradigm used in this study demonstrates a tractable model to study the potential neuroprotective effects of shifting microglial activation from M1 to M2 phenotype.
6. Conclusions
Taken in concert, the data reported here demonstrate the balance between pro- and anti-inflammatory microglial phenotypes influences the overall inflammatory state of the nigrostriatal system and its contribution to dopamine neuron loss following repeated LPS administration. This is the first study to examine microglial activation phenotypes in a model that shows some progressive loss of neurons and demonstrate that shifting from a pro- to an anti-inflammatory phenotype coincides with cessation of neuron loss. If these data are confirmed in additional ongoing studies in additional animal models of PD, it would provide the basis for future immune-modulatory therapies in PD aimed at promoting an inflammation-resolving microglial phenotype in order to promote neuroprotection and stopping the progression of the disease.
Supplementary Material
Figure S1 Photomicrographs of immunofluorescent staining for macrophages (CD169, green), activated microglia (Iba1, red), and DAPI (blue) in striatum of C57BL/6J mice on experimental days 5 and 19 after treatment with PBS or LPS. Bars: 50 μm.
Figure S2 Photomicrographs of immunofluorescent staining for activated microglia (Iba1, red) in the cortex, striatum, and midbrain of PBS or LPS-treated mice. Bars: 50 μm.
Highlights.
LPS caused progressive neurodegeneration and neuroinflammation
Overt time, microglia converted from a pro- to anti-inflammatory phenotype
Conversion to anti-inflammatory phenotype was associated with cessation of neurodegeneration
Acknowledgments
This work was supported by National Institutes of Health grants T32ES007148, P30ES005022, R01ES021800, R01NS088627, R21DE025689, R01NS088267 and the Michael J Fox Foundation for Parkinson's Disease Research. Additional support was provided through generous donations from the Allan and Janice Woll, the Richard Nicely and the Glenn and Karen Leppo Parkinson's Disease Research Funds. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding contributors. The funding sources had no role in the conduct and interpretation of the studies or decision to publish this study.
Footnotes
The authors further declare they have no competing financial conflicts or other conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clin Neuropharmacol. 2008;31:141–50. doi: 10.1097/WNF.0b013e3181342f32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam G, et al. Single low doses of MPTP decrease tyrosine hydroxylase expression in the absence of overt neuron loss. Neurotoxicology. 2017;60:99–106. doi: 10.1016/j.neuro.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, et al. A comparison of model-based (2D) and design-based (3D) stereological methods for estimating cell number in the substantia nigra pars compacta (SNpc) of the C57BL/6J mouse. Neuroscience. 2009;161:1082–90. doi: 10.1016/j.neuroscience.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnum CJ, Tansey MG. Modeling neuroinflammatory pathogenesis of Parkinson's disease. Prog Brain Res. 2010;184:113–32. doi: 10.1016/S0079-6123(10)84006-3. [DOI] [PubMed] [Google Scholar]
- Bodea LG, et al. Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci. 2014;34:8546–56. doi: 10.1523/JNEUROSCI.5002-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JM, et al. Exposure to the polybrominated diphenyl ether mixture DE-71 damages the nigrostriatal dopamine system: role of dopamine handling in neurotoxicity. Exp Neurol. 2013;241:138–47. doi: 10.1016/j.expneurol.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodacki B, et al. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci Lett. 2008;441:158–62. doi: 10.1016/j.neulet.2008.06.040. [DOI] [PubMed] [Google Scholar]
- Byler SL, et al. Systemic lipopolysaccharide plus MPTP as a model of dopamine loss and gait instability in C57Bl/6J mice. Behav Brain Res. 2009;198:434–9. doi: 10.1016/j.bbr.2008.11.027. [DOI] [PubMed] [Google Scholar]
- Castano A, et al. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem. 1998;70:1584–92. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- Chen Z, et al. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32:11706–15. doi: 10.1523/JNEUROSCI.0730-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, et al. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11:98. doi: 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DK, et al. Inhibitors of microglial neurotoxicity: focus on natural products. Molecules. 2011;16:1021–43. doi: 10.3390/molecules16021021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DY, et al. Striatal neuroinflammation promotes Parkinsonism in rats. PLoS One. 2009;4:e5482. doi: 10.1371/journal.pone.0005482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliburn RA, et al. Immunochemical localization of vesicular monoamine transporter 2 (VMAT2) in mouse brain. J Chem Neuroanat. 2016 doi: 10.1016/j.jchemneu.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins LM, et al. Contributions of central and systemic inflammation to the pathophysiology of Parkinson's disease. Neuropharmacology. 2012;62:2154–68. doi: 10.1016/j.neuropharm.2012.01.028. [DOI] [PubMed] [Google Scholar]
- Conde JR, Streit WJ. Effect of aging on the microglial response to peripheral nerve injury. Neurobiol Aging. 2006;27:1451–61. doi: 10.1016/j.neurobiolaging.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, et al. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation. 2012;9:179. doi: 10.1186/1742-2094-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65:304–12. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hooge R, et al. Neuromotor alterations and cerebellar deficits in aged arylsulfatase A- deficient transgenic mice. Neurosci Lett. 1999;273:93–6. doi: 10.1016/s0304-3940(99)00647-3. [DOI] [PubMed] [Google Scholar]
- Di Giovanni G, et al. Redox sensitivity of tyrosine hydroxylase activity and expression in dopaminergic dysfunction. CNS Neurol Disord Drug Targets. 2012;11:419–29. doi: 10.2174/187152712800792938. [DOI] [PubMed] [Google Scholar]
- Diguet E, et al. Deleterious effects of minocycline in animal models of Parkinson's disease and Huntington's disease. Eur J Neurosci. 2004;19:3266–76. doi: 10.1111/j.0953-816X.2004.03372.x. [DOI] [PubMed] [Google Scholar]
- Du Y, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98:14669–74. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta G, et al. The lipopolysaccharide Parkinson's disease animal model: mechanistic studies and drug discovery. Fundam Clin Pharmacol. 2008;22:453–64. doi: 10.1111/j.1472-8206.2008.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari CC, et al. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1beta in the substantia nigra. Neurobiol Dis. 2006;24:183–93. doi: 10.1016/j.nbd.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Fortin MC, et al. Alteration of the expression of pesticide-metabolizing enzymes in pregnant mice: potential role in the increased vulnerability of the developing brain. Drug Metab Dispos. 2013;41:326–31. doi: 10.1124/dmd.112.049395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Cannon TC, et al. Parkin deficiency increases vulnerability to inflammation- related nigral degeneration. J Neurosci. 2008;28:10825–34. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, et al. The neurotoxin MPTP causes degeneration of specific nucleus A8, A9 and A10 dopaminergic neurons in the mouse. Neurodegeneration. 1996;5:299–312. doi: 10.1006/neur.1996.0041. [DOI] [PubMed] [Google Scholar]
- Ghosh S, et al. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer's mouse model. J Neurosci. 2013;33:5053–64. doi: 10.1523/JNEUROSCI.4361-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, et al. Spinal Microgliosis Due to Resident Microglial Proliferation Is Required for Pain Hypersensitivity after Peripheral Nerve Injury. Cell Rep. 2016;16:605–14. doi: 10.1016/j.celrep.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday GM, Stevens CH. Glia: initiators and progressors of pathology in Parkinson's disease. Mov Disord. 2011;26:6–17. doi: 10.1002/mds.23455. [DOI] [PubMed] [Google Scholar]
- Hellwig S, et al. The brain's best friend: microglial neurotoxicity revisited. Front Cell Neurosci. 2013;7:71. doi: 10.3389/fncel.2013.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland IC, et al. Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation. 2015;12:114. doi: 10.1186/s12974-015-0332-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MM, et al. Hippocampal ER stress and learning deficits following repeated pyrethroid exposure. Toxicol Sci. 2015;143:220–8. doi: 10.1093/toxsci/kfu226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PF, et al. Behavior, neurochemistry and histology after intranigral lipopolysaccharide injection. Neuroreport. 2002;13:277–80. doi: 10.1097/00001756-200203040-00006. [DOI] [PubMed] [Google Scholar]
- Hunter RL, et al. Intrastriatal lipopolysaccharide injection induces parkinsonism in C57/B6 mice. J Neurosci Res. 2009;87:1913–21. doi: 10.1002/jnr.22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RL, et al. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem. 2007;100:1375–86. doi: 10.1111/j.1471-4159.2006.04327.x. [DOI] [PubMed] [Google Scholar]
- Jadidi-Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol. 2011;74:1–13. doi: 10.1111/j.1365-3083.2011.02536.x. [DOI] [PubMed] [Google Scholar]
- Joers V, et al. Microglial phenotypes in Parkinson's disease and animal models of the disease. Prog Neurobiol. 2016 doi: 10.1016/j.pneurobio.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang KH, et al. Protection of dopaminergic neurons by 5-lipoxygenase inhibitor. Neuropharmacology. 2013;73:380–7. doi: 10.1016/j.neuropharm.2013.06.014. [DOI] [PubMed] [Google Scholar]
- Klegeris A, McGeer PL. Cyclooxygenase and 5-lipoxygenase inhibitors protect against mononuclear phagocyte neurotoxicity. Neurobiol Aging. 2002;23:787–94. doi: 10.1016/s0197-4580(02)00021-0. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, et al. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–70. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Bing G. Lipopolysaccharide animal models for Parkinson's disease. Parkinsons Dis. 2011;2011 doi: 10.4061/2011/327089. 327089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XG, et al. Microglia in the aging brain: relevance to neurodegeneration. Mol Neurodegener. 2010;5:12. doi: 10.1186/1750-1326-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, et al. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J Neurosci. 2012;32:10117–28. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2010;9:156–67. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manev H, et al. Neurogenesis and neuroprotection in the adult brain. A putative role for 5-lipoxygenase? Ann N Y Acad Sci. 2001;939:45–51. doi: 10.1111/j.1749-6632.2001.tb03610.x. [DOI] [PubMed] [Google Scholar]
- McCormack AL, et al. Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–27. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev Immunol. 2012;32:463–88. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- Mittelbronn M, et al. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001;101:249–55. doi: 10.1007/s004010000284. [DOI] [PubMed] [Google Scholar]
- Moehle MS, West AB. M1 and M2 immune activation in Parkinson's Disease: Foe and ally? Neuroscience. 2015;302:59–73. doi: 10.1016/j.neuroscience.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, et al. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson's disease. Neurosci Lett. 1996;211:13–6. doi: 10.1016/0304-3940(96)12706-3. [DOI] [PubMed] [Google Scholar]
- Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol. 2014;88:594–604. doi: 10.1016/j.bcp.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounayar S, et al. A new model to study compensatory mechanisms in MPTP-treated monkeys exhibiting recovery. Brain. 2007;130:2898–914. doi: 10.1093/brain/awm208. [DOI] [PubMed] [Google Scholar]
- Noh H, et al. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem Int. 2014;69:35–40. doi: 10.1016/j.neuint.2014.02.008. [DOI] [PubMed] [Google Scholar]
- Orihuela R, et al. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173:649–65. doi: 10.1111/bph.13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, et al. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161–7. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Pisanu A, et al. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson's disease. Neurobiol Dis. 2014;71:280–91. doi: 10.1016/j.nbd.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, et al. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–21. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–62. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016a;19:987–91. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016b;353:777–83. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- Rappold PM, et al. Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter-3. Proc Natl Acad Sci U S A. 2011;108:20766–71. doi: 10.1073/pnas.1115141108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JR, Miller GW. Acute exposure to aroclor 1016 or 1260 differentially affects dopamine transporter and vesicular monoamine transporter 2 levels. Toxicol Lett. 2004;148:29–40. doi: 10.1016/j.toxlet.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Sanchez-Guajardo V, et al. Neuroimmunological processes in Parkinson's disease and their relation to alpha-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro. 2013;5:113–39. doi: 10.1042/AN20120066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada M, et al. Effects of aging on neuroprotective and neurotoxic properties of microglia in neurodegenerative diseases. Neurodegener Dis. 2008;5:254–6. doi: 10.1159/000113717. [DOI] [PubMed] [Google Scholar]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh RA, et al. Effects of the organochlorine pesticide methoxychlor on dopamine metabolites and transporters in the mouse brain. Neurotoxicology. 2009;30:274–80. doi: 10.1016/j.neuro.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheleg M, et al. Ephrin-A5 deficiency alters sensorimotor and monoaminergic development. Behav Brain Res. 2013;236:139–47. doi: 10.1016/j.bbr.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song DD, Haber SN. Striatal responses to partial dopaminergic lesion: evidence for compensatory sprouting. J Neurosci. 2000;20:5102–14. doi: 10.1523/JNEUROSCI.20-13-05102.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreq L, et al. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017;18:557–570. doi: 10.1016/j.celrep.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Federoff HJ. Immune responses in Parkinson's disease: interplay between central and peripheral immune systems. Biomed Res Int. 2014;2014 doi: 10.1155/2014/275178. 275178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sveinbjornsdottir S. The clinical symptoms of Parkinson's disease. J Neurochem. 2016;139(1):318–324. doi: 10.1111/jnc.13691. [DOI] [PubMed] [Google Scholar]
- Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2015 doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2016;53:1181–94. doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- Theriault P, et al. The dynamics of monocytes and microglia in Alzheimer's disease. Alzheimers Res Ther. 2015;7:41. doi: 10.1186/s13195-015-0125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillerson JL, et al. Detection of behavioral impairments correlated to neurochemical deficits in mice treated with moderate doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Exp Neurol. 2002;178:80–90. doi: 10.1006/exnr.2002.8021. [DOI] [PubMed] [Google Scholar]
- Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp (Warsz) 2012;60:251–66. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, et al. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19. doi: 10.1186/s40035-015-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka M, et al. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–31. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, et al. Minocycline enhances MPTP toxicity to dopaminergic neurons. J Neurosci Res. 2003;74:278–85. doi: 10.1002/jnr.10709. [DOI] [PubMed] [Google Scholar]
- Yochum C, et al. Prenatal cigarette smoke exposure causes hyperactivity and aggressive behavior: role of altered catecholamines and BDNF. Exp Neurol. 2014;254:145–52. doi: 10.1016/j.expneurol.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, et al. Intrapallidal lipopolysaccharide injection increases iron and ferritin levels in glia of the rat substantia nigra and induces locomotor deficits. Neuroscience. 2005;135:829–38. doi: 10.1016/j.neuroscience.2005.06.049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Photomicrographs of immunofluorescent staining for macrophages (CD169, green), activated microglia (Iba1, red), and DAPI (blue) in striatum of C57BL/6J mice on experimental days 5 and 19 after treatment with PBS or LPS. Bars: 50 μm.
Figure S2 Photomicrographs of immunofluorescent staining for activated microglia (Iba1, red) in the cortex, striatum, and midbrain of PBS or LPS-treated mice. Bars: 50 μm.