

Abstract
Exposure to petrogenic polycyclic aromatic hydrocarbons (PPAHs) is the major human health hazard associated with the Deepwater Horizon oil spill. Alkylated phenanthrenes are the most abundant PPAHs present in the crude oil and could contaminate the food chain. We describe the metabolism of a C1-phenanthrene regioisomer 1-methylphenanthrene (1-MP) and a C2-phenanthrene regioisomer 9-ethylphenanthrene (9-EP) in human HepG2 cells. The structures of the metabolites were identified by HPLC-UV-fluorescence detection and LC-MS/MS. Side chain hydroxylation of 1-MP and 9-EP was observed as the major metabolic pathway. The formation of 1-(hydroxymethyl)-phenanthrene was confirmed by reference to an authentic synthetic standard. However, formation of the bioactivated sulfate was not detected. Tetraols were also identified as signature metabolites of 1-MP and 9-EP, indicating that metabolic activation occurred via the diol-epoxide pathway. O-Monosulfonated-catechols were discovered as signature metabolites of the o-quinone pathway of metabolic activation of 1-MP and 9-EP, respectively. The identification of O-monosulfonated-catechols supports the metabolic activation of 1-MP and 9-EP by P450 and AKR isozymes followed by metabolic detoxification of the o-quinone through interception of redox cycling by phase II isozymes. The signature metabolites identified could be used as biomarkers of human exposure to 1-MP and 9-EP resulting from oil spills.
Graphical Abstract
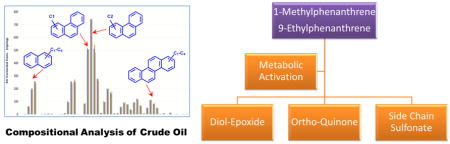
INTRODUCTION
The Deepwater Horizon oil spill in the Gulf of Mexico in 2010 was the largest release of crude oil in United States history.1,2 Over 200 million gallons of crude oil were released from the Macondo well in the Gulf of Mexico.3–5 Polycyclic aromatic hydrocarbons (PAHs) are one of the most toxic and persistent components among thousands of hydrocarbons present in crude oil.6 Benzo[a]pyrene, a representative pyrogenic PAH which results from the incomplete combustion of fossil fuels, is ranked as a Group 1 carcinogen by IARC7 raising concerns about exposure to the petrogenic PAHs (PPAHs) in oil. PPAHs that are uniquely present in crude oil differ from pyrogenic PAHs in that they are either extensively alkylated or oxygenated. Contamination of the food chain with PPAHs is considered the major hazard associated with human health.8
The toxicity of unsubstituted pyrogenic PAHs has been extensively studied.9–12 It is widely accepted that pyrogenic PAHs themselves are biologically inert and their carcinogenic effects require metabolic activation to generate biologically reactive intermediates and form DNA adducts that result in mutation.13 Two such reactive metabolites are diol-epoxides and o-quinones.9–12 However, information on the toxicological properties of alkylated or oxygenated PPAHs is limited so far. In order to fill in this knowledge gap, we have described metabolic activation of three representative alkylated PPAHs, including retene (1-methyl-7-isopropyl-phenanthrene; a C4-phenanthrene regioisomer), 5-methylchrysene (a C1-chrysene regioisomer), 6-ethylchrysene (a C2-chrysene regioisomer), and a representative oxygenated PPAH phenanthrene-9,10-quinone in human hepatoma (HepG2) cells.14–17 These studies showed that metabolic activation of retene, which cannot give rise to a diol-epoxide, due to its extensive alkylation, was metabolically activated to mono- and bis-o-quinones. By contrast, metabolic activation of 5-methyl-chrysene and 6-ethyl-chrysene led to the formation of bis-electrophiles, for example, bis-diol-epoxides, bis-o-quinones, and a mono-diol-epoxide and a mono-o-quinone observed within the same structure. The formation of these bis-electrophiles indicates that bioactivation to potential DNA and protein cross-linking agents can occur.
C1 and C2 phenanthrenes are the most abundant species of PPAHs, based on the compositional analysis of the crude oil released from the Deepwater Horizon oil spill.18 In the present study, 1-methylphenanthrene (1-MP), a representative C1-phenanthrene regioisomer, as well as 9-ethylphenanthrene (9-EP), a representative C2-phenanthrene regioisomer, were selected to elucidate their metabolism in human hepatoma (HepG2) cells. HepG2 cells were chosen in the present study so that data could be directly compared with that obtained in the previous studies on the metabolism of PPAHs. Importantly, HepG2 cells have inducible P4501A1, a major isoform responsible for the metabolic activation of pyrogenic PAH,19 and also contain aldo-keto reductases (AKRs) capable of converting trans-dihydrodiols to o-quinones.20
Three possible routes for the metabolic activation of 1-MP and 9-EP were considered to predict and identify their metabolites (Scheme 1). First, we predicted the diol-epoxide pathway, involving conversion of alkylated phenanthrenes to trans-dihydrodiols followed by formation of diol-epoxides, which can form DNA adducts or be hydrolyzed to the corresponding tetraols. Second, we predicted the o-quinone pathway, involving conversion of trans-dihydrodiols to catechols followed by either formation of phase II conjugates or their oxidation to o-quinones. Once formed, the o-quinones would be reduced back to catechols to establish futile redox cycles leading to oxidative DNA damage. The electrophilic o-quinone could also react with DNA. Third, we also predicted that hydroxylation on the alkyl side chain followed by sulfonation by SULTs could introduce a sulfate as a good leaving group and lead to DNA adduct formation.
Scheme 1.

Possible Metabolic Activation Pathways of 1-MP and 9-EP
It was found that the metabolism of 1-MP involved side-chain hydroxylation without subsequent sulfonation. The formation of diol-epoxides and the formation of o-quinones were also observed. Unlike C1 and C2 alkylated chrysenes, we did not detect the formation of bis-electrophiles. Similar metabolic activation pathways for 9-EP were observed. These signature metabolites represent potential human exposure biomarkers for 1-MP and 9-EP that may result from consuming seafood contaminated by crude oil spills.
MATERIALS AND METHODS
Caution: All PAHs are potentially hazardous and should be handled in accordance with the National Institutes of Health Guidelines for the Laboratory Use of Chemical Carcinogens.
Chemicals and Reagents
Cell culture media and reagents were all obtained from Invitrogen Co. (Carlsbad, CA) except fetal bovine serum (FBS) which was purchased from Hyclone (Logan, UT). 1-Methylphenanthrene was purchased from Chem Service, Inc. (West Chester, PA). 9-Ethylphenanthrene was purchased from Sigma-Aldrich Co. (St. Louis, MO). 1-(Hydroxymethyl)-phenanthrene and 6-hydroxy-1-MP were synthesized as described in the Supporting Information (Figure S1). All other chemicals used were of the highest grade available, and all solvents were HPLC grade.
Cell Culture
HepG2 (human hepatocellular carcinoma) cells were obtained from American Type Culture Collection and maintained as previously described.15 Cultured cells with a passage number of 10–20 were used in the experiments to reduce variability due to long-term culture conditions. Cultured cells were authenticated by short-terminal repeat DNA analysis and were mycoplasma free (DNA Diagnostics Center Medical, Fairfield, OH).
Detection and Identification of 1-MP and 9-EP Metabolites in HepG2 Cells
The confluent HepG2 cells were plated in a 6-well plate (~5 × 106). The cells were washed twice and then treated with MEM (without phenol red) containing 10 mM glucose and 1 μM 1-MP or 9-EP (DMSO, 0.2% v/v). These concentrations were chosen based on contamination of seafood with 1-methylphenanthrene (Cmax = 31 μg/kg wet weight oysters) and estimates of cumulative exposure.21 The culture media were collected at 0 and 24 h, respectively, and subsequently acidified with 0.1% formic acid before extraction as previously described.15 The extract from the cell culture media was reconstituted in 150 μL of methanol.
For HPLC-UV-FLR analysis, a 10 μL aliquot of the reconstituted extract was analyzed on a tandem Waters Alliance 2695 chromatographic system with a Waters 2996 photodiode array (PDA) detector and a Waters 2475 multi λ fluorescence (FLR) detector (Waters Corporation, Milford, MA). Separations were accomplished on a Zorbax-ODS C18 analytical column (5 μm, 4.6 mm × 250 mm) with a Zorbax-ODS analytical guard column (5 μm, 4.6 mm × 12.5 mm) (DuPont Co., Wilmington, DE) at ambient temperature. The mobile phase consisted of 5 mM ammonium acetate and 0.1% trifluoroacetic acid (TFA) (v/v) in H2O (solvent A) and 5 mM ammonium acetate and 0.1% TFA in acetonitrile (solvent B) and was delivered at a flow rate of 0.5 mL/min. The linear gradient elution program was as follows: 5% to 95% B over 30 min, followed by an isocratic hold at 95% B for another 10 min. At 40 min, B was returned to 5% in 1 min, and the column was equilibrated for 19 min before the next injection. The total run time for each analysis was 60 min. Eluants from the column were introduced sequentially into the PDA detector and the FLR detector. Excitation wavelength (λex) and emission wavelength (λem) for the FLR detector were set at 254 and 369 nm for 1-MP and at 252 and 369 nm for 9-EP, respectively, based on the spectral properties of each PPAH (Figures S2 and S3). The optimal pair of λex and λem for each PPAH was employed to detect its metabolites based on the assumption that most PPAH metabolites show fluorescence signals at these wavelengths.
For ion trap LC-MS/MS analysis, a 10 μL aliquot of the reconstituted extract was analyzed on a Waters Alliance 2690 HPLC system (Waters Corporation, Milford, MA) coupled to a Finnigan LTQ linear ion trap mass spectrometer (Thermo Scientific, San Jose, CA). The column, mobile phase, flow rate, and the linear gradient elution program were the same as described above. During LC-MS/ MS analysis, up to 10 min of the initial flow was diverted to the waste before evaluation of eluants. The mass spectrometer was operated in both the positive and negative ion modes with an electrospray ionization (ESI) source. Eluants were monitored on the LTQ using product ion scan (MS2), subsequent MS/MS/MS (MS3), and pseudo-selected reaction monitoring (SRM) modes. The mass spectrometry parameters included spray voltage (3 kV in positive ion mode, 5 kV in negative ion mode), sheath gas flow rate (40 arbitrary units in both ion modes), auxiliary gas flow rate (15 arbitrary units in both ion modes), capillary temperature (275 °C in both ion modes), capillary voltage (38 V in positive ion mode, –19 V in negative ion mode), and tube lens (20 V in positive ion mode, –22.05 V in negative ion mode). An isolation width of three bracketed around the m/z of interest, activation Q of 0.25, and activation time of 30 ms were used for data acquisition. Xcalibur software version 2.0 (Thermo Scientific, San Jose, CA) was used to control the LC-MS/MS system and to process the data. The information on analyte identity was obtained by interpreting the corresponding MS2 and MS3 spectra of 1-MP and 9-EP metabolites from ion trap LC-MS/MS.
In some instances, another 5 μL aliquot of the reconstituted extract was analyzed on a nano-Acquity ultraperformance liquid chromatography (UPLC) system (Waters Corporation, Milford, MA) coupled to a LTQ Orbitrap XL mass spectrometer (Thermo Scientific, San Jose, CA). Separations were accomplished on a nano-UPLC C18 column (1.7 μm BEH130, 150 μm × 100 mm) (Waters Corporation, Milford, MA) at 50 °C. The mobile phase consisted of 0.1% formic acid (v/v) in H2O (solvent A) and 0.1% formic acid (v/v) in acetonitrile (solvent B) and was delivered at a flow rate of 1.6 μL/min. The linear gradient elution program was as follows: an isocratic hold at 5% B for 5 min, 5% to 95% B over 30 min, followed by an isocratic hold at 95% B for another 10 min. At 46 min, B was returned to 5% in 2 min, and the column was equilibrated for 12 min before the next injection. The total run time for each analysis was 60 min. The mass spectrometer was operated in the positive and negative ion modes, respectively, with a nanoelectrospray ionization (nano-ESI) source after accurate calibration with the manufacturer’s calibration mixture. The ionization voltage was set to 1.5 kV, and the capillary temperature was set to 200 °C. Full scan spectra were acquired with a resolving power of 60,000 full width half-maximum (fwhm) in a mass range from m/z 100 to 600. Xcalibur software version 2.0 (Thermo Scientific, San Jose, CA) was used to control the Orbitrap mass spectrometry, process data, and generate exact mass.
RESULTS
Detection of 1-MP and 9-EP Metabolites in HepG2 Cells by HPLC-UV-FLR
Comparison of UV chromatograms at 0 and 24 h showed that 16 metabolites of 1-MP (Figure 1) and 9 metabolites of 9-EP (Figure S4) were detected in the organic phase of the ethyl acetate-extracted acidified media from HepG2 cells. The peaks attributed to 1-MP and 9-EP at 0 h were almost completely absent at 24 h, suggesting that 1-MP and 9-EP were rapidly metabolized by HepG2 cells over this time course. The corresponding UV spectra of the 1-MP metabolites and 9-EP metabolites were extracted from the PDA detector and are shown in Figures S5 and S6, respectively.
Figure 1.

HPLC detection of 1-MP metabolites in human HepG2 cells. (A) UV chromatogram at λmax 255 nm at 0 h. (B) UV chromatogram at λmax 255 nm at 24 h. (C) FLR chromatogram at λex 254 nm and λem 369 nm at 0 h. (D) FLR chromatogram at λex 254 nm and λem 369 nm at 24 h. Human HepG2 cells (~5 × 106) were treated with 1-MP (1 μM, 0.2% (v/v) DMSO) in MEM (without phenol red) containing 10 mM glucose. The cell media were collected at 0 and 24 h, respectively, and subsequently acidified with 0.1% formic acid before extraction with ethyl acetate. The extracts were analyzed on a HPLC-UV-FLR.
Comparison of FLR chromatograms for 1-MP metabolites at 0 and 24 h showed that 14 fluorescence peaks in FLR chromatogram corresponded to the 1-MP metabolites in UV chromatograms (Figure 2), and comparison of the FLR chromatograms for 9-EP metabolites at 0 and 24 h showed that 8 fluorescence peaks in FLR chromatograms corresponded to the 9-EP metabolites in UV chromatograms (Figure S6), thus validating the presence of a fluorophore for an aromatic ring system. Those metabolites detected in the FLR chromatogram using the same excitation and emission wavelengths as the parent hydrocarbon and which contained the same absorbance maximum as the parent hydrocarbon were assumed to contain the aromatic phenanthrene nucleus. Thus, 1-MP metabolites 5–14 and 16 retain the aromatic phenanthrene ring structure and 9-EP metabolites 3–9 retain the aromatic phenanthrene ring structure (Figures S5 and S6), whereas 1-MP metabolites 1–4 and 9-EP metabolites 1–2 do not.
Figure 2.

Identification of 1-(hydroxmethyl)-phenanthrene as a 1-MP metabolite in human HepG2 cells. (A) UV chromatogram at λmax 255 nm of synthetic 1-(hydroxymethyl)-phenanthrene. (B) UV chromatogram at λmax 255 nm of 1-MP metabolites in human HepG2 cells at 24 h. (C) FLR chromatogram at λex 254 nm and λem 369 nm of synthetic 1-(hydroxymethyl)-phenanthrene. (D) FLR chromatogram at λex 254 nm and λem 369 nm of 1-MP metabolites in human HepG2 cells at 24 h.
Evidence for Side Chain Hydroxylation
To aid in analyte identity, 1-(hydroxymethyl)-phenanthrene and 6-hydroxy-1-MP (phenolic metabolite) were synthesized as authentic standards (Figure S1 and Supporting Information) to determine if these metabolites were formed in human HepG2 cells. Detection of these metabolites would support hydroxylation either on the alkyl side chain or on the ring system of 1-MP. The major metabolite observed with 1-MP corresponded to metabolite 10 on the UV and fluorescence chromatogram. Comparison of the retention times and UV spectra of authentic 1-(hydroxymethyl)-phenanthrene with metabolite 10 on HPLC-UV-FLR in Figure 1 showed concordance. The extracted ion chromatogram of the Orbitrap full scan and MS spectrum confirmed that metabolite 10 was 1-(hydroxymethyl)-phenanthrene. This metabolite had the same exact mass as the authentic standard analyzed on a Q Exactive HF extracted ion chromatogram indicating that the [M – H]− of the metabolite in the negative ion mode had the same ion composition as the standard. Comparison of the retention times and UV spectra of 6-hydroxy-1-MP with metabolites 11–16 on HPLC-UV-FLR in Figure 1 showed similarity, suggesting that these metabolites were 1-MP-monophenols. Based on the retention times and UV spectra for 9-EP metabolites, metabolite 5 in Figure S4 is predicted to be a side chain hydroxylation product, and metabolites 6–9 in Figure S4 would be monophenols.
Evidence for the Diol-Epoxide Pathway
Identification of O-monosulfonated-dihydrodiols and tetraols indicated the occurrence of the diol-epoxide pathway for the metabolic activation of 1-MP and 9-EP in HepG2 cells.
Three isomers of monodehydrated O-monosulfonated-1-MP-dihydrodiols with the retention times of 18.81, 19.52, 20.06 min were detected by comparing the pseudo-SRM chromatograms (m/z 287 → 207) at 0 h (Figure 4A) and 24 h (Figure 4B) in the negative ion mode. The monodehydrated-O-monosulfonated 1-MP-dihydrodiol arises from the loss of H2O in the ion source. A similar phenomenon is observed when detecting the O-monosulfonated-retene dihydrodiol.17 The corresponding MS2 spectrum (m/z 287 → 207) of the metabolite at 18.81 min showed the characteristic neutral loss of the SO3 group (80 amu) from the deprotonated molecular ion (Figure 3C). Comparison of the retention times of these three isomers with metabolites 3, 4, and 5 on HPLC-UV-FLR in Figure 1 showed good agreement. The metabolites 3, 4, and 5 were identified as O-monodehydrated-O-monosulfonated-1-MP-dihydrodiols and are not phenol sulfate esters for the following reasons. First, metabolites 3 and 4 lack the UV and fluorescence characteristics present in phenanthrene aromatic ring system. Second, we detected the O-monosulfonated 1-MP catechol and its precursor would be the dihydrodiol. Third, the O-monosulfonated 1-MP catechol has a retention time of 20.85 min and would elute earlier than phenol sulfate esters due to the presence of the additional hydroxyl group. Thus, the retention times of the isomeric O-monodehydrated-O-monosulfonated-1-MP-dihydrodiols (18.81–20.06 min) are not consistent with those expected of the phenol sulfate esters.
Figure 4.

Detection of monodehydrated O-monosulfonated-1-MP-dihydrodiol in human HepG2 cells. (A) Extracted ion chromatogram of pseudo-SRM transition at 0 h. (B) Extracted ion chromatogram of pseudo-SRM transition at 24 h. (C) MS2 spectrum of the peak at 18.81 min. The samples were prepared as described the legend to Figure 1 and were subsequently analyzed on an ion trap LC-MS/MS.
Figure 3.

Detection of 1-(hydroxymethyl)-phenanthrene in human HepG2 cells by mass spectrometry. (A) Extracted ion chromatogram of Orbitrap full scan at 0 h. (B) Extracted ion chromatogram of Orbitrap full scan at 24 h and MS spectrum of the peak at 22.4 min showing exact mass for 1-(hydroxymethyl)-phenanthrene. (C) Extracted ion-chromatogram of Q-Exactive-HF full scan and MS spectrum of synthetic 1-(hydroxymethyl)-phenanthrene.
Similarly, a single isomer of monodehydrated O-monosulfonated-9-EP-dihydrodiol with the retention time of 20.06 min was detected by comparing the pseudo-SRM chromatograms (m/z 301 → 221) at 0 and 24 h in the negative ion mode (Figure S7). As with the monodehydrated O-monosulfonated 1-MP dihydrodiol, the monodehydrated-O-monosulfonated 9-EP dihydrodiol is formed in the ion source. The corresponding MS2 spectrum (m/z 301 → 221 → 206 → 188) of this metabolite showed the sequential neutral loss of the SO3 group (80 amu), loss of the CH3 group (15 amu), and loss of H2O (18 amu) from the deprotonated molecular ion in the negative ion mode (Figure S7).
A single isomer of a monodehydrated 1-MP-tetraol at 14.53 min was detected by monitoring the extracted ion chromatogram (m/z 243) at 0 and 24 h in the positive ion mode (Table 1). This metabolite was not identified on the metabolite chromatogram (Figure 1) since it was present in low abundance. The corresponding MS2 spectrum (m/z 243 → 225 → 198) of this metabolite showed the sequential loss of H2O (18 amu) and CO (28 amu) from the protonated molecular ion (Table 1). A single isomer of a monodehydrated 1-MP-tetraol was detected in HepG2 cells by monitoring the extracted ion chromatograms of the Orbitrap full scan at 0 and 24 h in the negative ion mode (Figure 5). HRMS of this isomer provided the accurate masses and molecular formula of a monodehydrated 1-MP-tetraol with acceptable ppm values (Figure 5). Because the C1 position of 1-MP is occupied with a methyl group, there is only one possible 1-MP-tetraol formed at positions 5, 6, 7 and 8 and would thus correspond to 5,6,7,8-tetrahydroxy-1-MP.
Table 1.
Mass Transitions for 1-MP Metabolites in HepG2 Cells
| metabolite no. | 1-MP metabolites | retention time (min) | mode | m/z |
|---|---|---|---|---|
| 3, 4, 5 | dehydrated O-sulfonated dihydrodiol | 18.81, 19.52, 20.06 | negative | 287 [M – H]−, 207 [M – H – SO3] |
| – | monodehydrated tetraol | 14.53 | positive | 243 [M + H]+, 225 [M + H – H2O]+, 198 [M + 2H – H2O – CO]+ |
| 7 | O-sulfonated catechol | 20.85 | negative | 303 [M – H]−, 223 [M – H – SO3]−, 180 [M – H – SO3 – CH3 – CO]− |
| – | quinone | 17.26 | positive | 223 [M + H]+, 204 [M + H – H2O –H]+, 176 [M + H – H2O – H – CO]+ |
| – | monohydroxyquinone | 16.74 | positive | 239 [M + H]+, 220 [M + H – H2O –H]+, 192 [M + H – H2O – H – CO]+, 163 [M + H – H2O – 2H – 2CO]+ |
Figure 5.
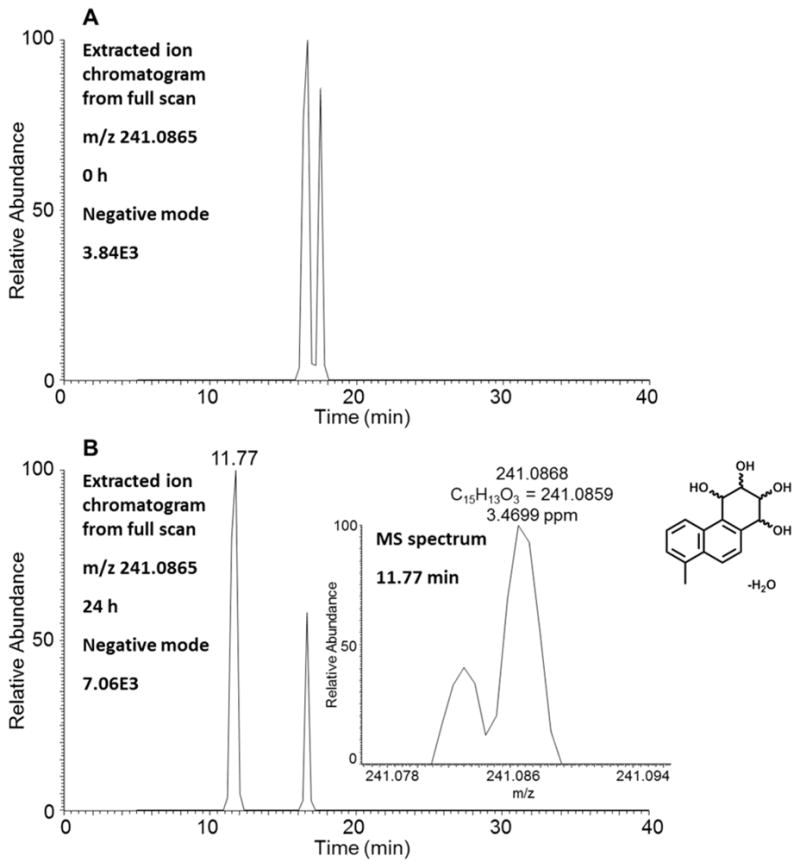
Detection of monodehydrated 1-MP-tetraol in human HepG2 cells. (A) Extracted ion chromatogram of Orbitrap full scan at 0 h. (B) Extracted ion chromatogram of Orbitrap full scan at 24 h and MS spectrum of the peak at 11.77 min.
Similarly, a single isomer of a bis-dehydrated 9-EP-tetraol at 16.70 min was detected by monitoring the extracted ion chromatograms (m/z 239) at 0 and 24 h in the positive ion mode (Figure S8). The corresponding MS2 spectrum (m/z 239 → 220 → 192) of this metabolite showed the sequential loss of H2O (18 amu) and CO (28 amu) from the protonated molecular ion (Figure S8). Because 9-EP is asymmetric, there are two possible regioisomeric 9-EP-tetraols: those that come from 9-EP-trans-1,2-dihydrodiol and those that come from 9-EP-trans-7,8-dihydrodiol. Comparison of the retention time of this tetraol isomer with metabolite 2 on HPLC-UV-FLR in Figure S4 showed good agreement.
Evidence for the O-Quinone Pathway
Identification of O-monosulfonated-dihydrodiol, O-monosulfonated-catechol, O-monomethyl-O-monosulfonated-catechol, O-monoglucuronosyl-catechol, and a monohydroxydione indicated the occurrence of the o-quinone pathway for the metabolic activation of 1-MP and 9-EP in HepG2 cells. The formation of similar catechol conjugates for B[a]P-7,8-catechol has been studied extensively.22–24
A single isomer at 20.85 min corresponding to either an O-monosulfonated-1-MP-catechol or an O-monosulfonated-1-MP-bis-phenol was detected by monitoring the pseudo-SRM chromatograms (m/z 303 → 223) at 0 h (Figure 6A) and 24 h (Figure 6B) in the negative ion mode. The corresponding MS2 spectrum (m/z 303 → 223) of this metabolite showed the characteristic neutral loss of the SO3 group (80 amu) from the deprotonated molecular ion (Figure 6C), and the MS3 spectrum (m/z 303 → 223 → 180) of this metabolite showed the subsequent loss of one CH3 group and one CO group (Figure 6D). This fragmentation pattern is consistent with that of O-monosulfonated-catechols of 5-methylchrysene, 6-ethyl-chrysene, and retene,14,15,17 strongly indicating the metabolite at 20.85 min was O-monosulfonated-1-MP-catechol. Comparison of the retention time of this peak with metabolite 7 on HPLC-UV-FLR in Figure 1 was in agreement and is consistent with the UV spectrum for 1-MP.
Figure 6.

Detection of an O-monosulfonated-1-MP-catechol in human HepG2 cells. (A) Extracted ion chromatogram of pseudo-SRM transition at 0 h. (B) Extracted ion chromatogram of pseudo-SRM transition at 24 h. (C) MS2 spectrum of the peak at 20.85 min. (D) MS3 spectrum of the peak at 20.85 min. The samples were prepared as described in Figure 1 legend and were subsequently analyzed on an ion trap LC-MS/MS.
Similarly, a single isomer at 22.10 min corresponding to an O-monosulfonated-9-EP-catechol was detected by monitoring the pseudo-SRM chromatograms (m/z 317 → 237) at 0 and 24 h in the negative ion mode (Figure S9). The corresponding MS2 spectrum (m/z 317 → 237) of this metabolite showed the characteristic neutral loss of the SO3 group (80 amu) from the deprotonated molecular ion and the subsequent loss of one CH3 group (Figure S9), ruling out the possibility of O-monosulfonated-9-EP-bis-phenol (no loss of H2O). Comparison of the retention time of this peak with metabolite 3 on HPLC-UV-FLR in Figure S3 was in agreement.
A single isomer of O-monomethyl-O-monosulfonated-1-MP-catechol was detected in HepG2 cells by monitoring the extracted ion chromatograms of the Orbitrap full scan at 0 and 24 h in the negative ion mode (Figure 7). The unique biotransformation of O-methylation strongly indicated the formation of the catechol, thus confirming the metabolic activation of 1-MP by the o-quinone pathway.9,10 Although LC/ MS cannot determine which regioisomer is formed, formation of a catechol on the 7,8 positions is favored due to the detection of the 5,6,7,8-tetrahydroxy-1-MP.
Figure 7.

Detection of O-monomethyl-O-monosulfonated-1-MP-catechols in human HepG2 cells. (A) Extracted ion chromatogram of Orbitrap full scan at 0 h. (B) Extracted ion chromatogram of Orbitrap full scan at 24 h and MS spectrum of the peak at 19.80 min. The samples were prepared as described in Figure 1 legend and were subsequently analyzed on an Orbitrap LC-MS/MS.
Similarly, two isomers of either O-monoglucuronosyl-9-EP-catechols or O-monoglucuronosyl-9-EP-bis-phenols were detected in HepG2 cells by monitoring the extracted ion chromatograms of the Orbitrap full scan at 0 and 24 h in the negative ion mode (Figure S10).
A single isomer of monohydroxy-1-MP-dione at 16.74 min was detected by monitoring the extracted ion chromatograms (m/z 239) at 0 and 24 h in the positive ion mode (Figure 8). The corresponding MS2 spectrum (m/z 239 → 220 → 192 → 163) of this metabolite (Figure 8C) showed the sequential loss of H2O (18 amu) and two CO (28 amu) from the protonated molecular ion. Monohydroxy-1-MP-dione could be derived from either an o-quinone or a remote quinone, and these alternatives cannot be distinguished based on mass spectrometry. However, the prospect that the O-monomethyl-O-monosulfonated 1-MP-catechol forms at the 7,8-positions suggests that the 7,8-dione is formed. As the loss of CH2OH was not observed, this ruled out activation of 1-MP by a combination of the o-quinone pathway and by side chain hydroxylation.
Figure 8.

Detection of monohydroxy-1-MP-dione in human HepG2 cells. (A) MS2 chromatogram at 0 h. (B) MS2 chromatogram at 24 h. (C) MS2 spectrum of the peak at 16.74 min. The samples were prepared as described in Figure 1 legend and were subsequently analyzed on an ion trap LC-MS/MS.
Similarly, a single isomer of monohydroxy-9-EP-dione was detected in HepG2 cells by monitoring the extracted ion chromatograms of the Orbitrap full scan at 0 and 24 h in the negative ion mode (Figure S11).
DISCUSSION
The Deepwater Horizon oil spill was the largest oil spill in United States history. Up to 47% of the total amount of oil discharged was not recovered from the spill and is found in ~110,000 km2, from coastal to deep-sea areas in the Gulf of Mexico.25 Filtering bivalves provide a conduit of the oil into the human food chain. In the initial federal response to seafood safety, pyrogenic PAHs were measured in >8000 samples of shell-fish and fin-fish using limits-of-concern for B[a]P and the seafood declared safe.8 However, the relative toxicity of PPAHs with respect to B[a]P is largely unknown. To inform this risk assessment, we have studied the metabolic fate of PPAHs in human liver cells.
1-MP and 9-EP are representative regioisomers of alkylated phenanthrenes, the major components with the highest abundance in crude oil. Considering human exposure pathways involve ingestion, the metabolism of 1-MP and 9-EP was examined in human HepG2 cells. HepG2 cells were chosen so that our findings could be directly compared to the metabolism of other PPAH studied in these cells in our laboratory. While HepG2 cells are transformed cells, they provide a source of reproducible metabolic profiles. We have shown that HepG2 cells express inducible P4501A1, a major isoform involved in PAH activation, and also express a complement of AKR isoforms known to involved in PAH activation.19,20 We considered the use of HepaRG cells since they retain the expression of multiple P450s, however, their expression of AKRs, a source of PPAHs metabolic activation, has not been documented. We also chose not to use pooled human hepatocytes since with a small n this is unlikely to be representative of a human population exposure. On balance, we believe that HepG2 cells are the best choice for our experiments.
On the basis of the peak areas of the metabolites on UV and FLR chromatograms, the major metabolic pathway for both 1-MP and 9-EP in human HepG2 cells involved side chain hydroxylation (Scheme 2). The hydroxylation of 1-MP was verified by comparison of the metabolite with an authentic synthetic standard. By contrast, side chain hydroxylation of 9-EP remains to be validated by authentic standards. Hydroxylation of the side chain could lead to bioactivation by sulfonation. However, 1-(hydroxymethyl)-phenanthrene-sulfonate was not detected in human HepG2 cells, which could be explained if the sulfonate group is rapidly lost before the formation of DNA adducts. It was challenging to obtain evidence for side chain hydroxylation of 9-EP due to the low ionization efficiency of monohydroxy-9-EP and the absence of authentic standards. Based on the similarity in metabolic profiles of 1-MP and 9-EP, the dominant metabolite 5 of 9-EP in Figure S4 is likely to be a side chain hydroxylation product. The minor metabolic activation pathways of both 1-MP and 9-EP involved formation of diol-epoxides and formation of o-quinones (Scheme 2). Although these pathways are relatively minor, they may be consequential due to the reactivity of the metabolites.
Scheme 2. Proposed Metabolic Pathways of 1-MP (Red) and 9-EP (Blue) in Human HepG2 Cellsa.

aThe red and blue numbers for each metabolite correspond to the metabolites labeled in the UV and fluorescence chromatograms in Figures 1 and S3, respectively. P = phenanthrene.
We proposed two additional pathways of 1-MP and 9-EP metabolic activation: formation of diol-epoxides and formation of o-quinones. Representative metabolites of these two pathways for 1-MP and 9-EP were detected and identified by HPLC-UV-FLR and LC-MS/MS (Tables 1 and S1). Metabolites 3, 4, and 5 in Figure 1 were identified as isomers of a monodehydrated- O-monosulfonated-1-MP-dihydrodiol. Evidence for a monodehydrated 1-MP-tetraol was obtained upon ion-trap LC/MS and confirmed by Orbitrap full scan. 1-MP has been shown to be mutagenic in the Ames test using an S9 activation system from Aroclor treated rats, suggesting that diol-epoxides were the mutagenic species. Interestingly, only the 3,4- and 5,6-dihydrodiol were mutagenic suggesting that a diol-epoxide might be responsible and would be consistent with the formation of the 5,6,7,8-tetrahydroxy-1-methyl phenanthrene.26,27 Metabolite 2 in Figure S4 was also identified as 9-EP-tetraol suggesting that 9-EP is also activated to a diol-epoxide.
In support of the o-quinone pathway, metabolite 7 in Figure 1 was identified as an O-monosulfonated-1-MP-catechol. Other metabolites in support of the o-quinone pathway for 1-MP include detection of the dihydrodiol precursor as a sulfonate as well as detection of the O-monomethyl-O-monosulfonated catechol and the O-monohydroxydione of 1-MP. Similarly, metabolite 3 in Figure S4 was identified as an O-monosulfonated-9-EP-catechol.
Examination of the metabolic profile for 9-EP suggests that metabolite 5 in Figure S4 could be a side chain hydroxylation product similar to 1-(hydroxymethyl)-phenanthrene. Evidence for the formation of monophenols was also obtained, and metabolites 6–9 in Figure S4 could be isomers of a 9-EP-monophenol.
The phase I and phase II isozymes responsible for the metabolic activation pathway of 1-MP and 9-EP in human HepG2 cells remain to be identified. The inducible cytochrome P4501A1 present in human HepG2 cells is probably responsible for the formation of diol-epoxides.28 Several members of the aldo-keto reductase (AKR) superfamily including AKR1C1, AKR1C2, and AKR1C3 present in human HepG2 cells would oxidize trans-dihydrodiols to o-quinones.20,29,30 AKRs, NAD(P)H: quinone oxidoreductase 1 (NQO1), and carbonyl reductase (CBR) can all catalyze the redox-cycling of o-quinones to form the intermediate catechols.31 Sulfotransferases (SULTs), uridine 5′-diphospho-glucuronosyltransferases (UGTs), and catechol-O-methyltransferase (COMT) would be responsible for the formation of O-monosulfonated-catechol, O-monomethyl-O-monosulfonated-catechol, and O-monoglucuronosyl-catechol.22–24 The SULTs that are expressed in human HepG2 cells and likely responsible for O-sulfonation are SULT1A1, 1A2, 1E1, and 2A1.32
Elucidation of the metabolic pathways of 1-MP and 9-EP in human liver cells can be used to identify exposure biomarkers that could be used for biomonitoring human urine. In our ongoing studies, we have established a cohort of human subjects that may have low, medium, and high exposures to contaminated seafood. It will be interesting to determine whether 1-MP and 9-EP signature metabolites of the diol-epoxide pathway, the o-quinone pathway, and the side chain sulfonation pathway can be detected in human urine and whether their levels vary by exposure group.
Supplementary Material
Acknowledgments
Funding
This publication was made possible by the Deepwater Horizon Research Consortia grant number U19 ES020676–05 and P30 ES013508 (T.M.P.) and by P30 ES006676 (R.P.H.) from the National Institute of Environmental Health Sciences (NIEHS), NIH, DHHS. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS or NIH.
ABBREVIATIONS
- AKR
aldo-keto reductase
- CBR
carbonyl reductase
- COMT
catechol-O-methyltransferase
- P450
cytochrome P450
- 9-EP
9-ethylphenanthrene
- ESI
electrospray ionization
- FBS
fetal bovine serum
- HPLC-UV-FLR
high performance liquid chromatography-ultraviolet-fluorescence
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- 1-MP
1-methyl-phenanthrene
- NQO1
NAD(P)H: quinone oxidoreductase 1
- PAH
polycyclic aromatic hydrocarbon
- PPAH
petrogenic PAH
- ROS
reactive oxygen species
- SULTs
sulfotransferases
- UGTs
uridine 5′-diphospho-glucuronosyltransferases
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.7b00232.
Mass transitions for 9-EP metabolites in HepG2 cells (Table S1); synthetic routes and details of synthesis of 1-(hydroxymethyl)-phenanthrene and 6-hydroxy-1-MP (Figure S1); excitation wavelength and emission wavelength spectra of 1-MP and 9-EP (Figure S2–S3); HPLC detection of 9-EP metabolites in human HepG2 cells (Figure S4); UV spectra of 1-MP and metabolite 1–16 in human HepG2 cells (Figure S5); UV spectra of 9-EP and metabolite 1–9 in human HepG2 cells (Figure S6); detection of monodehydrated O-monosulfonated-9-EP-dihydrodiol in human HepG2 cells (Figure S7); detection of bis-dehydrated 9-EP-tetraol in human HepG2 cells (Figure S8); detection of O-monosulfonated-9-EP-catechol in human HepG2 cells (Figure S9); detection of either O-monoglucuronosyl-9-EP-catechols or O-monoglucuronosyl-9-EP-bis-phenols in human HepG2 cells (Figure S10); detection of monohydroxy-9-EP-dione in human HepG2 cells (Figure S11) (PDF)
References
- 1.Joye SB, MacDonald IR, Leifer I, Asper V. Magnitude and oxidation potential of hydrocarbon gases released from the BP oil well blowout. Nat Geosci. 2011;4:160–164. [Google Scholar]
- 2.Atlas RM, Hazen TC. Oil Biodegradation and Bioremediation: A Tale of the Two Worst Spills in US History. Environ Sci Technol. 2011;45:6709–6715. doi: 10.1021/es2013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reddy CM, Arey JS, Seewald JS, Sylva SP, Lemkau KL, Nelson RK, Carmichael CA, McIntyre CP, Fenwick J, Ventura GT, Van Mooy BA, Camilli R. Composition and fate of gas and oil released to the water column during the Deepwater Horizon oil spill. Proc Natl Acad Sci U S A. 2012;109:20229–20234. doi: 10.1073/pnas.1101242108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McNutt MK, Camilli R, Crone TJ, Guthrie GD, Hsieh PA, Ryerson TB, Savas O, Shaffer F. Review of flow rate estimates of the Deepwater Horizon oil spill. Proc Natl Acad Sci U S A. 2012;109:20260–20267. doi: 10.1073/pnas.1112139108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryerson TB, Camilli R, Kessler JD, Kujawinski EB, Reddy CM, Valentine DL, Atlas E, Blake DR, de Gouw J, Meinardi S, Parrish DD, Peischl J, Seewald JS, Warneke C. Chemical data quantify Deepwater Horizon hydrocarbon flow rate and environmental distribution. Proc Natl Acad Sci U S A. 2012;109:20246–20253. doi: 10.1073/pnas.1110564109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldstein BD, Osofsky HJ, Lichtveld MY. The Gulf oil spill. N Engl J Med. 2011;364:1334–1348. doi: 10.1056/NEJMra1007197. [DOI] [PubMed] [Google Scholar]
- 7.IARC. Some Non-Heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures. Vol. 92. World Health Organization; Lyon France: 2010. [PMC free article] [PubMed] [Google Scholar]
- 8.Ylitalo GM, Krahn MM, Dickhoff WW, Stein JE, Walker CC, Lassitter CL, Garrett ES, Desfosse LL, Mitchell KM, Noble BT, Wilson S, Beck NB, Benner RA, Koufopoulos PN, Dickey RW. Federal seafood safety response to the Deepwater Horizon oil spill. Proc Natl Acad Sci U S A. 2012;109:20274–20279. doi: 10.1073/pnas.1108886109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Penning TM. Human aldo-keto reductases and the metabolic activation of polycyclic aromatic hydrocarbons. Chem Res Toxicol. 2014;27:1901–1917. doi: 10.1021/tx500298n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: generation of reactive and redox active o-quinones. Chem Res Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- 11.Levin W, Wood A, Chang R, Ryan D, Thomas P, Yagi H, Thakker D, Vyas K, Boyd C, Chu SY, Conney A, Jerina D. Oxidative metabolism of polycyclic aromatic hydrocarbons to ultimate carcinogens. Drug Metab Rev. 1982;13:555–580. doi: 10.3109/03602538209011087. [DOI] [PubMed] [Google Scholar]
- 12.Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogensis by polycyclic aromatic hydrocrabons. G.H.A Clowes Memorial Lecture. Cancer Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 13.Rothman N, Poirier MC, Baser ME, Hansen JA, Gentile C, Bowman ED, Strickland PT. Formation of polycyclic aromatic hydrocarbon-DNA adducts in peripheral white blood cells during consumption of charcoal-broiled beef. Carcinogenesis. 1990;11:1241–1243. doi: 10.1093/carcin/11.7.1241. [DOI] [PubMed] [Google Scholar]
- 14.Huang M, Mesaros C, Zhang S, Blair IA, Penning TM. Potential Metabolic Activation of a Representative C2-Alkylated Polycyclic Aromatic Hydrocarbon 6-Ethylchrysene Associated with the Deepwater Horizon Oil Spill in Human Hepatoma (HepG2) Cells. Chem Res Toxicol. 2016;29:991–1002. doi: 10.1021/acs.chemrestox.6b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang M, Zhang L, Mesaros C, Hackfeld LC, Hodge RP, Blair IA, Penning TM. Metabolism of an Alkylated Polycyclic Aromatic Hydrocarbon 5-Methylchrysene in Human Hepatoma (HepG2) Cells. Chem Res Toxicol. 2015;28:2045–2058. doi: 10.1021/acs.chemrestox.5b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang M, Zhang L, Mesaros C, Zhang S, Blaha MA, Blair IA, Penning TM. Metabolism of a representative oxygenated polycyclic aromatic hydrocarbon (PAH) phenanthrene-9,10-quinone in human hepatoma (HepG2) cells. Chem Res Toxicol. 2014;27:852–863. doi: 10.1021/tx500031p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang M, Mesaros C, Hackfeld LC, Hodge RP, Zang T, Blair IA, Penning TM. Potential Metabolic Activation of a Representative C4-Alkylated Polycyclic Aromatic Hydrocarbon Retene (1-Methyl-7-isopropyl-phenanthrene) Associated with the Deepwater Horizon Oil Spill in Human Hepatoma (HepG2) Cells. Chem Res Toxicol. 2017;30:1093–1101. doi: 10.1021/acs.chemrestox.6b00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.NIST. Standard Reference Material (SRM) 2779, Gulf of Mexico Crude Oil. National Institute of Standards and Technology; Gaithersburg, MD: 2012. pp. 1–7. [Google Scholar]
- 19.Burczynski ME, Lin HK, Penning TM. Isoform-specific induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: implications for the alternative pathway of PAH activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res. 1999;59:607–614. [PubMed] [Google Scholar]
- 20.Steckelbroeck S, Oyesanmi B, Jin Y, Lee SH, Kloosterboer HJ, Penning TM. Tibolone metabolism in human liver is catalyzed by 3alpha/3beta-hydroxysteroid dehydrogenase activities of the four isoforms of the aldo-keto reductase (AKR)1C subfamily. J Pharmacol Exp Ther. 2006;316:1300–1309. doi: 10.1124/jpet.105.091587. [DOI] [PubMed] [Google Scholar]
- 21.Xia K, Hagood G, Childers C, Atkins J, Rogers B, Ware L, Armbrust K, Jewell J, Diaz D, Gatian N, HF Polycyclic aromatic hydrocarbons (PAHs) in Mississippi seafood from areas affected by the Deepwater Horizon Oil Spill. Environ Sci Technol. 2012;46:5310–5318. doi: 10.1021/es2042433. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Huang M, Blair IA, Penning TM. Detoxication of benzo[a]pyrene-7,8-dione by sulfotransferases (SULTs) in human lung cells. J Biol Chem. 2012;287:29909–29920. doi: 10.1074/jbc.M112.386052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Huang M, Blair IA, Penning TM. Interception of benzo[a]pyrene-7,8-dione by UDP glucuronosyltransferases (UGTs) in human lung cells. Chem Res Toxicol. 2013;26:1570–1578. doi: 10.1021/tx400268q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L, Jin Y, Chen M, Huang M, Harvey RG, Blair IA, Penning TM. Detoxication of structurally diverse polycyclic aromatic hydrocarbon (PAH) o-quinones by human recombinant catechol-O-methyltransferase (COMT) via O-methylation of PAH catechols. J Biol Chem. 2011;286:25644–25654. doi: 10.1074/jbc.M111.240739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romero IC, Toro-Farmer G, Diercks AR, Schwing P, Muller-Karger F, Murawski S, Hollander DJ. Large-scale deposition of weathered oil in the Gulf of Mexico following a deep-water oil spill. Environ Pollut. 2017;228:179–189. doi: 10.1016/j.envpol.2017.05.019. [DOI] [PubMed] [Google Scholar]
- 26.LaVoie EJ, Tulley-Freiler L, Bedenko V, Hoffman D. Mutagenicity, tumor-initating activity and metabolism of methylphenanthrenes. Cancer Res. 1981;41:3441–3447. [PubMed] [Google Scholar]
- 27.LaVoie EJ, Tulley-Freiler L, Bedenko V, Hoffman D. Mutagenicity of substituted phenanthrenes in Salmonella typhimurium. Mutat Res, Genet Toxicol Test. 1983;116:91–102. doi: 10.1016/0165-1218(83)90100-3. [DOI] [PubMed] [Google Scholar]
- 28.Burczynski ME, Penning TM. Genotoxic polycyclic aromatic hydrocarbon ortho-quinones generated by aldo-keto reductases induce CYP1A1 via nuclear translocation of the aryl hydrocarbon receptor. Cancer Res. 2000;60:908–915. [PubMed] [Google Scholar]
- 29.Palackal NT, Burczynski ME, Harvey RG, Penning TM. The ubiquitous aldehyde reductase (AKR1A1) oxidizes proximate carcinogen trans-dihydrodiols to o-quinones: potential role in polycyclic aromatic hydrocarbon activation. Biochemistry. 2001;40:10901–10910. doi: 10.1021/bi010872t. [DOI] [PubMed] [Google Scholar]
- 30.Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J Biol Chem. 2002;277:24799–24808. doi: 10.1074/jbc.M112424200. [DOI] [PubMed] [Google Scholar]
- 31.Shultz CA, Quinn AM, Park JH, Harvey RG, Bolton JL, Maser E, Penning TM. Specificity of human aldo-keto reductases, NAD(P)H:quinone oxidoreductase, and carbonyl reductases to redox-cycle polycyclic aromatic hydrocarbon diones and 4-hydroxyequilenin-o-quinone. Chem Res Toxicol. 2011;24:2153–2166. doi: 10.1021/tx200294c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westerink WM, Schoonen WG. Phase II enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol In Vitro. 2007;21:1592–1602. doi: 10.1016/j.tiv.2007.06.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.