

Abstract
Previously, we showed that a major in vitro and in vivo mechanism of resistance to pyrazinoic acid (POA), the bioactive component of the critical tuberculosis (TB) prodrug pyrazinamide (PZA), involves missense mutations in the aspartate decarboxylase PanD, an enzyme required for coenzyme A biosynthesis. What is the mechanism of action of POA? Upon demonstrating that treatment of M. bovis BCG with POA resulted in a depletion of intracellular coenzyme A and confirming that this POA-mediated depletion is prevented by either missense mutations in PanD or exogenous supplementation of pantothenate, we hypothesized that POA binds to PanD and that this binding blocks the biosynthetic pathway. Here, we confirm both hypotheses. First, metabolomic analyses showed that POA treatment resulted in a reduction of the concentrations of all coenzyme A precursors downstream of the PanD-mediated catalytic step. Second, using isothermal titration calorimetry, we established that POA, but not its prodrug PZA, binds to PanD. Binding was abolished for mutant PanD proteins. Taken together, these findings support a mechanism of action of POA in which the bioactive component of PZA inhibits coenzyme A biosynthesis via binding to aspartate decarboxylase PanD. Together with previous works, these results establish PanD as a genetically, metabolically, and biophysically validated target of PZA.
Keywords: tuberculosis, pyrazinamide, pyrazinoic acid, coenzyme A, aspartate decarboxylase
Graphical Abstract
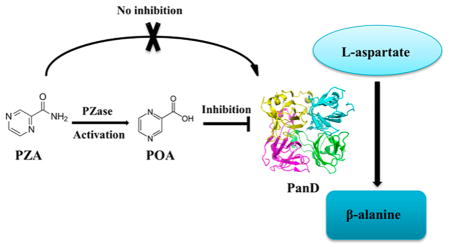
Of the first- and second-line tuberculosis (TB) drugs, pyrazinamide (PZA) is the most enigmatic in terms of its mechanism of action.1 Similar to other fragment-sized TB drugs, PZA is a prodrug that requires bioactivation to its active metabolite pyrazinoic acid (POA). Prodrug activation is performed by M. tuberculosis pyrazinamidase PncA2 and by host metabolism.3 Inactivation of amidase PncA causes resistance to PZA in vitro2 and in vivo4 as well as in the majority of clinical isolates.5
The mechanism of action of POA remains controversial. Until recently, it was believed that PZA inhibits M. tuberculosis growth only at acidic pH and that POA, being a low molecular weight carboxylic acid, acts as an ionophore affecting membrane energetics.6,7 This model has been questioned by recent evidence demonstrating that PZA can inhibit M. tuberculosis growth at neutral pH3,8 and that POA does not act as an ionophore.8 In addition, it turned out that the caseous cores of TB lesions may in fact not be acidic4,9,10 (however, see ref 11). Largely on the basis of biochemical evidence, two pathways have been proposed to be targeted by POA: fatty acid synthesis via targeting fatty acid synthetase FAS I12 and NAD+ biosynthesis via targeting quinolinic acid phosphoribosyltransferase QAPRTase.13 Furthermore, the ribosomal protein S114 and Rv2783, a protein involved in RNA and DNA metabolism,15 were proposed as targets. Recent evidence demonstrates that the antimycobacterial activity of PZA/POA is independent of trans-translation and RpsA.16 In addition, these targets could not be confirmed by the identification of respective resistance mutations through gene sequencing of clinical isolates17–19 or in vitro isolated POA-resistant M. tuberculosis/M. bovis BCG.20–22 Instead, genome sequencing of in vitro-isolated POA-resistant M. tuberculosis strains revealed two major mechanisms of resistance to PZA/POA: missense mutations in the aspartate decarboxylase PanD, involved in coenzyme A biosynthesis,20,22 and in the unfoldase/ATPase ClpC1, which is part of the degradative caseinolytic protease complex.21,23 The resistance mechanism associated with ClpC1 mutations may be indirect as overexpression of this protein did not result in resistance to POA.23 In addition to these two higher level resistance mechanisms, a third, lower level resistance mechanism was found to be associated with loss-of-function mutations in the phthiocerol dimycocerosate (PDIM) virulence factor producing polyketide synthases Mas and PpsA-E.20 Whether the Mas/PpsA-E related resistance mechanism is direct or indirect remains to be elucidated. Sequencing of POA-resistant M. tuberculosis strains isolated from lungs of mice treated with POA revealed that missense mutations in panD constitute the predominant mechanism of resistance to POA in vivo.24 Probing of the panD-related resistance mechanism in vitro revealed that treatment of M. bovis BCG with POA results in a depletion of intracellular coenzyme A levels.20,25 Our observation that resistance mutations in panD prevent POA-mediated depletion of intracellular coenzyme A levels20 and that this PanD-mediated resistance phenotype can be mimicked by supplementation of downstream metabolites of PanD such as β-alanine and pantothenate20,22,26 suggests a role of POA in disrupting the coenzyme A biosynthetic pathway.
The exact mechanism of the drug pertaining to the described observations remains largely a mystery. Shi et al.22 demonstrated that overexpression of wild-type PanD rendered M. tuberculosis resistant to POA, which indicates that POA may directly inhibit the enzyme. However, biochemical enzyme inhibition studies showed only a weak effect of POA on the conversion of aspartate to β-alanine (IC50 ~ 25 μg/mL22) and experiments using POA-resistance conferring mutant versions of aspartate decarboxylase still showed inhibition of enzyme activity by POA.22 In addition, Baughn and colleagues demonstrated that PZA could inhibit growth of an auxotrophic ΔpanCD strain of M. tuberculosis26 and proposed the intriguing hypothesis that the carboxylic acid POA may react directly with coenzyme A to form a thioester adduct.25
Taken together, it is evident that POA depletes intracellular coenzyme A and that missense mutations in PanD prevent that depletion and cause resistance. However, the exact molecular basis for these effects is not clear. Does POA bind to PanD and does this binding disrupt coenzyme A biosynthesis? Or, alternatively, does POA form an adduct with coenzyme A and directly deplete the cofactor? In this study, we provide evidence that PanD is a direct target of POA and that POA blocks coenzyme A synthesis at the aspartate decarboxylase step.
RESULTS
POA Treatment Reduces the Concentrations of Coenzyme A Pathway Metabolites Downstream of the PanD Catalytic Step
To determine the impact of POA treatment on mycobacterial metabolism, we carried out unbiased metabolomics analyses. A total of 343 metabolites were profiled upon treatment with different concentrations of POA at different time points. On the basis of our previous studies,20 we used concentrations corresponding to MIC90 (1 mM POA) and 4× MIC90 (4 mM POA) to treat M. bovis BCG and detected metabolites after 4 and 24 h to evaluate immediate POA-mediated on-target effects and follow-on metabolic consequences, respectively. We also show that POA is a slow-acting drug, whereby growth of M. bovis BCG remained unaffected by POA for 24 h after treatment (Figure S1A,B,20). These analyses were carried out with the attenuated, BSL2-compatible relative of M. tuberculosis, M. bovis BCG, for which we had previously shown that the mechanisms of POA resistance are conserved.20 To determine segregation of individual samples as a function of cellular metabolites upon treatment with POA at different time points, a principal component analysis was applied. This demonstrated overlap among all treatment groups at the 0 h time point. Segregation was observed between treatment groups at 4 and 24 h indicating that POA had a pronounced impact on global metabolism (Figure S1C).
Inspection of the metabolites involved in coenzyme A biosynthesis showed a dramatic reduction in the concentration of β-alanine, the product of the PanD-catalyzed step (>10-fold within 4 h, Figure 1B), as well as a reduction in the concentrations of all downstream metabolites including coenzyme A upon POA treatment (Figure 1). In contrast, the level of the substrate of PanD, aspartate, remained unaffected by POA treatment (Figure 1A). Overall, Figure 1 shows that POA exerted a time and concentration-dependent effect on all coenzyme A precursors downstream of the PanD-catalyzed step. These results suggest that POA disrupts coenzyme A biosynthesis by blocking PanD-mediated production of β-alanine.
Figure 1.

Effect of POA treatment on the levels of intermediates of the coenzyme A biosynthetic pathway. Intracellular levels of (A) aspartate, (B) β-alanine, (C) pantothenate, (D) phosphopantetheine, (E) 3′-dephospho coenzyme A, and (F) coenzyme A at time points 0, 4, and 24 h after treatment with no POA (black circles), 1 mM POA (blue squares), and 4 mM POA (purple triangles) are represented as scaled intensity, which are obtained by quantification of individual peaks from spectral data using area-under-the curve, normalized for the respective sample by total protein concentration as determined by the Bradford assay. Phosphopantothenoyl-L-cysteine, an intermediate in the production of 4′-phosphopantetheine (reaction catalyzed by CoaBC), was not detected in our experiments. Experiments were carried out in 4 independent biological replicates. Mean and standard deviations from these 4 replicate experiments are shown. Means were significantly different from levels of respective metabolites in untreated controls at respective time points at p <0.05 (*), <0.01 (**), <0.001 (***), one-way ANOVA multiple comparisons test, GraphPad Prism. Statistical significance is indicated by blue and purple asterisks for 1 and 4 mM POA treatment, respectively.
POA Treatment Results in Accumulation of Free Fatty Acids
Coenzyme A is a cofactor for the activation of fatty acids. Thus, upon depletion of coenzyme A by POA, one would expect an increase in concentration of free fatty acids. Figure 2 shows that this is the case: POA exposure resulted in increased concentrations of medium-chain (Figure 2A), branched-chain, dicarboxylate (Figure 2B), and long-chain fatty acids (Figure 2C). While this effect was observed after 24 h of POA treatment, free fatty acid concentrations remained largely unchanged at 4 h after POA treatment (Table S1). This kinetic is consistent with the accumulation of free fatty acids being a follow-on event from the disruption of the coenzyme A biosynthetic pathway by POA.20
Figure 2.

Effect of POA treatment on levels of intracellular free fatty acids. Levels of (A) medium-chain fatty acids, (B) branched-chain and dicarboxylate fatty acids, and (C) long-chain fatty acids after 24 h upon treatment with no POA (black bars), 1 mM POA (blue bars), or 4 mM POA (purple bars) are represented as scaled intensity, which are obtained by quantification of individual peaks from spectral data using area-under-the curve, normalized for the respective sample by total protein concentration as determined by the Bradford assay. Experiments were carried out in 4 biological replicates. Mean and standard deviations from these 4 replicate experiments are shown. Means were significantly different from levels of respective metabolites in untreated controls after 24 h at p <0.05 (*), <0.01 (**), <0.001 (***), one-way ANOVA multiple comparisons test, GraphPad Prism.
POA Treatment Affects Central Carbon Metabolism but Not NAD Biosynthesis
Previous studies have shown that several different classes of antibiotics modulating different cellular targets in M. tuberculosis redirect cellular carbon fluxes by remodelling the central carbon metabolism.27,28 Similarly, POA treatment results in an increase in concentrations of several glycolytic metabolites (Figure S2, Table S1). In addition to affecting glycolysis, POA treatment also remodelled the tricarboxylic acid (TCA) cycle (Figure S2, Table S1). This is accompanied by a significant decrease in glutamate pathway intermediates (Table S1). The observed POA-induced depletion of TCA cycle intermediates is in agreement with the metabolic signatures that were determined upon silencing of specific genes (panB, panC, coaBC, coaE) involved in the coenzyme A biosynthetic pathway.29
Table S1 also shows that POA treatment does not inhibit NAD biosynthesis. It was previously suggested, on the basis of biochemical studies by Kim et al., that PZA and POA inhibit QAPRTase, a key enzyme in the de novo synthesis of NAD in mycobacteria.13 We see here that POA treatment did not result in a decrease of metabolites downstream of QAPRTase. These results suggest that NAD biosynthesis is not a target of the drug in whole cells (Table S1).
POA but Not the Prodrug PZA Binds to PanD
Our metabolomics data show that POA blocks the pathway leading to the biosynthesis of coenzyme A at the PanD-catalyzed step, suggesting that PanD may be the direct target of POA. To determine whether POA indeed binds to PanD, wild-type (WT) PanD protein was generated (Figure 3) and isothermal titration calorimetry (ITC) experiments were carried out. Figure 4A shows the profile of a typical exothermic reaction, and the KD for POA was calculated to be 6.1 μM ± 0.88 μM. In contrast, no binding was detected for the prodrug PZA, consistent with PZA requiring bioactivation within M. tuberculosis to exert antimicrobial activity.2 Figure 4B shows that the force contributing to the free energy of this interaction comprises mainly attractive forces (electrostatic and hydrogen bonding) as suggested by the negative enthalpies (ΔH= −1.2 × 104 ± 1440 cal/mol) of the binding signature.
Figure 3.

Schematic structure and SDS PAGE analysis of PanD proteins used in this study. (A) Schematic structure of PanD proteins. The structures of PanD wild-type protein (PanDwt), PanDH21R (POA resistance conferring H21R substitution in the N-terminal region), PanD127TRASC131 (POA resistance conferring substitution of the C-terminal tail 127NAGELLDPRLGVG139 with 127TRASC131), and PanD114Stop (without C-terminal, i.e., with aspartate decarboxylase domain only) are shown. Numbers indicate amino acid residues. G24-S25 indicates position of the autocleavage site (1). Amino acid sequences at the C-terminal tail are indicated in single letter code.39 (B) Verification of purified recombinant PanD proteins. SDS gel (17% total acrylamide and 0.4% cross-linked acrylamide) of the purified recombinant proteins PanDWT, PanD127TRASC131, and PanDH21R and PanD114Stop. Lane M shows molecular weight markers. The observed larger bands are likely oligomers as reported previously and seen in the X-ray structure of the protein (1). PanD127TRASC131 migrates further compared to the other recombinant proteins, which may be caused by the charge difference at the accessible C-terminus of the protein.
Figure 4.

Binding affinity measurements for POA, PZA, and nicotinic acid (NA) with PanDwt using ITC. (A) Representative ITC profiles are shown. The top panel in the figures shows the injection profile after baseline correction. The bottom panel shows the integration (heat release) for each injection. The solid lines reveal the fit of the data to a function based on a one-site binding model. NA + POA + PanDwt shows the results of a competitive titration where PanD was incubated with nicotinic acid for 15 min before titration with POA. (B) Thermodynamic signature plots of POA and NA binding to PanDwt as deduced from the isothermal titration calorimetry profiles (see Materials and Methods).
POA Analogue Nicotinic Acid Binds Only Weakly to PanD but Shares Its Binding Epitope
To gain further insight into the interaction of POA with PanD, a close analogue of POA, nicotinic acid, was titrated against PanD. Nicotinic acid differs from POA only in its aromatic ring which contains one instead of two nitrogens. Similar to POA, nicotinic acid has been demonstrated to inhibit the growth of the tubercle bacillus. However, we showed previously that the mechanism of action of nicotinic acid is PanD/coenzyme A independent.20 Consistent with our genetic data, Figure 4A shows that nicotinic acid displayed greater than 5-fold lower affinity (KD = 34 μM ± 7.2) to PanD. By analyzing the signature plots of the respective interactions, it becomes clear that the polar elements in the aromatic ring of POA also contribute to binding affinity through hydrogen bonding. Reducing the number of nitrogen atoms in the ring to 1 results in a corresponding reduction in the enthalpic contributions to the free energy of the reaction, while the entropic contributions are significantly increased. To confirm that the analogue of POA binds to PanD via the same binding site as POA, a competitive titration was performed, where PanD was incubated with nicotinic acid for 15 min before titration with POA. As can be seen in Figure 4A, the binding affinity of POA was appreciably decreased, indicating that both ligands share the same binding site.
Missense Mutations in PanD Abolish Binding of POA
Multiple studies20,22,24,30 have established that amino acid sequence altering mutations in PanD constitute a key mechanism of resistance to POA. The majority of these mutations in PanD are clustered within the C-terminal region of PanD, outside its aspartate decarboxylase domain. In addition, these independent studies showed that mutations of H21 within the N-terminal region of the aspartate decarboxylase domain are also associated with POA resistance.20,22,24,30
After showing that POA binds PanD, suggesting that the enzyme is a direct target of POA, we hypothesized that the mechanistic basis for resistance is loss of binding. To determine whether POA resistance mutations indeed abolish binding to POA, we produced two recombinant proteins representing the N-terminal and the C-terminal resistance mutations and carried out isothermal titration calorimetry experiments. Figure 5 shows that PanDH21R, harboring the POA resistance mutation H21R (Figure 3; presented in POA resistant strains M. bovis BCG POA1.320), indeed did not show any binding to POA. Furthermore, Figure 5 shows that PanD127TRASC131, containing an altered C-terminal tail due to a frameshift mutation (Figure 3; presented in POA resistant strain M. bovis BCG POA2.220), also did not show any binding to POA. The finding that both mutations completely abolished binding of POA suggest that the mechanism of resistance is loss of POA binding and is consistent with PanD being a direct target for POA.
Figure 5.

Effect of N-terminal and C-terminal mutations in PanD on the binding affinity of POA. Representative isothermal titration calorimetry profiles are shown for POA with PanDH21R, PanD127TRASC131, and PanD114Stop, respectively. The top panel in each figure shows the injection profile after baseline correction. The corresponding bottom panel shows the integration (heat release) for each injection. The solid lines in the bottom panel reveal the fit of the data to a function based on a one-site binding model.
Our observation that both the N-terminal mutant PanDH21R as well as the C-terminal mutant PanD127TRASC131 lose their ability to bind POA suggests that both the N- and C-terminus are involved in POA–PanD binding. To confirm this dual binding model, we generated a truncated recombinant protein PanD114Stop which contained the aspartate decarboxylase domain without C-terminal tail (Figure 3). Figure 5 shows that, as expected, the PanD114Stop protein, which retained the H21 binding site, displayed a reduced but still significant binding affinity to POA.
Structural Insight of PanD and Its Mutants PanDH21R and PanD127TRASC131 in Solution
In order to characterize the structural features of PanD and the mutants PanDH21R and PanD127TRASC131 in solution, solution X-ray scattering (SAXS) measurements were carried out. SAXS analysis of a protein in solution provides information about the size (radius of gyration (Rg) and the maximum particle dimension (Dmax)), low-resolution shape, conformation, and assembly state. In the scattering curve, the size and shape information is at low angles (called Guinier region) and the surface-to-volume ratio is at higher angles (Porod region). SAXS patterns of PanDWT were recorded for four concentrations (1.5, 2.8, 5.5, and 7.4 mg/mL), which showed no concentration-dependent increase in size (Figure S4). The Guinier plots at low angles appeared linear and confirmed good data quality with no indication of protein aggregation (Figure S4, inset). The Rg value derived from the Guinier region was determined to be 35.40 ± 0.24 (Figure 6A) (Table S2). The extended scattering curve is converted using indirect Fourier transform to provide the distance distribution function (P(r)), which is a histogram of distances between all possible pairs of atoms within a particle. The P(r) of PanDWT showed a single peak and is right-skewed (Figure 6B) with the maximum particle dimension (Dmax) of 116 ± 10 Å. The Rg value extracted from the P(r) function was 35.69 ± 0.12 Å and is in agreement with the value extracted from the Guinier region. The scattering curve is transformed to a normalized Kratky plot ((qRg)2(I(q)/I(0)) vs qRg), which provides the information about the potential flexible behavior and/or extended shape of the particle. The normalized Kratky plot exhibited a very broad bell-shaped profile for PanDWT, indicating that the protein is well folded, and its peak is well shifted toward the right with respect to standard globular proteins, indicating the presence of motion and/or being extended (Figure 6C). On the basis of the volumes extracted from the higher angle of the scattering data, the Porod and excluded volumes (the scattering of the bulk solvent in the excluded volume of the solute with its ordered layer), and volume of correlation (defined as the ratio of I(0), to its total scattered intensity), the molecular weight of PanDWT was calculated to be approximately 104 ± 10 kDa reflecting that the recombinant protein exists as an octamer in solution (Table S2). The poor fitting (χ2 = 39.8) of the tetrameric crystal structure (PDB ID: 2C45; with Rg value of 21.09 Å and Dmax of 72 Å) to the experimental scattering data further confirmed the presence of a larger order oligomeric particle. An octamer model was generated using the SASREF program, based on the tetrameric crystal structure, and had a good fit to the experimental data with χ2 value of 1.2 (Figure 6E). The octamer model also superimposed nicely to the low resolution shape of PanDWT, which was reconstructed ab initio using DAMMIN, with a normalized spatial discrepancy (NSD) of 1.06 (Figure 6D).
Figure 6.

Solution X-ray scattering studies of PanD proteins. (A) SAXS pattern (○) of WT PanD (PanDwt) at 7.4 mg/mL (black), PanDH21R at 5.0 mg/mL (green), and PanD127TRASC131 at 6.5 mg/mL (orange) concentrations. (Inset) Guinier plots show linearity at the concentrations used, indicating no aggregation. The scattering profiles are offset for clarity by applying arbitrary scale factors. (B) Overlapping of pair-distance distribution function P(r) of PanDwt (—; black), PanDH21R (—; green), and PanD127TRASC131 (—; orange) show no difference. (C) Normalized Kratky plot of PanDwt (●; black) compared to the mutants PanDH21R (●; green) and PanD127TRASC131 (●; orange) and the compact globular lysozyme (●; gray) with a peak (---; gray), representing the theoretical peak and assuming an ideal Guinier region of a globular particle. The scattering pattern of wild-type and mutants of PanD are similar and exhibits a broad bell-shaped profile shifted toward the right with respect to standard globular protein. (D) The averaged and filtered ab initio DAMMIN envelope (NSD = 0.47 ± 0.01) of WT PanD (gray spheres) superimposed onto the cartoon representation of the octameric model of PanD generated using the tetrameric crystallographic structure (PDB ID: 2C45). The tetrameric subunits are colored green and cyan. Front (left) and side (right) views are displayed. (E) Fitting of the experimental scattering pattern of WT PanD (○; black) and calculated scattering profile of the octameric model (—; red) derived from crystal structure using SASREF.
The SAXS data for the mutants, PanDH21R and Pan-D127TRASC131, were collected at 5.0 and 6.5 mg/mL, respectively (Table S2). The scattering pattern and the derived structural parameters were very similar to the wild-type protein, with the Guinier Rg values of 35.31 ± 0.31 and 35.46 ± 0.40 Å for PanDH21R and PanD127TRASC131, respectively (Figure 6A). The profiles of the P(r) function and the normalized Kratky plot are similarly shaped (Figure 6B,C), demonstrating that the wild-type and mutant proteins do not differ in size and shape in solution and that the mutations did not alter the overall folding.
The tendency of oligomer formation in PanDWT as well as the mutants used in these studies is nicely confirmed by SDSPAGE where the semidenaturated proteins run higher than their predicted molecular mass. As demonstrated for other oligomeric proteins like the c-subunits of thermophilic31 and archaeal ATP synthases,32,33 only autoclaving of these proteins enabled a complete denaturation of the proteins. Therefore, PanDWT as well as PanDH21R and PanD127TRASC131 were autoclaved in the presence of 1 mM of DTT and subsequently applied onto an SDS gel. As revealed in Figure S5, all the recombinant proteins moved similarly inside the electric field and comparable to the respective marker protein. The fact that PanDWT, its single mutant, and truncated forms run differently in semidenaturated gels in the presence of the detergent SDS is not unusual as shown for the truncated ε subunit of the Escherichia coli F-ATP synthase34 or subunit d of the V-ATPase, 35 where changes of charges in amino acid composition alter the properties of the protein inside the electric field of the gel or where proteins of specific pKa values run higher than their respective predicted molecular weights.
POA–Coenzyme A Adducts Cannot Be Detected upon POA Treatment
Rosen et al. proposed an alternative mechanism of how POA may deplete coenzyme A. POA, being a low molecular weight carboxylic acid, may be recognized as a substrate by some acyl CoA ligases, resulting in an adduct formation of POA with coenzyme A, thus depleting the intracellular cofactor pool.25 To determine if treatment with POA results in the formation of POA–coenzyme A adducts, we employed an LC-MS approach using a synthesized POA–coenzyme A thioester as standard (Figure S6). We have demonstrated that treatment of M. bovis BCG with 1 or 4 mM POA (MIC90 of POA = 1 mM) results in a significant decrease in coenzyme A levels after 24 h.20 Under the same conditions, we sought to detect POA–coenzyme A adduct formation in wild-type M. bovis BCG. However, no POA–CoA was detectable in POA treated samples (Figure S6B). This result argues against POA–coenzyme A adduct formation as a mechanism of action of POA.
DISCUSSION AND CONCLUSION
Recently, missense mutations in the aspartate decarboxylase PanD, catalyzing the conversion of aspartate to β-alanine in the coenzyme A biosynthetic pathway, emerged as the major mechanism conferring resistance to POA in M. tuberculosis in vitro and in vivo,20–22,24 while also conferring resistance to the prodrug PZA. Furthermore, it was shown that POA depletes coenzyme A and pantothenate and that panD POA-resistance mutations prevent this depletion.20 However, whether PanD is actually a target of POA, i.e., whether POA interferes with coenzyme A production via binding to PanD and blocking this enzymatic step or whether resistance is caused by some indirect mechanism, remained controversial.22,26 Here, we first carried out an unbiased metabolome analysis to determine the effect of POA on the coenzyme A biosynthetic pathway. The results show that treatment with POA causes a collapse of all metabolite concentrations of the pathway immediately down-stream of the aspartate decarboxylase PanD catalyzed reaction (Figure 1). This suggests that POA interferes with PanD as its mechanism of action. Indeed, biophysical binding studies showed that POA (but not its prodrug PZA) binds to recombinant wild-type PanD with relatively high affinity (KD = 6.1 μM ± 0.88 vs MIC = 1 mM) (Figure 4). Binding was abolished for recombinant PanD proteins carrying POA-resistance conferring mutations (Figure 5), thus explaining the observed resistance mechanism. Taken together, these results establish PanD as the direct molecular target of POA and inhibition of coenzyme A biosynthesis as a mechanism of action of PZA/POA.
The analyses of the effect of POA on the metabolome also revealed accumulation of a large variety of free fatty acids (Figure 2), consistent with POA-mediated depletion of coenzyme A. It is interesting to note that accumulation of fatty acids can be cytotoxic by interfering with the cell membrane, thus disrupting processes such as oxidative phosphorylation and resulting in collapse of the membrane potential.36 Hence, POA induced “fatty acid cytotoxicity” may represent a follow-on event of the primary mechanism of action of POA via depletion of coenzyme A and therefore contribute to the antimicrobial whole cell activity of POA. Indeed, it was demonstrated previously that POA exposure results in disruption of the membrane potential.7 The authors attributed this effect to POA acting as an ionophore, a mechanism later put into question.8 The observed POA-induced fatty acid accumulation reported here provides a possible alternative explanation for the observed POA-induced collapse of the membrane potential. The secondary effect of POA treatment on the metabolome, accumulation of potentially toxic fatty acids, is also interesting in light of the recent discovery that loss-of-function mutations of the acyl coenzyme A ligase FadD2 confer hyper-susceptibility to POA.25 FadD2, mediating intrinsic resistance to POA, is involved in the detoxification of fatty acids and the POA-dependent increase in fatty acids uncovered here may explain FadD2’s POA-related phenotype. Finally, blocking of coenzyme A synthesis and the follow-on effect on fatty acid synthesis also provides an explanation for the previous observation that POA interferes with fatty acid biosynthesis.12 The proposed mechanism, direct inhibition of fatty acid synthetase I,12 was later put into question.37 The accumulation of a wide range of fatty acids presented here could be interpreted as evidence that POA interferes with the synthesis of this class of molecules, suggesting an alternative explanation via an indirect mechanism for the impact of POA on fatty acid synthesis. It is noteworthy that the metabolome did not show a significant effect on NAD biosynthesis upon POA treatment (Table S1), suggesting that QAPRTase, proposed as a POA target based on biochemical studies,13 may not be of relevance in intact cells. Finally, our mass spectrometric analysis of metabolites in POA treated M. bovis BCG did not provide any evidence for the formation of a POA adduct with coenzyme A (Figure S6). POA–coenzyme A thioester formation was proposed as a possible PanD-independent mechanism of POA.25 Our data suggest that this alternative is unlikely. One should note that the interpretation of the effects of POA treatment on metabolite concentrations is limited by the fact that POA exposure also slowed down growth of the cultures; i.e., changes we see may be due to changes in growth rate as opposed to being POA-specific. In future works, additional controls need to be carried out, including metabolome studies of cultures treated with other growth inhibitors and studies employing silencing of coenzyme A pathway genes while confirming these effects in virulent M. tuberculosis, which is different from M. bovis BCG with regard to some specific steps in central carbon metabolism.38
What can we learn from the POA binding studies with several recombinant forms of the PanD protein regarding the binding epitope? The mycobacterial PanD protein consists of a canonical aspartate decarboxylase domain and a 25-amino acid C-terminal tail, with the last 13 amino acids being specific to mycobacteria (Figure 3A).39 The function of this C-terminal region is unknown.39 Aspartate decarboxylases are synthesized as pro-enzymes and undergo an autocatalytic cleavage in the N-terminal region between G24 and S25 to produce the active form of the enzyme (Figure 3A).40 An X-ray structural analysis showed that the mycobacterial PanD forms tetrameric complexes (Figure S7).39 The terminal tail of the C-terminal region appears to be located in proximity to the autocleavage site of the preceding subunit. However, atomic resolution was not achievable, suggesting that the tail may be partially disordered (Figure S7).39
Amino acid sequence-altering mutations occur in two discrete locations in PanD (Figure S8).20,22,24,30 One location is a single amino acid position, H21, close to the autocatalytic cleavage site G24-S25 in the N-terminal part of the aspartate decarboxylase domain (Figure 3A). H21 was found to be substituted by the residues R, N, or Q in several POA-resistant strains (Figure S8).30 In contrast to the very site-specific resistance mutations in the N-terminus, a large number of different amino acid sequence changes scattered along the 25 amino acid C-terminal confer POA resistance (Figures 3A and S8). Mutation types in this region include single amino acid substitutions as well as frameshift mutations (Figure S8). The largest number of missense mutations is observed in the 13-amino acid terminal tail of the C-terminus (Figure S8), i.e., the region of the tail that is closest to the autocatalytic cleavage site in the oligomeric PanD complex (Figure S7).
POA–PanD binding studies employing PanDH21R, a representative of N-terminal resistance mutations at H21, and PanD127TRASC131, a representative of C-terminal resistance mutations, showed that both mutation types abolished POA binding (Figure 5). The tetrameric crystal structure of PanD [2C45] shows that the C-terminus of PanD appears to come close to N-terminal residues including H21 (Figure S7). On the basis of the genetic data, together with the biophysical and available structural data, we propose a speculative dual binding model in which the imidazole ring of H21 allows π–π interactions and/or interactions between the NH groups of the imidazole and the ring of POA, while the carboxyl group of POA interacts with one of the polar C-terminal residues. These interactions would be consistent with the attractive forces observed in the isothermal titration calorimetry data, suggesting a large negative enthalpy for POA binding to the protein (Figure 4B). Consistent with our speculations on the dual binding model, a truncated PanD, in which the last 25 amino acids were deleted, still showed binding to POA, albeit with a more than 10-fold reduced affinity (Figure 5). It is interesting to note that, while the truncated PanD114Stop without C-terminal tail retained weak POA binding, POA binding of PanD127TRASC131 harboring an altered C-terminus was not detectable. This suggests that the C-terminal mutations alter the structure and flexibility of the tail which in turn prevents access of POA to the H21 binding site in the N-terminus.
It is interesting to note that the proposed binding model of POA also provides a plausible mechanism for how the compound may block the aspartate–β-alanine conversion step. As mentioned earlier (see the introduction), biochemical studies of PanD showed only weak inhibition of its aspartate decarboxylase activity, which was not abolished for PanD protein harboring POA resistance mutations.22 This could indicate that inhibition of PanD’s enzymatic activity may not be the primary or only mechanism of action of this compound. An additional possibility is that POA interferes with, and prevents, autocatalytic formation of enzymatically active aspartate decarboxylase from the inactive pro-enzyme.
In conclusion, the data presented here, together with other recent findings, reveal PanD as the first genetically, metabolically, and biophysically validated target of POA/PZA. How exactly POA binding to the aspartate decarboxylase blocks its function remains to be determined. We propose a model in which POA binds to the PanD complex at a dual epitope comprised of the N-terminal part of the aspartate decarboxylase domain near its autocatalytic cleavage site and the mycobacterium-specific C-terminal tail of the protein (of the neighboring subunit). It remains to be determined whether POA binding to PanD causes inhibition of autocatalysis releasing the active form of the enzyme, inhibition of the active form, or both. The identification of PanD as the molecular target of PZA/POA offers new testable mechanistic models and paves the way for a more detailed understanding of how POA interferes with the PanD target. Importantly, our findings provide the basis for a target-based, rational optimization of this critical drug and the discovery of the next generation of PZA with improved efficacy.
MATERIALS AND METHODS
Bacterial Strains, Culture Medium, and Chemicals
M. bovis BCG (ATCC 35734) and M. tuberculosis H37Rv (ATCC 27294) strains were maintained in complete Middlebrook 7H9 medium (BD Difco) supplemented with 0.05% (v/v) Tween 80 (Sigma), 0.5% (v/v) glycerol (Fisher Scientific), and 10% (v/v) Middlebrook albumin-dextrosecatalase (BD Difco) at 37 °C with agitation at 80 rpm. POA-resistant strains used for this study, M. bovis BCG POA2.2 and M. bovis BCG POA1.3, were isolated and described in ref 20. Pyrazinamide, pyrazinoic acid, and nicotinic acid were purchased from Sigma-Aldrich. Pyrazinoic acid was freshly dissolved in 90% DMSO at a concentration of 0.5 M and sterilized using 0.2 μm PTFE membrane filters (Acrodisc PALL).
Sample Preparation for Metabolomic Analyses
M. bovis BCG cultures were grown in complete 7H9 liquid medium to mid-log phase in 2 L Roller bottles (Corning) in 4 independent biological replicates (4 independent precultures were generated from 4 independent seed stocks). Each of these cultures was separately adjusted to an OD600 of 0.4 and treated with 0, 1, or 4 mM POA, respectively. For each replicate, at each time point (0, 4, or 24 h, respectively), an equivalent of 150 mL of OD600 = 0.4 was pelleted down at 3200 rpm for 10 min. The cells were pooled together into a single 1.5 mL microcentrifuge tube (Axygen), and the centrifugation step was repeated for each sample. The supernatant was removed using a fine-bore white tip (Eppendorf). The pellet was immediately frozen by placing on a cryorack (Thermo Fisher) and stored at −80 °C. The samples were shipped to Metabolon, Inc., USA. The metabolomics analysis was performed by Metabolon, Inc., CA, USA (details are described in Supplemental Methods, Text S1). To evaluate corresponding mycobacterial growth upon treatment of POA, growth curve analysis of M. bovis BCG was performed under identical conditions that were used to obtain samples for the metabolomics experiment by measuring optical density (OD600) at 600 nm (Ultrospec 10, GE Amersham) and plating samples on 7H10 agar to determine CFU/mL at each time point.
Cloning, Production, and Purification of M. tuberculosis PanD and the Resistant Mutants
The coding region of M. tuberculosis PanD (139 amino acids) was amplified by polymerase chain reaction (PCR) using the forward primer 5′-GCGCCATGGCCATGTTACGGACGATG-3′, the reverse primer 5′-GTAGAGCTCCTATCCCACACCGAGCC-3′, and the reverse primer 5′-CCATGAGCTCCTACGGTTTGTTGTAAGCGTCGAC-3′ for PanD114Stop using genomic DNA from M. tuberculosis H37Rv as a template. The sequences of panD in M. bovis BCG (ATCC 35734) and M. tuberculosis H37Rv (ATCC 27294) are 100% identical as determined by whole genome sequencing and PCR confirmation.20 The panD from POA-resistant mutant M. bovis BCG POA1.3 (panD: H21R) was amplified by PCR using the forward and reverse primers of the M. tuberculosis PanD and by using the resistant strain DNA as template. The other resistant strain M. bovis BCG POA2.2 (panD: Δ380A) contained a mutant PanD with 131 residues with the last five residues altered to 127TRASC131 when compared to the wild-type protein. This mutant was amplified using the forward primer 5′-GCGCCATGGCCATGTTACGGACGATG-3′ and the reverse primer 5′-GCCGAGCTCCTA GCAGCTCGCCCGCGTTTC-3′ and the respective resistant strain DNA as template. In all cases, the restriction sites (bold) for NcoI and SacI enzymes were used for the forward and reverse primers, respectively. The amplified products were ligated into the pET9-d1-His6 vector.41 The respective coding sequences were verified by DNA sequencing. The final plasmids were subsequently transformed into E. coli BL21 (DE3) cells (Stratagene). To express the individual proteins, liquid cultures were shaken in LB medium containing kanamycin (30 μg/mL) for about 6 h at 37 °C until an optical density (OD600) of 0.6–0.7 was reached. To induce the production of proteins, cultures were supplemented with isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, followed by incubation for 16 h at 20 °C. The cells were harvested by centrifugation at 8500g for 15 min.
The cells were lysed on ice by sonication with an ultrasonic homogenizer (Bandelin, KE76 tip) for 3 × 1 min in buffer A (50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 2 mM phenylmethane sulfonyl fluoride or phenylmethylsulfonyl fluoride (PMSF), 1 mM PefablocSC (4-(2-aminoethyl)-benzene-sulfonyl fluoride), and 0.8 mM DTT). After sonication, the cell lysate was centrifuged at 10 000g for 35 min at 4 °C. The resulting supernatant was passed through a filter (0.45 μm; Millipore) and supplemented with Ni2+-NTA resin pre-equilibrated in the same buffer. The His-tagged proteins were allowed to bind to the matrix for 1.5 h at 4 °C by mixing on a sample rotator (Neolab) and subsequently eluted with an imidazole gradient (0–350 mM). The fractions containing the wild-type PanD and the mutant PanD proteins were identified by SDS-PAGE42 and further purified by gel filtration chromatography using a Superdex 75 HR 10/30 column (GE Healthcare) with buffer containing 50 mM Tris-HCl (pH 7.5), 200 mM NaCl. The purity and homogeneity of the protein samples were analyzed by a 17% SDS gel. The gels were stained with Coomassie Brilliant Blue G250. Protein concentrations were determined using a BioSpec-nano spectrophotometer (Shimadzu, USA).
Isothermal Titration Calorimetry
ITC experiments were carried out with an ITC200 microcalorimeter (MicroCal, Northampton, UK), to study the binding of wild-type PanD, PanDH21R, PanD127TRASC131, or the recombinant PanD114Stop with PZA, POA, and their analogue nicotinic acid. Samples were desalted with water, centrifuged, and degassed before ITC studies. All the experiments were performed using 100 μM of protein and 2 mM of respective compound at 20 °C. Since the drugs POA, PZA, and NA used for ITC titrations released more heat upon titration with buffer, the drugs were dissolved in water and the protein was desalted with water using centricons. The titration of POA against water and the raw data of titration of POA against PanD in water are shown in the Figure S3. It is revealed as representative data, and the heat released from the water vs drug titration is always subtracted from the titration of PanD against test compounds. In all cases, control experiments were performed with the test compound and water. All experiments were repeated at least three times with different batches of protein. The dissociation constant was determined by the least-squares method, and the binding isotherm was fitted using the in-built Origin v7.0 (MicroCal), assuming a single-site binding model. The binding signature plots were obtained using thermodynamic parameters (ΔH, ΔS) obtained from ITC measurement. Gibbs-free energy (ΔG) was calculated from ΔG = ΔH − TΔS.
SAXS Studies
Solution X-ray scattering data were collected on a BRUKER NANOSTAR SAXS instrument equipped with a Metal-Jet X-ray source (Excillum, Germany) and VÅNTEC 2000-detector system as described in refs 43–45. The scattering patterns were measured using a sample detector distance of 0.67 m and a wavelength of λ = 1.3414 Å, covering the range of momentum transfer of 0.016 < q < 0.4 Å−1 (q = 4π sin(θ)/λ, where 2θ is the scattering angle). To monitor for radiation damage, six 5 min exposures were collected for each protein sample and no radiation effect was observed. WT PanD was measured at a concentration of 1.5, 2.8, 5.5, and 7.4 mg/mL, while PanDH21R and PanD127TRASC131 were measured at 5.0 and 6.5 mg/mL, respectively. The data were normalized to the intensity of the transmitted beam, and the scattering of the buffer was subtracted. The difference curves were scaled for concentration, and the data processing steps were performed using the program package PRIMUS from ATSAS package version 2.7.1.46 The forward scattering I(0) and the radius of gyration, Rg, were computed using the Guinier approximation. The pair distribution functions were calculated by GNOM47 providing also the maximum particle size Dmax. The low-resolution shape of PanDWT was determined ab initio by DAMMIN.48 Qualitative particle motion was inferred by plotting the scattering patterns in the normalized Kratky plot ((qRg)2(I(q)/I(0)) vs qRg).49 The theoretical scattering curves from atomic structure were generated and evaluated against experimental scattering curves using CRYSOL.50 The oligomeric state of the protein was confirmed from the molecular mass calculation based on Porod volume (Vp), excluded volume (Vex), and volume of correlation (Vc).51 An octameric model was generated using SASREF.52
Detection of POA–CoA
M. bovis BCG cultures were grown to mid-log phase in 1 L roller bottles (Corning) at 37 °C and pelleted in 50 mL tubes at 3200 rpm for 10 min. Pellets were resuspended in the specific growth medium after adjusting to an OD600 of 0.2 and incubated with or without POA (1 or 4 mM). At each time point for each treatment (0 or 24 h), the samples were plated on 7H10 to determine CFU, and an equivalent of 2 × 109 CFU were pelleted by centrifugation. The pellet was resuspended in 500 μL of sterile phosphate buffer saline (PBS) and homogenized by bead beating (Precellys 24 homogenizer). The lysate was pelleted by centrifugation, and the supernatant was subsequently used for analysis after being shipped to PHRI. POA–CoA quantification was achieved by using liquid chromatography coupled to mass spectrometry (LC-MS) methods as described in the Supplemental Methods.
Supplementary Material
Acknowledgments
This research was supported by the Singapore Ministry of Health’s National Medical Research Council under its TCR Flagship grant NMRC/TCR/011-NUHS/2014 and Centre Grant “MINE”, Research Core number 4, NMRC/CG/013/2013 to T.D. and is part of Singapore Programme of Research Investigating New Approaches to Treatment of Tuberculosis (SPRINT-TB; www.sprinttb.org) led by Nick Paton. T.D. receives institutional core funding from the Public Health Research Institute. We thank Priya Ramamoorthy, Kirk Pappan, and Rob Mahoney of Metabolon for the metabolomics data and analyses and Martin Gengenbacher, NUS BSL3 Core Facility, for discussions. P.G. received a scholarship from Yong Loo Lin School of Medicine.
ABBREVIATIONS
- ITC
isothermal titration calorimetry
- TB
tuberculosis
- PZA
pyrazinamide
- POA
pyrazinoic acid
- SAXS
solution X-ray scattering
Footnotes
Author Contributions
P.G., C.S., and M.Y. carried out microbiological work. W.N., P.R., and M.S.S.M. carried out protein biochemical and biophysical experiments. J.S. and F.K. carried out the POA–coenzyme A adduct related works. P.G., V.D., W.N., G.G., and T.D. wrote the paper.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis. 7b00079.
Principal component analysis (PCA) of metabolite profiles in M. bovis BCG, schematic representation of the glycolytic pathway and tricarboxylic acid cycle indicating metabolites altered upon POA treatment, binding affinity measurements for POA with PanDWT using ITC, solution X-ray scattering pattern of PanDWT protein, verification of purified recombinant PanD proteins, LC/MS/MS chromatograms displaying peaks (and mass transitions) for CoA, acetyl-CoA, and POA–CoA, structure of the PanD tetramer with the position of His21 indicated by spheres, mapping of POA-resistance mutations in aspartate decarboxylase PanD, supplemental methods, list of 343 detected metabolites and their fold change over untreated controls in POA-treated M. bovis BCG, and data collection and scattering derived parameters for PanD proteins (PDF)
References
- 1.Gopal P, Dick T. Reactive dirty fragments: implications for tuberculosis drug discovery. Curr Opin Microbiol. 2014;21:7–12. doi: 10.1016/j.mib.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 2.Scorpio A, Zhang Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat Med. 1996;2:662–667. doi: 10.1038/nm0696-662. [DOI] [PubMed] [Google Scholar]
- 3.Via LE, Savic R, Weiner DM, Zimmerman MD, Prideaux B, Irwin SM, Lyon E, O’Brien P, Gopal P, Eum S, Lee M, Lanoix JP, Dutta NK, Shim T, Cho JS, Kim W, Karakousis PC, Lenaerts A, Nuermberger E, Barry CE, Dartois V. Host-Mediated Bioactivation of Pyrazinamide: Implications for Efficacy, Resistance, and Therapeutic Alternatives. ACS Infect Dis. 2015;1:203–214. doi: 10.1021/id500028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lanoix JP, Ioerger T, Ormond A, Kaya F, Sacchettini J, Dartois V, Nuermberger E. Selective Inactivity of Pyrazinamide against Tuberculosis in C3HeB/FeJ Mice Is Best Explained by Neutral pH of Caseum. Antimicrob Agents Chemother. 2016;60:735–743. doi: 10.1128/AAC.01370-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Njire M, Tan Y, Mugweru J, Wang C, Guo J, Yew W, Tan S, Zhang T. Pyrazinamide resistance in Mycobacterium tuberculosis: Review and update. Adv Med Sci. 2016;61:63–71. doi: 10.1016/j.advms.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Scorpio A, Nikaido H, Sun Z. Role of Acid pH and Deficient Efflux of Pyrazinoic Acid in Unique Susceptibility of Mycobacterium tuberculosis to Pyrazinamide. J Bacteriol. 1999;181:2044–2049. doi: 10.1128/jb.181.7.2044-2049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Wade MM, Scorpio A, Zhang H, Sun Z. Mode of action of pyrazinamide: disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J Antimicrob Chemother. 2003;52:790–795. doi: 10.1093/jac/dkg446. [DOI] [PubMed] [Google Scholar]
- 8.Peterson ND, Rosen BC, Dillon NA, Baughn AD. Uncoupling Environmental pH and Intrabacterial Acidification from Pyrazinamide Susceptibility in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2015;59:7320–7326. doi: 10.1128/AAC.00967-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irwin SM, Prideaux B, Lyon ER, Zimmerman MD, Brooks EJ, Schrupp CA, Chen C, Reichlen MJ, Asay BC, Voskuil MI, Nuermberger EL, Andries K, Lyons MA, Dartois V, Lenaerts AJ. Bedaquiline and Pyrazinamide Treatment Responses Are Affected by Pulmonary Lesion Heterogeneity in Mycobacterium tuberculosis Infected C3HeB/FeJ Mice. ACS Infect Dis. 2016;2:251–267. doi: 10.1021/acsinfecdis.5b00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenaerts A, Barry CE, Dartois V. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol Rev. 2015;264:288–307. doi: 10.1111/imr.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kempker RR, Heinrichs MT, Nikolaishvili K, Sabulua I, Bablishvili N, Gogishvili S, Avaliani Z, Tukvadze N, Little B, Bernheim A, Read TD, Guarner J, Derendorf H, Peloquin CA, Blumberg HM, Vashakidze S. Lung Tissue Concentrations of Pyrazinamide among Patients with Drug-Resistant Pulmonary Tuberculosis. Antimicrob Agents Chemother. 2017;61:e00226–17. doi: 10.1128/AAC.00226-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimhony O, Cox JS, Welch JT, Vilcheze C, Jacobs WR. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat Med. 2000;6:1043–1047. doi: 10.1038/79558. [DOI] [PubMed] [Google Scholar]
- 13.Kim H, Shibayama K, Rimbara E, Mori S. Biochemical Characterization of Quinolinic Acid Phosphoribosyl-transferase from Mycobacterium tuberculosis H37Rv and Inhibition of Its Activity by Pyrazinamide. PLoS One. 2014;9:e100062. doi: 10.1371/journal.pone.0100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE, Wang H, Zhang W, Zhang Y. Pyrazinamide Inhibits Trans-Translation in Mycobacterium tuberculosis. Science. 2011;333:1630. doi: 10.1126/science.1208813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Njire M, Wang N, Wang B, Tan Y, Cai X, Liu Y, Mugweru J, Guo J, Hameed HMA, Tan S, Liu J, Yew WW, Nuermberger E, Lamichhane G, Liu J, Zhang T. Pyrazinoic Acid Inhibits a Bifunctional Enzyme in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2017;61:e00070–17. doi: 10.1128/AAC.00070-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dillon NA, Peterson ND, Feaga HA, Keiler KC, Baughn AD. Anti-tubercular Activity of Pyrazinamide is Independent of trans-Translation and RpsA. Sci Rep. 2017;7:6135. doi: 10.1038/s41598-017-06415-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander DC, Ma JH, Guthrie JL, Blair J, Chedore P, Jamieson FB. Gene Sequencing for Routine Verification of Pyrazinamide Resistance in Mycobacterium tuberculosis: a Role for pncA but Not rpsA. J Clin Microbiol. 2012;50:3726–3728. doi: 10.1128/JCM.00620-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia Q, Zhao L-l, Li F, Fan Y-m, Chen Y-y, Wu B-b, Liu Z-w, Pan A-z, Zhu M. Phenotypic and Genotypic Characterization of Pyrazinamide Resistance among Multidrug-Resistant Mycobacterium tuberculosis Isolates in Zhejiang, China. Antimicrob Agents Chemother. 2015;59:1690–1695. doi: 10.1128/AAC.04541-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akhmetova A, Kozhamkulov U, Bismilda V, Chingissova L, Abildaev T, Dymova M, Filipenko M, Ramanculov E. Mutations in the pncA and rpsA genes among 77 Mycobacterium tuberculosis isolates in Kazakhstan. Int J Tuberc Lung Dis. 2015;19:179–184. doi: 10.5588/ijtld.14.0305. [DOI] [PubMed] [Google Scholar]
- 20.Gopal P, Yee M, Sarathy J, Low JL, Sarathy JP, Kaya F, Dartois V, Gengenbacher M, Dick T. Pyrazinamide Resistance Is Caused by Two Distinct Mechanisms: Prevention of Coenzyme A Depletion and Loss of Virulence Factor Synthesis. ACS Infect Dis. 2016;2:616–626. doi: 10.1021/acsinfecdis.6b00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yee M, Gopal P, Dick T. Missense Mutations in the Unfoldase ClpC1 of the Caseinolytic Protease Complex Are Associated with Pyrazinamide Resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2017;61:e02342–16. doi: 10.1128/AAC.02342-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi W, Chen J, Feng J, Cui P, Zhang S, Weng X, Zhang W, Zhang Y. Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerging Microbes Infect. 2014;3:e58. doi: 10.1038/emi.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang S, Chen J, Shi W, Cui P, Zhang J, Cho S, Zhang W, Zhang Y. Mutation in clpC1 encoding an ATP-dependent ATPase involved in protein degradation is associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerging Microbes Infect. 2017;6:e8. doi: 10.1038/emi.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gopal P, Tasneen R, Yee M, Lanoix JP, Sarathy J, Rasic G, Li L, Dartois V, Nuermberger E, Dick T. In Vivo-Selected Pyrazinoic Acid-Resistant Mycobacterium tuberculosis Strains Harbor Missense Mutations in the Aspartate Decarboxylase PanD and the Unfoldase ClpC1. ACS Infect Dis. 2017;3:492. doi: 10.1021/acsinfecdis.7b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosen BC, Dillon NA, Peterson ND, Minato Y, Baughn AD. Long-Chain Fatty Acyl-CoA Ligase FadD2Mediates Intrinsic Pyrazinamide Resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2016:e02130-16. doi: 10.1128/AAC.02130-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dillon NA, Peterson ND, Rosen BC, Baughn AD. Pantothenate and Pantetheine Antagonize the Antitubercular Activity of Pyrazinamide. Antimicrob Agents Chemother. 2014;58:7258–7263. doi: 10.1128/AAC.04028-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nandakumar M, Nathan C, Rhee KY. Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat Commun. 2014;5:4306. doi: 10.1038/ncomms5306. [DOI] [PubMed] [Google Scholar]
- 28.Baek SH, Li AH, Sassetti CM. Metabolic Regulation of Mycobacterial Growth and Antibiotic Sensitivity. PLoS Biol. 2011;9:e1001065. doi: 10.1371/journal.pbio.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans JC, Trujillo C, Wang Z, Eoh H, Ehrt S, Schnappinger D, Boshoff HIM, Rhee KY, Barry CE, Mizrahi V. Validation of CoaBC as a Bactericidal Target in the Coenzyme A Pathway of Mycobacterium tuberculosis. ACS Infect Dis. 2016;2:958–968. doi: 10.1021/acsinfecdis.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S, Chen J, Shi W, Liu W, Zhang W, Zhang Y. Mutations in panD encoding aspartate decarboxylase are associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerging Microbes Infect. 2013;2:e34. doi: 10.1038/emi.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coskun Ü, Chaban YL, Lingl A, Müller V, Keegstra W, Boekema EJ, Grüber G. Structure and Subunit Arrangement of the A-type ATP Synthase Complex from the Archaeon Methanococcus jannaschii Visualized by Electron Microscopy. J Biol Chem. 2004;279:38644–38648. doi: 10.1074/jbc.M406196200. [DOI] [PubMed] [Google Scholar]
- 32.Brandt K, Müller DB, Hoffmann J, Hübert C, Brutschy B, Deckers-Hebestreit G, Müller V. Functional production of the Na+ F1FO ATP synthase from Acetobacterium woodii in Escherichia coli requires the native AtpI. J Bioenerg Biomembr. 2013;45:15–23. doi: 10.1007/s10863-012-9474-8. [DOI] [PubMed] [Google Scholar]
- 33.Brandt K, Müller DB, Hoffmann J, Langer JD, Brutschy B, Morgner N, Müller V. Stoichiometry and deletion analyses of subunits in the heterotrimeric F-ATP synthase c ring from the acetogenic bacterium Acetobacterium woodii. FEBS J. 2016;283:510–520. doi: 10.1111/febs.13606. [DOI] [PubMed] [Google Scholar]
- 34.Mendel-Hartvig J, Capaldi RA. Nucleotidedependent and dicyclohexylcarbodiimide-sensitive conformational changes in the epsilon subunit of Escherichia coli ATP synthase. Biochemistry. 1991;30:10987–10991. doi: 10.1021/bi00109a025. [DOI] [PubMed] [Google Scholar]
- 35.Thaker YR, Roessle M, Grüber G. The boxing glove shape of subunit d of the yeast V-ATPase in solution and the importance of disulfide formation for folding of this protein. J Bioenerg Biomembr. 2007;39:275–289. doi: 10.1007/s10863-007-9089-7. [DOI] [PubMed] [Google Scholar]
- 36.Desbois AP, Smith VJ. Antibacterial free fatty acids: activities, mechanisms of action and biotechnological potential. Appl Microbiol Biotechnol. 2010;85:1629–1642. doi: 10.1007/s00253-009-2355-3. [DOI] [PubMed] [Google Scholar]
- 37.Boshoff HI, Mizrahi V, Barry CE. Effects of Pyrazinamide on Fatty Acid Synthesis by Whole Mycobacterial Cells and Purified Fatty Acid Synthase I. J Bacteriol. 2002;184:2167–2172. doi: 10.1128/JB.184.8.2167-2172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keating LA, Wheeler PR, Mansoor H, Inwald JK, Dale J, Hewinson RG, Gordon SV. The pyruvate requirement of some members of the Mycobacterium tuberculosis complex is due to an inactive pyruvate kinase: implications for in vivo growth. Mol Microbiol. 2005;56:163–174. doi: 10.1111/j.1365-2958.2005.04524.x. [DOI] [PubMed] [Google Scholar]
- 39.Gopalan G, Chopra S, Ranganathan A, Swaminathan K. Crystal structure of uncleaved L-aspartate-α-decarboxylase from Mycobacterium tuberculosis. Proteins: Struct, Funct, Genet. 2006;65:796–802. doi: 10.1002/prot.21126. [DOI] [PubMed] [Google Scholar]
- 40.Albert A, Dhanaraj V, Genschel U, Khan G, Ramjee MK, Pulido R, Sibanda BL, Delft Fv, Witty M, Blundell TL, Smith AG, Abell C. Crystal structure of aspartate decarboxylase at 2.2 A resolution provides evidence for an ester in protein selfprocessing. Nat Struct Biol. 1998;5:289–293. doi: 10.1038/nsb0498-289. [DOI] [PubMed] [Google Scholar]
- 41.Grüber G, Godovac-Zimmermann J, Link TA, Coskun Ü, Rizzo VF, Betz C, Bailer SM. Expression, purification, and characterization of subunit E, an essential subunit of the vacuolar ATPase. Biochem Biophys Res Commun. 2002;298:383–391. doi: 10.1016/s0006-291x(02)02468-3. [DOI] [PubMed] [Google Scholar]
- 42.Laemmli UK. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 43.Balakrishna AM, Basak S, Manimekalai MSS, Grüber G. Crystal Structure of Subunits D and F in Complex Gives Insight into Energy Transmission of the Eukaryotic V-ATPase from Saccharomyces cerevisiae. J Biol Chem. 2015;290:3183–3196. doi: 10.1074/jbc.M114.622688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dip PV, Kamariah N, Subramanian Manimekalai MS, Nartey W, Balakrishna AM, Eisenhaber F, Eisenhaber B, Gruber G. Structure, mechanism and ensemble formation of the alkylhydroperoxide reductase subunits AhpC and AhpF from Escherichia coli. Acta Crystallogr, Sect D: Biol Crystallogr. 2014;D70:2848–2862. doi: 10.1107/S1399004714019233. [DOI] [PubMed] [Google Scholar]
- 45.Saw WG, Tria G, Grüber A, Subramanian Manimekalai MS, Zhao Y, Chandramohan A, Srinivasan Anand G, Matsui T, Weiss TM, Vasudevan SG, Grüber G. Structural insight and flexible features of NS5 proteins from all four serotypes of Dengue virus in solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2015;D71:2309–2327. doi: 10.1107/S1399004715017721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- 47.Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- 48.Svergun DI. Restoring Low Resolution Structure of Biological Macromolecules from Solution Scattering Using Simulated Annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Durand D, Vivés C, Cannella D, Pérez J, Pebay-Peyroula E, Vachette P, Fieschi F. NADPH oxidase activator p67phox behaves in solution as a multidomain protein with semi-flexible linkers. J Struct Biol. 2010;169:45–53. doi: 10.1016/j.jsb.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 50.Svergun D, Barberato C, Koch MHJ. CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- 51.Mylonas E, Svergun DI. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J Appl Crystallogr. 2007;40:s245–s249. [Google Scholar]
- 52.Petoukhov MV, Svergun DI. Global Rigid Body Modeling of Macromolecular Complexes against Small-Angle Scattering Data. Biophys J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.