

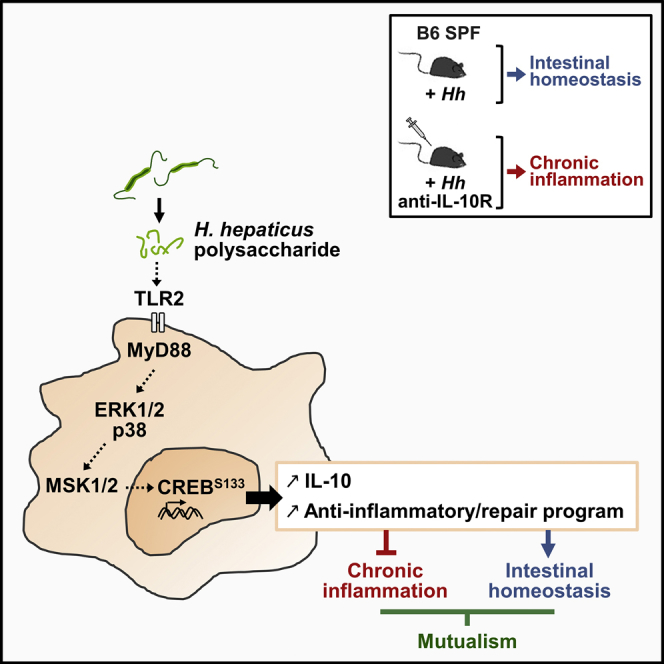
Summary
Interactions between the host and its microbiota are of mutual benefit and promote health. Complex molecular pathways underlie this dialog, but the identity of microbe-derived molecules that mediate the mutualistic state remains elusive. Helicobacter hepaticus is a member of the mouse intestinal microbiota that is tolerated by the host. In the absence of an intact IL-10 signaling, H. hepaticus induces an IL-23-driven inflammatory response in the intestine. Here we investigate the interactions between H. hepaticus and host immune cells that may promote mutualism, and the microbe-derived molecule(s) involved. Our results show that H. hepaticus triggers early IL-10 induction in intestinal macrophages and produces a large soluble polysaccharide that activates a specific MSK/CREB-dependent anti-inflammatory and repair gene signature via the receptor TLR2. These data identify a host-bacterial interaction that promotes mutualistic mechanisms at the intestinal interface. Further understanding of this pathway may provide novel prevention and treatment strategies for inflammatory bowel disease.
Keywords: inflammatory bowel disease, host-microbe interactions, mutualism, Helicobacter hepaticus, macrophage, anti-inflammatory gene signature, polysaccharide, TLR2, CREB, MSK1/2
Graphical Abstract
Highlights
-
•
Helicobacter hepaticus (Hh) activates a specific anti-inflammatory program in macrophages
-
•
This activity is driven by an Hh polysaccharide inducing high IL-10/IL-6 ratio in BMDMs
-
•
The polysaccharide-specific response is dependent on the TLR2/MSK/CREB pathway
Host-microbiota interactions are of mutual benefit, and chronic intestinal inflammation develops when this dialog is altered. Danne et al. identified a polysaccharide produced by Helicobacter hepaticus that induces a specific anti-inflammatory and repair program in macrophages by activating the TLR2/MSK/CREB pathway. Further understanding may provide prevention and treatment strategies for IBD.
Introduction
Shaped by a long history of co-evolution, the relationship between mammalian hosts and their intestinal commensal bacteria is of mutual benefit and promotes health. Complex molecular pathways underlie this dialog; however, the identity of microbe-derived molecules that contribute to the mutualistic state remain elusive. In inflammatory bowel disease (IBD), maladaptation of the host-microbe interface results in aberrant inflammatory responses to the intestinal microbiota. Recent evidence suggests that a complex interplay of host genetic, environmental, and microbial factors contributes to disease development (Kaser et al., 2010). Studies both in mouse models and in human disease have highlighted the role of appropriate intestinal epithelial barrier function, host defense, and immune regulation for maintaining intestinal homeostasis (Maloy and Powrie, 2011).
Infection of mice with the mouse pathogen Helicobacter hepaticus has provided important insights into host-microbe interactions in the gut. H. hepaticus inhabits the lower intestine, primarily the caecum, but does not induce immune pathology in normal mice (Kullberg et al., 1998). However, infection of genetically susceptible T and B cell-deficient 129SvEvRag−/− mice results in colitis (Erdman et al., 2003, Maloy et al., 2003), and H. hepaticus exacerbates T cell transfer colitis in C.B17 severe combined immunodeficient mice (Cahill et al., 1997). Appropriate host immune regulatory responses are crucial, as H. hepaticus infection of lymphocyte-replete mice with genetic- or pharmacological-induced deficiencies of the interleukin-10/interleukin-10 receptor (IL-10/IL-10R) pathway also results in colitis and typhlitis (Kullberg et al., 1998, Kullberg et al., 2006, Morrison et al., 2013). Under these circumstances, there is an aberrant IL-23-driven inflammatory response in the intestine with accumulation of pro-inflammatory granulocytes and monocytes that contributes to disease pathology (Arnold et al., 2016, Griseri et al., 2012, Hue et al., 2006, Krausgruber et al., 2016). Early studies showed that infection with H. hepaticus induces colonic regulatory T cells that prevent inflammation in an IL-10-dependent manner (Kullberg et al., 2002) and, more recently, that large amounts of IL-23 impede this response (Schiering et al., 2014). Relying both on a bacterial trigger and on an immune defect, H. hepaticus-induced colitis in the presence of IL-10/IL-10R pathway deficiency shares many features of human IBD. Indeed, mutations in Il10 or Il10R result in severe early-onset forms of IBD (Glocker et al., 2009, Kotlarz et al., 2012, Uhlig et al., 2014), indicating that IL-10 signaling is critical to prevent colitis both in humans and in mice. To date, little is known about the interaction of H. hepaticus with the innate immune compartment and its capacity to induce IL-10 production by these cells.
Strategically located at the mucosal barrier, intestinal lamina propria-resident macrophages function as immune sentinels. Essential for maintaining homeostatic responses, they are both sensors and interpreters of the intestinal tissue microenvironment, which comprises microbes and their products, and of immune mediators such as cytokines and chemokines (Ginhoux et al., 2016). The loss of IL-10R in tissue-resident macrophages results in spontaneous colitis, highlighting the importance of the macrophage response to IL-10 (Zigmond et al., 2014). Microbial products from both commensals and pathogens are recognized by pattern recognition receptors (PRRs), including Toll-like receptors (TLRs). The activation of PRRs triggers the production of pro-inflammatory mediators, such as IL-6 and tumor necrosis factor-α (TNFα), via several signaling pathways including the transcription factor NF-κB (Cohen, 2014). To prevent chronic inflammation and tissue damage, PRRs also activate anti-inflammatory signals, such as IL-10, together with negative feedback mechanisms, including the transcriptional repressor cAMP response element-binding protein (CREB) (Wen et al., 2010). However, the signaling mechanisms employed by particular gut inhabitants to actively promote intestinal homeostasis are poorly understood, as is their interaction with intestinal macrophages.
Here, we have investigated the interactions between H. hepaticus and macrophages that may promote tolerance and mutualism. Our results show that H. hepaticus induces an early IL-10 response by intestinal macrophages and produces a large soluble polysaccharide that activates a specific MSK/CREB-dependent anti-inflammatory and repair gene signature via the receptor TLR2.
Results
H. hepaticus Induces IL-10 in Gut-Resident Macrophages
As blockade of IL-10 signaling induces colitis in mice colonized with H. hepaticus (Kullberg et al., 2006, Morrison et al., 2013), we first assessed whether H. hepaticus interacts directly with the innate immune compartment within the intestinal mucosa to trigger IL-10 production. In order to investigate early responses to H. hepaticus at the cellular level, lamina propria leukocytes (LPL) from the colonic and caecal mucosa were isolated from Specific Pathogen Free (SPF) mice 3 days after infection with H. hepaticus strain ATCC51449 (Figure 1A). Flow cytometric analysis revealed that the frequency of CD4+ IL-10high T cells did not change at this time point (Figure S1A). Similarly, the frequency of MHCII+CD11b+CD11cint/highCD64+ resident macrophages among total CD45+ cells was unaffected by H. hepaticus infection (Figure 1B). However, the percentage of IL-10high resident macrophages increased significantly in the caecum following H. hepaticus colonization (Figures 1C and S1B). In whole tissue, no difference in levels of Il10 mRNA could be detected 3 days following infection (Figure 1D). However, there was a marked increase in the amount of Il10 mRNA 5 days after infection in both caecum and colon, whereas mRNA levels of the pro-inflammatory cytokines IL-6 and TNFα were unchanged or moderately increased, respectively (Figure 1D). These data indicate that H. hepaticus selectively promotes IL-10 production without a corresponding increase in pro-inflammatory cytokines from intestinal macrophages.
Figure 1.

H. hepaticus Induces IL-10 in Gut-Resident Macrophages
(A–D) SPF WT mice were infected with Helicobacter hepaticus (Hh) for 3 or 5 days.
(A) Experimental design.
(B and C) FACS analysis of caecum and colon LPL after 3 days of infection with Hh. (B) Frequency of resident macrophages among total CD45+ cells. (C) Frequency of IL-10high cells among resident macrophages, using an anti-mouse IL-10 antibody or its isotype control.
(D) Expression level of Il10, Il6, and Tnf mRNA in caecum or colon tissue after 3 or 5 days of infection with Hh.
Each symbol represents an individual mouse (two to three independent experiments). Mann-Whitney test (B and C) or one-way ANOVA and Tukey’s multiple comparisons test (D), p < 0.05. See also Figure S1.
H. hepaticus Produces a Large Soluble Polysaccharide that Can Induce IL-10 Production by Macrophages
To investigate whether H. hepaticus directly induces IL-10 in macrophages, we generated M-CSF differentiated bone marrow-derived macrophages (BMDMs) and subjected them to stimulation with whole bacteria (Hh, strain ATCC51449), a filtered supernatant of H. hepaticus culture medium (SNHh) or the control culture medium (TSB). Measurement of cytokine gene transcription and protein expression after 3 hr stimulation showed that Hh induced high amounts of IL-10 and moderate IL-6 and TNFα (Figures 2A and S2A). Interestingly, SNHh was sufficient to induce equivalent amounts of IL-10 in BMDMs, but a markedly diminished amount of IL-6 and TNFα compared to whole bacteria (Figures 2A and S2A). Treatment of SNHh (SNHht) with DNase, RNase, and proteinase K followed by heat (2 hr, 95°C) had no effect on its capacity to induce IL-10 production in BMDMs, but significantly attenuated both IL-6 and TNFα production (Figures 2B, 2C, S2B, and S2C). This suggests that the IL-10-inducing factor produced by H. hepaticus is not a nucleic acid or a protein, and therefore likely a polysaccharide. Fractionation by size using Vivaspin concentrator columns showed that the ability to induce IL-10 in BMDMs was restricted to a high molecular weight component (SNHht > 30 kDa), indicating that the active molecule is a large polysaccharide (Figure 2B). Stimulation with SNHht and SNHht > 30 kDa resulted in significantly higher IL-10/IL-6 and IL-10/TNFα ratios compared to whole H. hepaticus and non-treated SNHh (Figures 2D and S2C). Comparison with a known bacteria-derived immunostimulatory ligand, ultra-pure lipopolysaccharide from Escherichia coli (LPS UP), showed that the latter produced a more mixed cytokine response with large amounts of IL-10, IL-6, and TNFα, resulting in lower IL-10/IL-6 and IL-10/TNFα ratios compared to SNHht (Figures 2B–2D and S2C). SNHht induced IL-10 in a dose-dependent manner, but the IL-10/IL-6 and IL-10/TNFα ratios remained constant (Figure S2D). This suggests that the high ratios induced by SNHht are not a function of the stimulus concentration and reflect a qualitative response.
Figure 2.

H. hepaticus Produces a Large Soluble Polysaccharide-Inducing IL-10 Production in Macrophages
(A–G) M-CSF-differentiated BMDMs were stimulated for 3 hr with different culture fractions of H. hepaticus.
(A) mRNA and protein induction of the cytokines IL-10 and IL-6 after stimulation with control medium (TSB), H. hepaticus whole bacteria (Hh), or H. hepaticus-filtered cultured supernatant (SNHh).
(B–D) Induction of (B) IL-10, (C) IL-6, and (D) IL-10/IL-6 ratio after 3 hr stimulation with SNHh treated with enzymes and heat (SNHht), SNHht fractionated by size (SNHht > 30 kDa and SNHht < 30 kDa), and LPS UP.
(E) Induction of IL-10 after stimulation with TSBt or SNHht, treated with buffer (−) or sodium metaperiodate (+, NaIO4). Stimulation with TSBt ± NaIO4 and LPS ST is used as a positive control for IL-10 production by BMDMs.
(F) Induction of IL-10 after stimulation with TSBt, LPS ST, SNHht, or SNHht depleted using increasing concentrations of ConA-lectin beads (0.8%, 1.5%, 3%, and 6% v/v).
(G) Induction of IL-10 and IL-6 after stimulation with SNHht crude polysaccharide fraction extracted by a cold-ethanol precipitation method (CPHht).
Data from three independent experiments. Mann-Whitney test, p < 0.05. Mean ± SD. LPS UP, LPS ultrapure from E. coli O111:B4; LPS ST, LPS standard from E. coli O55:B5. See also Figure S2.
To further characterize the active molecule released into H. hepaticus culture supernatant that differentially stimulates IL-10 production, we oxidized SNHht using sodium metaperiodate (NaIO4) and dialyzed it against water to remove traces of reagent. This treatment cleaves sugars in the polysaccharide chains. Strikingly, after NaIO4 oxidation, SNHht lost its capacity to induce IL-10 (Figure 2E), supporting the idea that the active molecule in SNHht is a polysaccharide. This result was not a consequence of cellular toxicity of residual NaIO4 as simultaneous stimulation with both TSBt treated with NaIO4 and standard E. coli LPS (LPS ST) triggered IL-10 production by BMDMs (Figure 2E). To complement this approach, we subjected SNHht to incubation with different lectin-coated agarose beads. Lectins are carbohydrate-binding proteins with high specificity for mono- and oligosaccharides that also bind a diversity of complex polysaccharides. Our results showed that Concanavalin A from Canavalia ensiformis (ConA)-coated beads could deplete the IL-10-inducing factor from SNHht in a dose-dependent manner (Figure 2F), whereas other lectin-coated beads (lectins from Arachis hypogea, Peanut Agglutinin (PNA) or Lens Culinaris (LcH) had little effect on SNHht-induced IL-10 (Figure S2E). The lectins did not directly affect the capacity of BMDMs to produce IL-10, as shown by the simultaneous stimulation with both TSBt treated with ConA-coated beads and LPS ST (Figure 2F). Depletion by ConA suggests that SNHht is rich in α-mannose and α-glucose sugars. Finally, we used cold-ethanol precipitation to isolate the crude polysaccharide fraction (CPHht) from the treated supernatant (SNHht). CPHht recapitulated SNHht activity on BMDMs, with induction of high IL-10 but low IL-6 production (Figures 2G and S2B).
Collectively, these data indicate that H. hepaticus releases a large polysaccharide into the culture medium that induces macrophages to produce a selective response with large amounts of IL-10 and smaller amounts of pro-inflammatory cytokines such as IL-6 and TNFα.
H. hepaticus Supernatant Is Sufficient to Induce IL-10 In Vivo
To assess the activity of H. hepaticus polysaccharide in vivo, we orally administered TSBt, SNHht, or live H. hepaticus to SPF mice daily for 2 days and analyzed the colon and caecum LPL by flow cytometry on day 3. Like H. hepaticus infection (Figure 1), SNHht treatment similarly increased the frequency of IL-10high intestinal macrophages in the caecum, but not in the colon (Figures 3A, S3A, and S3B).
Figure 3.

SNHht Is Sufficient to Induce IL-10 In Vivo
(A) Frequency of IL-10high cells among resident macrophages from caecum and colon LPL. SPF WT mice were infected with Hh or orally gavaged with TSBt or SNHht for 3 days.
(B and C) Il10, Il6, and Tnf mRNA expression levels in the peritoneal cell fraction after 2 days (B) or Il10 and Il6 mRNA transcripts after 6 hr (C) challenge.
Ligands (TSBt and SNHht, 200 μL; LPS UP and Pam3, 50 μg) were injected into the intraperitoneal cavity of SPF WT mice. Each symbol represents an individual mouse (two to three independent experiments). Pam3, Pam3CSK4. Mann-Whitney test, p < 0.05. See also Figure S3.
To test the activity of SNHht in a different immune compartment, we injected TSBt or SNHht intraperitoneally either daily for 2 days with analysis 48 hr after the first administration or as a single injection with analysis after 6 hr. In both conditions, SNHht induced a significant increase in Il10 mRNA in total peritoneal cells compared to TSBt controls, with no change in Il6 or Tnf (Figures 3B, 3C, and S3C), similar to the in vitro observations. Pam3CSK4 (Pam3) and LPS UP injections induced similar Il10 mRNA but higher amounts of Il6 (Figure 3C) and Tnf (non-significant; Figure S3C) in total peritoneal cells after 6 hr.
SNHht Signals through TLR2 and MyD88 to Induce IL-10
To determine which sensing pathways were used by macrophages for IL-10 induction, we stimulated a panel of knockout BMDMs deficient in signaling proteins or PRRs—such as TLRs, NOD-like receptors (NLRs), and C-type lectin receptors (CLRs)—with SNHht or TSBt. In the response to bacterial components, TLR activation leads to recruitment of the adaptor molecules MyD88 (myeloid differentiation primary response gene 88) and/or TRIF (Toll-interleukin receptor domain containing adaptor-inducing interferon-γ), and switches on signaling networks that induce the production of various cytokines including IL-10, IL-6 and TNFα. Interestingly, Myd88−/− and Myd88−/−Trif−/−, but not Trif−/− BMDMs, lost their capacity to induce IL-10 at both transcriptional and protein levels in response to a 3 hr stimulation with SNHht (Figures 4A and 4B). Among the TLR-deficient cells tested (Tlr1−/−, Tlr2−/−, Tlr3−/−, Tlr4−/−, Tlr6−/−, and Tlr9−/−), only Tlr2−/− BMDMs failed to produce IL-10 after stimulation with SNHht (Figures 4A and 4B), suggesting that SNHht signals through the TLR2/MyD88 pathway. As expected, the crude polysaccharide fraction CPHht and the canonical TLR2/1 ligand Pam3 were not able to induce IL-10 in Tlr2−/− BMDMs, contrary to the TLR4 ligand LPS UP (Figure 4C). By contrast, BMDMs deficient for CLRs (MINCLE, CLEC4E; DECTIN-1, CLEC7A; MCL, CLECSF8; DNGR1, CLEC9A; MICL, CLEC12A), other receptors (IFNAR, interferon-α/β receptor; MR, Mannose Receptor; SRA, Scavenger Receptor A; MARCO; NOD2) or signaling proteins (RIP2; CARD9) were still able to produce IL-10 after stimulation with SNHht (Figure 4A).
Figure 4.

SNHht Signals through TLR2 and MyD88 to Induce IL-10
(A) Induction of IL-10 production in different knockout C57BL/6 BMDMs stimulated for 3 hr with SNHht normalized to WT BMDMs (three independent experiments). Two-way ANOVA and Bonferroni’s multiple comparisons test, p < 0.05. Ifnar, interferon-a/b receptor; Mr, mannose receptor; Sra, scavenger receptor A.
(B) Il10 mRNA and protein induction in different knockout BMDMs stimulated for 3 hr with SNHht (one of three independent experiments). One-way ANOVA and Tukey’s multiple comparisons test, p < 0.05.
(C) Induction of IL-10 production in WT and Tlr2−/− BMDMs stimulated for 3 hr with TSBt, SNHht, CPHht (SNHht crude polysaccharide fraction), LPS UP (TLR4 ligand), or Pam3 (TLR2/1) (one of three independent experiments). Two-way ANOVA and Sidak’s multiple comparisons test, p < 0.05.
(D) Induction of IL-10 production in WT BMDMs pre-treated for 2 hr with a blocking antibody specifically inhibiting TLR2 signaling (monoclonal α-mTLR2-IgG) or its isotype control before stimulation for 3 hr with TSBt, SNHht, Pam2 (TLR2/6), Pam3, or LPS UP (three independent experiments). Two-way ANOVA and Sidak’s multiple comparisons test, p < 0.05.
(E) Western blot showing the phosphorylation of ERK1/2 and p38 and the total ERK1/2 and p38 protein amounts in WT and Tlr2−/− BMDMs after stimulation with TSBt, SNHht, Pam3, or LPS UP for 15 min, 30 min, and 3 hr. Mean ± SD. See also Figure S4.
As a complementary approach, a TLR2 blocking monoclonal antibody or an isotype control was added prior to BMDM stimulation with SNHht or different canonical TLR ligands. TLR2 blockade inhibited IL-10 induction by SNHht and Pam3 (TLR2/1 ligand) but not by Pam2CSK4 (Pam2, TLR2/6) or LPS UP (TLR4) (Figure 4D). Downstream of TLR2 and MyD88, the mitogen-activated protein kinase (MAPK) TAK1 acts as an activator of several kinases, including the MAPKs ERK1/2, p38, and c-jun N-terminal kinase (JNK), which converge on transcription factors such as NF-κB. Pharmacological blockade of these downstream kinases showed that IL-10 induction by SNHht involves TAK1, MEK1/2 (ERK pathway), and p38, with comparatively weak contributions by JNK and NF-κB (Figure S4A). Immunoblot analysis showed marked phosphorylation of ERK1/2 (pT202/Y204) and p38 (pT180/Y182) MAPKs in WT BMDMs stimulated with SNHht at early time points (15 and 30 min) compared to Pam3 and LPS UP, with no change in total protein amounts (Figure 4E). As expected, the phosphorylation pattern induced by SNHht and Pam3 was lost in Tlr2−/− BMDMs (Figure 4E). In addition, as SNHht signals through TLR2, we compared its capacity to induce IL-10, IL-6, and TNFα in BMDMs to various canonical TLR2 and TLR4 ligands. Interestingly, SNHht induced higher IL-10/IL-6 and IL-10/TNFα ratios compared to Pam2 (TLR2/6 ligand), Pam3 (TLR2/1), FSL-1 (TLR2/6), zymosan from Saccharomyces cerevisiae (TLR2), lipomannan from Mycobacterium smegmatis (TLR2), LPS UP (TLR4), and LPS ST (TLR4 and TLR2) (Figure S4B).
These data indicate that TLR2/MyD88 is the pathway required for SNHht signaling, and furthermore that SNHht is a stronger driver of anti-inflammatory activity in macrophages than canonical TLR2 agonists.
Differential Regulation of Transcription by SNHht and Pam3
In order to assess whole genome differences in the macrophage transcriptional response to SNHht or to the canonical TLR2/1 agonist Pam3, we performed a microarray analysis of TSBt, SNHht, or Pam3-stimulated M-CSF differentiated BMDMs. Principle components analysis (PCA) demonstrated clear gene expression differences in macrophages stimulated by SNHht compared to Pam3 (Figure 5A). Hierarchical clustering analysis of differentially regulated genes identified four distinct clusters of co-regulated genes (Figure 5B). Each gene was assigned to a distinct cluster using k-means clustering (k = 4), representing genes that were (1) induced by Pam3 only (red cluster 1, n = 75); (2) repressed by both SNHht and Pam3 (green cluster 2, n = 10); (3) induced by both SNHht and Pam3 (blue cluster 3, n = 68); and (4) induced by SNHht only (purple cluster 4, n = 19). A large repertoire of pro-inflammatory genes was associated with the Pam3-specific transcriptional signature (Figure 5C). These included a number of genes associated with M1 pro-inflammatory macrophages (Il6, Saa3, Ccl5, Lcn2, and Fpr2) and others involved in the recruitment, activation, and proliferation of T cells (Cd40 or Tnfrsf5, Tnfrsf9, Icam1) (Chen and Flies, 2013, Lebedeva et al., 2005, Liu et al., 2013, Zigmond et al., 2014). By contrast, SNHht specifically induced the transcription of a number of genes that are highly expressed in tissue resident or M2 macrophages (Ccl7, Mmp13, Ptpn22) (Chang et al., 2013, Shi and Pamer, 2011, Toriseva et al., 2012), transcription factors known to repress the NF-κB pathway (Rcan1, Atf3) and T cell activation (Egr3), and genes involved in tissue repair and wound healing (Edn1, Mmp13, Hbegf) (Figure 5C) (Cheluvappa et al., 2014, De Nardo et al., 2014, Junkins et al., 2013, Safford et al., 2005, Toriseva et al., 2012, Wei et al., 2015). Of note, we observed that although the anti-inflammatory cytokine IL-10 was induced by both SNHht and Pam3 (compared to TSBt), the strength of induction was greater for SNHht, consistent with a lower capacity to induce a pro-inflammatory response upon SNHht stimulation (Figure 5C).
Figure 5.

Differential Regulation of Transcription by SNHht and Pam3
(A–F) Microarray analysis performed on M-CSF BMDMs differentiated from 5 WT mice and stimulated for 3 hr.
(A) Principle components analysis (PCA) of transcriptional profiles (empirical Bayes normalized expression values from LIMMA) across three conditions (TSBt, Pam3, and SNHht, n = 5/condition, 16,692 probes).
(B) Hierarchical clustering heatmap (Manhattan distance with Ward clustering of empirical Bayes normalized expression values) of the union of differentially expressed probes between any condition-condition contrast (i.e., TSBt versus Pam3, TSBt versus SNHht, or Pam3 versus SNHht) (n = 172 probes, n = 146 genes). Probes were assigned to clusters using k-means clustering (k = 4) and cluster assignments are annotated as colors to the left of the heatmap. The number of probes assigned to each cluster were: cluster 1 = 75, cluster 2 = 10, cluster 3 = 68, and cluster 4 = 19.
(C) Scatterplot displaying the relationship between log2 (fold changes) for each gene shown in (B) obtained when comparing either Pam3 (x axis) or SNHht (y axis) with the TSBt control condition. Colors represent the cluster assignments.
(D and E) Transcription factor (TF) motif enrichment analysis among genes assigned to (D) cluster 4 (i.e., specifically induced by SNHht) and (D) cluster 1 (i.e., specifically induced by Pam3). Motif names correspond to Transfac motif identifiers derived from the Molecular Signatures Database (MSigDB). Motifs significantly overrepresented at an adjusted p < 0.05 (permutation test) are shown.
(F) mRNA levels of CREB (Il10, Fosb, Egr3, Rcan1) and NF-κB (Il6, Ccl5, Cd40, Icam1) target genes were validated by qRT-PCR.
We reasoned that the differential transcriptional programs activated in response to these two TLR2 agonists were driven by alternative transcription factor profiles. To test this, we performed enrichment analysis of transcription factor motif gene sets from the Molecular Signatures Database (MSigDB) among genes that were specifically induced by either SNHht (purple cluster 4) or Pam3 (red cluster 1). Specific targets induced by SNHht were enriched for genes (Rcan1, Egr3, Fosb) with predicted binding sites for multiple transcription factors that included two motifs for the cyclic AMP response-element binding protein (CREB) (Figure 5D). This is of interest as CREB is a key transcriptional regulator known to promote anti-inflammatory signaling and repair (Lawrence and Natoli, 2011, Wen et al., 2010). Therefore, CREB activation might play a role in the suppression of TLR2-induced pro-inflammatory responses by SNHht. In addition, the observation of a more pro-inflammatory transcriptional program in response to Pam3 was confirmed through the identification of a significant enrichment of genes with predicted NF-κB binding sites among genes that were specifically induced by Pam3 stimulation (Figure 5E). Differential induction of each gene of interest, including targets of CREB (Il10, Fosb, Egr3, Rcan1) and NF-κB (Il6, Ccl5, Cd40, Icam1), was validated by qRT-PCR (Figure 5F).
Taken together, these data suggest that TLR2 activation in BMDMs results in markedly different transcriptional programs depending on the agonist. The increased anti-inflammatory properties of SNHht are likely dependent on its ability to activate CREB and repress the pro-inflammatory cytokine response.
SNHht Stimulates CREB Phosphorylation and Induction of an Anti-inflammatory and Repair Gene Signature in Macrophages
Immunoblot analysis of pS133-CREB1 and pS536-RelA in BMDMs revealed differential induction of CREB and NF-κB pathways after 30 min stimulation with SNHht, Pam3, or LPS UP (Figure 6A). Consistent with the microarray data, SNHht induced stronger CREB but weaker RelA phosphorylation compared to Pam3, and this pattern was lost in Tlr2−/− cells (Figure 6A). To test the functional role of CREBS133 phosphorylation in the induction of the CREB target genes identified in Figure 5, we differentiated BMDMs from conditional CREBS133A KI (Serin replaced by Alanin in position 133 of the CREB protein) and CREBWT mice. These mice were both generated using a minigene strategy (Wingate et al., 2009) with a CRE recombinase under the vav promoter, expressed throughout the hematopoietic compartment. CREB could not be phosphorylated at Ser133 in conditional Vav-cre CREBS133A KI BMDMs after stimulation with SNHht (Figure S5A). Stimulation of conditional Vav-cre CREBS133A KI and CREBWT BMDMs with SNHht for 1 hr showed significant induction of the CREB target genes Il10, Fosb, and Egr3 relative to stimulation with whole H. hepaticus or Pam3 (Figure 6B). The induction of Il10, Fosb, and Egr3 was partially dependent on CREB phosphorylation, whereas Il6, Tnf, or Cd40 induction was unaffected (Figures 6B and S5B). Antibody blockade of the IL-10R showed that induction of CREB target genes, specifically Fosb, Egr3, and Rcan1, was not due to an IL-10 autocrine pathway (Figure S5C). Interestingly, after 10 hr stimulation, CREBS133A KI BMDMs produced less IL-10 but more IL-6 and TNFα in response to SNHht compared to CREBWT BMDMs (Figure 6C). This led to strongly reduced IL-10/IL-6 and IL-10/TNFα ratios, reaching levels similar to those observed with whole H. hepaticus or Pam3 (Figure 6C). Importantly, the induction of anti-inflammatory CREB targets (Fosb, Egr3, Rcan1) but not pro-inflammatory NF-κB targets (Il6, Tnf, Cd40) was also observed in total caecum tissue from mice gavaged for 2 days with SNHht (Figure 6D). These data indicate that CREB phosphorylation promotes the anti-inflammatory properties of SNHht and that this pathway is operational in the intestine.
Figure 6.

SNHht Stimulates CREB Phosphorylation and Induction of an Anti-inflammatory and Repair Gene Signature in Macrophages
(A) Western blot showing the phosphorylation of CREB1 S133 and RelA S536 and the total CREB1 and RelA protein amounts in WT and Tlr2−/− BMDMs after 30 min stimulation with TSBt, SNHht, Pam3, or LPS UP. β-actin used as a loading control.
(B) mRNA levels of Il10, Fosb, and Egr3 genes in BMDMs from conditional vav-cre CREBWT and CREBS133A KI mice after 1 hr stimulation with TSBt, Hh, SNHht, or Pam3.
(C) Induction of IL-10, IL-6, and TNFα; IL-10/IL-6; and IL-10/TNFα protein ratios in BMDMs from conditional vav-cre CREBWT and CREBS133A KI mice after 10 hr stimulation with TSBt, Hh, SNHht, or Pam3. One of three independent experiments. Two-way ANOVA and Sidak’s and/or Tukey’s multiple comparisons tests, p < 0.05.
(D) mRNA levels of Il10, Fosb, Egr3, Rcan1, Il6, Tnf, and Cd40 genes in caecum total tissue from WT mice gavaged with TSBt or SNHht for 2 days. Each symbol represents an individual mouse (two independent experiments). Mann-Whitney test, p < 0.05.
Mean ± SD. See also Figure S5.
MSK1/2 is Essential for the Anti-inflammatory Properties of SNHht
The mitogen and stress-activated protein kinase (MSK) isoforms, MSK1 and MSK2, are activated downstream of p38 and ERK1/2 in vivo and phosphorylate CREB, triggering the rapid transcription of CREB target genes (Ananieva et al., 2008). We stimulated BMDMs differentiated from WT and Msk1/2−/− mice with SNHht. Immunoblot analysis of pS133-CREB1 and pT581-MSK1 revealed that SNHht phosphorylates MSK1 and that SNHht-mediated CREB phosphorylation requires MSK1/2 (Figure 7A). The stimulation of WT and Msk1/2−/− BMDMs with SNHht for 1 hr confirmed that the induction of CREB target genes (Il10, Fosb, and Egr3) is highly dependent on MSK1/2, contrary to the induction of Il6, Tnf, or Cd40 (Figures 7B and S6). As expected, Msk1/2−/− BMDMs produced less IL-10 but more IL-6 and TNFα in response to 10 hr stimulation with SNHht. The Msk1/2 deletion led to a strong reduction of both IL-10/IL-6 and IL-10/TNFα ratios, such that they were indistinguishable from the ratios induced by whole H. hepaticus or Pam3 (Figure 7C). Together, these data indicate that both MSK1/2 and CREB are required for the anti-inflammatory properties of SNHht.
Figure 7.

MSK1/2 Is Essential to the Anti-inflammatory Properties of SNHht
(A) Western blot showing the phosphorylation of CREB1 S133 and MSK1 T581 and the total CREB1 and MSK1 protein amounts in WT and Msk1/2−/− BMDMs after 30 min stimulation with TSBt, SNHht, Pam3, or LPS UP.
(B) mRNA levels of Il10, Fosb, and Egr3 genes in BMDMs from WT or Msk1/2−/− mice after 1 hr stimulation.
(C) Induction of IL-10, IL-6, and TNFα; IL-10/IL-6; and IL-10/TNFα protein ratios in BMDMs from WT or Msk1/2−/− mice after 10 hr stimulation.
One of three independent experiments. Two-way ANOVA and Sidak’s and/or Tukey’s multiple comparisons tests, p < 0.05. Mean ± SD. See also Figure S6.
Discussion
The intestine is home to billions of bacteria, and this complex ecosystem plays an important role in health and disease. It is well appreciated that microbial induction of IL-10 in the intestine is a non-redundant feature of tolerance to the microbiota, but the molecular dialog between host and microbe that determines tolerance and immunity remains elusive. Here, we show that H. hepaticus strain ATCC51449 produces a large polysaccharide that shapes the macrophage response. This polysaccharide triggers a high IL-10/IL-6 ratio compared to canonical TLR2 agonists and selectively activates the transcription factor CREB that, in turn, induces a panel of anti-inflammatory and repair mediators in the intestine.
H. hepaticus can be considered as a pathosymbiont based on its ability to live in harmony with its murine host in the presence of an IL-10 response, and its potential to drive IL-23-driven intestinal inflammation in IL-10 deficiency. The bacterial-sensing pathways that mediate these distinct responses are poorly understood. Our current understanding derives from in vitro studies in BMDMs showing that the activation of ERK inhibits the induction of IL-12 p40 by H. hepaticus (Tomczak et al., 2006). Moreover, epithelial cell lines transfected with human TLR2 could recognize the whole bacteria (Mandell et al., 2004). A key role for the adaptor MyD88 has been shown in vivo in the inflammatory response to H. hepaticus in IL-10 deficiency, but the upstream sensors were not identified (Asquith et al., 2010, Boulard et al., 2010). Our results show that H. hepaticus produces a polysaccharide that induces a specific CREB-dependent anti-inflammatory and repair gene signature in macrophages via the TLR2 pathway. CREB targets include Il10 as well as transcription factors that downregulate NF-κB and T cell activation, and genes involved in tissue repair. The transcription factor CREB is known to limit pro-inflammatory signaling downstream of TLRs and stimulate Il-10 transcription (Cohen, 2014, Wen et al., 2010). Previous studies have shown TLR-mediated activation of the CREB pathway. For example, LPS stimulation of BMDMs induced anti-inflammatory genes in an MSK/CREB-dependent manner (Ananieva et al., 2008, Darragh et al., 2010). Similarly, the TLR2/DECTIN-1 ligand zymosan and TLR2/4-signaling extracts from Schistosoma mansoni cercariae were able to activate CREB in BMDMs and induce various CREB target genes that are associated with a “regulatory” macrophage phenotype or cell metabolism, respectively (Elcombe et al., 2013, Sanin et al., 2015). Here, we identified a bacterial factor able to activate a CREB-dependent anti-inflammatory program both in vitro and in vivo, suggesting that the CREB pathway might play a crucial role in host-microbiota mutualism.
IL-10 itself inhibits the production of pro-inflammatory cytokines in macrophages through Jak–STAT3 activation (Moore et al., 2001, Murray, 2006); however, we found that the early induction of CREB targets by SNHht is independent of IL-10R signaling but rather relies on MSK1/2 kinase activity. Activated by p38 and ERK1/2, MSK1/2 phosphorylates multiple substrates, including CREB, ATF1, and Histone H3, and predominantly has anti-inflammatory roles (Reyskens and Arthur, 2016). Here, the deletion of MSK1/2 has a larger effect on SNHht-induced anti-inflammatory ratios compared to the loss of CREBS133 phosphorylation. This suggests that MSK1/2 activates both CREB-dependent and independent anti-inflammatory pathways in response to SNHht, potentially involving Histone H3-mediated epigenetic changes. As SNHht also induces this CREB/MSK-dependent immunomodulatory program in intestinal tissue in vivo, it is likely that it contributes to mutualistic relationships in the H. hepaticus-infected gut. Further studies are required to test this hypothesis.
IL-10 was previously shown to be critical for macrophage conditioning within the intestinal tissue, but IL-10 production by macrophages per se is not sufficient to explain their homeostatic activity (Shouval et al., 2014, Zigmond et al., 2014). The ability of SNHht to induce a CREB-driven anti-inflammatory response that is IL-10 independent may reveal additional checkpoints that control the macrophage inflammatory response in the intestine. H. hepaticus also promotes IL-10-producing regulatory T cells (Kullberg et al., 2002) raising the possibility that SNHht-conditioned macrophages promote this response.
By contrast with SNHht, the canonical TLR2 ligand Pam3 induced a broad NF-κB-dependent pro-inflammatory response in BMDMs. Microbe-induced TLR2 signaling promotes both pro- and anti-inflammatory responses (Nemati et al., 2017, Oliveira-Nascimento et al., 2012). However, it is not known whether this is a consequence of the structural features of particular ligands or whether it reflects the activities of additional sensors and receptors. CLRs, such as DECTIN-1 and DC-SIGN, have been shown to collaborate with TLRs to select specific responses to infectious agents (Ferwerda et al., 2008, Gantner et al., 2003). Here, we have ruled out the involvement of several CLRs and the common downstream CLR-signaling protein CARD9 in IL-10 induction by SNHht. Progress in this area is hampered by the often complex bacterial cell wall structures. Despite considerable effort, we were not able to analyze the structure and composition of the polysaccharide produced by H. hepaticus. The genome of H. hepaticus ATCC51449 does not contain a canonical polysaccharide biosynthesis operon, but many polysaccharide glycosyltransferase and LPS synthesis genes (Lombard et al., 2014). The LPS from H. hepaticus CCUG33637 was described as a low molecular weight molecule (Hynes et al., 2004), making a similar molecule an unlikely candidate. However, we cannot rule out that the high molecular weight polysaccharide produced by H. hepaticus ATCC51449 also contains lipid moieties. Additional analyses are required to decipher the structure and the conservation of this molecule across species. One approach is to sequence different strains of H. hepaticus to determine whether this activity is unique to a particular strain or a more general feature. In preliminary studies, we found that H. hepaticus strain ATCC51448 did not possess the same immunomodulatory activity as strain ATCC51449 (data not shown). Comparative analyses of the sequences of these two stains could help identify gene candidates associated with polysaccharide biosynthesis. However, these studies are in their infancy as only strain ATCC51449 has been fully sequenced (Suerbaum et al., 2003).
The host response to H. hepaticus shares some features with that elicited by Bacteroides fragilis, a member of the human gut microbiota that can induce abscesses and bacteraemia (Wexler, 2007). Challenge of II-10−/− mice with B. fragilis also leads to higher inflammation and mortality compared to WT mice, emphasizing the important role of IL-10 in host-microbe interactions (Cohen-Poradosu et al., 2011). Interestingly, B. fragilis polysaccharide A (PSA, strain NCTC 9343) was able to limit colitis in mice (Mazmanian et al., 2008). PSA activates TLR2 on plasmacytoid dendritic cells and regulatory T cells to induce immune regulatory functions, including IL-10 production and suppression of Th17 responses (Dasgupta et al., 2014, Round et al., 2011). PSA is a capsular zwitterionic polysaccharide (ZPS). ZPSs contain positive and negative repeating charges that are crucial to their immunomodulatory activity (Surana and Kasper, 2012). Recently, Lozupone et al. performed a genomic screen for human gut bacteria encoding ZPSs but did not identify Helicobacter species (Neff et al., 2016). Furthermore, in the genome of H. hepaticus strain ATCC51449, we did not find any significant BLAST hits to ZPS biosynthesis genes from B. fragilis, suggesting that these two bacteria produce different types of polysaccharide or employ different mechanisms of production. These results, together with our own, illustrate that the host utilizes TLR pathways not only to initiate host defense, but to bolster immune regulatory pathways that promote intestinal homeostasis (Rakoff-Nahoum et al., 2004).
In summary, our studies show that H. hepaticus produces an immunomodulatory polysaccharide that conditions the macrophage response via a TLR2/CREB-driven anti-inflammatory pathway. A better understanding of the molecular crosstalk between host cells and commensal bacteria is of major importance to decipher mutualistic mechanisms. Enhancement of these could help maintain homeostasis following disruptive challenges such as antibiotic treatment, enteric infection, stress, and food allergy, as well as aid restoration of a balanced dialog in the face of chronic inflammation. Identification of immunomodulatory ligands produced by commensals will require an integrated approach combining immunology, microbiology, bioinformatics, and biochemistry. This field of investigation is challenging but highly valuable as it could ultimately provide new preventive and therapeutic strategies for infectious and inflammatory diseases.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-IL-10 APC antibody (clone JES5-16E3) | eBioscience | clone JES5-16E3, RRID: AB_469502 |
| Mouse IgG2k isotype control (Rat, clone A95-1) | BD Pharmingen | clone A95-1, Cat# 556924 |
| Anti-mTLR2-IgG (clone C9A12) monoclonal antibody | InvivoGen | clone C9A12, Cat# mabg-mtlr2 |
| Mouse IgG2A isotype control (clone 20102) | Biotechne (R&D systems) | clone 20102, MAB003 |
| CD11b (clone M1/70) | BioLegend, London, UK | clone M1/70 RRID: AB_312791 |
| CD11c (clone N418) | BioLegend, London, UK | clone N418 RRID: AB_313771 |
| MHCII (clone M5/114.15.2) | BioLegend, London, UK | clone M5/114.15.2, RRID: AB_313321 |
| CD64 (clone X54-5/7.1) | BioLegend, London, UK | clone X54-5/7.1 RRID:AB_10612740 |
| CD45 (clone 30-F11) | BD Biosciences, Oxford, UK | clone 30-F11 |
| Total ERK1/2 | Cell Signaling Technology | #4695 |
| pT202/Y204-ERK1/2 (clone D13.14.4E) | Cell Signaling Technology | clone D13.14.4E, #4370 |
| Total p38 | Cell Signaling Technology | #9212 |
| pT180/Y182-p38 (clone 3D7) | Cell Signaling Technology | clone 3D7, #9215 |
| pS133-CREB1 (clone 87G3) | Cell Signaling Technology | clone 87G3, #9198 |
| pT581-MSK1 | Cell Signaling Technology | #9595 |
| Total CREB1 (clone E306) | Abcam | clone E306, #ab32515 |
| Total MSK1 (#AF2518) | R&D systems | Accession #O75582 |
| Bacterial and Virus Strains | ||
| Helicobacter hepaticus strain ATCC51449 | ATCC | ATCC51449 |
| Helicobacter hepaticus strain ATCC51448 | ATCC | ATCC51448 |
| Biological Samples | ||
| Pam2CSK4 | InvivoGen | Cat# tlrl-pm2s-1 |
| Pam3CSK4 | InvivoGen | Cat# tlrl-pms |
| FSL-1 | InvivoGen | Cat# tlrl-fsl |
| Zymosan, cell wall from Saccharomyces cerevisiae | InvivoGen | Cat# tlrl-zyn |
| LM-MS, Lipomannan from Mycobacterium smegmatis | InvivoGen | Cat# tlrl-lmms1 |
| LPS ultrapure from E. coli O111:B4 | InvivoGen | Cat# tlrl-3pelps |
| LPS standard from E. coli O55:B5 | InvivoGen | Cat# tlrl-b5lps |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sodium metaperiodate | Alfa Aesar, Heysham, UK | CAS# 7790-28-5 |
| Concanavalin A from Canavalia ensiformis (Jack bean), agarose conjugate | Sigma | L7555 |
| Lectin from Arachis hypogaea (peanut) agarose conjugate |
Sigma | L2507 |
| Lectin from Lens culinaris (lentil), agarose conjugate | Sigma | L4018 |
| U0126 | Merck Chemicals | 662005-1MG |
| p38 MAP Kinase Inhibitor VIII, EO 1428 | Merck Chemicals | 506163-5MG |
| NF-kB Activation Inhibitor II, JSH-23 | Merck Chemicals | 481408-5MG |
| (5Z)-7-Oxozeaenol | Calbiochem | 499610 |
| sp600125 | Sigma | S5567 |
| Critical Commercial Assays | ||
| DuoSet ELISA kits | R&D Systems Europe – Biotechne | N/A |
| High-Capacity cDNA Reverse Transcriptase | Applied Biosystems, Life Technologies, Paisley, UK | N/A |
| PrecisionFAST mastermix | Primerdesign | N/A |
| TaqMan gene expression assays | Life Technologies | N/A |
| RNeasy Mini kit (for tissue and microarray) | QIAGEN, Manchester, UK | N/A |
| Quick-RNA MiniPrep kit (for primary cell cultures) | Zymo Research, Cambridge Bioscience, UK | N/A |
| Illumina MouseWG-6-V2 microarrays | Illumina | N/A |
| Deposited Data | ||
| Microarray data | ArrayExpress | E-MTAB-5780 |
| Experimental Models: Organisms/Strains | ||
| Mus musculus, NCBI Taxonomy ID:10090, C57BL/6 | Charles River | RRID:IMSR_JAX:000664 |
| Oligonucleotides | ||
| Hprt | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm01545399_m1 |
| Il10 | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm00439614_m1 |
| Il6 | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm00446190_m1 |
| Tnf | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm00443258_m1 |
| Fosb | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm00500401_m1 |
| Egr3 | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm00516979_m1 |
| Rcan1 | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm01213406_m1 |
| Cd40 | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm00441891_m1 |
| Icam1 | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm00516023_m1 |
| Ccl5 | TaqMan Gene Expression Assays for mouse, Life Technologies | Mm01302427_m1 |
| Software and Algorithms | ||
| FlowJo software | Tree Star, Ashland, OR | N/A |
| GraphPad Prism 7.02 | GraphPad Software, CA | N/A |
| Lumi | R/Bioconductor | N/A |
| LIMMA | R/Bioconductor | N/A |
| Molecular Signatures Database (MSigDB, C3, motif gene sets) | http://software.broadinstitute.org/gsea/msigdb/collections.jsp | N/A |
| Transfac identifier | http://www.gene-regulation.com/pub/databases.html | N/A |
| runGO.py from the Computational Genomics Analysis Toolkit (CGAT) | https://github.com/CGATOxford/cgat | N/A |
| Other | ||
| Fortessa | BD Biosciences | N/A |
| ViiA 7 Real-Time PCR System | Life Technologies | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Fiona Powrie (fiona.powrie@kennedy.ox.ac.uk).
Experimental Model Details
Mice
Wild-type C57BL/6 mice were bred and maintained under specific pathogen-free (SPF) conditions in accredited animal facilities at the University of Oxford. Experiments were conducted in accordance with the UK Scientific Procedures Act (1986) under a Project License (PPL) authorized by the UK Home Office. Mice were routinely screened and negative for Helicobacter spp. and other known intestinal pathogens and were over 6 week-old when experiments were started. Both males and females were used. Co-housed littermates of same sex were randomly assigned to experimental groups with the same number of males and females in the different experimental groups. Experimental groups were then kept in separated cages during treatment with TSB/SNHht or infection with H. hepaticus. H. hepaticus infection was performed by oral administration. Colonization with live H. hepaticus was controlled by qRT-PCR performed on total tissue mRNA using probes for the gene cdtB. 250 μL of 1mg/ml brefeldin A (from Penicillium Breffeldianu, Sigma-Aldrich Co) was injected intra-peritoneally 12 h before harvest for FACS staining experiments.
Bacterial Culture
The Gram-negative mouse pathobiont Helicobacter hepaticus strain ATCC51449 was grown in tryptone soya broth (TSB, Fisher) supplemented with 10% Fetal Calf Serum (FCS) and Skirrow Campylobacter supplements (Oxoid) in microaerophilic conditions (1%–3% oxygen). After 24 h at 37°C under agitation, the culture broth was centrifuged at 4500 rpm for 50 min and filtrated through a 0.2 mm filter. The supernatants were collected and stored at −20°C.
Culture of Mouse Bone-Marrow Derived Macrophages
Bone-marrow cells from SPF WT or knockout C57BL/6 mice (any sex) were cultured for 8 days in complete RPMI with 50 ng/ml murine M-CSF (Peprotech) and seeded at 100,000 cells on 96-well tissue culture plates overnight before stimulation. After differentiation with M-CSF, 90%–95% of bone-marrow derives macrophages (BMDMs) expressed CD11b and CD64, 15% MHCII and 5% CD11c by flow cytometry. Bone-marrow from knock-out and knock-in mice was provided by the collaborators listed in the Acknowledgments section.
Methods Details
Isolation of Leukocytes from the Lamina Propria
For lamina propria leukocyte (LPL) isolation, the colon and the caecum of C57BL/6 mice were opened longitudinally, washed, and cut into pieces. Pieces were washed twice in RPMI-1640 medium supplemented with 5% fetal bovine serum (FBS) and 5mM EDTA at 37°C with shaking to remove epithelial cells. Tissue was then digested at 37°C for 1 h in a shaking incubator with 1 mg/ml type VIII collagenase (Sigma-Aldrich, Gillingham, UK) and 40 μg/ml DNase I in RPMI supplemented with 5% FBS. Cells were then layered on a 40/80% Percoll gradient, centrifuged, and the interface was recovered.
Flow Cytometry and Intracellular Staining
For surface staining, cells were first incubated with Fc block (anti-CD16/CD32, eBioscience) to minimize nonspecific antibody binding, and then stained for 20 min in phosphate-buffered saline / 0.1% bovine serum albumin / 5 mM EDTA buffer with a fixable viability dye and a combination of the following antibodies: CD11b (M1/70), CD11c (N418), MHCII (M5/114.15.2), CD64 (X54-5/7.1) (all from BioLegend, London, UK) and CD45 (30-F11) (from BD Biosciences, Oxford, UK). Following surface staining, cells were fixed with PFA 2%, permeabilized with saponin 0.05% and incubated with an anti-IL-10 APC antibody (clone JES5-16E3, eBioscience) at dilution 1/50 for 20 min. All cells were analyzed on a Fortessa (BD Biosciences) and analysis was performed using FlowJo software (Tree Star, Ashland, OR).
Treatment of H. hepaticus Culture Supernatant
SNHht was obtained by treating the filtered culture supernatant with 50 μg/ml DNase I (Roche) and RNase A (Roche) for 2 hours at 37°C, then with 40 μg/ml proteinase K (Sigma) overnight at 56°C, and finally with heat for 2 h in a water bath at 95°C. Protein and nucleic acid digestions were controlled using Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and agarose gels, respectively. To obtain SNHht > 30kDa and SNHht < 30kDa fractions, SNHht was concentrated on Vivaspin 20 MWCO 30 000 columns (GE healthcare Biosciences) for 30 min at 3000 rpm. Sodium metaperiodate oxidation was performed at 1 mM NaIO4 98% (Alfa Aesar, Heysham, UK) in 0.1 M sodium acetate pH 5.5 for 6 h at RT in foil, followed by dialysis against distilled water for 2 days at 4°C to remove NaIO4 traces using Pur-A-Lyzer™ Maxi 3500 tubes (Sigma). Lectin agarose conjugates (Concanavalin A from Canavalia ensiformis (ConA), lectins from Arachis hypogaea (peanut - PNA) and Lens culinaris (lentil - LcH) (Sigma) were added to SNHht or TSBt to deplete the polysaccharide and incubated for 5 h on a wheel at RT, followed by 30 min rest and 10 min centrifugation at 13 000 rpm.
Ethanol Precipitation of the Crude Polysaccharide
CPHht was precipitated from SNHht by the addition of 4 volumes of ice cold ethanol and incubated at −20°C overnight. Crude polysaccharide fractions from TSBt (CPTSBt) were extracted using the same method as a control. Precipitated polysaccharides were recovered by centrifugation at 5000 rpm for 10 min, the pelleted precipitate was re-suspended in water and dialysed against 5 L of water for 48 h with two water changes to remove residual ethanol before being dried in a freeze dryer.
Stimulation of BMDMs
BMDM cultures were infected with 0.5 OD H. hepaticus in RPMI (from frozen stock of live culture, Multiplicity Of Infection = 10), H. hepaticus culture supernatant, TSBt control medium or different TLR ligands (Pam2CSK4 (85 ng/ml), Pam3CSK4 (75 ng/ml), FSL-1 (100 ng/ml), Zymosan, cell wall from Saccharomyces cerevisiae (100 ng/ml); LM-MS, Lipomannan from Mycobacterium smegmatis (1 ng/ml), LPS ultrapure from E. coli O111:B4 (20 ng/ml), LPS standard from E. coli O55:B5 (1 ng/ml) - all from InvivoGen) for 3 h at 37°C in a humidified incubator with 5% CO2. Blocking antibodies specifically inhibiting TLR2 signaling (Anti-mTLR2-IgG (C9A12) monoclonal antibody, InvivoGen) or a mouse IgG2A isotype control (Biotechne) were added at 0.3 μg/ml 1 h prior stimulation. Kinase inhibitors were used at 10 μM (UO126 (MEK1/2), JSH-23 (NFkb), Merck Chemicals; Sp600125 (JNK), Sigma), 5 μM (EO 1428 (p38), Merck Chemicals) or 1 μM (5z-7-oxozeanol (Tak1), Calbiochem) and added 30 min prior stimulation. Stimulation assays were performed in duplicates and repeated three or more times.
ELISA Assay
For measurement of secreted cytokines using Mouse IL-10, IL-6 and TNF-α DuoSet ELISA kits (R&D Systems Europe – Biotechne), culture supernatants were collected and stored at −20°C.
RNA Extraction
BMDMs or lamina propria cells were lysed in RLT buffer (QIAGEN, Manchester, UK) or Zymo Lysis Buffer (Zymo Research, Cambridge Bioscience, UK) and stored at −80°C. RNA was isolated from snap-frozen samples using the RNeasy Mini kit (QIAGEN, Manchester, UK) or the Quick-RNA MiniPrep kit (Zymo Research, Cambridge Bioscience, UK) according to the manufacturer’s instructions, including an on-column DNase I digestion step. For microarray analysis on BMDMs, the RNeasy Mini kit (QIAGEN, Manchester, UK) was used.
Quantitative RT-PCR
Complementary DNA synthesis was performed using High-Capacity cDNA Reverse Transcriptase (Applied Biosystems, Life Technologies, Paisley, UK). Quantitative PCR reactions for the candidate genes were performed using PrecisionFAST mastermix (Primerdesign) and TaqMan gene expression assays (Life Technologies). Complementary DNA samples were analyzed in duplicate using ViiA 7 Real-Time PCR System (Life Technologies), and gene expression levels for each sample were normalized to HPRT. Mean relative gene expression was determined, and the differences were calculated using the 2ΔC(t) method. Primer pairs and probes were as follows: TaqMan Gene Expression Assays for mouse Hprt (Mm01545399_m1), Il10 (Mm00439614_m1), Il6 (Mm00446190_m1), Tnf (Mm00443258_m1), Fosb (Mm00500401_m1), Egr3 (Mm00516979_m1), Rcan1 (Mm01213406_m1), Cd40 (Mm00441891_m1), Icam1 (Mm00516023_m1) and Ccl5 (Mm01302427_m1).
Immunoblot Analysis
Cells were washed with PBS and lysed using Laemmli sample loading buffer (2% SDS, 10% glycerol, 50 mM DTT, 0.002% bromphenol blue and 62.5 mM Tris HCl, pH 6.8). Equal amounts of proteins were resolved by SDS–PAGE using NuPAGE Novex Bis-Tris Gels (Thermo Fisher Scientific) and analyzed with antibodies against total ERK1/2, pT202/Y204-ERK1/2 (D13.14.4E), total p38, pT180/Y182-p38 (3D7), pS133-CREB1 (87G3), pT581-MSK1 (all from Cell Signaling Technology), total CREB1 (E306, ab32515, Abcam) and total MSK1 (R&D systems) followed by detection with horseradish peroxidase (HRP)–conjugated secondary antibodies and the chemiluminescent substrate solution ECL (GE Healthcare)
Quantification and Statistical Analysis
Microarray Analysis of Stimulated BMDMs
Expression profiles of M-CSF differentiated BMDMs stimulated for 3 h with either TSBt (1/10), SNHht (1/10) or Pam3CSK4 (75 ng/ml) were obtained using Illumina MouseWG-6-V2 microarrays (n = 5 for each condition). Probes were used for downstream processing if they were expressed significantly above background (detection P value < 0.05) in at least three samples. This resulted in the analysis of 16, 692 probes (out of a total of 45, 289). Array signal intensities were background adjusted, transformed using the variance-stabilizing transformation and quantile normalized using Lumi from R/Bioconductor. Differential expression analysis was performed for each condition contrast using the empirical Bayes method in LIMMA (Linear Models for Microarray and RNA-seq Data from R/Bioconductor). Significance was defined as a Benjamini-Hochberg adjusted P value < 0.05 and fold change > 2. The union of significantly different probes was visualized in a heatmap and probes were clustered into distinct sets using k-means clustering (k = 4) as implemented in R (k means function in R3.1.0). Sequencing data was generated by the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z).
Transcription Factor Motif Enrichment Analysis
We tested for the enrichment of transcription factor motifs among genes that were observed to be specifically upregulated in either SNHht or Pam3CSK4 conditions. Transcription factor motif annotations were downloaded from the Molecular Signatures Database (MSigDB, C3, motif gene sets at http://software.broadinstitute.org/gsea/msigdb/collections.jsp). These comprise sets of genes that are predicted to be bound by a given transcription factor based on the presence of a conserved motif in their promoter or 3′ UTR. As we were interested in motifs that could readily be attributed to a particular transcription factor, we filtered the database to retain only those motifs that had a designated Transfac (http://www.gene-regulation.com/pub/databases.html) identifier. This resulted in the analysis of 499 motifs (from a total of 836 in the full database). Enrichment analysis was performed using the hypergeometric test implemented in runGO.py from the Computational Genomics Analysis Toolkit (CGAT; https://github.com/CGATOxford/cgat) and gene sets were considered significant at an empirically derived adjusted p value < 0.05.
Statistical Analysis
Statistical tests were performed using GraphPad Prism 7.02 and specified in figure legends. Differences were considered to be significant when p < 0.05. All bar charts represent means ± SD.
Data and Software Availability
Accession Numbers/Data Availability Statement
The accession number for the microarray data reported in this paper is ArrayExpress: E-MTAB-5780.
Acknowledgments
We thank Claudia Monaco, Thierry Roger, Caetano Reis e Sousa, Kevin Maloy, Siamon Gordon, David Sancho, Paul-Albert König, and Gordon Brown for providing tissues from knockout mice. We thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics for the generation of the Sequencing data. F.F. was supported by Cancer Research UK (OCRC-DPhil13-FF) and N.E.I. by the Kennedy Trust (KENN 15 16 03). M.M.-L. received a fellowship from the Spanish Ministry of Education, Culture, and Sport. This work was funded by the Wellcome Trust UK (095688/Z/11/Z), an ERC grant (Advanced Grant Ares(2013)3687660), and the Fondation Louis Jeantet.
Author Contributions
Conceptualization, C.D., F.P.; Investigation, C.D., G.R., M.M.-L., F.F., F.C., E.L., S.B.; Formal analysis, N.E.I.; Writing – Original Draft, C.D., N.E.I.; Writing – Review & Editing, C.D., G.R., M.M.-L., F.F., F.C., E.L., J.S.A., F.P.; Supervision, C.D., F.P.; Resources, J.S.A., F.P.; Funding Acquisition, F.P.
Published: December 13, 2017
Footnotes
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.chom.2017.11.002.
Contributor Information
Camille Danne, Email: camille.danne@gmail.com.
Fiona Powrie, Email: fiona.powrie@kennedy.ox.ac.uk.
Supplemental Information
References
- Ananieva O., Darragh J., Johansen C., Carr J.M., McIlrath J., Park J.M., Wingate A., Monk C.E., Toth R., Santos S.G. The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat. Immunol. 2008;9:1028–1036. doi: 10.1038/ni.1644. [DOI] [PubMed] [Google Scholar]
- Arnold I.C., Mathisen S., Schulthess J., Danne C., Hegazy A.N., Powrie F. CD11c(+) monocyte/macrophages promote chronic Helicobacter hepaticus-induced intestinal inflammation through the production of IL-23. Mucosal Immunol. 2016;9:352–363. doi: 10.1038/mi.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asquith M.J., Boulard O., Powrie F., Maloy K.J. Pathogenic and protective roles of MyD88 in leukocytes and epithelial cells in mouse models of inflammatory bowel disease. Gastroenterology. 2010;139:519–529. doi: 10.1053/j.gastro.2010.04.045. e511–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulard O., Asquith M.J., Powrie F., Maloy K.J. TLR2-independent induction and regulation of chronic intestinal inflammation. Eur. J. Immunol. 2010;40:516–524. doi: 10.1002/eji.200939669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill R.J., Foltz C.J., Fox J.G., Dangler C.A., Powrie F., Schauer D.B. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect. Immun. 1997;65:3126–3131. doi: 10.1128/iai.65.8.3126-3131.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.H., Miaw S.C., Tseng W., Sun Y.W., Liu C.C., Tsao H.W., Ho I.C. PTPN22 modulates macrophage polarization and susceptibility to dextran sulfate sodium-induced colitis. J. Immunol. 2013;191:2134–2143. doi: 10.4049/jimmunol.1203363. [DOI] [PubMed] [Google Scholar]
- Cheluvappa R., Eri R., Luo A.S., Grimm M.C. Endothelin and vascular remodelling in colitis pathogenesis–appendicitis and appendectomy limit colitis by suppressing endothelin pathways. Int. J. Colorectal Dis. 2014;29:1321–1328. doi: 10.1007/s00384-014-1974-z. [DOI] [PubMed] [Google Scholar]
- Chen L., Flies D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. The TLR and IL-1 signalling network at a glance. J. Cell Sci. 2014;127:2383–2390. doi: 10.1242/jcs.149831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Poradosu R., McLoughlin R.M., Lee J.C., Kasper D.L. Bacteroides fragilis-stimulated interleukin-10 contains expanding disease. J. Infect. Dis. 2011;204:363–371. doi: 10.1093/infdis/jir277. [DOI] [PubMed] [Google Scholar]
- Darragh J., Ananieva O., Courtney A., Elcombe S., Arthur J.S. MSK1 regulates the transcription of IL-1ra in response to TLR activation in macrophages. Biochem. J. 2010;425:595–602. doi: 10.1042/BJ20091062. [DOI] [PubMed] [Google Scholar]
- Dasgupta S., Erturk-Hasdemir D., Ochoa-Reparaz J., Reinecker H.C., Kasper D.L. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe. 2014;15:413–423. doi: 10.1016/j.chom.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo D., Labzin L.I., Kono H., Seki R., Schmidt S.V., Beyer M., Xu D., Zimmer S., Lahrmann C., Schildberg F.A. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat. Immunol. 2014;15:152–160. doi: 10.1038/ni.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcombe S.E., Naqvi S., Van Den Bosch M.W., MacKenzie K.F., Cianfanelli F., Brown G.D., Arthur J.S. Dectin-1 regulates IL-10 production via a MSK1/2 and CREB dependent pathway and promotes the induction of regulatory macrophage markers. PLoS One. 2013;8:e60086. doi: 10.1371/journal.pone.0060086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdman S.E., Poutahidis T., Tomczak M., Rogers A.B., Cormier K., Plank B., Horwitz B.H., Fox J.G. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am. J. Pathol. 2003;162:691–702. doi: 10.1016/S0002-9440(10)63863-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferwerda G., Meyer-Wentrup F., Kullberg B.J., Netea M.G., Adema G.J. Dectin-1 synergizes with TLR2 and TLR4 for cytokine production in human primary monocytes and macrophages. Cell. Microbiol. 2008;10:2058–2066. doi: 10.1111/j.1462-5822.2008.01188.x. [DOI] [PubMed] [Google Scholar]
- Gantner B.N., Simmons R.M., Canavera S.J., Akira S., Underhill D.M. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F., Schultze J.L., Murray P.J., Ochando J., Biswas S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 2016;17:34–40. doi: 10.1038/ni.3324. [DOI] [PubMed] [Google Scholar]
- Glocker E.O., Kotlarz D., Boztug K., Gertz E.M., Schäffer A.A., Noyan F., Perro M., Diestelhorst J., Allroth A., Murugan D. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griseri T., McKenzie B.S., Schiering C., Powrie F. Dysregulated hematopoietic stem and progenitor cell activity promotes interleukin-23-driven chronic intestinal inflammation. Immunity. 2012;37:1116–1129. doi: 10.1016/j.immuni.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue S., Ahern P., Buonocore S., Kullberg M.C., Cua D.J., McKenzie B.S., Powrie F., Maloy K.J. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes S.O., Ferris J.A., Szponar B., Wadström T., Fox J.G., O’Rourke J., Larsson L., Yaquian E., Ljungh Å., Clyne M., Andersen L.P., Moran A.P. Comparative chemical and biological characterization of the lipopolysaccharides of gastric and enterohepatic Helicobacters. Helicobacter. 2004;9:313–323. doi: 10.1111/j.1083-4389.2004.00237.x. [DOI] [PubMed] [Google Scholar]
- Junkins R.D., MacNeil A.J., Wu Z., McCormick C., Lin T.J. Regulator of calcineurin 1 suppresses inflammation during respiratory tract infections. J. Immunol. 2013;190:5178–5186. doi: 10.4049/jimmunol.1203196. [DOI] [PubMed] [Google Scholar]
- Kaser A., Zeissig S., Blumberg R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlarz D., Beier R., Murugan D., Diestelhorst J., Jensen O., Boztug K., Pfeifer D., Kreipe H., Pfister E.D., Baumann U. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143:347–355. doi: 10.1053/j.gastro.2012.04.045. [DOI] [PubMed] [Google Scholar]
- Krausgruber T., Schiering C., Adelmann K., Harrison O.J., Chomka A., Pearson C., Ahern P.P., Shale M., Oukka M., Powrie F. T-bet is a key modulator of IL-23-driven pathogenic CD4(+) T cell responses in the intestine. Nat. Commun. 2016;7:11627. doi: 10.1038/ncomms11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullberg M.C., Ward J.M., Gorelick P.L., Caspar P., Hieny S., Cheever A., Jankovic D., Sher A. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 1998;66:5157–5166. doi: 10.1128/iai.66.11.5157-5166.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullberg M.C., Jankovic D., Gorelick P.L., Caspar P., Letterio J.J., Cheever A.W., Sher A. Bacteria-triggered CD4(+) T regulatory cells suppress Helicobacter hepaticus-induced colitis. J. Exp. Med. 2002;196:505–515. doi: 10.1084/jem.20020556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullberg M.C., Jankovic D., Feng C.G., Hue S., Gorelick P.L., McKenzie B.S., Cua D.J., Powrie F., Cheever A.W., Maloy K.J., Sher A. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T., Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- Lebedeva T., Dustin M.L., Sykulev Y. ICAM-1 co-stimulates target cells to facilitate antigen presentation. Curr. Opin. Immunol. 2005;17:251–258. doi: 10.1016/j.coi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Liu Y., Chen K., Wang C., Gong W., Yoshimura T., Liu M., Wang J.M. Cell surface receptor FPR2 promotes antitumor host defense by limiting M2 polarization of macrophages. Cancer Res. 2013;73:550–560. doi: 10.1158/0008-5472.CAN-12-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V., Golaconda Ramulu H., Drula E., Coutinho P.M., Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy K.J., Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- Maloy K.J., Salaun L., Cahill R., Dougan G., Saunders N.J., Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 2003;197:111–119. doi: 10.1084/jem.20021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell L., Moran A.P., Cocchiarella A., Houghton J., Taylor N., Fox J.G., Wang T.C., Kurt-Jones E.A. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect. Immun. 2004;72:6446–6454. doi: 10.1128/IAI.72.11.6446-6454.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian S.K., Round J.L., Kasper D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- Moore K.W., de Waal Malefyt R., Coffman R.L., O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Morrison P.J., Bending D., Fouser L.A., Wright J.F., Stockinger B., Cooke A., Kullberg M.C. Th17-cell plasticity in Helicobacter hepaticus-induced intestinal inflammation. Mucosal Immunol. 2013;6:1143–1156. doi: 10.1038/mi.2013.11. [DOI] [PubMed] [Google Scholar]
- Murray P.J. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 2006;6:379–386. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Neff C.P., Rhodes M.E., Arnolds K.L., Collins C.B., Donnelly J., Nusbacher N., Jedlicka P., Schneider J.M., McCarter M.D., Shaffer M. Diverse intestinal bacteria contain putative Zwitterionic capsular polysaccharides with anti-inflammatory properties. Cell Host Microbe. 2016;20:535–547. doi: 10.1016/j.chom.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemati M., Larussa T., Khorramdelazad H., Mahmoodi M., Jafarzadeh A. Toll-like receptor 2: An important immunomodulatory molecule during Helicobacter pylori infection. Life Sci. 2017;178:17–29. doi: 10.1016/j.lfs.2017.04.006. [DOI] [PubMed] [Google Scholar]
- Oliveira-Nascimento L., Massari P., Wetzler L.M. The role of TLR2 in infection and immunity. Front. Immunol. 2012;3:79. doi: 10.3389/fimmu.2012.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S., Paglino J., Eslami-Varzaneh F., Edberg S., Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Reyskens K.M., Arthur J.S. Emerging roles of the mitogen and stress activated kinases MSK1 and MSK2. Front. Cell Dev. Biol. 2016;4:56. doi: 10.3389/fcell.2016.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round J.L., Lee S.M., Li J., Tran G., Jabri B., Chatila T.A., Mazmanian S.K. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safford M., Collins S., Lutz M.A., Allen A., Huang C.T., Kowalski J., Blackford A., Horton M.R., Drake C., Schwartz R.H., Powell J.D. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat. Immunol. 2005;6:472–480. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- Sanin D.E., Prendergast C.T., Mountford A.P. IL-10 production in macrophages is regulated by a TLR-driven CREB-mediated mechanism that is linked to genes involved in cell metabolism. J. Immunol. 2015;195:1218–1232. doi: 10.4049/jimmunol.1500146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiering C., Krausgruber T., Chomka A., Fröhlich A., Adelmann K., Wohlfert E.A., Pott J., Griseri T., Bollrath J., Hegazy A.N. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C., Pamer E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval D.S., Biswas A., Goettel J.A., McCann K., Conaway E., Redhu N.S., Mascanfroni I.D., Al Adham Z., Lavoie S., Ibourk M. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–719. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerbaum S., Josenhans C., Sterzenbach T., Drescher B., Brandt P., Bell M., Droge M., Fartmann B., Fischer H.P., Ge Z. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc. Natl. Acad. Sci. USA. 2003;100:7901–7906. doi: 10.1073/pnas.1332093100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana N.K., Kasper D.L. The yin yang of bacterial polysaccharides: lessons learned from B. fragilis PSA. Immunol. Rev. 2012;245:13–26. doi: 10.1111/j.1600-065X.2011.01075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomczak M.F., Gadjeva M., Wang Y.Y., Brown K., Maroulakou I., Tsichlis P.N., Erdman S.E., Fox J.G., Horwitz B.H. Defective activation of ERK in macrophages lacking the p50/p105 subunit of NF-kappaB is responsible for elevated expression of IL-12 p40 observed after challenge with Helicobacter hepaticus. J. Immunol. 2006;176:1244–1251. doi: 10.4049/jimmunol.176.2.1244. [DOI] [PubMed] [Google Scholar]
- Toriseva M., Laato M., Carpén O., Ruohonen S.T., Savontaus E., Inada M., Krane S.M., Kähäri V.M. MMP-13 regulates growth of wound granulation tissue and modulates gene expression signatures involved in inflammation, proteolysis, and cell viability. PLoS One. 2012;7:e42596. doi: 10.1371/journal.pone.0042596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlig H.H., Schwerd T., Koletzko S., Shah N., Kammermeier J., Elkadri A., Ouahed J., Wilson D.C., Travis S.P., Turner D. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 2014;147:990–1007.e3. doi: 10.1053/j.gastro.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Zhou Y., Besner G.E. Heparin-binding EGF-like growth factor and enteric neural stem cell transplantation in the prevention of experimental necrotizing enterocolitis in mice. Pediatr. Res. 2015;78:29–37. doi: 10.1038/pr.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen A.Y., Sakamoto K.M., Miller L.S. The role of the transcription factor CREB in immune function. J. Immunol. 2010;185:6413–6419. doi: 10.4049/jimmunol.1001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler H.M. Bacteroides: the good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007;20:593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingate A.D., Martin K.J., Hunter C., Carr J.M., Clacher C., Arthur J.S. Generation of a conditional CREB Ser133Ala knockin mouse. Genesis. 2009;47:688–696. doi: 10.1002/dvg.20548. [DOI] [PubMed] [Google Scholar]
- Zigmond E., Bernshtein B., Friedlander G., Walker C.R., Yona S., Kim K.W., Brenner O., Krauthgamer R., Varol C., Müller W., Jung S. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40:720–733. doi: 10.1016/j.immuni.2014.03.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.