

Abstract
Lysine acetylation has emerged as a global protein regulation system in all domains of life. Sirtuins, or Sir2-like enzymes, are a family of histone deacetylases characterized by their employing NAD+ as a co-substrate. Sirtuins can deacetylate several acetylated proteins, but a consensus substrate recognition sequence has not yet been established. Product inhibition of many eukaryotic sirtuins by nicotinamide and its analogues has been studied in vitro due to their potential role as anticancer agents. In this work, the kinetics of CobB, the main Escherichia coli deacetylase, have been characterized. To our knowledge, this is the first kinetic characterization of a sirtuin employing a fully acetylated and natively folded protein as a substrate. CobB deacetylated several acetyl-CoA synthetase acetylated lysines with a single kinetic rate. In addition, in vitro nicotinamide inhibition of CobB has been characterized, and the intracellular nicotinamide concentrations have been determined under different growth conditions. The results suggest that nicotinamide can act as a CobB regulator in vivo. A nicotinamidase deletion strain was thus phenotypically characterized, and it behaved similarly to the ΔcobB strain. The results of this work demonstrate the potential regulatory role of the nicotinamide metabolite in vivo.
Introduction
Protein lysine acetylation is a post-translational modification (PTM) in which the scientific community is increasingly interested [1–3]. Lysine acetylation consists of the transfer of an acetyl group from a donor molecule such as acetyl-CoA or acetyl-phosphate to a lysine amino acid of a protein. This transfer can be catalysed by an acetyltransferase enzyme (KAT) [4,5] or occur in a non-enzymatic or chemical manner [6,7]. The number of proteins that are modified post-translationally by acetylation is continuously discovered in all types of organisms. This PTM has traditionally been associated with transcriptional regulation (histone modifications) [8], although in the past 10 years, many metabolic routes have been demonstrated to be regulated by lysine acetylation [9–15].
Protein acetylation can be reversed by a family of enzymes known as the lysine deacetylases (KDACs). KDACs can be classified into two main groups. The first group acts in a Zn2+-dependent manner, while the members of the second group, also known as sirtuins or Sir2-like enzymes, deacetylate lysines in a reaction that employs oxidized nicotinamide adenine dinucleotide (NAD+) [16,17] as a co-substrate. Sirtuins remove an acetyl group from the ɛ-amino group of a lysine in a reaction that consumes NAD+ and releases nicotinamide (NAM), O-acetyl-ADP-ribose, and the deacetylated protein [18]. Many eukaryotic species possess multiple sirtuin paralogs (human sirt1-7) that differ in structure and cell localization. Archaea and bacteria also produce sirtuins, e.g., the bacterial CobB [19–21]. In Escherichia coli (E. coli), the most studied deacetylase is CobB, a bacterial sirtuin that was first characterized in Salmonella enterica [21]. Other E. coli proteins such as acetyl-CoA synthetase (Acs) and phosphotransacetylase (Pta) have been reported to functionally act as deacetylases of the chemotaxis signalling molecule CheY, although, to our knowledge, there is not a proteomic study that shows a deacetylase activity with other substrates [22,23]. In addition, it has recently been demonstrated that YcgC deacetylates several acetylated lysines from different proteins without needing NAD+ or Zn2+ as co-substrates [24]. Sirtuins share a common structural core formed by a Rossman domain, a Zn2+-finger domain and a connector loop. The N- and C-terminal regions differ in sequence and length, which might determine differences in substrate specificity [25].
With respect to sirtuin substrates, eukaryotic sirtuins have been linked to cell survival, apoptosis, and stress resistance [26,27]. The typical substrates of archaea and bacteria sirtuins are DNA regulatory proteins (such as the chromatin protein Alba) and metabolic enzymes (such as acyl-CoA synthetases) [2,21,28–33]. In E. coli, Acs regulation by acetylation-deacetylation has been studied in vitro [15,34,35]. Thus, Acs is acetylated at K609 by the acetyltransferase YfiQ/Pka/PatZ inhibiting its catalytic activity, while enzymatic activity is recovered by CobB deacetylation [34–36]. The CheY chemotaxis response regulator [37,38], the Nhoa protein [39] and the RcsB transcription factor [15,40] have also been identified as CobB substrates. Recent proteomic studies have identified as many as 69 acetyl-lysine residues among 51 proteins to be CobB substrates [7] and have also found 183 proteins that interact with this sirtuin [41]. Several studies have tried to identify a consensus recognition sequence for CobB and other sirtuins [18,42–48], but the results are not clear and do not always correspond to proteomic studies performed in vivo [7,15,49,50]. Due to the interest in sirtuins as targets for the treatment of metabolic diseases [51,52], several studies have addressed the regulation and inhibition of these enzymes. Nicotinamide, a product of the sirtuin catalytic reaction, is a potent inhibitor for all of these enzymes. Although the mechanism of inhibition by NAM is not completely clear, most of the in vitro studies carried out with eukaryotic sirtuins point to non-competitive inhibition via a base-exchange mechanism that involves reacting with an intermediate to reform NAD+ (transglycosidation mechanism) [53–56].
Herein we carry out a CobB sirtuin study by kinetically characterizing acetylated-Acs deacetylation. To our knowledge, this report is the first kinetic characterization of a sirtuin deacetylation reaction with a complete natively folded substrate. In addition, we studied CobB inhibition by nicotinamide in vitro and in vivo. The results of this work represent an advance in the study of sirtuin specificity and substrate recognition; in addition, our data suggest a new level of CobB regulation in vivo via inhibition by nicotinamide, a poorly studied metabolite with a central metabolic role.
Material and methods
Molecular biology
To generate overexpression plasmids, the cobB and nicotinamidase (pncA) genes of E. coli BW25113 were PCR-amplified and cloned into pBAD24-MBP (Maltose Binding Protein) (kindly transferred by Dr. Antonio Sánchez-Amat, University of Murcia) and pRSETA (Invitrogen) plasmids, respectively. To overexpress Acs protein, the correspondent ASKA plasmid was employed [57]. Single amino acid mutant Acs K609A was obtained by site-directed mutagenesis from acsASKA plasmid. To carry out the complementation assays, the pncA and cobB genes were PCR-amplified from E. coli BW25113 genomic DNA and cloned into the pBAD24 plasmid [58]. All molecular biology enzymes that were used were purchased from Thermo Fisher Scientific. The strains, plasmids and primers used are listed in S1 Table.
Escherichia coli strains and culture conditions
Escherichia coli BW25113 and its deletion strains (S1 Table) were grown in batch mode in minimal M9 medium or in complex TB7 medium [59] (10 g/l tryptone buffered at pH 7.0 with 100 mM potassium phosphate) supplemented with glucose (20 mM) or glycerol (40 mM) as the carbon source. Cell growth was monitored spectrophotometrically by determining the optical density at 600 nm (OD600). Kanamycin (50 mg/ml), ampicillin (100 mg/ml), and chloramphenicol (30 mg/ml) were added when needed.
Overexpression and purification of proteins
Chemically competent E. coli BL21 (DE3) (CobB and nicotinamidase overexpression) or E. coli BL21 (DE3) ΔcobB (Acs and Acs K609A overexpression) strains were transformed by heat shock at 42°C with overexpression plasmids. Cultures were grown overnight at 30°C with orbital shaking (200 rpm). The culture medium that was used was Luria-Bertani broth (LB). Expression was induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) (cobBpBAD24-MBP and pncApRSETA) or 0.1 mM IPTG (acsASKA) when the culture OD600 reached 0.5–0.6. Cells were harvested by centrifugation, thoroughly washed with 0.9% NaCl and resuspended in binding buffer (50 mM potassium phosphate, pH 7.5, containing 500 mM NaCl and 20 mM imidazole for IMAC purification and 20 mM Tris-HCl, pH 7.5, containing 200 mM NaCl for amylose purification) that was supplemented with an EDTA-free protease inhibitor (SigmaFast Protease Inhibitor Cocktail Tablet, from Sigma Aldrich). Cells were lysed on ice by sonication for 2 min (20 s each pulse) using a Vibra Cell sonicator (Sonicator Sonics & Materials). The lysates were clarified by centrifugation at 10000 x g for 15 min at 4°C. Recombinant Acs, Acs K609A and nicotinamidase proteins were purified by using immobilized metal affinity chromatography (IMAC). The cell-free extract was loaded onto a 5 ml His GraviTrap column (GE Healthcare), and the column was washed with washing buffer (50 mM potassium phosphate buffer, pH 7.5, containing 500 mM NaCl and 50 mM imidazole). The His6-tagged proteins were eluted with an elution buffer (50 mM potassium phosphate buffer, pH 7.5, containing 500 mM NaCl and 500 mM imidazole). CobB sirtuin was purified using an amylose resin (New England Biolabs). The resin was washed with a binding buffer, and CobB was eluted using an amylose elution buffer (20 mM Tris-HCl, pH 7.5, containing 200 mM NaCl and 10 mM maltose). MBP CobB tag was cleaved by incubating the protein with H3V 3C protease (Pierce) over night at 4°C. MBP-tag and protease were removed with a His GraviTrap column. Purified proteins were dialyzed (Pur-a-lyzer dialysis kits) into 50 mM potassium phosphate buffer, pH 7.5, containing 100 mM NaCl and 10% v/v glycerol. Proteins were stored at -80°C until used. Finally, Spin-X UF concentrators (Corning) were employed to concentrate the proteins. Proteins were analysed by using SDS-PAGE electrophoresis with 10% acrylamide gels in a Mini-Protean cell (Bio-Rad).
Western blot analysis
Proteins with acetylated lysines were separated by using SDS-PAGE and transferred to PVDF membranes using a semi-dry transfer unit (Trans-Blot® SD Semi-Dry Transfer Cell, Bio-Rad). The membranes were incubated with a rabbit monoclonal primary anti-acetyl-lysine antibody (anti-Ac-K) (ImmuneChem) and a goat anti-rabbit IgG secondary antibody (Santa Cruz Biotechnology). Finally, the membrane was incubated for 10 min with Amersham ECL western blotting detection reagent (Thermo Scientific). ImageJ Gel Analyzer software was used for densitometric quantification.
Liquid chromatography–tandem mass spectrometry assay (LC-MS)
Samples were alkylated by incubation with 100 mM iodoacetamide (IAA) for 30 min at room temperature in the dark. Proteins were digested with 0.5–1 μg of proteomics grade Trypsin Gold (Promega) for 3 h at 37°C. The reaction was stopped by the addition of 0.1% formic acid, and the samples were dried using a vacuum evaporator. Tryptic peptides generated from the samples were separated and analysed by LC-MS. An Agilent 1100 (Agilent Technologies) was equipped with a Zorbax SB-C18 HPLC column (Agilent Technologies) and connected to an Agilent Ion Trap XCT Plus mass spectrometer (Agilent Technologies) that had an electrospray (ESI) interface. Two mobile phases were used: phase A was composed of water/acetonitrile/formic acid (94.9:5:0.1, v/v), and phase B consisted of water/acetonitrile/formic acid (10:89.9:0.1, v/v). The digested peptides were resuspended in 20 μl of phase A and eluted using a linear gradient of 0–80% phase B for 180 min at a flow rate of 10 μl min-1. The mass spectrometer was operated in positive mode with a capillary spray voltage of 3500 V, at a scan speed of 8100 (m/z)/sec from 50 to 2200 m/z, with a target mass of 1000 m/z, and three scans were averaged. The nebulizer gas pressure was set at 15 psi, and the drying gas flowed at 5 l min-1 at a temperature of 350°C. MS/MS data were collected in an automated data-dependent mode (AutoMS mode). Data processing was performed with the Data Analysis program for LC/MSD Trap Version 3.3 (Bruker Daltonik) and the Spectrum Mill MS Proteomics Workbench (Agilent Technologies) [60,61]. After automated validation of the results, the identified proteins and the sequences of the digested peptides were compiled. Peptides with a score threshold of 8 and a percentage-scored peak intensity higher than 70% were considered valid.
Acs enzyme assay
The acetyl-CoA synthetase assay was based on the coupled assay reported by Williamson and Corkey [62]. AMP production was detected via a coupled enzyme assay in which myokinase (MK), pyruvate kinase (PK) and lactate dehydrogenase (LDH) couple AMP production to NADH oxidation. Standard acetyl-CoA synthetase assays (0.1 ml) were performed at 37°C in 50 mM potassium phosphate buffer at pH 7.5 and contained 3 mM PEP (phosphoenolpyruvate), 5 units of MK, 1 unit of PK, 1.5 units of LDH, 5 mM MgCl2, 1 mM ATP, 1.5 mM CoA, 0.1 mM NADH, 5 mM acetate and 1 mM dithiothreitol. The reaction was started by the addition of Acs. All reactions were performed in triplicate. Specific activity was calculated using the extinction coefficient of NADH (6.22 mM-1 cm-1) and was based on the oxidation of two molecules of NADH for each AMP molecule released. One unit of Acs activity is defined as 1 μmole of acetyl-CoA formed per minute at pH 7.5 and 37°C.
CobB and nicotinamidase enzymatic assay
The continuous CobB assay that was employed was based on the coupled assay published by Denu et al., [63]. In this assay, NAM released by CobB is converted into nicotinamidic acid by nicotinamidase catalysis. A coupled reaction based on glutamate dehydrogenase uses the ammonia and α-ketoglutarate (αKG) to oxidize NADH. Standard CobB assays (0.1 ml) were performed at 37°C in 50 mM potassium phosphate buffer at pH 7.5 and contained 3 mM αKG, 1 mM dithiothreitol, 3 units of GDH (glutamate dehydrogenase), 0.4 mM NADH, 1 μM nicotinamidase and 5 mM NAD+. The reaction was started by the addition of CobB. All reactions were performed in triplicate. Specific activity was calculated using the extinction coefficient of NADH (6.22 mM-1 cm-1) and was based on one molecule of NADH being oxidized for each NAM molecule released. One unit of CobB activity is defined as 1 μmole of NAM formed per minute at pH 7.5 and 37°C.
Intracellular NAM measurement
Cultures (50 ml) were harvested during the exponential (OD600 = 1) or stationary growth phases, and the collected pellets were washed with 0.9% NaCl and stored at -80°C. Nicotinamide extraction was carried out by using a freeze-thaw method with methanol [64]. Cell pellets were resuspended in 1 ml of pure methanol at -20°C with 15 μM d4-nicotinamide (deuterated nicotinamide), which was employed as an internal standard. The suspensions were put in liquid nitrogen for 5 min to ensure a completely frozen sample and then on ice for 10 min to thaw them. This freeze-thaw cycle was repeated three times. Then, samples were centrifuged at 16000 rpm and 0°C, and the supernatants were evaporated to dryness. To quantify intracellular metabolites, samples were resuspended in 0.3 ml of HPLC water. The analyses were carried out on an HPLC-MS/MS system consisting of an Agilent 1100 Series HPLC (Agilent Technologies) that was connected to an Agilent Ion Trap XCT Plus mass spectrometer (Agilent Technologies) and used an electrospray (ESI) interface. Samples were injected onto an Agilent Zorbax SB-Aq HPLC column (5 μm, 150 x 4.6 mm) that was thermostatted at 40°C and eluted at a flow rate of 200 μl/min. Mobile phase A, consisting of water with 0.1% formic acid, and mobile phase B, consisting of acetonitrile with 0.1% formic acid, were used for the chromatographic separation. The elution program consisted of 10% phase B for 10 minutes and then a gradient from 10 to 100% of mobile phase B in 15 minutes. Finally, 100% solvent B was maintained for 5 additional minutes. The mass spectrometer was operated in positive polarity mode with a capillary spray voltage of 3500 V and a scan speed of 22000 (m/z)/sec from 50–250 m/z, with the target mass located at 125 m/z. The Smart ICC target was set to 200.000 counts, whereas the maximum accumulation time was 20 msec. The nebulizer gas pressure was set to 30 psi, whereas the drying gas was set to a flow rate of 8 l min-1 and the temperature was set to 350°C. The ion chromatograms were extracted, and the peak areas were quantified using the Data Analysis program in LC/MSD Trap Version 3.2 (Bruker Daltonik). The peak area data of the compound in the standards were used to calculate the calibration curve, which was then used to determine the concentrations of NAM and d4-nicotinamide in the samples. To determine intracellular NAM concentrations a cell volume of 0.7 μm3 was employed and viable cell mass was determined using a linear calibration curve relating optical density at 600 nm and dry cell weight.
Electron microscopy
Escherichia coli strains were grown in TB7-glycerol complex medium. The cells were harvested in mid-exponential phase (OD600 = 1) and fixed with 3% glutaraldehyde for 30 minutes. After two washes with 0.9% NaCl, each cell suspension was placed on electron microscopy grids and stained for 15 s with 2% uranyl acetate before their flagella were examined in a Philips Tecnai 12 electron microscope operating at 120 kV.
Migration assays
Strains were grown TB7-glycerol complex medium until mid-exponential phase (OD600 = 1). One microliter of these cultures was inoculated into semisolid agar (10 g/l tryptone, 5 g/l NaCl and 0.25% agar). The migration diameter was measured after 16h at 30°C.
Extracellular metabolite quantification
Extracellular acetate, glucose and glycerol were analysed by using an HPLC (Shimadzu Scientific Instruments) equipped with differential refractive and UV detectors and a cation-exchange column (HPX- 87H, Bio-Rad). The mobile phase was 5 mM H2SO4 at 0.5 ml min-1 flow rate, and the temperature was 65°C.
Results
Kinetics of Acs deacetylation by CobB are monophasic
Acetyl-CoA synthetase protein was purified from an E. coli BL21 (DE3) ΔcobB strain to achieve a high Acs acetylation level. To ensure that Acs was acetylated, an LC-MS/MS assay was carried out to determine the Acs acetylation state. In total, 15 acetylated lysines were found in the Acs sequence, namely, K68, K111, K130, K200, K207, K221, K226, K348, K370, K400, K555, K585, K604, K609 and K617. These 15 acetylated lysines had been previously detected by other authors [7,15,35,49,65,66]. Acetylated Acs (10 μM) was incubated for 4 h with CobB (800 nM) (12:1 ratio) and NAD+ (5 mM). Aliquots were taken and processed for western blot and Acs activity measurements. Acs activity increased about twenty five-fold after 2 hours of incubation with CobB. This increase coincided with a decrease in chemiluminescent signal from the western blot. However, the reaction was not completed to 2 h of incubation. We therefore knew that Acs deacetylation was being partially inhibited, likely by nicotinamide reaction product. The results are shown in Fig 1.
Fig 1. Acs is deacetylated and activated by CobB.
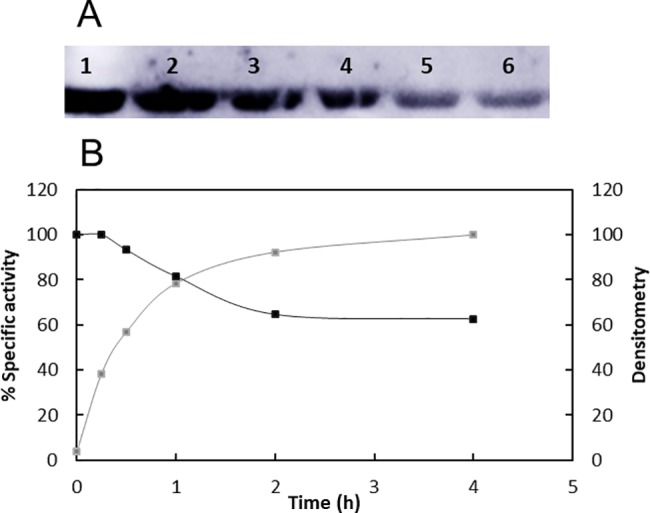
(A) Western blot with anti-Ac-K of Acs incubated with CobB and NAD+ at 0 (lane 1), 0.25 (lane 2), 0.5 (lane 3), 1 (lane 4), 2 (lane 5) and 4 (lane 6) hours of agitation at 37°C. (B) Acs specific activity at 0, 0.25, 0.5, 1, 2 and 4 hours (grey curve) and densitometry of Acs western blot (black curve).
Nicotinamide inhibition of sirtuins is well known and has been studied in many sirtuins [53–56,67,68]. To study the effect of NAM inhibition, E. coli nicotinamidase or PncA (an enzyme that consumes nicotinamide to generate ammonium and nicotinate) was coupled to the reaction. Acetylated Acs (10 μM) was incubated with CobB (800 nM) with and without E. coli nicotinamidase (1 μM), and Acs activity was measured for 4 hours. Acs activation rate was 8.5 times higher when nicotinamidase was present (8.95 U/mg in the absence of nicotinamidase and 76.62 U/mg in the presence of nicotinamidase). Thus, Acs total activation time was reduced from 2 hours to 15 minutes when nicotinamide was continuously degraded by nicotinamidase.
To study the kinetics of Acs deacetylation by CobB using a continuous assay, the sirtuin reaction was coupled to nicotinamidase and glutamate dehydrogenase activities. It is important to highlight that NAM product inhibition was eliminated with this assay, since nicotinamide is continuously degraded. NADH oxidation was studied at different acetylated Acs concentrations with a constant concentration of NAD+ (5 mM, saturated). The CobB concentration was 0.7 μM in the reactions. CobB exhibited hyperbolic Michaelis-Menten kinetics when the acetylated Acs concentration was varied from 0 to 60 μM (Fig 2A). The calculated kinetic parameters are shown in Table 1.
Fig 2. CobB activity on acetylated Acs.
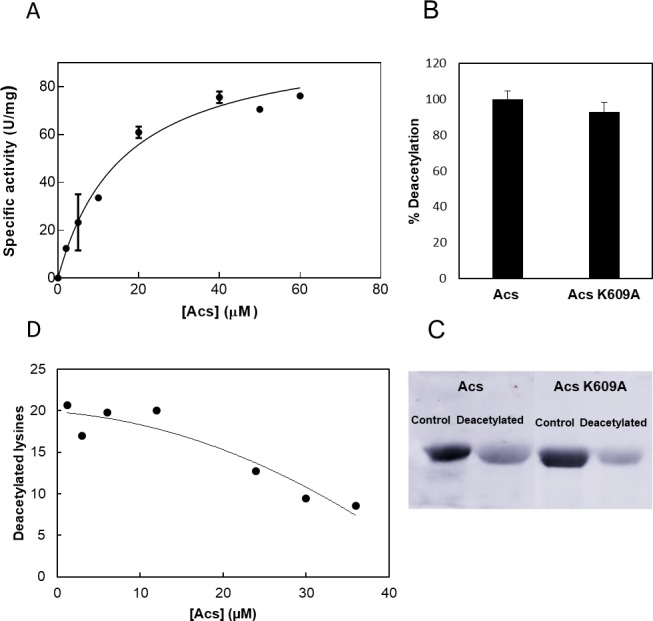
(A) Substrate saturation curve of CobB at different Acs concentrations. (B) Acs and Acs K609A mutant deacetylation (%). (C) Western blot with anti-Ac-K of Acs (lanes 1 and 2) and Acs K609A mutant (lanes 3 and 4) before (control) and after CobB deacetylation (deacetylated). (D) Number of lysines deacetylated by CobB per Acs molecule at different Acs concentrations.
Table 1. Kinetic parameters of CobB activity with acetylated Acs as a substrate.
| Km (μM) | Vmax (U/mg) | kcat (s-1) | kcat/Km (s-1 μM-1) |
|---|---|---|---|
| 16.19 ± 3.23 | 101 ± 7.66 | 2404,76 | 148,53 |
To know if the results observed corresponded only with K609 deacetylation or with several acetylated lysines, the recombinant protein Acs K609A (lysine K609 was substituted by an alanine residue) was overexpressed and purified from an E. coli BL21 (DE3) ΔcobB strain to achieve a high acetylation level. Acs K609A deacetylation was studied kinetically at a protein concentration of 20 μM with a CobB and NAD+ concentrations of 0.7 μM and 5 mM, respectively. The resultant deacetylation activity was compared with the obtained for Acs wt protein. Data are expressed in deacetylation percentage in Fig 2B (100% deacetylation was fixed for Acs wt deacetylation). A western blot assay of the deacetylation reactions was carried out using an anti-acetyl lysine antibody (Fig 2C). The Acs K609A kinetics and western blot assays demonstrated that the kinetics of Acs wt deacetylation by CobB studied are the result of the deacetylation of several Acs acetylated lysines, not only of K609. However, Acs activity was not recovered if K609 was acetylated or mutated, as has been reported for Salmonella enterica Acs and other acyl-CoA synthetases [21,31,69,70].
Since enzymatic CobB activity (μmol NAM produced/min), the number of deacetylated lysines per Acs molecule was calculated at each Acs concentration that was assayed (1 NAM produced per an acetylated lysine deacetylated by CobB). The results are shown in Fig 2D. Approximately 20 lysines were deacetylated by CobB when deacetylation showed a linear dependence on Acs concentration (0–12 μM).
CobB is non-competitively inhibited by nicotinamide
Nicotinamide is a sirtuin reaction product and a well-known inhibitor of sirtuins [53–56,67,68]. NAM inhibition of yeast and mammalian sirtuins has been biochemically and kinetically studied [54,55,67,71,72], but to our knowledge, it has not been studied in depth in a bacterial sirtuin. To study CobB nicotinamide inhibition, acetylated Acs was incubated in a 12:1 ratio with CobB. Acs activity was measured after 4 hours of reaction. Nicotinamide concentrations were quantified in a parallel assay employing coupled nicotinamidase and glutamate dehydrogenase reactions. The highest CobB activity (0% inhibition) was assured by coupling the nicotinamidase enzyme. The value was determined by studying CobB activity at 0.5 mM NAD+ and different NAM concentrations (Fig 3A). The IC50 value was approximately 52 μM. To determine the inhibition constant (ki), NAD+ concentration was varied from 0 to 1 mM (10, 50, 500 and 1000 μM) at different NAM concentrations and a Dixon plot (1/v vs NAM concentrations at different [NAD+]) was made (Fig 3B). The calculated value of ki was approximately 108 μM NAD+. To complete the inhibition study, a double-reciprocal plot (1/v versus 1/ [NAD+]) at different NAM concentrations was carried out (Fig 3C). These plots suggest that the activity of the CobB sirtuin has a non-competitive inhibition mechanism. This result is in concordance with the transglycosidation mechanism proposed for most examples of sirtuin nicotinamide inhibition [53,56,73].
Fig 3. Analysis of CobB inhibition by nicotinamide.

(A) CobB activity on acetylated Acs at different NAM concentrations. (B) Dixon plot ((1/v) vs [NAM]) of CobB deacetylation at different NAD+ concentrations: 10 (—), 50 (–∙), 500 (--) and 100 μM (---). (C) Double-reciprocal plot (1/v versus 1/ [NAD+]) of CobB deacetylation at different NAM concentrations: 0 (-∙∙), 40 (∙∙∙∙∙∙) and 50 (—) μM. Presented data are average of triplicates. Standard deviations were always less than 10%.
Nicotinamide concentrations are high enough to regulate CobB in vivo
Nicotinamide is an E. coli metabolite in the NAD+ salvage pathway, an important route that allows NAD+ recycling. NAM is synthetized from NAD+ by the protein NMN nucleosidase and is released by nicotinamidase (PncA) to form nicotinamidic acid. In this study, E. coli PncA has been biochemically characterized. To kinetically characterize PncA, kinetics assays were performed in which the nicotinamide concentration was varied from 0 to 5 mM and the PncA concentration was 0.76 μM. The enzyme showed Michaelis-Menten kinetics with values of 0.175 ± 0.027 mM and 2.05 ± 0.07 U/mg for Km and Vmax, respectively (Fig 4). This Km value is higher than the value reported for nicotinamidase enzymes from other organisms (2–110 μM) [74,75]. PncA inhibition by 3-pyridinecarbonitrile and 3-pyridinecarboxaldehide, a potent irreversible and a reversible competitive inhibitors of Mycobacterium tuberculosis nicotinamidase [74], respectively, was evaluated. PncA activity was measured at different nicotinamide concentrations after incubating the enzyme with 1 mM inhibitor for 10 minutes at 37°C. Nicotinamidase activity was undetectable in both assays. This result confirms PncA inhibition by 3-pyridinecarbonitrile and 3-pyridinecarboxaldehide in vitro. The relatively high nicotinamidase Km determined in this work could suggest, based on this inhibition study (Fig 3), that E. coli intracellular nicotinamide concentrations are high enough to regulate CobB activity in vivo.
Fig 4. Nicotinamidase kinetics characterization.
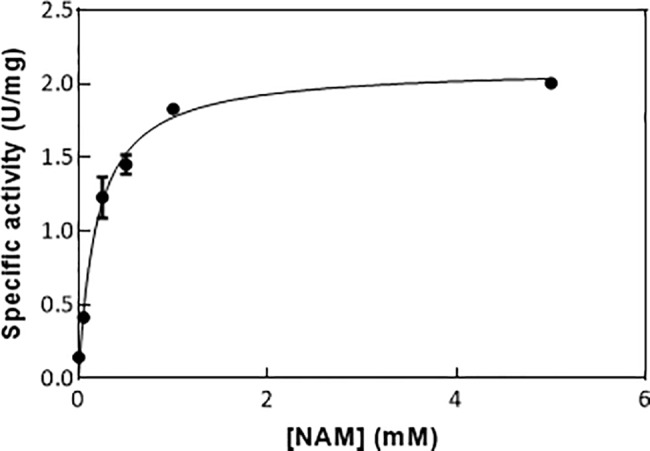
Substrate saturation curve of nicotinamidase at different nicotinamide concentrations.
To study in vivo CobB regulation by nicotinamide, intracellular nicotinamide concentrations were measured during the exponential and stationary growth phases for both the E. coli wt strain and a ΔpncA mutant growing in minimal and complex media with glucose or glycerol as their carbon sources. The resultant concentrations are summarized in Table 2.
Table 2. Intracellular nicotinamide concentrations.
Nicotinamide concentrations measured during exponential (X) and stationary (S) growth phases of E. coli wt and ΔpncA mutant strains growing in minimal and complex media with glucose or glycerol as the carbon source.
| wt X | wt S | ΔpncA X | ΔpncA S | |
|---|---|---|---|---|
| TB7 + Glucose | 36,51 ± 7,52 | 38,15 ± 1,66 | 91,95 ± 4 | 31,96 ±0,93 |
| TB7 + Glycerol | 67,89 ± 20,02 | 28,31 ± 5,03 | 198,45 ± 24,65 | 77,65 ±12,87 |
| MM9 + Glucose | 21,20 ± 2,81 | 63,82 ± 8,33 | 33,55 ± 2,3 | |
| MM9 + Glycerol | 33,98 ± 4,26 | 14,35 ± 2,60 | 47,03 ± 0,1 | 46,86 ± 3,06 |
Nicotinamide concentrations in cells were higher in complex than in minimal medium and higher concentrations were generally found during exponential growth. The ΔpncA mutant showed higher nicotinamide concentrations than the wt strain (approximately 2.5 times higher in complex TB7 medium). The maximum concentration was quantified for the pncA mutant during the exponential growth phase growing in TB7 complex medium supplemented with glycerol.
CobB could be regulated in vivo by nicotinamide
To study in vivo CobB regulation by nicotinamide, physiological characterization of E. coli wt, ΔcobB and ΔpncA strains was carried out. Specific growth rates, glucose and glycerol consumption and acetate overflow were analysed for the strains growing in minimal and complex media with glucose or glycerol as carbon source. Specific growth rates in MM9 medium with glucose were lower in the cobB mutant than in the wt strain, while significant differences were not observed in the rest of media. The pncA mutant also grew slower than E. coli wt, with a value similar to that observed for ΔcobB. The results are shown in Table 3.
Table 3. Specific growth rate.
Specific growth rate (h-1) of E. coli wt, ΔcobB and ΔpncA strains growing in minimal and complex media with glucose or glycerol as the carbon source.
| Medium | wt | ΔcobB | ΔpncA |
|---|---|---|---|
| MM9 + Glucose | 0.72 ± 0.02 | 0.58 ± 0.02 | 0.56 ± 0.02 |
| MM9 + Glycerol | 0.49 ± 0.01 | 0.48 ± 0.01 | 0.53 ± 0.02 |
| TB7 + Glucose | 1.01 ± 0.14 | 1.07 ± 0.05 | 0.85 ± 0.03 |
| TB7 + Glycerol | 0.95 ± 0.13 | 0.84 ± 0.06 | 0.77 ± 0.04 |
Glucose and glycerol consumption were not affected by cobB or pncA depletion; however, extracellular acetate levels, as a consequence of E. coli acetate overflow from bacteria growing in MM9 with glucose, were higher in the mutant strains (Fig 5). Observing similar behaviour in the deletion strains suggests total or partial CobB inhibition in the ΔpncA strain due to a higher intracellular nicotinamide concentration.
Fig 5. Acetate overflow.

Extracellular acetate concentrations (mM) per OD (600 nm) of E. coli wt (black line), ΔcobB (green line) and ΔpncA (blue line) strains grown in minimal medium with 20 mM glucose. E. coli wt growth curve is shown as a discontinuous red line.
To analyse the acetylation levels in the strains, a western blot of total protein extract, employing an anti-acetyl-lysine antibody, was carried out. The strains were grown in TB7 supplemented and non-supplemented with glucose, and cells were harvested during the mid-exponential (OD600 = 1) and stationary phases. The western blot did not show differences in the acetylation of the protein extracts. The similar acetylation patterns in wt and ΔcobB strains have been previously detected by other authors [7,15].
To complete the physiological characterization of the E. coli wt, ΔcobB and ΔpncA strains, the presence of their flagella was analysed using electron microscopy. CobB regulates the RcsB transcription factor by deacetylating its K154 and the absence of CobB causes an increase in the number of flagella [7,15,40]. We hypothesized that a CobB-inhibited strain would also show an increase in the number of flagella, so electron microscope imaging of the three strains during the exponential growth phase in TB7 with glucose as carbon source was carried out. The results are shown in Fig 6 (A1-C1). The cobB-deficient strain had significantly more and longer flagella than the wt strain. E. coli lacking the nicotinamidase protein also showed a phenotype that was similar to but less pronounced than that of ΔcobB. The ΔpncA strain had more flagella than the E. coli wt, but fewer than the ΔcobB strain. A migration assay using semi-solid agar plates was employed to quantify cells migration diameter. The assay was carried out on triplicate and the results are shown in Fig 6 (A2-C2). A migration diameter of 0.43 ± 0.05 cm was observed on E. coli wt plates, while a 5.30 ± 0.14 cm and 4.40 ± 0.26 cm was measured for ΔcobB and ΔpcnA strains. To verify the effect of cobB and pncA deletions, the mutant strains ΔcobB and ΔpncA were transformed with cobBpBad24 and pncApBad24, respectively. Cells phenotype was evaluated through electron microscopy and migration assays. The results are shown in Fig 6 (D1, D2, E1, E2). Both of mutant strains (ΔcobB and ΔpncA) recovered the wt phenotype when were complemented with the expression plasmids. These assays prove that the migration and flagella expression observed in ΔcobB and ΔpncA are consequence of genes deletion. Finally, an epistasis experiment was carried out to determine if the effect of PncA depends upon CobB. Thus, PncA was overexpressed in the cob mutant and phenotype was evaluated with semi-solid agar plates and electron microscopy. Results are shown in Fig 6 (F1 and F2). Cells showed a ΔcobB phenotype and no effect was observed with PncA overexpression. This result points out that PncA has not a CobB-independent effect on Escherichia coli migration.
Fig 6. Electron microscopy and migration assays.

Electron microscope images of (A1) E. coli wt, (B1) ΔcobB, (C1) ΔpncA, (D1) ΔcobB + cobBpBAD24, (E1) ΔpncA+ pncApBAD24 and (F1) ΔcobB + pncApBAD24 strains during the exponential growth phase in TB7 medium supplemented with glycerol. Semi-solid agar plates of (A2) E. coli wt, (B2) ΔcobB, (C2) ΔpncA strains, (D2) ΔcobB + cobBpBAD24, (E2) ΔpncA+ pncApBAD24 and (F2) ΔcobB + pncApBAD24 strains. Migration distances (diameter) and standard deviation from the mean migration of three biological replicates are shown.
Discussion
Sirtuins are an important family of enzymes that are widely distributed all domains of life. They are involved in many metabolic processes such as glucose or lipid metabolism, DNA repair, transcription regulation and tumour proliferation [76]. The kinetics of sirtuin activity against acetylated peptides have been studied and most of these studies have found a sirtuin selectivity for acetylated peptides substrates [43–48]; however there is no clear evidence of this sequence specificity when native proteins are used as substrates. Thus, one of the biggest challenges in sirtuin research is to determine how specificity is determined in natively folded substrate proteins. In recent years, a few studies have focused on complete and native protein specificity of sirtuins [42,77]. The results suggest that structural protein components are the main determinants of sirtuin specificity. The kinetics study carried out in this work, examining natively folded Acs deacetylation by CobB, is in concordance with this hypothesis. The results have shown a single kinetic phase for several acetylated lysines of Acs, indicating the same binding affinity or specificity for acetylated lysines at different positions (Fig 2). The results of this study and the lack of understanding of the reasons for the discrepancy between the in vitro and in vivo results suggest that the results of assays carried out using peptides as substrates should be contrasted with new results, which use whole native proteins.
Nicotinamide inhibition of several eukaryotic sirtuins has been studied, and the mechanism of inhibition has been characterized. The mechanism of NAM inhibition is based on nicotinamide intercepting the ADP-ribosyl-enzyme-acetyl-peptide intermediate, resulting in regeneration of the NAD+ substrate (transglycosidation) [53]. Several crystal structures of sirtuins with NAM have been determined. In these structures, NAM binds to the peptide-sirtuin reaction intermediate, but the binding location is not clear. In some structures, NAM appears to be bound to the pocket where the NAD+ substrate binds (C pocket) despite this binding not being consistent with non-competitive inhibition [67]. In other structures, NAM binds in a pocket other than the NAD+-binding pocket [56,72]. The characterization of CobB inhibition suggests that non-competitive nicotinamide inhibition occurs, which would agree with the transglycosidation mechanism. The kinetic parameters (Ki and IC50) that describe CobB inhibition by nicotinamide have also been determined. The obtained values (IC50 = 52 μM and Ki = 108 μM) are very similar to those found when nicotinamide inhibits other sirtuins. The Ki parameters of hSIRT2 and HST2 are 33.9 and 298 μM [53], respectively, while the IC50 values for Sirt1, Sirt2 and Sirt3 have been recently established at 175, 80 and 72 μM [71]. The Ki value determined for CobB is also in agreement with the only inhibition study of a bacterial sirtuin (Sir2Af2 from the sulfate-reducing Archaea Archeaoglobus fulgidus), which reported a Ki value of 26 μM [78]. These inhibition parameters, including the CobB Ki calculated in this study, are in the range from 20–300 μM.
Nicotinamidase is a relatively unstudied enzyme in E. coli. PncA participates in the NAD+ salvage pathway in E. coli, which is a route to NAD+ recycling. In this study, nicotinamidase has been kinetically characterized for the first time, and the physiology of the deletion strain ΔpncA has been studied. The results showed that a deficiency in nicotinamidase causes several differences between the mutant strain and wt E. coli and point to a clear similarity between the ΔpncA and ΔcobB strains. Physiological characterization of wt, ΔcobB and ΔpncA strains showed that when the mutant strains grew in minimal M9 medium supplemented with glucose, their specific growth rate (Table 3) was much lower than that of the wt strain in the same conditions. The levels of extracellular acetate, a result of E. coli acetate overflow, were also different in the ΔcobB and ΔpncA strains than in the wt (Fig 5), being higher in the mutant stains. This fact could be due to low activity by acetylated Acs in these mutants. Acs activity constitutes one of the two routes to assimilate acetate from the extracellular E. coli medium, where it was excreted to as a consequence of overflow metabolism [79,80]. The absence or inhibition of CobB would cause an increase in the acetylation of Acs that would totally or partially block this acetate pathway [21]. In addition, the flagella and motility in the three studied strains were analysed by electron microscopy and semi-solid agar assays (Fig 6). It has been reported that the transcription factor RcsB is acetylated by acetyl-phosphate and deacetylated by CobB at K154 [15,40,81]. Besides, RcsB K154 acetylation increases the expression of the flagella regulon, provoking an increase in motility and number of flagella. The ΔcobB mutant also shows this phenotype when RcsB is acetylated [15]. In this study, the pncA deletion strain showed more flagella and motility than the wt strain (a ten-fold increase), but flagella expression and motility were less pronounced than those of ΔcobB. This result suggests that in the ΔpncA mutant, the transcription factor RcsB could be partially inhibited by lysine acetylation as a consequence of low or no CobB deacetylase activity.
Intracellular nicotinamide concentrations in wt and ΔpncA strains have been determined under different conditions. In the wt E. coli strain, nicotinamide concentrations ranged between 30 and 70 μM during exponential growth phase. Stationary NAM concentrations were always lower (wt MM9 stationary NAM concentrations were not detected). Based on the in vitro inhibition of CobB (Fig 3), these concentrations could regulate CobB activity in vivo. To our knowledge, this example is the first time that an intracellular bacterial nicotinamide concentration has been determined to date. Previous studies have quantified nicotinamide concentrations in yeast (10–150 μM) [82] and in mammalian tissues (11–400 μM) [83–85]. These concentrations are very similar to those determined in this study in E. coli. Intracellular nicotinamide concentrations were higher in the ΔpncA strain than in wt E. coli (approximately two times higher). These higher concentrations could justify the physiological behaviour of the pncA deletion strain, i.e., conditions in which the CobB protein is totally or partially inhibited. This result suggests that CobB is regulated in vivo by nicotinamide concentration. Previous studies have demonstrated that yeast processes regulated by Sir2 are altered by additional copies of the NAD+-salvage route genes such as npt1 (nicotinate phosphoribosyltransferase), pnc1 (nicotinamidase), nma1, and nma2 (nicotinate mononucleotide adenylyltransfereases 1 and 2) [86], and exogenous nicotinamide [68]. In addition, pnc1 overexpression activate Sir2 in vivo, altering Saccharomyces longevity [87]. In addition, human breast cancer cell treatment with nicotinamide induces cellular apoptosis in vivo through Sirt1 inhibition [88], although a recent study points to the fact that nicotinamide addition to cells can stimulate sirtuins due to rapid in vivo conversion of nicotinamide to NAD+ [89].
The role of nicotinamide as an in vivo inhibitor of CobB activity, as suggested in this study, identifies this metabolite and the nicotinamidase PncA as potential regulators of central E. coli metabolism due to the global role of CobB (Fig 7). Thus, we have suggested that nicotinamide concentrations could regulate very different aspects of E. coli, specifically, acetate consumption and flagellum expression. The results of this work open the way for future studies on the regulatory roles of the NAD+ salvage route and nicotinamide, and how they affect post-translational acetylation in E. coli.
Fig 7. Central role of the nicotinamide metabolite in E. coli metabolism.

Supporting information
Restriction nuclease sites are in grey. Bold typeface indicates the modified codon during site-directed-mutagenesis.
(PDF)
Acknowledgments
Dr. Antonio Sánchez-Amat (Dept. Genetics and Microbiology, University of Murcia) is acknowledged for pBAD24-MBP plasmid transference. Dr. Alejandro Torrecillas (CAID, University of Murcia) is acknowledged for technical support.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported Ministerio de Industria, Economía y Competitividad (which includes ERDF European co-funding) (España) (http://www.mineco.gob.es/portal/site/mineco/), Grant number: BIO2014-54411-C2-1-R to MCD and Fundación Séneca (CARM, Murcia, España) (https://fseneca.es/#), Grant number: 19236/PI/14 to MCD. JG and AE are recipients of a PhD fellowship from Fundación Séneca (CARM, Murcia). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. Nature Publishing Group; 2014;16: 258–264. doi: 10.1038/nrm3931 [DOI] [PubMed] [Google Scholar]
- 2.Bernal V, Castaño-Cerezo S, Gallego-Jara J, Écija-Conesa A, de Diego T, Iborra JL, et al. Regulation of bacterial physiology by lysine acetylation of proteins. N Biotechnol. 2014;31: 586–95. doi: 10.1016/j.nbt.2014.03.002 [DOI] [PubMed] [Google Scholar]
- 3.Hentchel KL, Escalante-Semerena JC. Acylation of biomolecules in prokaryotes: a widespread strategy for the control of biological function and metabolic stress. Microbiol Mol Biol Rev. 2015;79: 321–46. doi: 10.1128/MMBR.00020-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedmann DR, Marmorstein R. Structure and mechanism of non-histone protein acetyltransferase enzymes. FEBS J. 2013;280: 5570–81. doi: 10.1111/febs.12373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vetting MW, S de Carvalho LP, Yu M, Hegde SS, Magnet S, Roderick SL, et al. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433: 212–26. doi: 10.1016/j.abb.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 6.Weinert BT, Iesmantavicius V, Wagner SA, Schölz C, Gummesson B, Beli P, et al. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol Cell. 2013;51: 265–72. doi: 10.1016/j.molcel.2013.06.003 [DOI] [PubMed] [Google Scholar]
- 7.Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, Minasov G, et al. Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS One. 2014;9: e94816 doi: 10.1371/journal.pone.0094816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64: 435–459. doi: 10.1128/MMBR.64.2.435–459.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okanishi H, Kim K, Masui R, Kuramitsu S. Acetylome with structural mapping reveals the significance of lysine acetylation in Thermus thermophilus. J Proteome Res. 2013;12: 3952–3968. doi: 10.1021/pr400245k [DOI] [PubMed] [Google Scholar]
- 10.Kim D, Yu BJ, Kim JA, Lee YJ, Choi SG, Kang S, et al. The acetylproteome of Gram-positive model bacterium Bacillus subtilis. Proteomics. 2013;13: 1726–1736. doi: 10.1002/pmic.201200001 [DOI] [PubMed] [Google Scholar]
- 11.Xie L, Wang X, Zeng J, Zhou M, Duan X, Li Q, et al. Proteome-wide lysine acetylation profiling of the human pathogen Mycobacterium tuberculosis. Int J Biochem Cell Biol. 2015;59: 193–202. doi: 10.1016/j.biocel.2014.11.010 [DOI] [PubMed] [Google Scholar]
- 12.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23: 607–18. doi: 10.1016/j.molcel.2006.06.026 [DOI] [PubMed] [Google Scholar]
- 13.Wagner GR, Payne RM. Widespread and enzyme-independent Nε-acetylation and Nε-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. 2013;288: 29036–45. doi: 10.1074/jbc.M113.486753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science (80-). 2010;327: 1000–4. doi: 10.1126/science.1179689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castaño-Cerezo S, Bernal V, Post H, Fuhrer T, Cappadona S, Sánchez-Díaz NC, et al. Protein acetylation affects acetate metabolism, motility and acid stress response in Escherichia coli. Mol Syst Biol. 2014;10: 762 doi: 10.15252/msb.20145227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gregoretti I V, Lee Y-M, Goodson H V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338: 17–31. doi: 10.1016/j.jmb.2004.02.006 [DOI] [PubMed] [Google Scholar]
- 17.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273: 793–798. doi: 10.1006/bbrc.2000.3000 [DOI] [PubMed] [Google Scholar]
- 18.Cosgrove MS, Bever K, Avalos JL, Muhammad S, Zhang X, Wolberger C. The structural basis of sirtuin substrate affinity. Biochemistry. 2006;45: 7511–7521. doi: 10.1021/bi0526332 [DOI] [PubMed] [Google Scholar]
- 19.Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999;260: 273–279. doi: 10.1006/bbrc.1999.0897 [DOI] [PubMed] [Google Scholar]
- 20.Landry J, Sutton a, Tafrov ST, Heller RC, Stebbins J, Pillus L, et al. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A. 2000;97: 5807–11. doi: 10.1073/pnas.110148297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science (80-). 2002;298: 2390–2392. doi: 10.1126/science.1077650 [DOI] [PubMed] [Google Scholar]
- 22.Baron S, Eisenbach M. CheY acetylation is required for ordinary adaptation time in Escherichia coli chemotaxis. FEBS Lett. 2017;591: 1958–1965. doi: 10.1002/1873-3468.12699 [DOI] [PubMed] [Google Scholar]
- 23.Barak R, Prasad K, Shainskaya A, Wolfe AJ, Eisenbach M. Acetylation of the chemotaxis response regulator CheY by acetyl-CoA synthetase purified from Escherichia coli. J Mol Biol. 2004;342: 383–401. doi: 10.1016/j.jmb.2004.07.020 [DOI] [PubMed] [Google Scholar]
- 24.Tu S, Guo SJ, Chen CS, Liu CX, Jiang HW, Ge F, et al. YcgC represents a new protein deacetylase family in prokaryotes. Elife. 2015;4: 1–17. doi: 10.7554/eLife.05322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan H, Marmorstein R. Structural basis for Sirtuin activity and inhibition. J Biol Chem. 2012;287: 42428–42435. doi: 10.1074/jbc.R112.372300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleszcz R, Paluszczak J, Baer-Dubowska W. Targeting aberrant cancer metabolism—The role of sirtuins. Pharmacol Reports. Institute of Pharmacology, Polish Academy of Sciences; 2015;67: 1068–1080. doi: 10.1016/j.pharep.2015.03.021 [DOI] [PubMed] [Google Scholar]
- 27.Kim E-J, Um S-J. SIRT1: roles in aging and cancer. BMB Rep. 2008;41: 751–756. doi: 10.5483/BMBRep.2008.41.11.751 [DOI] [PubMed] [Google Scholar]
- 28.Bell SD. The interaction of Alba, a conserved archaeal chromatin protein, with Sir2 and its regulation by acetylation. Science (80-). 2002;296: 148–151. doi: 10.1126/science.1070506 [DOI] [PubMed] [Google Scholar]
- 29.Starai VJ, Escalante-Semerena JC. Acetyl-coenzyme A synthetase (AMP forming). Cell Mol Life Sci. 2004;61: 2020–30. doi: 10.1007/s00018-004-3448-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardner JG, Escalante-Semerena JC. In Bacillus subtilis, the sirtuin protein deacetylase, encoded by the srtN gene (formerly yhdZ), and functions encoded by the acuABC genes control the activity of acetyl coenzyme a synthetase. J Bacteriol. 2009;191: 1749–1755. doi: 10.1128/JB.01674-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crosby HA, Heiniger EK, Harwood CS, Escalante- JC. Reversible N-Lysine acetylation regulates the activity of Acyl-CoA synthetases involved in anaerobic benzoate catabolism in Rhodopseudomonas palustris. Mol Microbiol. 2010;76: 874–888. doi: 10.1111/j.1365-2958.2010.07127.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayden JD, Brown LR, Gunawardena HP, Perkowski EF, Chen X, Braunstein M. Reversible acetylation regulates acetate and propionate metabolism in Mycobacterium smegmatis. Microbiology. 2013;159: 1986–1999. doi: 10.1099/mic.0.068585-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103: 10224–10229. doi: 10.1073/pnas.0603968103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao K, Chai X, Marmorstein R. Structure and substrate binding properties of cobB, a Sir2 homolog protein deacetylase from Escherichia coli. J Mol Biol. 2004;337: 731–41. doi: 10.1016/j.jmb.2004.01.060 [DOI] [PubMed] [Google Scholar]
- 35.De Diego Puente T, Gallego-Jara J, Castaño-Cerezo S, Sánchez VB, Espín VF, De La Torre JG, et al. The protein acetyltransferase PatZ from Escherichia coli is regulated by autoacetylation-induced oligomerization. J Biol Chem. 2015;290: 23077–23093. doi: 10.1074/jbc.M115.649806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Starai VJ, Escalante-Semerena JC. Identification of the protein acetyltransferase (Pat) enzyme that acetylates Acetyl-CoA synthetase in Salmonella enterica. J Mol Biol. 2004;340: 1005–12. doi: 10.1016/j.jmb.2004.05.010 [DOI] [PubMed] [Google Scholar]
- 37.Yan J, Barak R, Liarzi O, Shainskaya A, Eisenbach M. In vivo acetylation of CheY, a response regulator in chemotaxis of Escherichia coli. J Mol Biol. 2008;376: 1260–71. doi: 10.1016/j.jmb.2007.12.070 [DOI] [PubMed] [Google Scholar]
- 38.Liarzi O, Barak R, Bronner V, Dines M, Sagi Y, Shainskaya A, et al. Acetylation represses the binding of CheY to its target proteins. Mol Microbiol. 2010;76: 932–43. doi: 10.1111/j.1365-2958.2010.07148.x [DOI] [PubMed] [Google Scholar]
- 39.Zhang Q-F, Zhang Q, Gu J, Gong P, Wang X-D, Wang X, et al. Reversibly acetylated lysine residues play important roles in the enzymatic activity of Escherichia coli N-hydroxyarylamine O-acetyltransferase. FEBS J. 2013;280: 1966–79. doi: 10.1111/febs.12216 [DOI] [PubMed] [Google Scholar]
- 40.Hu LI, Chi BK, Kuhn ML, Filippova E V, Walker-Peddakotla AJ, Bäsell K, et al. Acetylation of the response regulator RcsB controls transcription from a small RNA promoter. J Bacteriol. 2013;195: 4174–86. doi: 10.1128/JB.00383-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu CX, Wu FL, Jiang HW, He X, Guo SJ, Tao SC. Global identification of CobB interactors by an Escherichia coli proteome microarray. Acta Biochim Biophys Sin (Shanghai). 2014;46: 548–555. doi: 10.1093/abbs/gmu038 [DOI] [PubMed] [Google Scholar]
- 42.Abouelfetouh A, Kuhn ML, Hu LI, Scholle MD, Sorensen DJ, Sahu AK, et al. The E. coli sirtuin CobB shows no preference for enzymatic and nonenzymatic lysine acetylation substrate sites. Microbiol Open. 2015;4: 66–83. doi: 10.1002/mbo3.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garske AL, Denu JM. SIRT1 top hits: Use of one-bead, one-compound acetyl-peptide libraries and quantum dots to probe deacetylase specificity. Biochemistry. 2006;10: 94–101. doi: 10.1038/nature13314.A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blander G, Olejnik J, Krzymanska-Olejnik E, McDonagh T, Haigis M, Yaffe MB, et al. SIRT1 shows no substrate specificity in vitro. J Biol Chem. 2005;280: 9780–9785. doi: 10.1074/jbc.M414080200 [DOI] [PubMed] [Google Scholar]
- 45.Gurard-Levis ZA, Kilian KA, Kim J, Bähr K, Mrksich M. Peptide arrays identify isoform-selective substrates for profiling endogenous lysine deacetylase activity. ACS Chem Biol. 2010;5: 863–873. doi: 10.1021/cb100088g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rauh D, Fischer F, Gertz M, Lakshminarasimhan M, Bergbrede T, Aladini F, et al. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat Commun. Nature Publishing Group; 2013;4: 2327 doi: 10.1038/ncomms3327 [DOI] [PubMed] [Google Scholar]
- 47.Smith BC, Settles B, Hallows WC, Craven MW, Denu JM. SIRT3 substrate specificity determined by peptide arrays and machine learning. ACS Chem Biol. 2011;6: 146–157. doi: 10.1021/cb100218d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khan AN, Lewis PN. Unstructured conformations are a substrate requirement for the Sir2 family of NAD-dependent protein deacetylases. J Biol Chem. 2005;280: 36073–36078. doi: 10.1074/jbc.M508247200 [DOI] [PubMed] [Google Scholar]
- 49.Baeza J, Dowell JA, Smallegan MJ, Fan J, Amador-Noguez D, Khan Z, et al. Stoichiometry of site-specific lysine acetylation in an entire proteome. J Biol Chem. 2014;289: 21326–21338. doi: 10.1074/jbc.M114.581843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bheda P, Jing H, Wolberger C, Lin H. The substrate specificity of Sirtuins. Annu Rev Biochem. 2016;85: 405–29. doi: 10.1146/annurev-biochem-060815-014537 [DOI] [PubMed] [Google Scholar]
- 51.Guarente L. Sirtuins in Aging and Diseases In: Walker JM, editor. Methods in Molecular Biology. Humana Press, Totowa, NJ; 2013. pp. 3–10. doi: 10.1007/978-1-62703-637-5_1 [DOI] [PubMed] [Google Scholar]
- 52.Sebastián C, Satterstrom FK, Haigis MC, Mostoslavsky R. From sirtuin biology to human diseases: An update. J Biol Chem. 2012;287: 42444–42452. doi: 10.1074/jbc.R112.402768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J Biol Chem. 2003;278: 50985–50998. doi: 10.1074/jbc.M306552200 [DOI] [PubMed] [Google Scholar]
- 54.Guan X, Lin P, Knoll E, Chakrabarti R. Mechanism of inhibition of the human sirtuin enzyme SIRT3 by nicotinamide: computational and experimental studies. PLoS One. 2014;9 doi: 10.1371/journal.pone.0107729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fischer F, Gertz M, Suenkel B, Lakshminarasimhan M, Schutkowski M, Steegborn C. Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao K, Harshaw R, Chai X, Marmorstein R. Structural basis for nicotinamide cleavage and ADP-ribose transfer by NAD(+)-dependent Sir2 histone/protein deacetylases. Proc Natl Acad Sci U S A. 2004;101: 8563–8. doi: 10.1073/pnas.0401057101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, et al. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12: 291–9. doi: 10.1093/dnares/dsi012 [DOI] [PubMed] [Google Scholar]
- 58.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177: 4121–4130. doi: 10.1128/JB.177.14.4121–4130.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Christensen DG, Orr JS, Rao C V, Wolfe J. Increasing growth yield and decreasing acetylation in Escherichia coli by optimizing the carbon-to-magnesium ratio in peptide-based media. Appl Environ Microbiol. 2017;83: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moulder R, Filén J-J, Salmi J, Katajamaa M, Nevalainen OS, Oresic M, et al. A comparative evaluation of software for the analysis of liquid chromatography-tandem mass spectrometry data from isotope coded affinity tag experiments. Proteomics. 2005;5: 2748–60. doi: 10.1002/pmic.200401187 [DOI] [PubMed] [Google Scholar]
- 61.Kapp EA, Schütz F, Connolly LM, Chakel JA, Meza JE, Miller CA, et al. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 2005;5: 3475–90. doi: 10.1002/pmic.200500126 [DOI] [PubMed] [Google Scholar]
- 62.Williamson J, Corkey B. Assays of intermediates of the citric acid In: Lowenstein J, Kaplan N, Colowick N, editors. Methods of Enzymology. Academic P. New York: Academic Press; 1969. pp. 494–497. [Google Scholar]
- 63.Smith BC, Hallows WC, Denu JM. A continuous microplate assay for sirtuins and nicotinamide producing enzymes. Anal Biochem. 2009;394: 101–109. doi: 10.1016/j.ab.2009.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duportet X, Bastos R, Aggio M, Granato S. The biological interpretation of metabolomic data can be misled by the extraction method used. Metabolomics. 2012;8: 410–421. doi: 10.1007/s11306-011-0324-1 [Google Scholar]
- 65.Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, et al. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics. 2009;8: 215–25. doi: 10.1074/mcp.M800187-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schilling B, Christensen D, Davis R, Sahu AK, Hu LI, Walker-Peddakotla A, et al. Protein acetylation dynamics in response to carbon overflow in Escherichia coli. Mol Microbiol. 2015;98: 847–863. doi: 10.1111/mmi.13161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: Altering the NAD + cosubstrate specificity of a Sir2 enzyme. Mol Cell. 2005;17: 855–868. doi: 10.1016/j.molcel.2005.02.022 [DOI] [PubMed] [Google Scholar]
- 68.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast Sir2 and human SIRT1. J Biol Chem. 2002;277: 45099–45107. doi: 10.1074/jbc.M205670200 [DOI] [PubMed] [Google Scholar]
- 69.Horswill AR, Escalante-Semerena JC. Characterization of the propionyl-CoA synthetase (PrpE) enzyme of Salmonella enterica: Residue lys592 is required for propionyl-AMP synthesis. Biochemistry. 2002;41: 2379–2387. doi: 10.1021/bi015647q [DOI] [PubMed] [Google Scholar]
- 70.Nambi S, Gupta K, Bhattacharya M, Ramakrishnan P, Ravikumar V, Siddiqui N, et al. Cyclic AMP-dependent protein lysine acylation in mycobacteria regulates fatty acid and propionate metabolism. J Biol Chem. 2013;288: 14114–14124. doi: 10.1074/jbc.M113.463992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feldman JL, Dittenhafer-Reed KE, Kudo N, Thelen JN, Ito A, Yoshida M, et al. Kinetic and structural basis for Acyl-group selectivity and NAD+ dependence in sirtuin-catalyzed deacylation. Biochemistry. 2015;54: 3037–3050. doi: 10.1021/acs.biochem.5b00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanders BD, Zhao K, Slama JT, Marmorstein R. Structural basis for nicotinamide inhibition and base exchange in Sir2 enzymes. Mol Cell. 2007;25: 463–472. doi: 10.1016/j.molcel.2006.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feldman JL, Dittenhafer-Reed KE, Denu JM. Sirtuin catalysis and regulation. J Biol Chem. 2012;287: 42419–42427. doi: 10.1074/jbc.R112.378877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seiner DR, Hegde SS, Blanchard JS. Kinetics and inhibition of nicotinamidase from Mycobacterium tuberculosis. Biochemistry. 2010;49: 9613–9619. doi: 10.1021/bi1011157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.French JB, Cen Y, Vrablik TL, Xu P, Allen E, Hana-Rose W, et al. Characterization of nicotinamidases: steady-state kinetic parameters, class-wide inhibition by nicotinaldehydes and catalytic mechanism. Biochemistry. 2011;49: 10421–10439. doi: 10.1021/bi1012518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mei Z, Zhang X, Yi J, Huang J, He J, Tao Y. Sirtuins in metabolism, DNA repair and cancer. J Exp Clin Cancer Res. Journal of Experimental & Clinical Cancer Research; 2016;35: 182 doi: 10.1186/s13046-016-0461-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knyphausen P, De Boor S, Kuhlmann N, Scislowski L, Extra A, Baldus L, et al. Insights into lysine deacetylation of natively folded substrate proteins by sirtuins. J Biol Chem. 2016;291: 14677–14694. doi: 10.1074/jbc.M116.726307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sauve AA, Schramm VL. Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry. 2003;42: 9249–9256. doi: 10.1021/bi034959l [DOI] [PubMed] [Google Scholar]
- 79.Renilla S, Bernal V, Fuhrer T, Castaño-Cerezo S, Pastor JM, Iborra JL, et al. Acetate scavenging activity in Escherichia coli: interplay of acetyl–CoA synthetase and the PEP–glyoxylate cycle in chemostat cultures. Appl Microbiol Biotechnol. 2012;93: 2109–24. doi: 10.1007/s00253-011-3536-4 [DOI] [PubMed] [Google Scholar]
- 80.Wolfe AJ. The Acetate Switch. Microbiol Mol Biol Rev. 2005;69: 12–50. doi: 10.1128/MMBR.69.1.12-50.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, Minasov G, et al. Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS One. 2014;9: e94816 doi: 10.1371/journal.pone.0094816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang T, Sauve A a. NAD metabolism and sirtuins: metabolic regulation of protein deacetylation in stress and toxicity. AAPS J. 2006;8: E632–E643. doi: 10.1208/aapsj080472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hagino Y, Lan SJ, Ng CY. Metabolism of pyridinium precursors of pyridine nucleotides in perfused rat Liver. J Biol Chem. 1968;10: 4980–4989. [PubMed] [Google Scholar]
- 84.Hoshino J, Schlüter U, Kröger H. Nicotinamide methylation and its relation to NAD synthesis in rat liver tissue culture. Biochemical basis for the physiological activities of 1-methylnicotinamide. BBA—Gen Subj. 1984;801: 250–258. doi: 10.1016/0304-4165(84)90074-6 [DOI] [PubMed] [Google Scholar]
- 85.Smythe G a., Braga O, Brew BJ, Grant RS, Guillemin GJ, Kerr SJ, et al. Concurrent quantification of quinolinic, picolinic, and nicotinic acids using electron-capture negative-ion gas chromatography–mass spectrometry. Anal Biochem. 2002;301: 21–26. doi: 10.1006/abio.2001.5490 [DOI] [PubMed] [Google Scholar]
- 86.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Cohen H, Lin SS, et al. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J Biol Chem. 2002;277: 18881–18890. doi: 10.1074/jbc.M111773200 [DOI] [PubMed] [Google Scholar]
- 87.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423: 181–185. doi: 10.1038/nature01578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang T, Cui H, Ma N, Jiang Y. Nicotinamide-mediated inhibition of SIRT1 deacetylase is associated with the viability of cancer cells exposed to antitumor agents and apoptosis. Oncol Lett. 2013;6: 600–604. doi: 10.3892/ol.2013.1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hwang ES, Song SB. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol Life Sci. Springer International Publishing; 2017; 1–16. doi: 10.1007/s00018-017-2527-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Restriction nuclease sites are in grey. Bold typeface indicates the modified codon during site-directed-mutagenesis.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.