

Abstract
Proteostasis is essential in the mammalian brain where post-mitotic cells must function for decades to maintain synaptic contacts and memory. The brain is dependent on glucose and other metabolites for proper function and is spared from metabolic deficits even during starvation. In this review, we outline how the nutrient sensitive nucleocytoplasmic posttranslational modification O-linked N-acetylglucosamine (O-GlcNAc) regulates protein homeostasis. The O-GlcNAc modification is highly abundant in the mammalian brain, and has been linked to proteopathies, including neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s. C. elegans, Drosophila, and mouse models harboring O-GlcNAc transferase and O-GlcNAcase knockout (KO) alleles have helped define the role O-GlcNAc plays in development as well as age-associated neurodegenerative disease. These enzymes add and remove the single monosaccharide from protein serine and threonine residues, respectively. Blocking O-GlcNAc cycling is detrimental to mammalian brain development and interferes with neurogenesis, neural migration, and proteostasis. Findings in C. elegans and Drosophila model systems indicate that the dynamic turnover of O-GlcNAc is critical for maintaining levels of key transcriptional regulators responsible for neurodevelopment cell fate decisions. In addition, pathways of autophagy and proteasomal degradation depend on a transcriptional network that is also reliant on O-GlcNAc cycling. Like the quality control system in the endoplasmic reticulum which uses a “mannose-timer” to monitor protein folding, we propose that cytoplasmic proteostasis relies on an “O-GlcNAc timer” to help regulate the lifetime and fate of nuclear and cytoplasmic proteins. O-GlcNAc-dependent developmental alterations impact metabolism and growth of the developing mouse embryo and persist into adulthood. Brain-selective KO mouse models will be an important tool for understanding the role of O-GlcNAc in the physiology of the brain and its susceptibility to neurodegenerative injury.
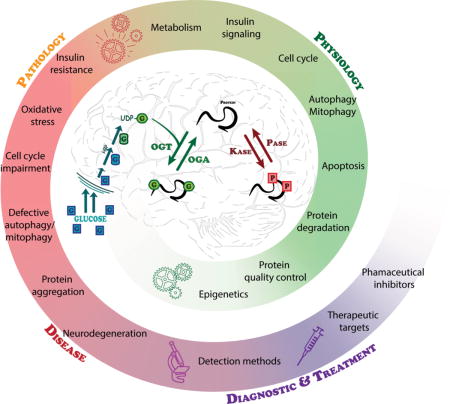
Graphical abstract
Proteostasis is essential in the mammalian brain where post-mitotic cells must function for decades to maintain synaptic contacts and memory. The brain is dependent on glucose and other metabolites for proper function and is spared from metabolic deficits even during starvation. In this review, we outline how the nutrient sensitive nucleocytoplasmic posttranslational modification O-linked N-acetylglucosamine (O-GlcNAc) regulates protein homeostasis. This cyclic modification is coordinately regulated with other PTMs such as phosphorylation to regulate the required intricacies of cellular processes. Deregulation of PTMs including O-GlcNAc leads to several pathologies that are associated with neurodegeneration.
Introduction
George Cahill, in his groundbreaking article “Starvation in Man” (Cahill, 1970) emphasized that the brain is the last organ to succumb to starvation owing to its strong dependence on glucose as a source of energy. The mammalian brain has evolved such that it has an unusually high requirement for glucose and does not easily tolerate its absence. In humans, the brain accounts for only about 2% of the total body weight, yet it consumes about 20% of glucose-derived energy making it the main consumer of glucose (Erbsloh et al., 1958). In addition, specialized centers in the brain sense central and peripheral glucose levels and regulate glucose metabolism through the vagal nerve as well as neuroendocrine signals. Glucose supply to the brain is under tight regulation by neurovascular coupling and enters the brain from the blood by crossing the blood-brain-barrier through glucose transporters (e.g., GLUT1). Glucose and other metabolites such as lactate are distributed through a highly coupled metabolic network of brain cells. The brain’s dependence on glucose metabolism to maintain neurotransmitter release, axonal transport, and cell survival has important implications for understanding human disease. In diabetics, dangerously high or low glucose can lead to diabetic coma. The high flux of glucose required for maintenance of the central nervous system may also be linked to the formation of reactive oxygen species causing oxidative damage in the brain. Since most neurons are post-mitotic, this is thought to impact the brain disproportionately. Moreover, there is a growing awareness that adult neurogenesis may be more important than once recognized, particularly in the dentate gyrus, hippocampus, and olfactory bulb. Since neurogenesis requires significant amount of energy from glucose, abnormal levels of glucose supply could perturb healthy neurogenesis in these regions.
Neurodegenerative diseases, including Alzheimer’s Disease (AD) have been directly linked with altered brain glucose consumption. It has been estimated that 5.4 million people in the US currently have AD. The risk of AD increases with age, and so by 2050, the Alzheimer’s Association estimates that between 11 and 16 million Americans will have the disease, with one new case appearing every 33 seconds (Association, 2017). The economic cost of AD is already immense and disproportionally high in countries with longer life expectancies. While some estimates suggest that the total worldwide economic cost of dementias represents about 1% of world gross domestic product, the human cost of these diseases is much greater. In this review, we evaluate the accumulating body of evidence suggesting that a signaling pathway that integrates the flux of nutrients including glucose, glutamine, and acetyl-CoA, and terminates in O-linked N-acetylglucosamine (O-GlcNAc) addition to proteins, may contribute to the pathophysiology of neurodegenerative diseases. With a central role in the regulation of gene expression, cellular signaling, protein synthesis, and degradation, the nutrient-responsive O-GlcNAc pathway may represent a unique therapeutic option for diagnosis and management of these disorders. But what makes the brain uniquely sensitive to deregulation of O-GlcNAc metabolism?
A. Protein aggregates in neurodegenerative disease
Human neurodegenerative diseases are associated with deposits of aggregated proteins. These aggregates and their designations are summarized in Figure 1 (right side). As Figure 1 suggests, the proteostasis networks that maintain protein folding involve every aspect of the life cycle of proteins involved. This includes transcription, translation, folding, posttranslational modification, targeting and degradation. It has been noted that many of the proteins that are involved in aggregation in neurodegenerative diseases have been found to be O-GlcNAc modified. The known consequences of these proteins’ modification are discussed below in sections relevant to the individual OGT substrates. However, the question of whether these protein aggregates are pathogenic or simply represent a cellular response to stress is currently unknown. Parkinson’s disease (PD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and other polyglutamine diseases each have characteristic deposits of protein aggregates in the brain (Ross and Poirier, 2005). These deposits can be cytoplasmic, nuclear, or even extracellular. In some cases, this protein aggregation results from a mutation in a disease-related protein, processing of those proteins, or elevation in their levels. Even proteins that are not associated with disease can aggregate in inclusion bodies and cause toxicity.
Figure 1. The potential roles of O-GlcNAcylation in maintaining the cellular proteostasis network and relationship to protein aggregates in neurodegeneration.
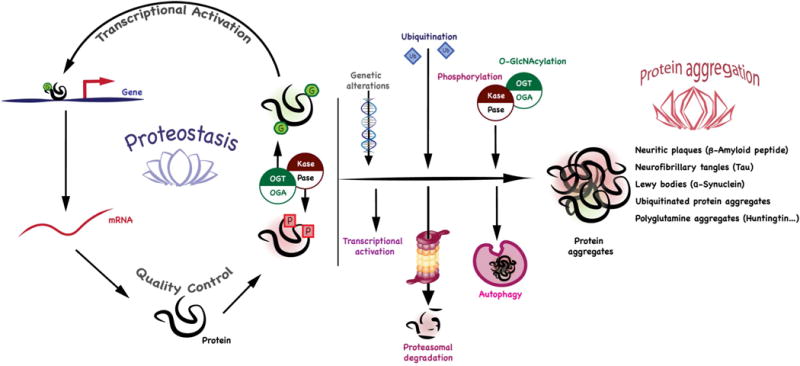
O-GlcNAcylation has been shown to be a key regulator of transcription, translation, protein folding and degradation that may be regulated in in neurodegenerative disease. The protein aggregates that are associated with human disease are listed at the right. O-GlcNAcylation can influence aggregation by modulating processes including transcription, protein folding, the cellular quality control, and stress pathways. Acting in consort with other post-translational modifications, O-GlcNAc may influence signaling pathways and the regulation of protein degradation including proteasomal degradation, autophagy, and the cellular stress response.
Neuritic plaques in AD contain mainly Aβ peptide, a proteolytic product of the amyloid precursor protein (APP). AD is also characterized by the accumulation of intracellular aggregates of the microtubule-associated protein tau, termed neurofibrillary tangles (NFTs). Tauopathies such as fronto-temporal dementia with Parkinsonism can be caused by mutations in the tau protein itself. PD, which is characterized by tremor, loss of motor function, and autonomic instability, is caused by degeneration of dopaminergic neurons in the substantia nigra of the midbrain. Point mutations or increased gene expression of the α-synuclein gene cause an autosomal dominant form of PD (Levine et al., 2017), whereas recessive PD can be caused by mutations in the genes encoding several proteins including parkin, DJ-1, or PINK1. Mutations in parkin and PINK1, key regulators of neuronal mitophagy, are associated with cell death correlated with immobile mitochondria unable to be sequestered (Wang et al., 2011). These recessive forms of PD are presumably induced by a loss-of-function of the associated proteins. The aggregates that form in PD are Lewy bodies, cytoplasmic perinuclear inclusion bodies in neurons, in which the α-synuclein protein is a major constituent (Ross and Poirier, 2004; Ross and Poirier, 2005). ALS is characterized by degeneration of motor neurons leading to progressive motor weakness with ubiquitinated protein aggregates present in patient brains. HD is caused by genomic expansion of a triplet CAG repeat coding for polyglutamine near the N-terminus of the huntingtin protein. Protein inclusions containing huntingtin and other proteins are present in regions of the brain that degenerates and there is a good correlation between the extent of triplet expansion and inclusion density. Other polyglutamine diseases such as spino-cerebellar ataxia (SCA) present with similar aggregates containing expanded polyglutamine repeats in the protein Atx-1. The Prion diseases are also associated with amyloid plaques which form both intra- and extra-cellularly by self-propagation of an abnormal protein conformation (Ross and Poirier, 2004; Ross and Poirier, 2005).
Domains common to neurodegenerative protein aggregates
Many of the proteins that are prone to aggregation in neurodegenerative diseases have domains that can be characterized as intrinsically disordered proteins (IDPs). These intrinsically disordered segments are also often the sites of posttranslational modification. Such domains are dynamic structures that interconvert between collapsed and extended structures on a timescale that differs from globular proteins. IDPs are relatively depleted in bulky hydrophobic amino acids and enriched in polar residues and structure-breaking amino acids like Pro and Gly. Aβ is about 20% disordered, tau is greater than 80% disordered, α-synuclein is greater than 90% disordered, and the ataxins range from 50–90% disordered (Uversky, 2015). Not surprisingly, many of the chaperones and other proteins involved in maintaining proteostasis are also extensively disordered (Uversky, 2015). Some of these proteins have an intrinsic propensity to adopt pathological conformations and persistently high concentrations, interaction with chaperones, or point mutations can initiate the aggregation cascade. Disruption of protein folding or protein degradation can contribute to aggregation and posttranslational modifications such as advanced glycation products, deamidation, and phosphorylation may influence the aggregation process (Ross and Poirier, 2005) (Figure 1). O-GlcNAcylation, like glycation, may be elevated under conditions of hyperglycemia and other forms of stress, although this is not always the case (Yang and Qian, 2017). The present review focuses on the many influences of O-GlcNAcylation on neurodegeneration through its role on pathways including protein folding, phosphorylation, and cellular protein degradation pathways. O-GlcNAc transferase, the enzyme responsible for the addition of O-GlcNAc to target proteins, has a propensity to interact with IDPs including many of those proteins that form aggregates in neurodegenerative disease (Trinidad et al., 2012; Bond and Hanover, 2015). In the following sections, we summarize the accumulating evidence for the involvement of O-GlcNAc metabolism in neurodegenerative toxicity. A timeline detailing the sequence of findings leading to our current understanding of the importance of O-GlcNAc cycling in neurodegeneration is shown in Figure 2.
Figure 2. A timeline summarizing the major advancements in understanding O-GlcNAc as a modulator of neurodegeneration.
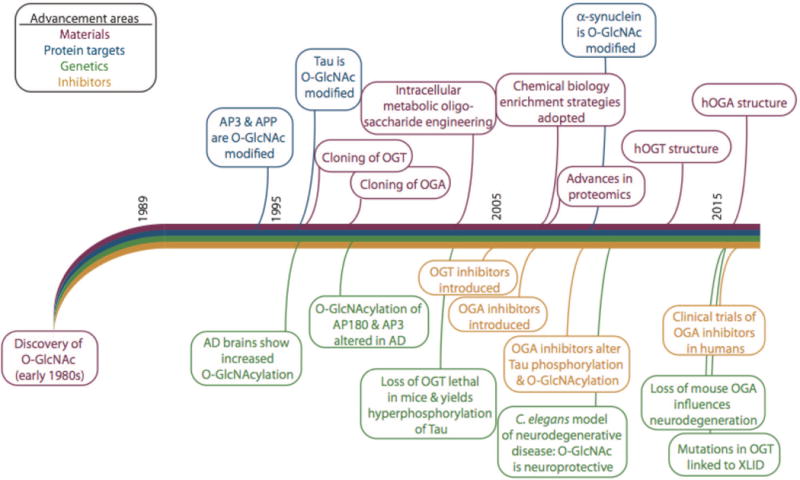
The timeline was derived from publication dates of these references and references therein: (Torres and Hart, 1984; Holt and Hart, 1986; Davis and Blobel, 1987; Hanover et al., 1987; Holt et al., 1987; Park et al., 1987; Snow et al., 1987; D’Onofrio et al., 1988; Caillet-Boudin et al., 1989; Datta et al., 1989; Inaba and Maede, 1989; Haltiwanger et al., 1990; Starr and Hanover, 1990; Starr et al., 1990; Kearse and Hart, 1991a; Kearse and Hart, 1991b; Lüthi et al., 1991; Chou et al., 1992; Hagmann et al., 1992; Haltiwanger et al., 1992; Reason et al., 1992; Roquemore et al., 1992; Dong et al., 1993; Elliot et al., 1993; Kelly et al., 1993; Murphy et al., 1994; Bailer et al., 1995; Chou et al., 1995; Griffith and Schmitz, 1995; Griffith et al., 1995; Dong et al., 1996; Roquemore et al., 1996; Kreppel et al., 1997; Lubas et al., 1997; Roos et al., 1997; Haltiwanger et al., 1998; Yao and Coleman, 1998a; Yao and Coleman, 1998b; Akimoto et al., 1999; Griffith and Schmitz, 1999; Hanover et al., 1999; Akimoto et al., 2000; Lubas and Hanover, 2000; Ross et al., 2000; Cole and Hart, 2001; Gao et al., 2001; Hanover, 2001; Rex-Mathes et al., 2001; Zachara et al., 2001; McClain et al., 2002; Vosseller et al., 2002; Wells et al., 2002; Zachara et al., 2002; Akimoto et al., 2003; Gao et al., 2003; Hanover et al., 2003; Iyer et al., 2003; Lefebvre et al., 2003a; Lefebvre et al., 2003b; Love et al., 2003; Marshall et al., 2003; Whelan and Hart, 2003; Zhang et al., 2003; Cieniewski-Bernard et al., 2004; Jínek et al., 2004; Khidekel et al., 2004; Liu et al., 2004a; Robertson et al., 2004; Brickley et al., 2005; Gross et al., 2005; Hanover et al., 2005; Lefebvre et al., 2005; Dorfmueller et al., 2006; Forsythe et al., 2006; März et al., 2006; Nandi et al., 2006; Andrali et al., 2007; Deng et al., 2008; Rexach et al., 2008; Bleckmann et al., 2009; Gambetta et al., 2009a; Lazarus et al., 2009; Liu et al., 2009; Sinclair et al., 2009; Yanagisawa and Yu, 2009; Yuzwa and Vocadlo, 2009; Di Domenico et al., 2010; Rexach et al., 2010; Srikanth et al., 2010; Kim, 2011; Lazarus et al., 2011; Smet-Nocca et al., 2011; Yuzwa et al., 2011; Nakamura et al., 2012; Wang et al., 2012; Yu et al., 2012; Yuzwa et al., 2012; Cameron et al., 2013; Diwu et al., 2013a; Diwu et al., 2013b; Hanover and Wang, 2013; Wang and Hanover, 2013; Graham et al., 2014; Skorobogatko et al., 2014; Taylor et al., 2014; Cha et al., 2015; Mao et al., 2015; Marotta et al., 2015; Akan et al., 2016; Gatta et al., 2016; Gong et al., 2016; Lagerlöf et al., 2016; Lagerlöf et al., 2017; Lewis et al., 2017; Olivier-Van Stichelen et al., 2017)
B. The enzymes of O-GlcNAc cycling and roles in neurophysiology
1. Hexosamine signaling pathway in the brain
Besides its function as an energy source, glucose is also an important precursor for the synthesis of downstream metabolites. Through metabolic pathways including glycolysis and the hexosamine biosynthetic pathway (HBP), the brain takes advantage of glucose as its primary energy source and converts this simple sugar to a myriad of critical metabolites. The HBP is an essential player in brain physiology with 2 to 3% of glucose (Simpson et al., 2008) converted to the pathway’s ultimate product, UDP-GlcNAc (Bouché et al., 2004) . The synthesis of UDP-GlcNAc integrates lipid, energy, and nucleotide metabolism making the nucleotide sugar sensitive to cellular metabolite flux (Hanover et al., 2012).
The concentration of nutrient-sensitive UDP-GlcNAc impacts many glycosylation processes, with those that are cyclic arguably the most affected by changes in metabolic flux. Glycosyltransferases utilize nucleotide sugars such as UDP-GlcNAc as substrates to add carbohydrates to proteins that ultimately reside both intra- and extra-cellularly. OGT utilizes UDP-GlcNAc to post translationally modify intracellular serine and threonine residues with O-GlcNAc. Tight regulation of O-GlcNAc cycling relies not only on the concentrations of UDP-GlcNAc and OGT but also on O-GlcNAcase (OGA), the enzyme responsible for the modification’s removal. While modification of some proteins is sub-stoichiometric, others are more-or-less permanently modified by O-GlcNAc supporting a varied role in protein structure, localization, and function In this way, this single monosaccharide post translational modification (PTM) can dynamically respond to changes in glucose-dependent cellular metabolism and modify it’s target proteins thereby potentially acting as an “O-GlcNAc timer” for the processes it influences.
Along with the pancreas, the brain is the most heavily O-GlcNAcylated tissue, consistent with its high glucose consumption. In fact, 40% of all neuronal proteins and 19% of synaptosomal proteins are O-GlcNAc modified. Moreover, mRNAs encoding OGT and OGA are highly enriched in the brain (Kreppel et al., 1997; Lubas et al., 1997; Gao et al., 2001). O-GlcNAc modifies thousands of proteins including those correlated with neurodegenerative disease such as amyloid precursor protein and tau (associated with pathophysiology of AD), α-synuclein (component of Lewy bodies in Parkinson’s Disease, PD)(Spillantini et al., 1997; Wang et al., 2010a), superoxide dismutase (Sprung et al., 2005), and neurofilament proteins (Dong et al., 1993) (involved in amyotropic lateral sclerosis, ALS). How is O-GlcNAc regulated in the brain? Do O-GlcNAc levels change globally or only on a subset of proteins upon neurodegeneration? Which of the many roles of O-GlcNAc are important in neurodegeneration? These are some of the questions that have come under scrutiny and are summarized in (Table 1).
Table I. The major targets and pathways shown to be altered by O-GlcNAc cycling in Neurodegenerative Disease.
The target, manipulation and cell type or organism are listed on the left. The basic finding and reference are in the two right panels. The Table is not an exhaustive compilation of the published literature but is meant to illustrate the approaches that have been taken
| Molecule/Protein | Agent Used | Tissue or cell line | Relevant result | Ref. |
|---|---|---|---|---|
| Tau | OGA inhibitor Thiamet-G | tau mutant (Pro301->Leu) mouse model of AD | Systemic administration of Thiamet-G reduced neurofibrillary tangles (NFT) | (Yuzwa et al., 2012) |
| Tau, APP | AD mouse model (TAPP) carrying tau (Pro301->Leu) and APP (APPSwe) mutations | Decreased tau phosphorylation and neurite plaques | (Yuzwa et al., 2014) | |
| Tau | Post mortem AD brain frontal cerebral cortex, and cerebellum | O-GlcNAc levels reduced | (Liu et al., 2004) | |
| Isolated proteins from AD patients brains | Normal tau entangles with hyperphosphorylated tau | (Alonso et al., 1996) | ||
| OGT Knockout mouse brain | Tau phosphorylation increased | (O’Donnell et al., 2004) | ||
| Tau, APP or polyglutamine expansion | C. elegans: OGT-1 and OGA-1 mutants | Neurodegeneration worsened in OGA-1 mutants, and ameliorated in OGT-1 mutant | (Wang et al., 2012) | |
| APP | SH-SY5Y neuroblastoma cells | O-GlcNAcylation induced α-secretase mediated cleavage of APP, resulting in neuroprotective sAPPα | (Jacobsen and Iverfeldt, 2011) | |
| Milton | Rat Hippocampal neurons | Increased O-GlcNAcylation decrease mitochondrial motility | (Pekkurnaz et al., 2014) | |
| Ogg1 | Murine neonatal cardiac myocytes | O-GlcNAcylation decreased Ogg1 activity that could lead to mDNA damage | (Cividini et al., 2016) | |
| MIBP1 | HEK293 cells | MIBP1 activity is suppressed by O-GlcNAcylation | (Iwashita et al., 2012) | |
| Electron Transport Chain (ETC) | Diabetic rat hearts | Increased O-GlcNAcylation could reduce ATP production by inhibiting the activity of ETC complexes | (Ma et al., 2016) | |
| TCA cycle enzymes | Diabetic rat hearts | Increased O-GlcNAc could reduce TCA activity | (Ma et al., 2016) | |
| Insulin Signaling Pathway | 3T3-L1 adipocytes | Increased O-GlcNAcylation inhibits insulin signaling pathway activation | (Vosseller et al., 2002) | |
| SNAP-29 | HeLa cells | Increased O-GlcNAcylation reduced autophagy by decreasing fusion between autophagosomes and endosome/lysosomes | (Guo et al., 2014) | |
| BCL2 | Diabetic db/db mice cardiomyocytes | (Marsh et al., 2013) | ||
| Atg7 | Mouse brain lysate | (Park et al., 2015) | ||
| α-synuclein | In vitro recombinant protein | O-GlcNAcylation prevents α-synuclein aggregation | (Marotta et al., 2015) | |
| Period | D. melanogaster | O-GlcNAcylation of regulate circadian timing by altering dPER subcellular localization | (Kim et al., 2012; Kaasik et al., 2013) | |
| KV7.3 (Kcnq3) | AgRP neurons in mouse | O-GlcNAcylation is essential for excitability of AgRP neurons by modifying Kcnq3 | (Ruan et al., 2014) |
2. OGT: isoforms and localization
It is intriguing that O-GlcNAc addition is regulated by a single, ubiquitously expressed, essential enzyme, OGT. Elimination of OGT is lethal in most organisms and in dividing cells: generation of Ogt KO stem cells and Ogt KO mice has been unsuccessful. Interestingly, two recent papers have successfully removed OGT in post-mitotic adult neurons, thus opening a new area to examine how O-GlcNAcylation of proteins regulates normal brain function in viable animals.
OGT expression, localization, and regulation
The Ogt gene produces several splice variants differing at the amino (N) terminal region in mammals with splicing and multiple start codons (Hanover et al., 2012; Abramowitz et al., 2014). Indeed, in Drosophila, levels of the genes encoding OGT and a small number of other transcripts are directly modulated by the function of spliceosome (Ashton-Beaucage et al., 2010; Hanover et al., 2012; Abramowitz et al., 2014).
The three canonic isoforms that are produced from alternative start codons in the splice variants of mammalian Ogt are found in the nucleus (ncOGT and sOGT), cytoplasm (ncOGT and sOGT), or mitochondria (mOGT). The largest isoform, ncOGT, is ~110 kDa and contains 12 tetratricopeptide (TPR) repeats at the N-terminus. The structure of the dimeric ncOGT with its 12 TPR repeats is shown in Figure 3A. The TPR repeats form a structure like the armadillo repeats of importin alpha in which Asn residues line a concave surface of the molecule and interact with target proteins as demonstrated by recent crystallographic findings (Lazarus et al., 2013). For example, the TPR repeats of OGT interact with regions of the unstructured domains of nucleoporins (Cordes and Krohne, 1993) suggesting that OGT will also have an affinity for unstructured regions of other substrates (Cordes and Krohne, 1993; Lubas and Hanover, 2000; Bond and Hanover, 2015). The 102 kDa mOGT has a unique mitochondrial targeting sequence and 9 TPR domains. And, the smallest isoform, sOGT, is about 75 kDa with only 2.5 TPR domains (Lazarus et al., 2006; Lazarus et al., 2009). Given that the sOGT isoform lacks the dimerization motif found in ncOGT and mOGT, it is likely to exist in complexes either as a monomer or higher order multimers (Jinek et al., 2004).
Figure 3. The structures of OGT, OGA, and the sites of interaction with some of the binding partners of the enzymes of O-GlcNAc cycling.
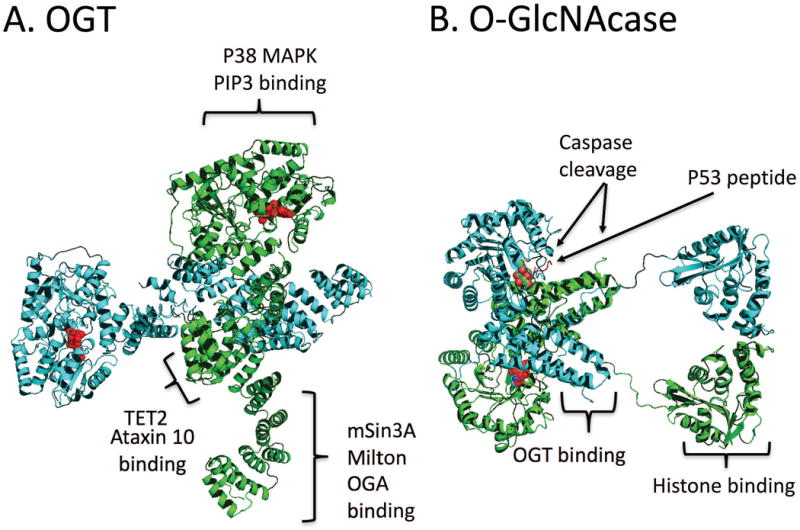
The crystal structures pictured are derived from crystal structures 1W3B and 3PE4 (OGT) (Jínek et al., 2004; Lazarus et al., 2011) and 5UN8 and 3ZJ0 (OGA) (Rao et al., 2013; Li et al., 2017). (A) Structure and interaction partners of dimer ncOGT. OGT has many binding partners that are crucial for cellular processes mediating neuronal homeostasis. The substrate UDP-GlcNAc bound to the active site is shown in red. The positions in the structure corresponding the interaction domains between OGT and these molecules are indicated in brackets B) Structure and binding partners of the long isoform of O-GlcNAcase. The O-GlcNAcylated p53 peptide binds in the active site is highlighted in red. The sites of caspase 3 cleavage are indicated by the arrows. This form of OGA has been shown to interact with OGT and histones. For discussion of these interactions, see associated portions of the text.
N-terminal variation in OGT’s TPR domains influences substrate recognition (Lubas and Hanover, 2000) and, therefore, enzyme localization, and cellular activity. Indeed, while all splice variants include the same linker region and the C-terminal catalytic domain responsible for transferase activity, the shortest isoform, sOGT, has not been shown to add O-GlcNAc to proteins in vivo (Lazarus et al., 2006). mOGT targets the inner mitochondrial membrane and displays different substrate specificity from ncOGT (Love et al., 2003; Lazarus et al., 2006). Some literature suggests that ncOGT, rather than mOGT, can catalyze O-GlcNAcylation of the mitochondrial proteome (Trapannone et al., 2016); however, recent work identified that mOGT was required for glycosylation of specific mitochondrial proteins (Sacoman et al., 2017). Interestingly, overexpression of mOGT caused apoptosis in a variety of cell lines (Shin et al., 2011) supporting that tight regulation of OGT and its downstream functions is essential for cellular homeostasis.
Modulation of OGT expression or activity through UDP-GlcNAc concentration, transcription, and post-translational modification not only influences O-GlcNAc addition but also other properties. For example, the OGT C-terminal domain was shown to have an unusual UDP-GlcNAc-dependent protease activity against epigenetic cell cycle regulator HCF-1 (Capotosti et al., 2011). The posttranslational modification of OGT itself is critical for modulating OGT activity in cells, such as neuronal cells. Phosphorylation of serine 3 or 4 by GSK3β is necessary for OGT activity and phosphorylation of Thr144 by AMPK plays a role in OGT substrate selectivity. Interestingly, both GSK3β and AMPK are O-GlcNAc modified by OGT showing a feedback loop in activity modulation (Kreppel et al., 1997; Kaasik et al., 2013; Bullen et al., 2014). Cysteine (S) nitrosylation inhibits OGT activity in resting immune cells and in neuroblastoma cells. S-nitrosylation of OGT is increased by β-amyloid peptide treatment, which results in reduced O-GlcNAcylation and increased tau phosphorylation in human neuroblastoma cells (Ryu and Do, 2011; Ryu et al., 2016)
OGT and its binding partners in the brain
Recent advances in proteomics have accelerated the discovery of protein-interaction networks and OGT has emerged as a common binding partner for proteins implicated in diseases associated with human neurodegeneration. Interactions that have been mapped on the OGT protein are summarized in Figure 3A and described below.
There is evidence that an abnormal cell cycle precedes the other known hallmarks of neurodegenerative diseases and cell cycle aberrations are a known part of AD etiology and will be discussed in more depth in later sections. OGT interacts with proteins required for regulation of gene expression and cell fate decisions including HCF-1, mSin3A, and TETs (Wysocka et al., 2003; Ruan et al., 2012; Deplus et al., 2013). Among these proteins, HCF-1 is a transcriptional co-regulator necessary for cell cycle progression. Mature HCF-1 is O-GlcNAcylated and proteolytically cleaved by OGT. Following its cleavage by OGT, the N and C termini of HCF-1 functions in G1 and M phase of cell cycle, respectively (Capotosti et al., 2011). Moreover, the OGT interaction with HCF-1 regulates gene expression during G1-S progression. In addition, transcriptional regulation through either histone modification or direct modification of DNA appears to be influenced by OGT, which interacts with the histone deacetylase complex through corepressor Sin3A to promote effective, specific gene silencing (Yang et al., 2002). Further, CARM1 histone methyltransferase interacts with OGT and is O-GlcNAc modified for correct CARM1 localization during mitosis (Sakabe and Hart, 2010). Finally, ten-eleven translocation (TET) 5-mC hydroxylases promote DNA demethylation thereby functioning to modulate transcriptional activation and repression. The importance of the OGT and TET interaction is largely unknown, especially in the nervous system where one of the earliest reports about TET mediated hydroxymethylation was discovered (Kriaucionis and Heintz, 2009).
Autosomal dominant cerebellar ataxia is directly linked to an ATTCT pentanucleotide expansion in intron 9 of ATXN10 (SCA10). The ataxin-10 (Atx-10) protein is predicted to have a helical armadillo-like three-dimensional structure like the Huntingtin protein. This motif is thought to regulate protein-protein interactions, including the interaction of Atx-10 with ncOGT in the cytoplasm. In tissues, such as the pancreas, this interaction modulates OGT activity as Atx-10 competes with known ncOGT inhibitor, sOGT. Interestingly, while sOGT expression is high in the pancreas, it is absent in the brain (Andrali et al., 2005; März et al., 2006) suggesting that the Atx-10 interaction with ncOGT in neuronal tissue may alter OGT activity or protein binding in some other way. Indeed, both OGT and Atx-10 are known to modulate cytokinesis through interaction with Aurora-B, which phosphorylates Atx-10. Intriguingly, knockdown of either Atx-10 or OGT causes cytokinesis defects (Slawson et al., 2005; Li et al., 2011). The OGT/Atx-10 protein interaction is promising for understanding these proteins’ role in neuronal maintenance and neurodegeneration. What is learned about this axis may be applied to better understand the role OGT plays in interactions with other proteins containing the aforementioned secondary structure.
Along with the dynamic regulation of the enzyme in neuronal cells and its high level of expression in brain tissue, the OGT gene maps to the locus associated with the Parkinsonian dystonia (DYT3) region in mammals (Németh et al., 1999; Shafi et al., 2000), supporting its foundational importance for ensuring efficacy of neuronal nutrient-sensitive processes. Gene mutations in an important cell cycle related transcription factor, THAP1, have been linked with DYT6 dystonia (Fuchs et al., 2009). THAP1 is an atypical DNA-binding zinc finger protein known to regulate cell proliferation by binding to specific DNA regions and is part of a family of proteins that affect pRB/E2F, proposed to be involved in PD cell death (Houlden et al., 2010). Like OGT, which is highly expressed in the nervous system, THAP1 protein is found in the cerebral cortex, thalamus, substantia nigra and hypothalamus (Zhao et al., 2013). The binding of OGT to THAP1 is critical for proper timing of the G1-S cell cycle progression (Cayrol et al., 2007; Mazars et al., 2010; Zhao et al., 2013) Together, these connections suggest that understanding the interaction between OGT and THAP1 may lead us to better understand dystonias.
OGT, NF-kB, and MIBP1 are evolving as potential theraputic targets for treatment of neuronal pathologies. c-MYC intron binding protein (MIBP1) is highly expressed in postmitotic neurons and its appropriate expression is required for perinatal development of the brain as well as immunological processes. This transcription factor binds to various genomic regions including the first intron of the c-MYC gene and its most prominent binding partner, OGT, is responsible for its O-GlcNAcylation. MIBP1, whose activity is suppressed by O-GlcNAcylation, binds and decreases of the transcription of NF-kB target genes. Loss of the MIBP1 154-amino acid region mapped to bind OGT yields an increase in MIBP1-dependent suppression of NF-kB pathway target genes (Iwashita et al., 2012). Interestingly, NF-kB activation is dependent on O-GlcNAc modification of the c-Rel subunit (Ramakrishnan et al., 2013). Expressed in many cells including neurons in the cortex and hippocampus (O’Neill and Kaltschmidt, 1997), NF-kB is a critical mediator of numerous physiological processes including inflammation and the cell cycle and plays a role in neurological disorders (Shih et al., 2015). OGT, through its interaction with the proteins above, has been shown to be a key regulator of cellular homeostasis via its influence on gene expression. Thus, deregulation of OGT and related mechanisms may be associated with the cascade of events leading to neurodegeneration.
3. OGA: isoforms and localization
OGA was first identified as a hyaluronidase triggering an immune response in a meningioma patient, therefore named as MGEA5 (Meningioma-Expressed Antigen 5), (Heckel et al., 1998). Subsequently, purified OGA was shown to be the enzyme previously described as hexosaminidase C, a glycosidase acting at neutral pH (Braidman et al., 1974). Oga is located on human chromosome 10q24 (mouse Chr 19) (Gao et al., 2001) in a locus associated with AD (Myers et al., 2000) and a predisposition to type II diabetes in the Mexican American subpopulation (Farook et al., 2002; Lehman et al., 2005). While ACEVIEW and other public databases annotate multiple isoforms, only two have been studied extensively and have varied levels of activity in vitro (Kim et al., 2006; Macauley and Vocadlo, 2009). OGA-L (OGA long, 130 kDa) is localized to the cytoplasm. OGA-S (OGA short, 75 kDa) is predominantly found in the nucleus (Comtesse et al., 2001) and associated with lipid droplets (Keembiyehetty et al., 2011) with the short 11- amino acid extension of the linker domain important for targeting to lipid droplets.
Figure Figure 3B depicts a model summarizing the structure of human OGA, which was derived from three X-Ray density maps (Elsen et al., 2017; Li et al., 2017; Roth et al., 2017). The protein has an interlocking dimer interface composed of helices from the opposing monomer. Both OGA isoforms harbor the N-terminal catalytic domain responsible for glycosidase activity, but the C-terminal domains are different. Indeed, while OGA-L was originally published to have C-terminal Histone Acetyl Transferase (HAT) activity in vitro, recent work demonstrates that the domain is a pseudo-HAT as OGA-L does not function this way in vivo (Toleman et al., 2004; Rao et al., 2013). However, the pseudo-HAT domain extends from the interlocking helices and could potentially alter the conformation of the catalytic domains and is critical for binding chromatin. The linker region between catalytic and pseudo-HAT domains of OGA-L is a caspase 3 site cleaved during apoptosis (Butkinaree et al., 2008).
While OGA is thought to interact with at least 90 proteins, (Groves et al., 2017) further studies will be required to understand the consequences of OGA deregulation on individual proteins. Importantly, it has been shown that the two canonic isoforms of OGA are expressed differentially during rat development; OGA-S seems more abundant in the first 19 days and then disappears whereas the OGA-L levels are maintained throughout development (Liu et al., 2012). Further, recent studies knocking out OGA in mice revealed critical roles for OGA in development – including development of the brain (Yang et al., 2012; Keembiyehetty et al., 2015; Olivier-Van Stichelen et al., 2017). Thus, OGA – and by extension O-GlcNAc cycling – plays a critical role in neuronal homeostasis.
C. Genetic approaches targeting O-GlcNAc in neurodegenerative disease
Manipulation of OGT and OGA have been explored in isolated tissues, cultured cells, and model organisms (Summarized in Table 1). To date, the data from different neurodegenerative disease models suggest that elevated O-GlcNAc levels can both increase or attenuate symptoms. These conflicting viewpoints call for a critical review of the literature, examination of experimental conditions, and an appropriate level of weight for findings in animals other than humans. Importantly, proteins associated with neurodegenerative disease including amyloid precursor peptide (APP) (Griffith et al., 1995), tau, and α-synuclein are O-GlcNAc modified (Arnold et al., 1996; Cuervo et al., 2004) with varied consequences. In this section, we focus on results from studies in which OGT and OGA were globally manipulated in whole organisms. These data support that the enzymes of O-GlcNAc cycling and O-GlcNAc itself impact brain development and neuronal plasticity.
1. C. elegans: O-GlcNAc as a regulator of proteostasis and proteotoxicity
OGT-1 and OGA-1 were identified as regulators of proteotoxicity in multiple C. elegans models of neurodegenerative disease (Wang et al., 2012). Phenotypes in transgenic models of tauopathy, amyloid beta-peptide, and polyglutamine expansion were ameliorated in animals lacking OGT-1 while loss of OGA-1 enhanced some phenotypes. These findings appear to contradict some of the findings from rodent studies wherein increases in O-GlcNAc through OGA inhibition appear to improve tau-driven neurodegeneration phenotypes (see section below) (Yuzwa et al., 2008; Yuzwa et al., 2012) . While developmental regulation may explain some differences in findings from mammals to C. elegans, the variation in phenotypes scored and the life stage at which O-GlcNAc was perturbed may also contribute.
Perturbation in O-GlcNAc alters proteostasis in C. elegans (see section on autophagy), a process which is critical for neuronal health. Transcription of proteasome subunit genes is induced in response to proteasome dysfunction caused by pathogen or proteasome inhibitor exposure. This pathway may also be induced by proteotoxic challenge and that response requires SKN-1, a transcription factor related to mammalian Nrf1/2. Intriguingly, Nrf1/2 is negatively regulated by O-GlcNAcylation and is thus an ideal target for pharmacological intervention (Chen et al., 2015).
2. Drosophila: O-GlcNAc and epigenetic regulation of development, cell cycle, and circadian rhythm
The Drosophila ogt(sxc) was discovered as a homeotic gene about 3 decades ago (Ingham, 1984). Subsequent studies underscored its importance as a member of the Polycomb repressor complex (PRC). Polyhomeotic (Ph) is O-GlcNAc modified and in the absence of OGT and, Ph forms large aggregates resulting in a non-functional PRC. Consequently, Hox genes were inappropriately expressed throughout the embryo (Gambetta et al., 2009b; Gambetta and Muller, 2014) resulting in ogt mutant embryos arresting during development.
Epigenetic regulation of gene activation is also partly regulated by O-GlcNAcylation. Trithorax (TRX), Absent Small or Homeotic 1 (ASH1), and SET1 O-GlcNAc modified. In Drosophila lacking OGA, increased O-GlcNAc results in changes in the expression of cell cycle related genes (Akan et al., 2016). Cell cycle dysregulation is an early indicator of neurodegeneration and these findings support O-GlcNAc as critical for appropriate developmental and cell cycle dynamics. In addition, work in Drosophila has revealed a role for O-GlcNAc in regulating circadian rhythm (Kim et al., 2012; Kaasik et al., 2013). Reduction in OGT levels in clock cells shortened circadian rhythm. Drosophila PERIOD (dPER) is O-GlcNAc modified this modification and stabilize and locates dPER in cytoplasm, where it is inactive. Upon reduction in O-GlcNAc levels, dPER could go to nucleus where it functions to repress its own transcription by inhibiting the activity of CLOCK/CYCLE heterodimer (Kim et al., 2012). Based on these results, O-GlcNAcylation seems to regulate proper circadian timing. Such disturbance in daily rhythms involing sleep, activity and hormone release have been linked to human neurodegenerative disease and therefore may represent a possible contributor to neurodegenerative disease progression (Association, 2017).
3. Mouse: neurodegenerative phenotypes associated with perturbation of O-GlcNAc
To decipher the role of O-GlcNAc and the proteins responsible for its cycling, numerous studies have used genetics to alter OGT and OGA expression in mice. Although full body knockout of Ogt and Oga are lethal in mice (Shafi et al., 2000; Keembiyehetty et al., 2015), tissue specific approaches have been fruitful. These studies have allowed researchers to better understand the role of O-GlcNAc cycling in the brain.
Ogt knockout in mouse models
Knockout of Ogt in mouse embryonic stem (ES) cells is lethal requiring knockout of Ogt in selected tissues (Shafi et al., 2000; Hanover et al., 2003; O’Donnell et al., 2004). Using Syn1-Cre driver, O’Donnell et al. triggered Ogt deletion in neuronal cells (O’Donnell et al., 2004). While heterozygote animals exhibited no phenotypes, homozygous knockout of Ogt was rare and animals rarely survived past nursing. Further, those that survived were considerably smaller with reduced motility compared to wild type (WT) littermates. Biochemical analysis demonstrated that tau expression and phosphorylation was increased in animals lacking Ogt in neurons. These findings suggest a possible mechanistic link between O-GlcNAc and neurodegeneration given that tau phosphorylation and O-GlcNAcylation are linked in vivo.
More recently, excitatory neuron-specific knockouts have been generated in mice by multiple groups who took advantage of an αCamKII-Cre driver (Lagerlöf et al., 2016; Wang et al., 2016). These two studies looked at animal development, glucose tolerance, and later physiological phenotypes. Surprisingly, the findings from the studies and interpretations are divergent. Wang, et al. demonstrated that animals without Ogt in excitatory neurons had reduced weight at 7 weeks of age (Wang et al., 2016). In contrast, mice in the related study showed increased weight gain and a significant amount of fat 3 weeks after birth (Lagerlöf et al., 2016). The weight gain in the latter was attributed to satiety defects and could be regulated by food restriction. Major loss of OGT was localized to the paraventricular nucleus (PVN) of the hippocampus and Lagerlof and colleagues concluded that loss of OGT regulates the excitatory synaptic transmission in PVN neurons (Lagerlöf et al., 2016). Conversely, Wang et al. showed significant increases in neuronal cell death by 6 months after Ogt ablation (Wang et al., 2016). This coincided with decreases in brain size, cortical thickness, and neuronal density. Loss of OGT yielded mice that recapitulate neurodegenerative phenotypes with increased cell cycle and immune response, phosphorylation of tau, appearance of tau aggregates, and soluble oligomeric Aβ peptide. Ogt loss also increased anxiety, another characteristic feature of neurodegeneration. While these studies used the same promoter to drive Cre expression, it is important to note that the approaches differed in other details. Lagerlof, et al. used a tamoxifen-inducible system, knocking out the Ogt in αCamKII expressing cells at 6 weeks of age whereas the Wang, et al. knockout of Ogt is initiated earlier (4 weeks) at the onset of αCamKII expression and synaptic maturation (Lagerlöf et al., 2016; Wang et al., 2016). Some common findings emerged. Both studies demonstrated the importance of OGT in excitatory neurons and provide clear evidence for O-GlcNAc cycling involvement in metabolism and neurodegeneration. However, the contrasting findings underscore the importance of taking developmental timing into account when analyzing the results of such knockout models. It is also critically important to examine toxicity and cell viability at every stage in such experiments.
Knockout of OGT in sensory neurons cause axonal defects, leading to thermal and mechanical hyposensitivity (Su and Schwarz, 2017). The phenotype is most likely due to the decrease in axonal endings. Moreover, OGT knockout in AgRP neurons results in a loss in neuronal excitability (Ruan et al., 2014). Possibility of OGT knockout in the nervous system is promised to highlight many essential functions of O-GlcNAc in the cell.
Oga knockout in mouse models
Previously, we generated a full body knockout of Oga (MGEA5) in mice using an oocyte-expressing Cre-line (MMTV-Cre) and demonstrated that Oga KO mice are largely perinatal lethal with few offspring surviving more than 3 weeks (Keembiyehetty et al., 2015). The perinatal lethality was associated with defects in neonatal liver glycogen storage, possibly linked to altered autophagy (Keembiyehetty et al., 2015). A similar perinatal lethality phenotype was observed in non-conditional, genetrap knockout model targeting Oga (Yang et al., 2012). The surviving Oga KO mice we generated were obese, and even heterozygous animals showed evidence of insulin-resistance. In addition to gross structural and developmental defects, Oga KO animals exhibit metabolic deregulation including impaired insulin and glucose homeostasis. A key feature of the Oga heterozygous mice included insulin resistance when challenged with a high fat diet. Due to their short lifespan and poor survival at birth, full body Oga KO animals are difficult to study and tissue-specific knockout models were used for further interrogation.
Using a highly neuron specific Nestin-Cre promoter to knockout Oga in the brain (Olivier-Van Stichelen et al., 2017), we found that Oga brain KO animals show significant increases in brain O-GlcNAc levels but little change in other tissues such as liver, heart, and muscle. Intriguingly, the levels of brain OGT is also diminished suggesting a feedback-regulation of OGT in response to elevated O-GlcNAc levels. The mechanism of this reduction in OGT levels is not known but may be due to changes in chromatin structure or splicing since the decreases occurred principally to the ncOGT isoform. The brain Oga KO animals exhibit several anatomical, behavioral, and metabolic phenotypes. Compared to WT, brain Oga KO animals were smaller, obese, and have a visibly flattened forehead. Closer neuroanatomic analysis revealed enlarged ventricles (lateral ventricles, 3rd and 4th), smaller forebrain structures, cortical layering, and a very small olfactory bulb. Examining neurogenesis, we found that brain Oga KO animals showed a dramatic developmental delay associated with persistent neurogenesis long after neurogenesis has ended in WT animals. The Oga brain knockout animals also exhibited striking metabolic imbalances including increased body fat, circulating leptin, triglycerides, and insulin as well as decreased insulin-like growth factor 1 (IGF-1) levels. Consistent with lower levels of GH and IGF-1, the brain Oga KO animals have a pituitary that is much smaller than in WT animals. These findings reinforce that O-GlcNAc cycling is important for maintaining the hypothalamus-pituitary axis and, thus, the maintenance of metabolic homeostasis (Olivier-Van Stichelen et al., 2017). Despite the physiological changes cataloged above, loss of Oga in the brain did not increase neurodegenerative damage or physiological apoptosis (Olivier-Van Stichelen et al., 2017).
Pharmacological findings have suggested that inhibitors of OGA may have therapeutic potential by reducing protein aggregation (Yuzwa et al., 2012; Yuzwa et al., 2014; Hastings et al., 2017). As Oga brain KO animals show reduced levels of OGT in the brain, it is unclear whether interference with O-GlcNAc cycling may provide neuroprotective effects by reducing OGA levels, by lowering OGT levels, or altering protein O-GlcNAcylation on specific substrates. While the brain specific Oga KO has provided essential information about the role of O-GlcNAc in neuronal development and function, neuron-specific Oga KO in a neurodegenerative disease mouse model will be required to examine the potential neuroprotective effects of interfering with O-GlcNAc cycling.
4. O-GlcNAc in neurodevelopment: impact of the intrauterine environment
Human neurodevelopment can be influenced by environmental perturbations in the intrauterine environment. Prenatal development represents a vulnerable period where the fetal and maternal compartments exchange nutrients and neuroendocrine signals. The mammalian placenta plays a critical role in this exchange and environmental factors including nutrients can influence developmental programming. Based on evolutionary and transcriptional evidence, we proposed a role for the enzymes of O-GlcNAc cycling as epigenetic regulators in the intrauterine environment (Love et al., 2010; Hanover et al., 2012). Subsequent work demonstrated that the enzymes of O-GlcNAc cycling are likely to play a crucial role in developmental programming (Howerton et al., 2013; Howerton and Bale, 2014; Nugent and Bale, 2015; Bale, 2016; Olivier-Van Stichelen et al., 2017; Pantaleon et al., 2017). As described above, disruption of OGT and OGA in the brain profoundly impacts brain development and function. Further, mutations in human OGT are associated with X-linked intellectual disability (XLID) providing direct evidence for an important role for this enzyme in neurodevelopment (Bouazzi et al., 2015; Niranjan et al., 2015; Vaidyanathan et al., 2017).
Several studies suggest that maternal stress increases the risk of neurodevelopmental diseases including autism, stuttering, dyslexia, and attention deficit/hyperactivity disorder and that males are more significantly affected than females (reviewed in (Bale, 2016)). These sex differences could be due to hormonal and metabolic influences, sex chromosomes, or a combination of these two factors. By examining the expression of X-linked genes differentially expressed in the placenta itself, OGT has emerged as a major biomarker of placental stress (Howerton et al., 2013; Howerton and Bale, 2014). Intriguingly, the increases observed are far more pronounced in the male offspring-specific placental stress response.
The presence of OGT on the mammalian X-chromosome near the Xist locus (lnRNA triggering X-inactivation) has important implications for OGT expression and subsequent functioning in both males and females (reviewed in (Love et al., 2010; Abramowitz et al., 2014; Olivier-Van Stichelen and Hanover, 2014; Olivier-Van Stichelen et al., 2014a)). OGT is one of the genes on the human X-chromosome with a low ratio of non-synonymous to synonymous substitution rates suggesting that it may represent a clinically significant gene that is subject to negative selection (Ge et al., 2015). In addition, OGT escapes X inactivation in the placenta resulting in transcript levels that are nearly 2-fold higher in female compared to male placenta (Howerton et al., 2013; Howerton and Bale, 2014). Knockout of OGT in the placenta to produce hemizygous and homozygous deletion of OGT in trophoblasts produced offspring with altered hypothalamus-pituitary-adrenal axis function. These changes largely recapitulate the early placental stress phenotype (Howerton and Bale, 2014). Thus, OGT is likely to play an important role in mediating the response to stress in the intrauterine environment, thereby impacting fetus development.
D. O-GlcNAc: A central role in cellular regulation of neuronal health and disease
There are several lines of evidence supporting that appropriate maintenance of O-GlcNAc at both global levels and on individual proteins is critical for mitigating neurodegenerative disease. To understand how changes in O-GlcNAc may impact brain function, researchers have focused on individual proteins associated with neurodegenerative disease noting that disease-associated proteins are O-GlcNAc modified (Griffith et al., 1995). Individual proteins’ roles are impacted by their O-GlcNAc status and thereby influence processes ranging from proteostasis and autophagy to insulin signaling and the cell cycle.
Insulin Signaling
Insulin signaling in the brain is considered a key player in brain homeostasis. The molecular response of the brain to insulin is analogous to the response in other organs. Insulin receptors (IR) are found throughout the brain (Marks et al., 1990) with internal or peripheral secretion of insulin triggering insulin signaling through PI3K activation and transcriptional activity. Both type 1 and 2 diabetic patients have increased risk of neurodegeneration (Kern et al., 2001) and altered expression of diabetes-related genes have been linked to AD pathology (Hokama et al., 2014). With insulin response impaired in AD brains, AD has been categorized as ‘type 3 diabetes’ without hyperglycemia. Moreover, insulin resistance in peripheral tissue reduces insulin uptake, increases amyloid beta levels (Baker et al., 2011), and accelerates the formation of neurite plaques (Matsuzaki et al., 2010).
While other tissues use the insulin pathway mainly for glucose metabolism, brain insulin signaling influences pathology by triggering multiple functions including glucose metabolism, reproduction, proliferation, differentiation, neuroprotection, cognition, and memory. Insulin in the brain does not induce a significant uptake of glucose likely due to low levels of the insulin responsive glucose transporter GLUT4. Nevertheless, brain insulin signaling participates in peripheral glucose homeostasis. Insulin signaling modulates glucose production by the liver (Obici et al., 2002), influences satiety (Kim et al., 2000), and regulates hormone secretion (Tanaka et al., 2000). Multiple studies have demonstrated the importance of insulin signaling for brain development, growth, neuronal outgrowth and maturation, and axon regeneration (Schubert et al., 2003; Toth et al., 2006; Parathath et al., 2008). Similarly, proliferation and differentiation of multipotent neural stem cells are regulated by insulin (Wozniak et al., 1993)
In addition to promoting proliferation, insulin signaling seems to protect neural cells against apoptosis though the PI3K/Akt/mTOR pathway (Ryu et al., 1999) and prevents beta-amyloid induced cell death though an undefined mechanism (Rensink et al., 2004). Cognitive function may also be influenced by insulin signaling in the brain as increasing insulin signaling in the hippocampus improved memory and learning (Zhao et al., 1999). Furthermore, it has been shown that insulin administration eliminates the loss of memory due to ischemic lesions (Voll et al., 1989).
Nutrient-driven O-GlcNAc may be an important regulator of brain insulin signaling. First, secretion of insulin is epigenetically regulated by O-GlcNAcylation in pancreatic beta cells (Durning et al., 2016). Major components of the insulin signaling pathway are O-GlcNAc modified including: the insulin receptor (beta-chain), IRS (Insulin Receptor Substrate), PI3K (Phosphatidylinositol 3 kinase), PDK1 (Phosphoinositide-Dependent Kinase 1), and Akt/PKB (Ball et al., 2006; Gandy et al., 2006; Klein et al., 2009; Whelan et al., 2010). It has been proposed that phosphorylation of IRS1, PI3K, and Akt/PKB activates the insulin signaling pathway while O-GlcNAcylation of the same proteins prevents pathway activation (Holman and Kasuga, 1997). In this model, O-GlcNAc competition with phosphorylation is accentuated by OGT recruitment to the plasma membrane through PPO domain (PIP-binding activity of OGT) (Yang et al., 2008b; Perez-Cervera et al., 2013). However, this proposed mechanism remains controversial as both supporting (Vosseller et al., 2002; Arias et al., 2004; Gandy et al., 2006; Yang et al., 2008b; Klein et al., 2009) and refuting data have been published (Macauley et al., 2008; Macauley et al., 2010). While O-GlcNAc influences insulin signaling, a direct connection with insulin resistance in neurodegenerative diseases has yet to be defined.
Co-translational O-GlcNAcylation and quality control
Quality control of newly synthesized proteins is an essential feature in cell homeostasis. Failure to detect and dispose of misfolded proteins results in proteotoxity and often correlates with disease. One of the key steps in cytosolic quality control is the co-translational ubiquitination of proteins targeted for proteasomal degradation (Bengtson and Joazeiro, 2010). For example, the generation of non-stop mRNA is a phenomenon common across all organisms and a major source of proteotoxicity. Non-stop mRNA encodes aberrant, non-stop proteins sequestrated in the ribosomal complex that cannot be recycled or corrected by a quality control chaperone (Doma and Parker, 2007). Interestingly, O-GlcNAcylation also occurs co-translationally (Starr and Hanover, 1990) and acts with ubiquitination to regulate the stability of the nascent proteins (Zhu et al., 2015b). Previous work supports that O-GlcNAc is a key player in protein stability (e.g., Sp1, beta-catenin) (Han and Kudlow, 1997; Olivier-Van Stichelen et al., 2014b) resulting from direct competition with phosphorylation or through O-GlcNAc modification of the proteasome itself (Sümegi et al., 2003; Zhang et al., 2003). O-GlcNAc addition was initially shown to occur on nascent chains and post-translationally by in vitro translation of the O-GlcNAc target nuclear pore protein p62 (Nup62) (Starr and Hanover, 1990). More recently Zhu et al. demonstrated that co-translational O-GlcNAcylation of Sp1 and Nup62 prevents the nascent proteins from precocious proteasomal degradation (Zhu et al., 2015b). O-GlcNAc addition and removal may then be regarded as an ‘O-GlcNAc timer’ serving a protective role at the time of synthesis on the ribosome and regulating the rate of degradation of nuclear and cytoplasmic proteins. This is similar to the so called ‘Mannose timer’ in the endoplasmic where the action of mannosidase I can trigger proteasomal degradation. It would appear that O-GlcNAc may play an analogous role in the nucleus and cytosol. We therefore suggest that an ‘O-GlcNAc’ timer’ may act in a similar manner to regulate protein stability. If O-GlcNAc addition and removal are both timed events in the life-cycle of the protein, then independent regulation of these activities could alter the stability of the target protein. When altered, O-GlcNAcylation may also significantly contribute to neurodegenerative pathologies.
Cell cycle regulation
Although neurons in the CNS are often viewed as post-mitotic and stable in number, a large body of recent evidence suggests that adult neurogenesis depends upon continued cell division of stem cell precursors. The major sites of adult neurogenesis in humans are two “neurogenic” brain regions. These sites are the subgranular zone (SGZ) in the dentate gyrus of the hippocampus and the subventricular zone (SVZ) of the lateral ventricles where new neurons are generated before migrating through the rostral migratory stream (RMS) to the olfactory bulb to become interneurons (Gage, 2000). The relationship between adult neurogenesis and neurodegenerative disease is complex and has been reviewed elsewhere (Winner et al., 2011; Winner and Winkler, 2015).
The cell cycle is essential for every organism to emerge, develop and survive. Divided in 4 major phases (G0, S, G1 and M), cell cycle progression is regulated by expression and action of cyclins (A, B, D, E) and their partners, the cyclin-dependent kinases (CDKs). In neurodegenerative diseases, there is growing evidence that abnormal cell cycle re-entry precedes other hallmarks of the disease and implicates an aberrant cell cycle in disease progression. Data suggest that at the onset of AD, neurons increase cell cycle re-entry but fail to complete the mitotic phase and thus die (Bonda et al., 2010). AD neurons exhibit elevated cell cycle markers (cyclin D, cdk4, Ki67, cyclin E/cdk4) (McShea et al., 1997; Nagy et al., 1997; Vincent et al., 1997). Indeed, no sign of a complete mitotic phase is found in AD neurons (Ogawa et al., 2003a; Ogawa et al., 2003b).
Inhibition of OGT and OGA prevents cell cycle progression indicating that O-GlcNAc levels are regulated to ensure proper cell cycle progression (Slawson et al., 2005; Dehennaut et al., 2007; Wang et al., 2010b; Drougat et al., 2012). Moreover, OGT and OGA are associated with the midbody and participate in cytokinesis (Slawson et al., 2008). In response to a mitogenic signal, protein O-GlcNAcylation increases as does localization of O-GlcNAc modified proteins to the nucleus (Kearse and Hart, 1991a). Functional studies of O-GlcNAc modified proteins such as p53 (Yang et al., 2006), p27 (Qiu et al., 2017), cyclin D1 (Olivier-Van Stichelen et al., 2012), CDK5 (Ning et al., 2017), c-myc (Chou et al., 1995), beta-catenin (Olivier-Van Stichelen et al., 2014b), NF-kB (Yang et al., 2008a; Kawauchi et al., 2009; Ramakrishnan et al., 2013), FoxM1 (Caldwell et al., 2010), HCF-1 (Capotosti et al., 2011), and Erk1/2 (Jiang et al., 2016) have been critical for furthering the understanding of O-GlcNAc in cell cycle progression. Key AD players are known to be O-GlcNAc modified including APP and tau, which have also been implicated in cell cycle regulation. Both APP and Aβ are mitogenic in vitro (Schubert et al., 1989; Milward et al., 1992). Interestingly, if APP is cleaved by α-secretase facilitated by O-GlcNAcylation, it becomes non-amyloidogenic and neuroprotective sAPPα (Jacobsen and Iverfeldt, 2011). Hyperphosphorylated tau is present in normal mitotic cells and has been linked to CDK activity (Brion et al., 1994; Preuss and Mandelkow, 1998). That O-GlcNAc cycling has been implicated as a key regulator of cell cycle progression and modifies proteins associated with neurodegeneration supports that O-GlcNAc might be involved in the aberrant cell cycles found in neurodegenerative diseases.
Autophagy and mitophagy
Recycling of cellular contents and elimination of damaged organelles/protein aggregates is required to ensure cellular homeostasis. Autophagy, the dynamic lysosomal degradation process, works in concert with endocytic mechanisms to process these proteins and organelles. Autophagy deregulation threatens neuronal health, especially as the brain ages. Mutations in autophagy pathway components are associated with neurodegenerative diseases as cataloged by Nixon (Nixon, 2013). Defects in autophagy induction, cargo recognition and sequestration, and efficient digestion of sequestered substrates yield accumulation of toxic proteins. Indeed, neurons accumulate waste over a lifetime and, while young neurons are effective and efficient at clearing waste, neurons that are compromised in autophagy accumulate protein aggregates and ultimately degenerate.
In multiple neurodegenerative diseases, there is increased autophagy induction (AD and ALS (Boland et al., 2008)), lysosome biogenesis (Cataldo et al., 1994; Cataldo et al., 1995; Cataldo et al., 1996; Ginsberg et al., 2010), and increased transcription of autophagy-related genes (Lipinski et al., 2010). Accumulation of autophagic structures are often noticeable in affected brain tissue (Kegel et al., 2000; Nixon et al., 2005; Boland et al., 2008). In Huntington’s disease, autophagosomes are unusually devoid of cargoes (Nixon, 2013). Mutation of α-synuclein in PD yields aggregates than cannot be degraded by chaperone-mediated autophagy (Cuervo et al., 2004). Recent evidence supports that the O-GlcNAc modification of calpain, responsible for α-synuclein cleavage, inhibits its action (Levine et al., 2017), thereby influencing processing of this key neurodegenerative protein. And, PINK1 and parkin mutations result in mitochondria that cannot undergo mitophagic disposal (Kitada et al., 1998; Valente et al., 2004; Park and Lee, 2006). Neurons in AD show strong phenotypic overlap with lysosomal storage disorders, but these findings have yet to be fully understood (Querfurth and LaFerla, 2010; Platt et al., 2012)
Given that neurons are particularly susceptible to dysfunction in the cellular waste stream, many researchers have endeavored to examine the regulators of autophagy and apoptosis. The dynamic O-GlcNAc is poised to provide crosstalk between autophagy and apoptosis by acting as rheostat to fine-tune these processes by modulating protein folding, localization, stability, etc (Fahie and Zachara, 2016). Loss of OGT and OGA lead to elevated induction of starvation-induced autophagy in the model organism C. elegans (Wang et al., 2012). This study also suggested that O-GlcNAc cycling plays a key role in regulating autophagy flux and proteasome activity when faced with a proteotoxic challenges such as pathological tau accumulation. Several studies in C. elegans noted that OGT is associated with phagosomes and protein O-GlcNAcylation influences autophagosome and lysosome fusion (Wang et al., 2012; Guo et al., 2014).
In vitro work in Neuro2A cells suggests that modulation of O-GlcNAc cycling affects autolysosome formation. Furthermore, the same study demonstrated that in cells and Drosophila, changes in O-GlcNAcylation modulate huntingtin toxicity (Kumar et al., 2014). Modulation of Atg proteins by O-GlcNAcylation is likely to alter autophagy (Jo et al., 2016; Wani et al., 2016b). It is not surprising that O-GlcNAc plays a role in autophagy as key autophagy regulators including Beclin1 (involved in autophagy initiation), SNAP-29 (SNARE complex member), (Guo et al., 2014) BCL2 (Marsh et al., 2013) and Atg7 (Park et al., 2015) are modified by O-GlcNAc. The interaction axis between SNAP-29, Syntaxin-17, and VAMP8 is thought to be controlled by OGT expression in HeLa cells. The authors argue that O-GlcNAcylation of SNAP-29 in both mammalian cells and C. elegans abrogates the complex’s formation thereby decreasing fusion between autophagosomes and endosomes/lysosomes (Guo et al., 2014). Further, they suggest this is intimately involved with nutrient status as starvation influences O-GlcNAcylation of SNAP-29. In Drosophila, a decrease in OGT is associated with an increase of Atg proteins and autolysosomes, and it has been shown that autophagy is regulated through Akt/dFOXO signaling (Park et al., 2015). Defective autophagy induction and processing are associated with accelerated pathology and increased neurotoxicity. Thus, we suggest that O-GlcNAc is poised to influence processing of autophagosome-targeted proteins, and thus neurodegeneration, given its key roles in autophagy.
Apoptosis
In rodent models of neurodegeneration, apoptotic cell death has been convincingly linked to neurodegeneration; although in humans, this connection is less clear (Radi et al., 2014). The regulation of apoptosis is tightly linked to cellular growth signals and, in the central nervous system where many post-mitotic cells must exist for decades, these control mechanisms are extraordinarily important (Frade and Ovejero-Benito, 2015). O-GlcNAcylation has long been known to influence apoptosis by modification of proteins linked to regulation of apoptotic cell death (Omary et al., 1998; Liu et al., 2004b; O’Donnell et al., 2004; Lazarus et al., 2009; Shin et al., 2011). More recently, the mechanisms by which this may occur on targets such as p53 have been examined (Yang et al., 2006; Kawauchi et al., 2009). There are numerous reports of O-GlcNAcylation altering mitochondrial function linked to apoptosis(Shin et al., 2011; Palaniappan et al., 2013; Bond and Hanover, 2015; Zhu et al., 2015a; Wani et al., 2016a; van der Harg et al., 2017). A link between apoptosis and neurodegenerative disease has been made in some studies (Liu et al., 2004b; Zhu et al., 2015a; Chen et al., 2017; Ning et al., 2017). Knockout of Ogt in excitatory neurons led to neurodegeneration with some indications that apoptosis-related genes might be altered (Wang et al., 2016), whereas another study showed no effect on apoptosis or neuron viability (Lagerlöf et al., 2016). Thus, the impact of OGT loss on apoptotic neuronal cell death is unclear. Deletion of Oga in the brain did not significantly increase the number of cortical neurons undergoing apoptosis (Olivier-Van Stichelen et al., 2017). Taken together, these findings suggest that O-GlcNAc may play limited role in neuronal cell death during neurodegeneration and that the modification may serve a protective role.
Mitochondrial movement & metabolism
Mitochondria, the cell’s primary source of ATP, is essential to sensing nutrient status as it utilizes glucose as its major carbon source. Neurons’ complex morphology results in a spatially heterogeneous glucose intake and, consequently, mitochondria need to rapidly re-localize to ensure rapid production of ATP (Detmer and Chan, 2007). Rapid ATP production is crucial for processes including synaptic assembly, transmission, and Ca2+ buffering with motor proteins including kinesin and dynein regulating mitochondrial motility (Schwarz, 2013). Deregulation of mitochondrial trafficking is associated with neurological disorders including AD, ALS, and PD. For example, the GTPase Miro (also called RhoT1/2) interacts with the adaptor protein Milton (also called TRAK1/2 and OIP106/98) thus coupling motor protein complexes to the mitochondria (Glater et al., 2006; Macaskill et al., 2009; van Spronsen et al., 2013). OGT interacts with Milton in both Drosophila and mammals (Iyer et al., 2003; Glater et al., 2006; Brickley et al., 2011). In response to an increase in glucose flux, O-GlcNAcylation of Milton1 (OIP106) decreases mitochondrial motility (Pekkurnaz et al., 2014). Furthermore, knockout of OGT or blockage of UDP-GlcNAc synthesis in both Drosophila and mouse models resulted in 60% of axonal mitochondria to remain stationary (Pekkurnaz et al., 2014). Finally, the Milton-like protein OIP106 forms a complex with OGT and RNA Pol II suggesting that OGT is also targeted to transcriptional machinery thereby potentially regulating downstream targets by O-GlcNAcylation. While Milton O-GlcNAcylation seems to be the main driver of mitochondrial trafficking, tubulin, kinesin, and dynein are also substrates of OGT and their O-GlcNAcylation may also contribute to mitochondrial dynamics (Ji et al., 2011; Ruan et al., 2012; Trinidad et al., 2012). Thus, impaired mitochondrial motility likely impacts the development of neurological disorders and OGT/ O-GlcNAcylation may play a critical role linking nutrient status to mitochondrial motility (MacAskill and Kittler, 2010).
Beyond proteins directly involved in mitochondrial motility, Hu et al. demonstrated that mitochondrial proteins including electron transport chain (ETC) complex members such as NDUFA9 and COX1 are O-GlcNAc modified (Hu et al., 2009). The activity of the ETC complexes was impaired by high glucose treatment mimicking diabetic conditions, with reduced ATP production. The authors interpreted this to suggest that protein O-GlcNAcylation of ETC complex members directly impacts mitochondrial function. Proteins including the TCA cycle enzymes (such as ACO2, IDH3A/3B, OGDH, etc.), redox enzymes (SOD2, PRDX3), and fatty acid oxidation enzymes are also modified by O-GlcNAc (Ma et al., 2016). Surprisingly, the diabetic conditions did not lead to an increase in protein O-GlcNAcylation across the board. In fact, 40% of the detected O-GlcNAc modified sites displayed a 20% reduction in O-GlcNAc levels under high glucose treatment (Ma et al., 2016).
Appropriate levels of protein O-GlcNAcylation are essential for mitochondrial function. Cividini et al. reported that mitochondrial 8-oxoguanine DNA glycosylase (Ogg1) activity is negatively regulated by O-GlcNAc (Cividini et al., 2016). Diabetic mice displayed increased Ogg1 O-GlcNAcylation in cardiac myopathy, leading to decreased Ogg1 activity and that could result in mDNA damage. In addition, O-GlcNAcylation of the voltage dependent anion channel (VDAC) is protective of mitochondrial function preventing the formation of calcium induced mitochondrial permeability transition pore (mPTP) (Ngoh et al., 2008). Upon formation, mPTP disrupts the mitochondrial membrane potential thereby preventing ETC mediated ATP production, triggering events in the apoptosis cascade by releasing pro-apoptotic proteins (Ngoh et al., 2008).
Combined, these studies support that the O-GlcNAc modification must be examined at the single protein – or even single site – level and that the regulation of mitochondrial function by O-GlcNAc is nuanced. This is particularly important for nervous system, and the brain, since most neurons are post mitotic and perturbed mitochondrial function could cause irreversible damage to the neurons. The ubiquity of O-GlcNAc in brain tissue and its dynamic nature on mitochondrial proteins support its role in mitochondrial motility as well as synaptic transmission.
E. Prospects for Imaging and Diagnostics
Modern brain imaging modalities exploit glucose uptake and differential blood flow as surrogates for neuronal activity allowing the monitoring of brain activity and probing neuropathology. While the brain is highly dependent on glucose under normal conditions, neurodegenerative diseases greatly change the brain’s metabolism of glucose. Therefore, glucose metabolism has been a surrogate for monitoring neuronal activity with hypo- and hyper-metabolism diagnostic for diseases (Zhu et al., 2014), including AD and PD (Mergenthaler et al., 2013). Indeed, loss of neurons can result in decreased localized glucose consumption but increased regional consumption elsewhere.
1. O-GlcNAc levels as a surrogate for neurodegenerative damage
Tantalizing evidence suggests that altered brain O-GlcNAc levels correlate with changes in neuronal cell death, neuroinflammation, production of hyperphosphorylated tau, amyloidogenic Aβ peptides, and memory deficits (Wang et al., 2016). A number of studies demonstrate that O-GlcNAcylation is decreased in AD brain tissue (Liu et al., 2004b) compared to control and OGT protein expression is reduced in AD human cortical brain tissue (Liu et al., 2009). The authors suggest that hypometabolism of glucose in AD coupled with the decreased nutrient-sensor O-GlcNAc correlates directly with neurodegeneration in these tissues. However, other work suggests that levels of O-GlcNAc increase in AD brain tissue (Förster et al., 2014). Experimental differences including choice of brain region likely influences the conclusions about global O-GlcNAc levels. Indeed, genetic and pharmacological manipulation in different organisms occasionally yields conflicting results and it is essential to continue profiling how O-GlcNAc levels are altered in neurodegenerative human tissue. There is great promise in being able to visualize O-GlcNAc, OGT, or OGA in patients in real time to give an indication of disease progression. Below, we discuss the use various imaging modalities to monitor glucose consumption and downstream nutrient-sensitive OGA in human patients in real time.
2. Can O-GlcNAc-based imaging improve diagnostics and patient outcomes?
With clinically overlapping features, neurodegenerative diseases including PD, multiple system atrophy, progressive supranuclear palsy, corticobasal degeneration, dementia with Lewy Bodies, AD, and frontotemporal dementia are difficult to differentiate at early stages. To improve patient care, the following strategies are employed: techniques that can assist in early diagnosis, define disease stage, stratify patients, and determine whether chosen therapies are successful. Thus, prediction of which patients will develop neurodegenerative diseases by use of non-invasive imaging techniques is of great interest (Weiner and Khachaturian, 2005) .
Methods including magnetic resonance imaging (MRI) and positron emission tomography (PET) have long been employed by physicians to monitor the structure and function of the brain, respectively. Briefly, MRI is a non-invasive method providing pictures of anatomy. PET imaging takes advantage of bio-available compounds tagged with radionuclides that are metabolized or bind to receptors/sites of drug action. These so-called radiotracers can be used to track a given compound’s biological utilization. Of note, PET scans are increasingly used in combination with imaging that provides anatomic information so that anatomy and function can be interpreted simultaneously.
Based on nuclear magnetic resonance, MRI utilizes magnetic fields, radio waves, and field gradients to generate contrast images as different tissues and fluids provide natural contrast. Topographic distribution of brain tissue can be characterized by neuroimaging and brain atrophy is a clear indicator of AD with loss of brain volume correlating with cognitive deterioration (Fox et al., 1999). While structural imaging can give an indication of pathological features prior to evident clinical symptoms, it is questionable whether this technique can be used early enough to improve outcomes (Johnson et al., 2012). Current optimal uses for MRI include patient stratification and measuring outcomes after treatment. As MRI cannot be used to assess function, functional MRI (fMRI) has become more common. fMRI monitors how the brain responds to an external stimulus or pharmacological agent by measurement of neuronal activity inferred from changes in signal and is often used to evaluate functional connectivity within brain networks (Johnson et al., 2012). This technique has great potential to assess treatment strategies in real-time. However, longitudinal fMRI studies in neurodegenerative patients is challenging and must be further explored to understand pitfalls and its potential impact.
Researchers have long used PET to monitor drug uptake, elimination, and bio-distribution in a living animal. Glucose hypo-metabolism is found in brain regions prone to neurodegeneration, thus profiling glucose utilization and associated molecules of interest to assess disease state and treatment efficacy is increasing in popularity. Several radiotracers have been developed to monitor brain function and the chemical arsenal of PET ligands includes compounds labeled with isotopes such as fluorine-18 (half-life of ~110 min) and carbon-11 (~20 min). For example, the biologically active glucose analog fluorodeoxyglucose (FDG) can be labeled with tracer fluorine-18 (FDG-PET) and used to measure glucose consumption. In patients with AD, FDG-PET measurements correlate with continued decline in cognitive function (Weiner and Khachaturian, 2005) and co-registered MRI and FDG-PET images have equivalent classification values (Santi et al., 2001) . Further, FDG-PET has shown efficacy in distinguishing frontotemporal dementia from AD (Weiner and Khachaturian, 2005) thereby demonstrating its utility beyond other diagnostic tools.
While monitoring FDG retention has proven useful, it is a non-specific indicator of metabolism and can indicate variation in glucose use for a number of reasons beyond neurodegeneration (i.e., ischemia and inflammation). Thus, PET ligands were developed to determine amyloid plaque formation. However, Aβ levels rise and fall depending on the neurodegenerative stage and the extent of plaques does not correlate with defined neurodegenerative stages (McLean et al., 1999) suggesting other targets must be tried. More recently, ligands for monitoring neurofibrillary tangles and neuritic plaques have also been in development and as tau deposition is more strongly linked with cognition in AD may prove more useful in the long-term (Shoghi-Jadid et al., 2002). It is hoped that with increasingly specific ligands and improved sensitivities, PET will increase accuracy of differential diagnosis of neurodegeneration at early clinical stages.
Given that O-GlcNAc appears critical for protein stability and, in some studies, increased O-GlcNAc has been shown to decrease tau aggregation; modulating global brain O-GlcNAc levels has become an attractive therapeutic target. To note, while compounds specifically targeting OGT would be of great interest, given this enzyme’s importance in global nutrient-sensing, targeting OGA has been a more achievable goal. Further, targeting OGT has been challenging because developed analogs often also target and inhibit other transferases, are polar thereby limiting cellular availability, and have been associated with cellular toxicity (Trapannone and Rafie…, 2016). Recently published crystal structures of OGT will give improved insight toward developing ligands (Ma et al., 2013; Janetzko and Walker, 2014). OGA inhibitors and complementary PET ligands to monitor inhibitor efficacy and outcomes have been developed with some rapidity and their use will be explored in the following paragraphs.
Therapeutic OGA inhibitors have been seen in model organism work as well as pre-clinical studies. Use of OGA PET ligands in clinical studies will give an indication of bioavailability (including passage of the blood brain barrier), retention, and potentially the efficacy of therapeutic agents. Companies including Merck and Alectos are pursuing at least one compound related to OGA imaging (Sean M. Smith1, 2016). The chemistries to generate these compounds must be robust, straightforward, and require minimal purification as they must be generated in a nearby cyclotron close to the imaging facility and used while active. The nature of many of the inhibitors heretofore generated gives options for potential clinical imaging modalities, as they are amenable to medicinal chemistry. The production of OGA inhibitors, with varying specificity, has spanned at least two decades (Macauley et al., 2010; Kim et al., 2013). With the publication of improved crystallography data, informed decisions about inhibitor/probe synthesis will only improve options. Thiazoline derivatives have proved of particular interest given that they are orally available, cross the blood brain barrier, and show decreased tau oligomerization in one mouse model (Yuzwa et al., 2012). Whether the data from mouse models will be recapitulated in human patients and ultimately quality of life is improved remains to be seen, but PET ligands targeting OGA, OGT, or O-GlcNAc have the potential to help us to understand treatment regimens.
3. O-GlcNAcase inhibitors targeting neurodegeneration
With accumulating evidence suggesting that the regulation of O-GlcNAcylation is tightly linked to neurodegenerative diseases, pharmaceutical inhibitors of O-GlcNAc cycling have emerged as a potential therapeutic solution to neurodegeneration.
Recent studies on Tau
Human tau is highly phosphorylated with nearly 85 phosphorylation sites and reported to have at least 12 sites of O-GlcNAcylation (Arnold et al., 1996; Liu et al., 2004b). Tau phosphorylation is 2–3 fold higher and O-GlcNAc levels are reduced in AD compared to healthy samples (Alonso et al., 1996; Liu et al., 2004b), as might be expected given the reciprocity between these PTMs (Alonso et al., 1996; Lazarus et al., 2009; Yuzwa et al., 2012). These changes are associated with a self-aggregating, toxic molecule that is detrimental to neuronal microtubule function and neuronal death (Alonso et al., 1996). Furthermore, the conditional mouse brain Ogt KO showed increased tau phosphorylation (O’Donnell et al., 2004) suggesting that these PTMs play key roles in maintaining appropriate levels of soluble protein.
It is interesting to note that regulation of tau modifications is intimately linked to OGT: the kinases GSK3B and CDK5 are responsible for tau phosphorylation and are known OGT substrates (Lubas and Hanover, 2000). GSK3B and CDK5 are part of a complex, in which phosphorylation of tau by CDK5 can prime tau phosphorylation by GSK3B (Mietelska-Porowska et al., 2014). GSK3B can then phosphorylate about 30 sites on tau and its activity is increased in AD. Furthermore, OGT activity is increased by GSK3B mediated phosphorylation under physiological conditions. OGT could then O-GlcNAc modify and increase GSK3B activity, thereby generating a feedback loop by which tau protein solubility is modulated (Yu et al., 2012). While the biological role of O-GlcNAcylation on tau function is still unclear, it has been suggested that O-GlcNAc is necessary to prevent tau from aggregation in mice and in vitro (Yuzwa et al., 2012; Yuzwa and Vocadlo, 2017). How this intricate relationship between OGT, GSK3B and tau plays a role in tauopathies remains to be determined.
With studies supporting that neurodegenerative diseases display lower O-GlcNAcylation in the brain, attempts have been made to increase O-GlcNAc levels in mouse models of neurodegeneration. These studies seek possible use of OGA inhibitors for future therapeutics. In one promising study, researchers have treated a neurodegenerative mouse model with the OGA inhibitor Thiamet-G and found that treatment limits tau aggregation (Yuzwa et al., 2012). In a separate study, Thiamet-G treatment by injection into lateral ventricle in the brain led to a decrease in tau phosphorylation at residues Thr181, Thr212, Ser214, Ser262/Ser356, Ser404 and Ser409, and an increase in tau phosphorylation at Ser199, Ser202, Ser396 and Ser422 in the mouse brain (Yu et al., 2012). In addition, Thiamet-G mediated increase in O-GlcNAcylation also upregulated the GSK3B activity. Since GSK3B is known to phosphorylate tau, its activation by O-GlcNAcylation may explain the increased tau phosphorylation on certain sites (Yu et al., 2012). Long term treatment with OGA inhibitor led to an increased O-GlcNAc and reduced pathologic tau levels (Yu et al., 2012). Another study using the same OGA inhibitor treatment in a tau mutant mouse model (rTg4510) reduced pathological tau phosphorylation levels (Graham et al., 2014). OGA inhibitor was also used in a mouse model with mutations in tau and APP (TAPP) and lead to reductions in levels of both neuritic plaques and amyloidogenic β-amyloid peptides in the TAPP mice (Yuzwa et al., 2014). However, one should consider the possible side effects of systemic use of OGA inhibitor. As we discussed throughout this review and other published excellent reviews about the importance of physiological levels of O-GlcNAc in cellular and organismal biology, increasing O-GlcNAc could lead to pathological conditions in other tissues or organs.
Systemic effects of altered O-GlcNAc cycling: A cautionary Tale
There are several cautionary notes regarding pharmacological manipulation of O-GlcNAc levels. Specifically, acute and chronic changes to O-GlcNAc may yield different cellular outcomes. Acute OGA inhibition correlates with decreased protein phosphorylation but longer-term inhibition does not affect phosphorylation on proteins such as tau suggesting that cells adapt (Zhu et al., 2014). Also, while tau is among the proteins modified by O-GlcNAc, there are thousands of other proteins whose O-GlcNAc status may be modulated by therapeutics targeting of OGT or OGA resulting in unintended consequences. Furthermore, off-target side effects in the brain and other tissues as well as disrupted glucose homeostasis can lead to complications and must be carefully considered for any treatment regimen. Concerns were raised based on the potential insulin resistance induction following increased O-GlcNAcylation. Studies in C. elegans and Drosophila have demonstrated that changes in O-GlcNAc cycling can induce changes in insulin-sensitivity (Hanover et al., 2003; Forsythe et al., 2006; Sekine et al., 2010). Previous studies had also suggested that overexpression of OGT in mice (McClain et al., 2002) or inhibition of OGA in the L1-3T3 cell model (Wells et al., 2003) could induce insulin resistance. However, Macauley, et al. demonstrated that the whole body treatment using OGA inhibitor NButGT does not trigger insulin resistance or alter glucose homeostasis in rats or in adipocytes (Macauley et al., 2008; Macauley et al., 2010). This is presumably due to compensation by other homeostatic physiological mechanisms that cannot occur in genetic experiments or experiments carried out in vitro. Importantly, treatment of mice with NButGT reduced Aβ production both in vitro and in vivo. Low γ-secretase activity mediated by increase of O-GlcNAcylation is suggested as a potential explanation. Since O-GlcNAc is a key epigenetic regulator, alteration in the levels or activities of O-GlcNAc cycling enzymes could have unforeseen effects on transcriptional programs and potentially transgenerational effects (Lewis and Hanover, 2014).
Summary and Conclusions
Genetic, pharmacological, and biochemical evidence suggests that intracellular protein O-GlcNAcylation is emerging as a major pathway contributing to age-dependent neurodegenerative damage. Yet, research in this area remains in its infancy. A summary of the major topics of this review appears in Figure 4. Nutrient-driven O-GlcNAcylation is likely to be a key player in maintaining normal proteostasis in the brain. Intriguingly, mutations in Ogt have been linked to X-linked intellectual deficiency suggesting a major role for O-GlcNAc metabolism in the normal functioning of the brain (Niranjan et al., 2015; Vaidyanathan et al., 2017). The brain’s heavy dependence on glucose metabolism renders postmitotic neurons particularly susceptible to age-dependent changes in proteostasis. Maintenance of synapses, axonal transport, neural mitochondrial metabolism, and trafficking are regulated by O-GlcNAcylation. In addition, nearly all the cellular degradation pathways are either directly or indirectly dependent upon O-GlcNAcylation. The enzymes of O-GlcNAc metabolism are known to partner with chaperones, transcriptional complexes, and other epigenetic regulators. These factors dictate the levels of proteins prone to aggregation in neurodegenerative disease. O-GlcNAcylation also acts on globular domains of kinases and phosphorylated substrates to directly alter signaling cascades. OGT acts co-translationally and post-translationally to modify IDPs prone to aggregation.
Figure 4. A summary of the role of O-GlcNAc in regulating essential brain functions.
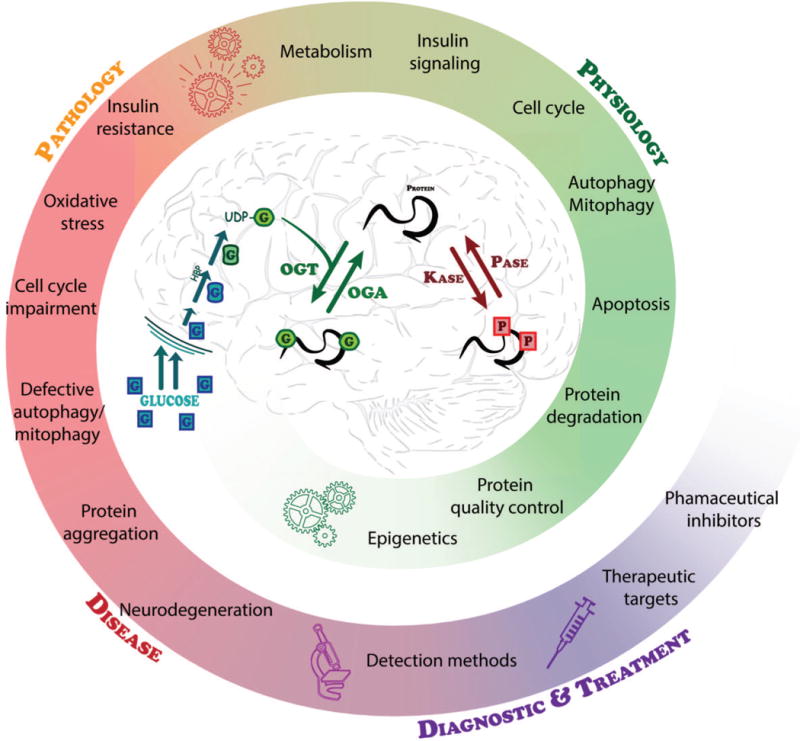
UDP-GlcNAc, the end product of the nutrient-sensitive HBP, is dynamically used by OGT to catalyze the addition of O-GlcNAc to substrate proteins. This cyclic modification is coordinately regulated with other PTMs such as phosphorylation to regulate the required intricacies of cellular processes. Deregulation of PTMs including O-GlcNAc leads to several pathologies that are associated with neurodegeneration. Methods for detecting O-GlcNAc and understanding its role in these pathologies continue to improve providing a strong foundation for considering targeted therapeutics based on individual or global protein O-GlcNAcylation and the activities of OGT and OGA.
The close association between O-GlcNAcylation and neurodegeneration provides unique opportunities for both diagnostics and therapeutics. Imaging paradigms based on monitoring O-GlcNAcylation or the enzymes modulating the modification’s addition and removal are currently in development. In addition, small molecule inhibitors of OGT and OGA may prove useful for therapy. These may act by directly preventing the formation of protein aggregates or by altering the signaling cascades leading to the onset of neurodegenerative damage. Yet, since O-GlcNAc metabolism is tightly regulated and ubiquitous, it may be difficult to target the pathway for inhibition using traditional pharmacological means. In addition, targeting therapies to the brain is complicated by the blood-brain barrier limiting the bioavailability of some therapeutic compounds. Finally, since all tissues are likely to exhibit a requirement for O-GlcNAc cycling, selective targeting of the brain may prove difficult. Such off-target effects may limit the usefulness of inhibitors that might otherwise prove useful. Despite these limitations, as one of the key cellular responses to stress and nutrient availability, O-GlcNAcylation represents a promising target for therapeutic strategies.
Acknowledgments
Supported by funds from Intramural NIDDK
ABBREVIATIONS
- O-GlcNAc
O-linked N-acetylglucosamine
- GLUT1
Glucose Transporter type 1
- GLUT4
Glucose Transporter type 4
- AD
Alzheimer’s Disease
- PD
Parkinson’s disease
- ALS
Amyotrophic lateral sclerosis
- HD
Huntington’s disease
- APP
Amyloid precursor protein
- NFT
Neurofibrillary tangles
- PINK1
PTEN induced kinase 1
- SCA
spino-cerebellar ataxia
- Atx-1
Ataxin-1
- IDPs
Intrinsically disordered proteins
- HBP
Hexosamine biosynthetic pathway
- UDP
Uridine Diphosphate
- PTM
Post translational modification
- OGT
O-GlcNAc Transferase
- ncOGT
Nuclear and Cytoplasmic OGT
- mOGT
Mitochondrial OGT
- sOGT
Short isoform of OGT
- OGA
O-GlcNacase
- OGA-L
OGA Long isoform
- OGA-S
OGA Short isoform
- KO
Knockout
- TPR
Tetratricopeptide Repeat
- HCF-1
Host Cell Factor-1
- GSK3B
Glycogen Synthase Kinase 3B
- AMPK
AMP activated Protein Kinase
- TET
Ten-eleven Translocation
- CARM1
Coactivator Associated Arginine Methyltransferase 1
- Atx-10
Ataxin-10
- DYT3
Parkinsonian dystonia
- THAP1
THAP Domain containing Protein 1
- pRB
Retinoblastoma protein
- E2F
Transcription Factor E2F1
- NF-kB
Nuclear Factor kappa Beta
- MIBP1
c-MYC intron binding protein
- MGEA5
Meningioma-Expressed Antigen 5
- HAT
Histone Acetyl Transferase
- SKN-1
Skinhead 1
- Nrf1/2
Nuclear respiratory factor 1/1
- PRC
Polycomb repressor complex
- Ph
Polyhomeotic
- TRX
Trithorax
- ASH1
Absent Small or Homeotic 1
- SET1
SET domain containing protein 1
- Syn1
Synapsin 1
- αCamKII
α-calcium–calmodulin-dependent kinase II
- MMTV-Cre
Cre Recombinase under mouse mammary tumor virus promoter
- PVN
Paraventricular nucleus
- IGF-1
Insulin-like growth factor 1
- GH
Growth Hormone
- XLID
X-linked intellectual disability
- Xist
lnRNA triggering X-inactivation
- IR
Insulin receptors
- PI3K
Phosphatidylinositol 3 kinase
- Akt/PKB
Protein Kinase B
- mTOR
Mammalian Target of Rapamycin
- PDK1
Phosphoinositide-Dependent Kinase 1
- IRS1
Insulin Receptor Substrate 1
- Sp1
Specificity Protein 1
- Nup62
Nuclear pore protein p62
- CNS
Central nervous system
- RMS
Rostral migratory stream
- SVZ
Subventricular zone
- SGZ
Subgranular zone
- CDK
Cyclin-dependent kinase
- SNAP-29
Synaptosome associated protein 29
- VAMP8
Vesicle associated membrane protein 8
- BCL2
B-cell lymphoma 2
- Atg7
Autophagy related 7
- ATP
Adenosine triphosphate
- TRAK1/2
Trafficking kinesin-binding protein 1
- OIP106/98
O-linked N-acetylglucosamine transferase interacting protein 106
- NDUFA9
NADH dehaydrogenase 1 aplha subunit A9
- COX1
Cyclooxygenase 1
- ETC
Electron transport chain
- TCA
Tricarboxylic acid
- ACO2
Aconitase 2
- IDH
Isocitrate dehydrogenase
- SOD
Superoxide dismutase
- PRDX3
Peroxiredoxin 3
- VDAC
Voltage dependent anion channel
- mPTP
Mitochondrial permeability transition pore
- Ogg1
8-oxoguanine DNA glycosylase
- MRI
Magnetic resonance imaging
- PET
Positron emission tomography
- PTM
Post translational modification
- GSK3B
Glycogen synthase kinase 3 beta
- CDK5
Cell division protein kinase 5
Footnotes
The authors have no conflicts to declare
References
- Abramowitz LK, Olivier-Van Stichelen S, Hanover JA. Chromosome imbalance as a driver of sex disparity in disease. J Genomics. 2014;2:77–88. doi: 10.7150/jgen.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akan I, Love DC, Harwood KR, Bond MR, Hanover JA. Drosophila O-GlcNAcase Deletion Globally Perturbs Chromatin O-GlcNAcylation. J Biol Chem. 2016;291:9906–9919. doi: 10.1074/jbc.M115.704783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto Y, Comer FI, Cole RN, Kudo A, Kawakami H, Hirano H, Hart GW. Localization of the O-GlcNAc transferase and O-GlcNAc-modified proteins in rat cerebellar cortex. Brain Res. 2003;966:194–205. doi: 10.1016/s0006-8993(02)04158-6. [DOI] [PubMed] [Google Scholar]
- Akimoto Y, Kreppel LK, Hirano H, Hart GW. Localization of the O-linked N-acetylglucosamine transferase in rat pancreas. Diabetes. 1999;48:2407–2413. doi: 10.2337/diabetes.48.12.2407. [DOI] [PubMed] [Google Scholar]
- Akimoto Y, Kreppel LK, Hirano H, Hart GW. Increased O-GlcNAc transferase in pancreas of rats with streptozotocin-induced diabetes. Diabetologia. 2000;43:1239–1247. doi: 10.1007/s001250051519. [DOI] [PubMed] [Google Scholar]
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nature medicine. 1996 doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- Andrali SS, März P, Ozcan S. Ataxin-10 interacts with O-GlcNAc transferase OGT in pancreatic beta cells. Biochem Biophys Res Commun. 2005;337:149–153. doi: 10.1016/j.bbrc.2005.09.026. [DOI] [PubMed] [Google Scholar]
- Andrali SS, Qian Q, Ozcan S. Glucose mediates the translocation of NeuroD1 by O-linked glycosylation. J Biol Chem. 2007;282:15589–15596. doi: 10.1074/jbc.M701762200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias EB, Kim J, Cartee GD. Prolonged incubation in PUGNAc results in increased protein O-Linked glycosylation and insulin resistance in rat skeletal muscle. Diabetes. 2004;53:921–930. doi: 10.2337/diabetes.53.4.921. [DOI] [PubMed] [Google Scholar]
- Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J Biol Chem. 1996;271:28741–28744. doi: 10.1074/jbc.271.46.28741. [DOI] [PubMed] [Google Scholar]
- Ashton-Beaucage D, Udell CM, Lavoie H, Baril C, Lefrançois M, Chagnon P, Gendron P, Caron-Lizotte O, Bonneil E, Thibault P, Therrien M. The exon junction complex controls the splicing of MAPK and other long intron-containing transcripts in Drosophila. Cell. 2010;143:251–262. doi: 10.1016/j.cell.2010.09.014. [DOI] [PubMed] [Google Scholar]
- Association, A. 2017 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 2017 [Google Scholar]
- Bailer SM, Berlin WK, Starr CM, Hanover JA. Characterization of nuclear pore protein p62 produced using baculovirus. Protein Expr Purif. 1995;6:546–554. doi: 10.1006/prep.1995.1072. [DOI] [PubMed] [Google Scholar]
- Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol. 2011;68:51–57. doi: 10.1001/archneurol.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale T. The placenta and neurodevelopment: sex differences in prenatal vulnerability. Dialogues Clin Neurosci. 2016;18:459–464. doi: 10.31887/DCNS.2016.18.4/tbale. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LE, Berkaw MN, Buse MG. Identification of the major site of O-linked beta-N-acetylglucosamine modification in the C terminus of insulin receptor substrate-1. Mol Cell Proteomics. 2006;5:313–323. doi: 10.1074/mcp.M500314-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CA. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010;467:470–473. doi: 10.1038/nature09371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleckmann C, Geyer H, Reinhold V, Lieberoth A, Schachner M, Kleene R, Geyer R. Glycomic analysis of N-linked carbohydrate epitopes from CD24 of mouse brain. J Proteome Res. 2009;8:567–582. doi: 10.1021/pr800729r. [DOI] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. 2015;208:869–880. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonda DJ, Lee HP, Kudo W, Zhu X, Smith MA, Lee HG. Pathological implications of cell cycle re-entry in Alzheimer disease. Expert Rev Mol Med. 2010;12:e19. doi: 10.1017/S146239941000150X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouazzi H, Lesca G, Trujillo C, Alwasiyah MK, Munnich A. Nonsyndromic X-linked intellectual deficiency in three brothers with a novel MED12 missense mutation [c.5922G>T (p.Glu1974His)] Clin Case Rep. 2015;3:604–609. doi: 10.1002/ccr3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouché C, Serdy S, Kahn CR, Goldfine AB. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr Rev. 2004;25:807–830. doi: 10.1210/er.2003-0026. [DOI] [PubMed] [Google Scholar]
- Braidman I, Carroll M, Dance N, Robinson D. Separation and properties of human brain hexosaminidase C. Biochem J. 1974;143:295–301. doi: 10.1042/bj1430295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley K, Pozo K, Stephenson FA. N-acetylglucosamine transferase is an integral component of a kinesin-directed mitochondrial trafficking complex. Biochim Biophys Acta. 2011;1813:269–281. doi: 10.1016/j.bbamcr.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Brickley K, Smith MJ, Beck M, Stephenson FA. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem. 2005;280:14723–14732. doi: 10.1074/jbc.M409095200. [DOI] [PubMed] [Google Scholar]
- Brion JP, Octave JN, Couck AM. Distribution of the phosphorylated microtubule-associated protein tau in developing cortical neurons. Neuroscience. 1994;63:895–909. doi: 10.1016/0306-4522(94)90533-9. [DOI] [PubMed] [Google Scholar]
- Bullen JW, Balsbaugh JL, Chanda D, Shabanowitz J, Hunt DF, Neumann D, Hart GW. Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK) J Biol Chem. 2014;289:10592–10606. doi: 10.1074/jbc.M113.523068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butkinaree C, Cheung WD, Park S, Park K, Barber M, Hart GW. Characterization of beta-N-acetylglucosaminidase cleavage by caspase-3 during apoptosis. J Biol Chem. 2008;283:23557–23566. doi: 10.1074/jbc.M804116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill GF. Starvation in man. N Engl J Med. 1970;282:668–675. doi: 10.1056/NEJM197003192821209. [DOI] [PubMed] [Google Scholar]
- Caillet-Boudin ML, Strecker G, Michalski JC. O-linked GlcNAc in serotype-2 adenovirus fibre. Eur J Biochem. 1989;184:205–211. doi: 10.1111/j.1432-1033.1989.tb15008.x. [DOI] [PubMed] [Google Scholar]
- Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, Vosseller K, Reginato MJ. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. 2010;29:2831–2842. doi: 10.1038/onc.2010.41. [DOI] [PubMed] [Google Scholar]
- Cameron A, Giacomozzi B, Joyce J, Gray A, Graham D, Ousson S, Neny M, Beher D, Carlson G, O’Moore J, Shearman M, Hering H. Generation and characterization of a rabbit monoclonal antibody site-specific for tau O-GlcNAcylated at serine 400. FEBS Lett. 2013;587:3722–3728. doi: 10.1016/j.febslet.2013.09.042. [DOI] [PubMed] [Google Scholar]
- Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, Conaway JW, Conaway RC, Herr W. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell. 2011;144:376–388. doi: 10.1016/j.cell.2010.12.030. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztajn S, Lippa C, Nixon RA. Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14:671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J Neurosci. 1996;16:186–199. doi: 10.1523/JNEUROSCI.16-01-00186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Hamilton DJ, Nixon RA. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res. 1994;640:68–80. doi: 10.1016/0006-8993(94)91858-9. [DOI] [PubMed] [Google Scholar]
- Cayrol C, Lacroix C, Mathe C, Ecochard V, Ceribelli M, Loreau E, Lazar V, Dessen P, Mantovani R, Aguilar L, Girard JP. The THAP-zinc finger protein THAP1 regulates endothelial cell proliferation through modulation of pRB/E2F cell-cycle target genes. Blood. 2007;109:584–594. doi: 10.1182/blood-2006-03-012013. [DOI] [PubMed] [Google Scholar]
- Cha MY, Cho HJ, Kim C, Jung YO, Kang MJ, Murray ME, Hong HS, Choi YJ, Choi H, Kim DK, Choi H, Kim J, Dickson DW, Song HK, Cho JW, Yi EC, Kim J, Jin SM, Mook-Jung I. Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum Mol Genet. 2015;24:6492–6504. doi: 10.1093/hmg/ddv358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Liu X, Lü F, Liu X, Ru Y, Ren Y, Yao L, Zhang Y. Transcription factor Nrf1 is negatively regulated by its O-GlcNAcylation status. FEBS Lett. 2015;589:2347–2358. doi: 10.1016/j.febslet.2015.07.030. [DOI] [PubMed] [Google Scholar]
- Chen R, Gong P, Tao T, Gao Y, Shen J, Yan Y, Duan C, Wang J, Liu X. O-GlcNAc Glycosylation of nNOS Promotes Neuronal Apoptosis Following Glutamate Excitotoxicity. Cell Mol Neurobiol. 2017 doi: 10.1007/s10571-017-0477-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CF, Smith AJ, Omary MB. Characterization and dynamics of O-linked glycosylation of human cytokeratin 8 and 18. J Biol Chem. 1992;267:3901–3906. [PubMed] [Google Scholar]
- Chou TY, Hart GW, Dang CV. c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J Biol Chem. 1995;270:18961–18965. doi: 10.1074/jbc.270.32.18961. [DOI] [PubMed] [Google Scholar]
- Cieniewski-Bernard C, Bastide B, Lefebvre T, Lemoine J, Mounier Y, Michalski JC. Identification of O-linked N-acetylglucosamine proteins in rat skeletal muscle using two-dimensional gel electrophoresis and mass spectrometry. Mol Cell Proteomics. 2004;3:577–585. doi: 10.1074/mcp.M400024-MCP200. [DOI] [PubMed] [Google Scholar]
- Cividini F, Scott BT, Dai A, Han W, Suarez J, Diaz-Juarez J, Diemer T, Casteel DE, Dillmann WH. O-GlcNAcylation of Ogg1 Impairs Oxidative Mitochondrial DNA Lesion Repair in Diabetic Hearts. J Biol Chem. 2016 doi: 10.1074/jbc.M116.754481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RN, Hart GW. Cytosolic O-glycosylation is abundant in nerve terminals. J Neurochem. 2001;79:1080–1089. doi: 10.1046/j.1471-4159.2001.00655.x. [DOI] [PubMed] [Google Scholar]
- Comtesse N, Maldener E, Meese E. Identification of a nuclear variant of MGEA5, a cytoplasmic hyaluronidase and a beta-N-acetylglucosaminidase. Biochem Biophys Res Commun. 2001;283:634–640. doi: 10.1006/bbrc.2001.4815. [DOI] [PubMed] [Google Scholar]
- Cordes VC, Krohne G. Sequential O-glycosylation of nuclear pore complex protein gp62 in vitro. Eur J Cell Biol. 1993;60:185–195. [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- D’Onofrio M, Starr CM, Park MK, Holt GD, Haltiwanger RS, Hart GW, Hanover JA. Partial cDNA sequence encoding a nuclear pore protein modified by O-linked N-acetylglucosamine. Proc Natl Acad Sci U S A. 1988;85:9595–9599. doi: 10.1073/pnas.85.24.9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta B, Ray MK, Chakrabarti D, Wylie DE, Gupta NK. Glycosylation of eukaryotic peptide chain initiation factor 2 (eIF-2)-associated 67-kDa polypeptide (p67) and its possible role in the inhibition of eIF-2 kinase-catalyzed phosphorylation of the eIF-2 alpha-subunit. J Biol Chem. 1989;264:20620–20624. [PubMed] [Google Scholar]
- Davis LI, Blobel G. Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc Natl Acad Sci U S A. 1987;84:7552–7556. doi: 10.1073/pnas.84.21.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehennaut V, Lefebvre T, Sellier C, Leroy Y, Gross B, Walker S, Cacan R, Michalski JC, Vilain JP, Bodart JF. O-linked N-acetylglucosaminyltransferase inhibition prevents G2/M transition in Xenopus laevis oocytes. J Biol Chem. 2007;282:12527–12536. doi: 10.1074/jbc.M700444200. [DOI] [PubMed] [Google Scholar]
- Deng Y, Li B, Liu F, Iqbal K, Grundke-Iqbal I, Brandt R, Gong CX. Regulation between O-GlcNAcylation and phosphorylation of neurofilament-M and their dysregulation in Alzheimer disease. FASEB J. 2008;22:138–145. doi: 10.1096/fj.07-8309com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, Shih AH, Levine RL, Bernard O, Mercher T, Solary E, Urh M, Daniels DL, Fuks F. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645–655. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F, Owen JB, Sultana R, Sowell RA, Perluigi M, Cini C, Cai J, Pierce WM, Butterfield DA. The wheat germ agglutinin-fractionated proteome of subjects with Alzheimer’s disease and mild cognitive impairment hippocampus and inferior parietal lobule: Implications for disease pathogenesis and progression. J Neurosci Res. 2010;88:3566–3577. doi: 10.1002/jnr.22528. [DOI] [PubMed] [Google Scholar]
- Diwu Y, Tian J, Shi J. Effect of xixin decoction on O-linked N-acetylglucosamine glycosylation of tau proteins in rat brain with sporadic Alzheimer disease. J Tradit Chin Med. 2013a;33:367–372. doi: 10.1016/s0254-6272(13)60180-6. [DOI] [PubMed] [Google Scholar]
- Diwu Y, Tian J, Shi J. [Effects of Xixin decoction on enzymes related to O-GlcNAc glycosylation of tau proteins in the brain of rats with sporadic Alzheimer’s disease] Nan Fang Yi Ke Da Xue Xue Bao. 2013b;33:1442–1447. [PubMed] [Google Scholar]
- Doma MK, Parker R. RNA quality control in eukaryotes. Cell. 2007;131:660–668. doi: 10.1016/j.cell.2007.10.041. [DOI] [PubMed] [Google Scholar]
- Dong DL, Xu ZS, Chevrier MR, Cotter RJ, Cleveland DW, Hart GW. Glycosylation of mammalian neurofilaments. Localization of multiple O-linked N-acetylglucosamine moieties on neurofilament polypeptides L and M. J Biol Chem. 1993;268:16679–16687. [PubMed] [Google Scholar]
- Dong DL, Xu ZS, Hart GW, Cleveland DW. Cytoplasmic O-GlcNAc modification of the head domain and the KSP repeat motif of the neurofilament protein neurofilament-H. J Biol Chem. 1996;271:20845–20852. doi: 10.1074/jbc.271.34.20845. [DOI] [PubMed] [Google Scholar]
- Dorfmueller HC, Borodkin VS, Schimpl M, Shepherd SM, Shpiro NA, van Aalten DM. GlcNAcstatin: a picomolar, selective O-GlcNAcase inhibitor that modulates intracellular O-glcNAcylation levels. J Am Chem Soc. 2006;128:16484–16485. doi: 10.1021/ja066743n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drougat L, Olivier-Van Stichelen S, Mortuaire M, Foulquier F, Lacoste AS, Michalski JC, Lefebvre T, Vercoutter-Edouart AS. Characterization of O-GlcNAc cycling and proteomic identification of differentially O-GlcNAcylated proteins during G1/S transition. Biochim Biophys Acta. 2012;1820:1839–1848. doi: 10.1016/j.bbagen.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Durning SP, Flanagan-Steet H, Prasad N, Wells L. O-Linked β-N-acetylglucosamine (O-GlcNAc) Acts as a Glucose Sensor to Epigenetically Regulate the Insulin Gene in Pancreatic Beta Cells. J Biol Chem. 2016;291:2107–2118. doi: 10.1074/jbc.M115.693580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliot SP, Schmied R, Gabel CA, Ambron RT. An 83 kDa O-GlcNAc-glycoprotein is found in the axoplasm and nucleus of Aplysia neurons. J Neurosci. 1993;13:2424–2429. doi: 10.1523/JNEUROSCI.13-06-02424.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsen NL, Patel SB, Ford RE, Hall DL, Hess F, Kandula H, Kornienko M, Reid J, Selnick H, Shipman JM, Sharma S, Lumb KJ, Soisson SM, Klein DJ. Insights into activity and inhibition from the crystal structure of human O-GlcNAcase. Nat Chem Biol. 2017;13:613–615. doi: 10.1038/nchembio.2357. [DOI] [PubMed] [Google Scholar]
- Erbsloh F, BERNSMEIER A, HILLESHEIM H. [The glucose consumption of the brain & its dependence on the liver] Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1958;196:611–626. doi: 10.1007/BF00344388. [DOI] [PubMed] [Google Scholar]
- Fahie K, Zachara NE. Molecular functions of glycoconjugates in autophagy. Journal of Molecular Biology. 2016 doi: 10.1016/j.jmb.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farook VS, Bogardus C, Prochazka M. Analysis of MGEA5 on 10q24.1-q24.3 encoding the beta-O-linked N-acetylglucosaminidase as a candidate gene for type 2 diabetes mellitus in Pima Indians. Mol Genet Metab. 2002;77:189–193. doi: 10.1016/s1096-7192(02)00127-0. [DOI] [PubMed] [Google Scholar]
- Förster S, Welleford AS, Triplett JC, Sultana R, Schmitz B, Butterfield DA. Increased O-GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochim Biophys Acta. 2014;1842:1333–1339. doi: 10.1016/j.bbadis.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ME, Love DC, Lazarus BD, Kim EJ, Prinz WA, Ashwell G, Krause MW, Hanover JA. Caenorhabditis elegans ortholog of a diabetes susceptibility locus: oga-1 (O-GlcNAcase) knockout impacts O-GlcNAc cycling, metabolism, and dauer. Proc Natl Acad Sci U S A. 2006;103:11952–11957. doi: 10.1073/pnas.0601931103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox NC, Scahill RI, Crum WR, Rossor MN. Correlation between rates of brain atrophy and cognitive decline in AD. Neurology. 1999;52:1687–1689. doi: 10.1212/wnl.52.8.1687. [DOI] [PubMed] [Google Scholar]
- Frade JM, Ovejero-Benito MC. Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle. 2015;14:712–720. doi: 10.1080/15384101.2015.1004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, Ozelius LJ. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41:286–288. doi: 10.1038/ng.304. [DOI] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Gambetta MC, Muller J. O-GlcNAcylation Prevents Aggregation of the Polycomb Group Repressor Polyhomeotic. Dev Cell. 2014;31:629–639. doi: 10.1016/j.devcel.2014.10.020. [DOI] [PubMed] [Google Scholar]
- Gambetta MC, Oktaba K, Müller J. Essential role of the glycosyltransferase sxc/Ogt in polycomb repression. Science. 2009a;325:93–96. doi: 10.1126/science.1169727. [DOI] [PubMed] [Google Scholar]
- Gambetta MC, Oktaba K, Muller J. Essential role of the glycosyltransferase sxc/Ogt in polycomb repression. Science. 2009b;325:93–96. doi: 10.1126/science.1169727. [DOI] [PubMed] [Google Scholar]
- Gandy JC, Rountree AE, Bijur GN. Akt1 is dynamically modified with O-GlcNAc following treatments with PUGNAc and insulin-like growth factor-1. FEBS Lett. 2006;580:3051–3058. doi: 10.1016/j.febslet.2006.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Miyazaki J, Hart GW. The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta-cells. Arch Biochem Biophys. 2003;415:155–163. doi: 10.1016/s0003-9861(03)00234-0. [DOI] [PubMed] [Google Scholar]
- Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem. 2001;276:9838–9845. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- Gatta E, Lefebvre T, Gaetani S, dos Santos M, Marrocco J, Mir AM, Cassano T, Maccari S, Nicoletti F, Mairesse J. Evidence for an imbalance between tau O-GlcNAcylation and phosphorylation in the hippocampus of a mouse model of Alzheimer’s disease. Pharmacol Res. 2016;105:186–197. doi: 10.1016/j.phrs.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Ge X, Kwok PY, Shieh JT. Prioritizing genes for X-linked diseases using population exome data. Hum Mol Genet. 2015;24:599–608. doi: 10.1093/hmg/ddu473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, Wuu J, Chao MV, Mufson EJ, Nixon RA, Che S. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry. 2010;68:885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong CX, Liu F, Iqbal K. O-GlcNAcylation: A regulator of tau pathology and neurodegeneration. Alzheimers Dement. 2016;12:1078–1089. doi: 10.1016/j.jalz.2016.02.011. [DOI] [PubMed] [Google Scholar]
- Graham DL, Gray AJ, Joyce JA, Yu D, O’Moore J, Carlson GA, Shearman MS, Dellovade TL, Hering H. Increased O-GlcNAcylation reduces pathological tau without affecting its normal phosphorylation in a mouse model of tauopathy. Neuropharmacology. 2014;79:307–313. doi: 10.1016/j.neuropharm.2013.11.025. [DOI] [PubMed] [Google Scholar]
- Griffith LS, Mathes M, Schmitz B. Beta-amyloid precursor protein is modified with O-linked N-acetylglucosamine. J Neurosci Res. 1995;41:270–278. doi: 10.1002/jnr.490410214. [DOI] [PubMed] [Google Scholar]
- Griffith LS, Schmitz B. O-linked N-acetylglucosamine is upregulated in Alzheimer brains. Biochem Biophys Res Commun. 1995;213:424–431. doi: 10.1006/bbrc.1995.2149. [DOI] [PubMed] [Google Scholar]
- Griffith LS, Schmitz B. O-linked N-acetylglucosamine levels in cerebellar neurons respond reciprocally to pertubations of phosphorylation. Eur J Biochem. 1999;262:824–831. doi: 10.1046/j.1432-1327.1999.00439.x. [DOI] [PubMed] [Google Scholar]
- Gross BJ, Kraybill BC, Walker S. Discovery of O-GlcNAc transferase inhibitors. J Am Chem Soc. 2005;127:14588–14589. doi: 10.1021/ja0555217. [DOI] [PubMed] [Google Scholar]
- Groves JA, Maduka AO, O’Meally RN, Cole RN, Zachara NE. Fatty acid synthase inhibits the O-GlcNAcase during oxidative stress. J Biol Chem. 2017;292:6493–6511. doi: 10.1074/jbc.M116.760785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nature cell. 2014 doi: 10.1038/ncb3066. [DOI] [PubMed] [Google Scholar]
- Hagmann J, Grob M, Burger MM. The cytoskeletal protein talin is O-glycosylated. J Biol Chem. 1992;267:14424–14428. [PubMed] [Google Scholar]
- Haltiwanger RS, Blomberg MA, Hart GW. Glycosylation of nuclear and cytoplasmic proteins. Purification and characterization of a uridine diphospho-N-acetylglucosamine:polypeptide beta-N-acetylglucosaminyltransferase. J Biol Chem. 1992;267:9005–9013. [PubMed] [Google Scholar]
- Haltiwanger RS, Grove K, Philipsberg GA. Modulation of O-linked N-acetylglucosamine levels on nuclear and cytoplasmic proteins in vivo using the peptide O-GlcNAc-beta-N-acetylglucosaminidase inhibitor O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenylcarbamate. J Biol Chem. 1998;273:3611–3617. doi: 10.1074/jbc.273.6.3611. [DOI] [PubMed] [Google Scholar]
- Haltiwanger RS, Holt GD, Hart GW. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine:peptide beta-N-acetylglucosaminyltransferase. J Biol Chem. 1990;265:2563–2568. [PubMed] [Google Scholar]
- Han I, Kudlow JE. Reduced O glycosylation of Sp1 is associated with increased proteasome susceptibility. Mol Cell Biol. 1997;17:2550–2558. doi: 10.1128/mcb.17.5.2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JA. Glycan-dependent signaling: O-linked N-acetylglucosamine. FASEB J. 2001;15:1865–1876. doi: 10.1096/fj.01-0094rev. [DOI] [PubMed] [Google Scholar]
- Hanover JA, Cohen CK, Willingham MC, Park MK. O-linked N-acetylglucosamine is attached to proteins of the nuclear pore. Evidence for cytoplasmic and nucleoplasmic glycoproteins. J Biol Chem. 1987;262:9887–9894. [PubMed] [Google Scholar]
- Hanover JA, Forsythe ME, Hennessey PT, Brodigan TM, Love DC, Ashwell G, Krause M. A Caenorhabditis elegans model of insulin resistance: altered macronutrient storage and dauer formation in an OGT-1 knockout. Proc Natl Acad Sci U S A. 2005;102:11266–11271. doi: 10.1073/pnas.0408771102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13:312–321. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- Hanover JA, Lai Z, Lee G, Lubas WA, Sato SM. Elevated O-linked N-acetylglucosamine metabolism in pancreatic beta-cells. Arch Biochem Biophys. 1999;362:38–45. doi: 10.1006/abbi.1998.1016. [DOI] [PubMed] [Google Scholar]
- Hanover JA, Wang P. O-GlcNAc cycling shows neuroprotective potential in C. elegans models of neurodegenerative disease. Worm. 2013;2:e27043. doi: 10.4161/worm.27043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JA, Yu S, Lubas WB, Shin SH, Ragano-Caracciola M, Kochran J, Love DC. Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Arch Biochem Biophys. 2003;409:287–297. doi: 10.1016/s0003-9861(02)00578-7. [DOI] [PubMed] [Google Scholar]
- Hastings NB, Wang X, Song L, Butts BD, Grotz D, Hargreaves R, Fred Hess J, Hong KK, Huang CR, Hyde L, Laverty M, Lee J, Levitan D, Lu SX, Maguire M, Mahadomrongkul V, McEachern EJ, Ouyang X, Rosahl TW, Selnick H, Stanton M, Terracina G, Vocadlo DJ, Wang G, Duffy JL, Parker EM, Zhang L. Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Mol Neurodegener. 2017;12:39. doi: 10.1186/s13024-017-0181-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckel D, Comtesse N, Brass N, Blin N, Zang KD, Meese E. Novel immunogenic antigen homologous to hyaluronidase in meningioma. Hum Mol Genet. 1998;7:1859–1872. doi: 10.1093/hmg/7.12.1859. [DOI] [PubMed] [Google Scholar]
- Hokama M, Oka S, Leon J, Ninomiya T, Honda H, Sasaki K, Iwaki T, Ohara T, Sasaki T, LaFerla FM, Kiyohara Y, Nakabeppu Y. Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb Cortex. 2014;24:2476–2488. doi: 10.1093/cercor/bht101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman GD, Kasuga M. From receptor to transporter: insulin signalling to glucose transport. Diabetologia. 1997;40:991–1003. doi: 10.1007/s001250050780. [DOI] [PubMed] [Google Scholar]
- Holt GD, Haltiwanger RS, Torres CR, Hart GW. Erythrocytes contain cytoplasmic glycoproteins. O-linked GlcNAc on Band 4.1. J Biol Chem. 1987;262:14847–14850. [PubMed] [Google Scholar]
- Holt GD, Hart GW. The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. J Biol Chem. 1986;261:8049–8057. [PubMed] [Google Scholar]
- Houlden H, Schneider SA, Paudel R, Melchers A, Schwingenschuh P, Edwards M, Hardy J, Bhatia KP. THAP1 mutations (DYT6) are an additional cause of early-onset dystonia. Neurology. 2010;74:846–850. doi: 10.1212/WNL.0b013e3181d5276d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howerton CL, Bale TL. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc Natl Acad Sci U S A. 2014;111:9639–9644. doi: 10.1073/pnas.1401203111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howerton CL, Morgan CP, Fischer DB, Bale TL. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci U S A. 2013;110:5169–5174. doi: 10.1073/pnas.1300065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Hu Y, Oyeleye MO, Dillmann WH. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem. 2009;284:547–555. doi: 10.1074/jbc.M808518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba M, Maede Y. O-N-acetyl-D-glucosamine moiety on discrete peptide of multiple protein 4.1 isoforms regulated by alternative pathways. J Biol Chem. 1989;264:18149–18155. [PubMed] [Google Scholar]
- Ingham PW. A gene that regulates the bithorax complex differentially in larval and adult cells of Drosophila. Cell. 1984;37:815–823. doi: 10.1016/0092-8674(84)90416-1. [DOI] [PubMed] [Google Scholar]
- Iwashita Y, Fukuchi N, Waki M, Hayashi K, Tahira T. Genome-wide repression of NF-kappaB target genes by transcription factor MIBP1 and its modulation by O-linked beta-N-acetylglucosamine (O-GlcNAc) transferase. J Biol Chem. 2012;287:9887–9900. doi: 10.1074/jbc.M111.298521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SP, Akimoto Y, Hart GW. Identification and cloning of a novel family of coiled-coil domain proteins that interact with O-GlcNAc transferase. J Biol Chem. 2003;278:5399–5409. doi: 10.1074/jbc.M209384200. [DOI] [PubMed] [Google Scholar]
- Jacobsen KT, Iverfeldt K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-β precursor protein (APP) Biochem Biophys Res Commun. 2011;404:882–886. doi: 10.1016/j.bbrc.2010.12.080. [DOI] [PubMed] [Google Scholar]
- Janetzko J, Walker S. The making of a sweet modification: structure and function of O-GlcNAc transferase. Journal of Biological Chemistry. 2014 doi: 10.1074/jbc.R114.604405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji S, Kang JG, Park SY, Lee J, Oh YJ, Cho JW. O-GlcNAcylation of tubulin inhibits its polymerization. Amino Acids. 2011;40:809–818. doi: 10.1007/s00726-010-0698-9. [DOI] [PubMed] [Google Scholar]
- Jiang M, Qiu Z, Zhang S, Fan X, Cai X, Xu B, Li X, Zhou J, Zhang X, Chu Y, Wang W, Liang J, Horvath T, Yang X, Wu K, Nie Y, Fan D. Elevated O-GlcNAcylation promotes gastric cancer cells proliferation by modulating cell cycle related proteins and ERK 1/2 signaling. Oncotarget. 2016 doi: 10.18632/oncotarget.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Rehwinkel J, Lazarus BD, Izaurralde E, Hanover JA, Conti E. The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin alpha. Nat Struct Mol Biol. 2004;11:1001–1007. doi: 10.1038/nsmb833. [DOI] [PubMed] [Google Scholar]
- Jínek M, Rehwinkel J, Lazarus BD, Izaurralde E, Hanover JA, Conti E. The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin alpha. Nat Struct Mol Biol. 2004;11:1001–1007. doi: 10.1038/nsmb833. [DOI] [PubMed] [Google Scholar]
- Jo YK, Park NY, Park SJ, Kim BG, Shin JH, Jo DS. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget. 2016 doi: 10.18632/oncotarget.11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Fox NC, Sperling RA, Klunk WE. Brain imaging in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006213. doi: 10.1101/cshperspect.a006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaasik K, Kivimae S, Allen JJ, Chalkley RJ, Huang Y, Baer K, Kissel H, Burlingame AL, Shokat KM, Ptacek LJ, Fu YH. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 2013;17:291–302. doi: 10.1016/j.cmet.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi K, Araki K, Tobiume K, Tanaka N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc Natl Acad Sci U S A. 2009;106:3431–3436. doi: 10.1073/pnas.0813210106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse KP, Hart GW. Lymphocyte activation induces rapid changes in nuclear and cytoplasmic glycoproteins. Proc Natl Acad Sci U S A. 1991a;88:1701–1705. doi: 10.1073/pnas.88.5.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse KP, Hart GW. Topology of O-linked N-acetylglucosamine in murine lymphocytes. Arch Biochem Biophys. 1991b;290:543–548. doi: 10.1016/0003-9861(91)90579-8. [DOI] [PubMed] [Google Scholar]
- Keembiyehetty C, Love DC, Harwood KR, Gavrilova O, Comly ME, Hanover JA. Conditional knock-out reveals a requirement for O-linked N-Acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J Biol Chem. 2015;290:7097–7113. doi: 10.1074/jbc.M114.617779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keembiyehetty CN, Krzeslak A, Love DC, Hanover JA. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J Cell Sci. 2011;124:2851–2860. doi: 10.1242/jcs.083287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Kim M, Sapp E, McIntyre C. Huntingtin expression stimulates endosomal–lysosomal activity, endosome tubulation, and autophagy. Journal of …. 2000 doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly WG, Dahmus ME, Hart GW. RNA polymerase II is a glycoprotein. Modification of the COOH-terminal domain by O-GlcNAc. J Biol Chem. 1993;268:10416–10424. [PubMed] [Google Scholar]
- Kern W, Peters A, Fruehwald-Schultes B, Deininger E, Born J, Fehm HL. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology. 2001;74:270–280. doi: 10.1159/000054694. [DOI] [PubMed] [Google Scholar]
- Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: direct identification of O-GlcNAc-modified proteins from the brain. Proc Natl Acad Sci U S A. 2004;101:13132–13137. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Nam DW, Park SY, Song H, Hong HS, Boo JH, Jung ES, Kim Y, Baek JY, Kim KS, Cho JW, Mook-Jung I. O-linked β-N-acetylglucosaminidase inhibitor attenuates β-amyloid plaque and rescues memory impairment. Neurobiol Aging. 2013;34:275–285. doi: 10.1016/j.neurobiolaging.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Kim EJ. Chemical arsenal for the study of O-GlcNAc. Molecules. 2011;16:1987–2022. doi: 10.3390/molecules16031987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Kang DO, Love DC, Hanover JA. Enzymatic characterization of O-GlcNAcase isoforms using a fluorogenic GlcNAc substrate. Carbohydr Res. 2006;341:971–982. doi: 10.1016/j.carres.2006.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Jeong EH, Park S, Jeong HJ, Edery I, Cho JW. A role for O-GlcNAcylation in setting circadian clock speed. Genes Dev. 2012;26:490–502. doi: 10.1101/gad.182378.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YB, Uotani S, Pierroz DD, Flier JS, Kahn BB. In vivo administration of leptin activates signal transduction directly in insulin-sensitive tissues: overlapping but distinct pathways from insulin. Endocrinology. 2000;141:2328–2339. doi: 10.1210/endo.141.7.7536. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Klein AL, Berkaw MN, Buse MG, Ball LE. O-linked N-acetylglucosamine modification of insulin receptor substrate-1 occurs in close proximity to multiple SH2 domain binding motifs. Mol Cell Proteomics. 2009;8:2733–2745. doi: 10.1074/mcp.M900207-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272:9308–9315. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Singh PK, Parihar R, Dwivedi V. Decreased O-linked GlcNAcylation protects from cytotoxicity mediated by huntingtin exon1 protein fragment. Journal of Biological …. 2014 doi: 10.1074/jbc.M114.553321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerlöf O, Hart GW, Huganir RL. O-GlcNAc transferase regulates excitatory synapse maturity. Proc Natl Acad Sci U S A. 2017;114:1684–1689. doi: 10.1073/pnas.1621367114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerlöf O, Slocomb JE, Hong I, Aponte Y, Blackshaw S, Hart GW, Huganir RL. The nutrient sensor OGT in PVN neurons regulates feeding. Science. 2016;351:1293–1296. doi: 10.1126/science.aad5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus BD, Love DC, Hanover JA. Recombinant O-GlcNAc transferase isoforms: identification of O-GlcNAcase, yes tyrosine kinase, and tau as isoform-specific substrates. Glycobiology. 2006;16:415–421. doi: 10.1093/glycob/cwj078. [DOI] [PubMed] [Google Scholar]
- Lazarus BD, Love DC, Hanover JA. O-GlcNAc cycling: implications for neurodegenerative disorders. Int J Biochem Cell Biol. 2009;41:2134–2146. doi: 10.1016/j.biocel.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S. HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science. 2013;342:1235–1239. doi: 10.1126/science.1243990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature. 2011;469:564–567. doi: 10.1038/nature09638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre T, Caillet-Boudin ML, Buée L, Delacourte A, Michalski JC. O-GlcNAc glycosylation and neurological disorders. Adv Exp Med Biol. 2003a;535:189–202. doi: 10.1007/978-1-4615-0065-0_12. [DOI] [PubMed] [Google Scholar]
- Lefebvre T, Ferreira S, Dupont-Wallois L, Bussière T, Dupire MJ, Delacourte A, Michalski JC, Caillet-Boudin ML. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins–a role in nuclear localization. Biochim Biophys Acta. 2003b;1619:167–176. doi: 10.1016/s0304-4165(02)00477-4. [DOI] [PubMed] [Google Scholar]
- Lefebvre T, Guinez C, Dehennaut V, Beseme-Dekeyser O, Morelle W, Michalski JC. Does O-GlcNAc play a role in neurodegenerative diseases. Expert Rev Proteomics. 2005;2:265–275. doi: 10.1586/14789450.2.2.265. [DOI] [PubMed] [Google Scholar]
- Lehman DM, Fu DJ, Freeman AB, Hunt KJ, Leach RJ, Johnson-Pais T, Hamlington J, Dyer TD, Arya R, Abboud H, Goring HH, Duggirala R, Blangero J, Konrad RJ, Stern MP. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-acetyl-beta-D glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes. 2005;54:1214–1221. doi: 10.2337/diabetes.54.4.1214. [DOI] [PubMed] [Google Scholar]
- Levine PM, De Leon CA, Galesic A, Balana A, Marotta NP, Lewis YE, Pratt MR. O-GlcNAc modification inhibits the calpain-mediated cleavage of α-synuclein. Bioorg Med Chem. 2017 doi: 10.1016/j.bmc.2017.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BA, Hanover JA. O-GlcNAc and the epigenetic regulation of gene expression. J Biol Chem. 2014;289:34440–34448. doi: 10.1074/jbc.R114.595439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis YE, Galesic A, Levine PM, De Leon CA, Lamiri N, Brennan CK, Pratt MR. O-GlcNAcylation of α-Synuclein at Serine 87 Reduces Aggregation without Affecting Membrane Binding. ACS Chem Biol. 2017;12:1020–1027. doi: 10.1021/acschembio.7b00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Li H, Lu L, Jiang J. Structures of human O-GlcNAcase and its complexes reveal a new substrate recognition mode. Nat Struct Mol Biol. 2017;24:362–369. doi: 10.1038/nsmb.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang J, Hou W, Jing Z, Tian C, Han Y, Liao J, Dong MQ, Xu X. Phosphorylation of Ataxin-10 by polo-like kinase 1 is required for cytokinesis. Cell Cycle. 2011;10:2946–2958. doi: 10.4161/cc.10.17.15922. [DOI] [PubMed] [Google Scholar]
- Lipinski MM, Zheng B, Lu T, Yan Z. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proceedings of the …. 2010 doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004a;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, Iqbal K, Gong CX. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain. 2009;132:1820–1832. doi: 10.1093/brain/awp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Paterson AJ, Zhang F, McAndrew J, Fukuchi K, Wyss JM, Peng L, Hu Y, Kudlow JE. Accumulation of protein O-GlcNAc modification inhibits proteasomes in the brain and coincides with neuronal apoptosis in brain areas with high O-GlcNAc metabolism. J Neurochem. 2004b;89:1044–1055. doi: 10.1111/j.1471-4159.2004.02389.x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Li X, Yu Y, Shi J, Liang Z, Run X, Li Y, Dai CL, Grundke-Iqbal I, Iqbal K, Liu F, Gong CX. Developmental regulation of protein O-GlcNAcylation, O-GlcNAc transferase, and O-GlcNAcase in mammalian brain. PLoS One. 2012;7:e43724. doi: 10.1371/journal.pone.0043724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love DC, Kochan J, Cathey RL, Shin SH, Hanover JA, Kochran J. Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J Cell Sci. 2003;116:647–654. doi: 10.1242/jcs.00246. [DOI] [PubMed] [Google Scholar]
- Love DC, Krause MW, Hanover JA. O-GlcNAc cycling: emerging roles in development and epigenetics. Semin Cell Dev Biol. 2010;21:646–654. doi: 10.1016/j.semcdb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas WA, Frank DW, Krause M, Hanover JA. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem. 1997;272:9316–9324. doi: 10.1074/jbc.272.14.9316. [DOI] [PubMed] [Google Scholar]
- Lubas WA, Hanover JA. Functional expression of O-linked GlcNAc transferase. Domain structure and substrate specificity. J Biol Chem. 2000;275:10983–10988. doi: 10.1074/jbc.275.15.10983. [DOI] [PubMed] [Google Scholar]
- Lüthi T, Haltiwanger RS, Greengard P, Bähler M. Synapsins contain O-linked N-acetylglucosamine. J Neurochem. 1991;56:1493–1498. doi: 10.1111/j.1471-4159.1991.tb02043.x. [DOI] [PubMed] [Google Scholar]
- Ma J, Banerjee P, Whelan SA, Liu T, Wei AC, Ramirez-Correa G, McComb ME, Costello CE, O’Rourke B, Murphy A, Hart GW. Comparative Proteomics Reveals Dysregulated Mitochondrial O-GlcNAcylation in Diabetic Hearts. J Proteome Res. 2016;15:2254–2264. doi: 10.1021/acs.jproteome.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Liu P, Yan H, Sun H, Liu X, Zhou F, Li L, Chen Y. Substrate specificity provides insights into the sugar donor recognition mechanism of O-GlcNAc transferase (OGT) PloS one. 2013 doi: 10.1371/journal.pone.0063452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010;20:102–112. doi: 10.1016/j.tcb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley MS, Bubb AK, Martinez-Fleites C, Davies GJ, Vocadlo DJ. Elevation of global O-GlcNAc levels in 3T3-L1 adipocytes by selective inhibition of O-GlcNAcase does not induce insulin resistance. J Biol Chem. 2008;283:34687–34695. doi: 10.1074/jbc.M804525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley MS, Shan X, Yuzwa SA, Gloster TM, Vocadlo DJ. Elevation of Global O-GlcNAc in rodents using a selective O-GlcNAcase inhibitor does not cause insulin resistance or perturb glucohomeostasis. Chem Biol. 2010;17:949–958. doi: 10.1016/j.chembiol.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley MS, Vocadlo DJ. Enzymatic characterization and inhibition of the nuclear variant of human O-GlcNAcase. Carbohydr Res. 2009;344:1079–1084. doi: 10.1016/j.carres.2009.04.017. [DOI] [PubMed] [Google Scholar]
- Mao X, Zhang D, Tao T, Liu X, Sun X, Wang Y, Shen A. O-GlcNAc glycosylation of p27(kip1) promotes astrocyte migration and functional recovery after spinal cord contusion. Exp Cell Res. 2015;339:197–205. doi: 10.1016/j.yexcr.2015.11.007. [DOI] [PubMed] [Google Scholar]
- Marks JL, Porte D, Stahl WL, Baskin DG. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology. 1990;127:3234–3236. doi: 10.1210/endo-127-6-3234. [DOI] [PubMed] [Google Scholar]
- Marotta NP, Lin YH, Lewis YE, Ambroso MR, Zaro BW, Roth MT, Arnold DB, Langen R, Pratt MR. O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson’s disease. Nat Chem. 2015;7:913–920. doi: 10.1038/nchem.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SA, Powell PC, Dell’italia LJ, Chatham JC. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci. 2013;92:648–656. doi: 10.1016/j.lfs.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall S, Duong T, Orbus RJ, Rumberger JM, Okuyama R. Measurement of UDP-N-acetylglucosaminyl transferase (OGT) in brain cytosol and characterization of anti-OGT antibodies. Anal Biochem. 2003;314:169–179. doi: 10.1016/s0003-2697(02)00686-3. [DOI] [PubMed] [Google Scholar]
- März P, Stetefeld J, Bendfeldt K, Nitsch C, Reinstein J, Shoeman RL, Dimitriades-Schmutz B, Schwager M, Leiser D, Ozcan S, Otten U, Ozbek S. Ataxin-10 interacts with O-linked beta-N-acetylglucosamine transferase in the brain. J Biol Chem. 2006;281:20263–20270. doi: 10.1074/jbc.M601563200. [DOI] [PubMed] [Google Scholar]
- Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, Sekita A, Suzuki SO, Kanba S, Kiyohara Y, Iwaki T. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75:764–770. doi: 10.1212/WNL.0b013e3181eee25f. [DOI] [PubMed] [Google Scholar]
- Mazars R, Gonzalez-de-Peredo A, Cayrol C, Lavigne AC, Vogel JL, Ortega N, Lacroix C, Gautier V, Huet G, Ray A, Monsarrat B, Kristie TM, Girard JP. The THAP-zinc finger protein THAP1 associates with coactivator HCF-1 and O-GlcNAc transferase: a link between DYT6 and DYT3 dystonias. J Biol Chem. 2010;285:13364–13371. doi: 10.1074/jbc.M109.072579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain DA, Lubas WA, Cooksey RC, Hazel M, Parker GJ, Love DC, Hanover JA. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc Natl Acad Sci U S A. 2002;99:10695–10699. doi: 10.1073/pnas.152346899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Annals of …. 1999 doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36:587–597. doi: 10.1016/j.tins.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietelska-Porowska A, Wasik U, Goras M. Tau protein modifications and interactions: their role in function and dysfunction. International journal of …. 2014 doi: 10.3390/ijms15034671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milward EA, Papadopoulos R, Fuller SJ, Moir RD, Small D, Beyreuther K, Masters CL. The amyloid protein precursor of Alzheimer’s disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9:129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- Murphy JE, Hanover JA, Froehlich M, DuBois G, Keen JH. Clathrin assembly protein AP-3 is phosphorylated and glycosylated on the 50-kDa structural domain. J Biol Chem. 1994;269:21346–21352. [PubMed] [Google Scholar]
- Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D, Shears S, Booth J, DeVrieze FW, Crook R, Hamshere M, Abraham R, Tunstall N, Rice F, Carty S, Lillystone S, Kehoe P, Rudrasingham V, Jones L, Lovestone S, Perez-Tur J, Williams J, Owen MJ, Hardy J, Goate AM. Susceptibility locus for Alzheimer’s disease on chromosome 10. Science. 2000;290:2304–2305. doi: 10.1126/science.290.5500.2304. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Esiri MM, Cato AM, Smith AD. Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol. 1997;94:6–15. doi: 10.1007/s004010050665. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Nakano S, Nishii M, Kaneko S, Kusaka H. Localization of O-GlcNAc-modified proteins in neuromuscular diseases. Med Mol Morphol. 2012;45:86–90. doi: 10.1007/s00795-011-0542-7. [DOI] [PubMed] [Google Scholar]
- Nandi A, Sprung R, Barma DK, Zhao Y, Kim SC, Falck JR, Zhao Y. Global identification of O-GlcNAc-modified proteins. Anal Chem. 2006;78:452–458. doi: 10.1021/ac051207j. [DOI] [PubMed] [Google Scholar]
- Németh AH, Nolte D, Dunne E, Niemann S, Kostrzewa M, Peters U, Fraser E, Bochukova E, Butler R, Brown J, Cox RD, Levy ER, Ropers HH, Monaco AP, Müller U. Refined linkage disequilibrium and physical mapping of the gene locus for X-linked dystonia-parkinsonism (DYT3) Genomics. 1999;60:320–329. doi: 10.1006/geno.1999.5929. [DOI] [PubMed] [Google Scholar]
- Ngoh GA, Watson LJ, Facundo HT, Dillmann W, Jones SP. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. J Mol Cell Cardiol. 2008;45:313–325. doi: 10.1016/j.yjmcc.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning X, Tao T, Shen J, Ji Y, Xie L, Wang H, Liu N, Xu X, Sun C, Zhang D, Shen A, Ke K. The O-GlcNAc Modification of CDK5 Involved in Neuronal Apoptosis Following In Vitro Intracerebral Hemorrhage. Cell Mol Neurobiol. 2017;37:527–536. doi: 10.1007/s10571-016-0391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjan TS, Skinner C, May M, Turner T, Rose R, Stevenson R, Schwartz CE, Wang T. Affected kindred analysis of human X chromosome exomes to identify novel X-linked intellectual disability genes. PLoS One. 2015;10:e0116454. doi: 10.1371/journal.pone.0116454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. Journal of …. 2005 doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Nugent BM, Bale TL. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol. 2015;39:28–37. doi: 10.1016/j.yfrne.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell N, Zachara NE, Hart GW, Marth JD. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol Cell Biol. 2004;24:1680–1690. doi: 10.1128/MCB.24.4.1680-1690.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. 2002;5:566–572. doi: 10.1038/nn0602-861. [DOI] [PubMed] [Google Scholar]
- Ogawa O, Lee HG, Zhu X, Raina A, Harris PL, Castellani RJ, Perry G, Smith MA. Increased p27, an essential component of cell cycle control, in Alzheimer’s disease. Aging Cell. 2003a;2:105–110. doi: 10.1046/j.1474-9728.2003.00042.x. [DOI] [PubMed] [Google Scholar]
- Ogawa O, Zhu X, Lee HG, Raina A, Obrenovich ME, Bowser R, Ghanbari HA, Castellani RJ, Perry G, Smith MA. Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a mitotic catastrophe. Acta Neuropathol. 2003b;105:524–528. doi: 10.1007/s00401-003-0684-3. [DOI] [PubMed] [Google Scholar]
- Olivier-Van Stichelen S, Abramowitz LK, Hanover JA. X marks the spot: does it matter that O-GlcNAc transferase is an X-linked gene. Biochem Biophys Res Commun. 2014a;453:201–207. doi: 10.1016/j.bbrc.2014.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier-Van Stichelen S, Dehennaut V, Buzy A, Zachayus JL, Guinez C, Mir AM, El Yazidi-Belkoura I, Copin MC, Boureme D, Loyaux D, Ferrara P, Lefebvre T. O-GlcNAcylation stabilizes β-catenin through direct competition with phosphorylation at threonine 41. FASEB J. 2014b;28:3325–3338. doi: 10.1096/fj.13-243535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier-Van Stichelen S, Drougat L, Dehennaut V, El Yazidi-Belkoura I, Guinez C, Mir AM, Michalski JC, Vercoutter-Edouart AS, Lefebvre T. Serum-stimulated cell cycle entry promotes ncOGT synthesis required for cyclin D expression. Oncogenesis. 2012;1:e36. doi: 10.1038/oncsis.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier-Van Stichelen S, Hanover JA. X-inactivation normalizes O-GlcNAc transferase levels and generates an O-GlcNAc-depleted Barr body. Front Genet. 2014;5:256. doi: 10.3389/fgene.2014.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier-Van Stichelen S, Wang P, Comly M, Love DC, Hanover JA. Nutrient-driven O-linked N-acetylglucosamine (O-GlcNAc) cycling impacts neurodevelopmental timing and metabolism. J Biol Chem. 2017;292:6076–6085. doi: 10.1074/jbc.M116.774042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omary MB, Ku NO, Liao J, Price D. Keratin modifications and solubility properties in epithelial cells and in vitro. Subcell Biochem. 1998;31:105–140. [PubMed] [Google Scholar]
- Palaniappan KK, Hangauer MJ, Smith TJ, Smart BP, Pitcher AA, Cheng EH, Bertozzi CR, Boyce M. A chemical glycoproteomics platform reveals O-GlcNAcylation of mitochondrial voltage-dependent anion channel 2. Cell Rep. 2013;5:546–552. doi: 10.1016/j.celrep.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleon M, Steane SE, McMahon K, Cuffe JSM, Moritz KM. Placental O-GlcNAc-transferase expression and interactions with the glucocorticoid receptor are sex specific and regulated by maternal corticosterone exposure in mice. Sci Rep. 2017;7:2017. doi: 10.1038/s41598-017-01666-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parathath SR, Mainwaring LA, Fernandez-L A, Campbell DO, Kenney AM. Insulin receptor substrate 1 is an effector of sonic hedgehog mitogenic signaling in cerebellar neural precursors. Development. 2008;135:3291–3300. doi: 10.1242/dev.022871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, D’Onofrio M, Willingham MC, Hanover JA. A monoclonal antibody against a family of nuclear pore proteins (nucleoporins): O-linked N-acetylglucosamine is part of the immunodeterminant. Proc Natl Acad Sci U S A. 1987;84:6462–6466. doi: 10.1073/pnas.84.18.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Lee Y, Pak JW, Kim H, Choi H, Kim J. O-GlcNAc modification is essential for the regulation of autophagy in Drosophila melanogaster. Cellular and molecular …. 2015 doi: 10.1007/s00018-015-1889-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SS, Lee D. Selective loss of dopaminergic neurons and formation of Lewy body-like aggregations in alpha-synuclein transgenic fly neuronal cultures. Eur J Neurosci. 2006;23:2908–2914. doi: 10.1111/j.1460-9568.2006.04844.x. [DOI] [PubMed] [Google Scholar]
- Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell. 2014;158:54–68. doi: 10.1016/j.cell.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Cervera Y, Dehennaut V, Aquino Gil M, Guedri K, Solórzano Mata CJ, Olivier-Van Stichelen S, Michalski JC, Foulquier F, Lefebvre T. Insulin signaling controls the expression of O-GlcNAc transferase and its interaction with lipid microdomains. FASEB J. 2013;27:3478–3486. doi: 10.1096/fj.12-217984. [DOI] [PubMed] [Google Scholar]
- Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. doi: 10.1083/jcb.201208152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss U, Mandelkow EM. Mitotic phosphorylation of tau protein in neuronal cell lines resembles phosphorylation in Alzheimer’s disease. Eur J Cell Biol. 1998;76:176–184. doi: 10.1016/S0171-9335(98)80032-0. [DOI] [PubMed] [Google Scholar]
- Qiu H, Liu F, Tao T, Zhang D, Liu X, Zhu G, Xu Z, Ni R, Shen A. Modification of p27 with O-linked N-acetylglucosamine regulates cell proliferation in hepatocellular carcinoma. Mol Carcinog. 2017;56:258–271. doi: 10.1002/mc.22490. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Radi E, Formichi P, Battisti C, Federico A. Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis. 2014;42(Suppl 3):S125–52. doi: 10.3233/JAD-132738. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. Activation of the Transcriptional Function of the NF-kappaB Protein c-Rel by O-GlcNAc Glycosylation. Sci Signal. 2013;6:ra75. doi: 10.1126/scisignal.2004097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao FV, Schüttelkopf AW, Dorfmueller HC, Ferenbach AT, Navratilova I, van Aalten DM. Structure of a bacterial putative acetyltransferase defines the fold of the human O-GlcNAcase C-terminal domain. Open Biol. 2013;3:130021. doi: 10.1098/rsob.130021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reason AJ, Morris HR, Panico M, Marais R, Treisman RH, Haltiwanger RS, Hart GW, Kelly WG, Dell A. Localization of O-GlcNAc modification on the serum response transcription factor. J Biol Chem. 1992;267:16911–16921. [PubMed] [Google Scholar]
- Rensink AA, Otte-Höller I, de Boer R, Bosch RR, ten Donkelaar HJ, de Waal RM, Verbeek MM, Kremer B. Insulin inhibits amyloid beta-induced cell death in cultured human brain pericytes. Neurobiol Aging. 2004;25:93–103. doi: 10.1016/s0197-4580(03)00039-3. [DOI] [PubMed] [Google Scholar]
- Rex-Mathes M, Werner S, Strutas D, Griffith LS, Viebahn C, Thelen K, Schmitz B. O-GlcNAc expression in developing and ageing mouse brain. Biochimie. 2001;83:583–590. doi: 10.1016/s0300-9084(01)01305-0. [DOI] [PubMed] [Google Scholar]
- Rexach JE, Clark PM, Hsieh-Wilson LC. Chemical approaches to understanding O-GlcNAc glycosylation in the brain. Nat Chem Biol. 2008;4:97–106. doi: 10.1038/nchembio.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach JE, Rogers CJ, Yu SH, Tao J, Sun YE, Hsieh-Wilson LC. Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags. Nat Chem Biol. 2010;6:645–651. doi: 10.1038/nchembio.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson LA, Moya KL, Breen KC. The potential role of tau protein O-glycosylation in Alzheimer’s disease. J Alzheimers Dis. 2004;6:489–495. doi: 10.3233/jad-2004-6505. [DOI] [PubMed] [Google Scholar]
- Roos MD, Su K, Baker JR, Kudlow JE. O glycosylation of an Sp1-derived peptide blocks known Sp1 protein interactions. Mol Cell Biol. 1997;17:6472–6480. doi: 10.1128/mcb.17.11.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roquemore EP, Chevrier MR, Cotter RJ, Hart GW. Dynamic O-GlcNAcylation of the small heat shock protein alpha B-crystallin. Biochemistry. 1996;35:3578–3586. doi: 10.1021/bi951918j. [DOI] [PubMed] [Google Scholar]
- Roquemore EP, Dell A, Morris HR, Panico M, Reason AJ, Savoy LA, Wistow GJ, Zigler JS, Earles BJ, Hart GW. Vertebrate lens alpha-crystallins are modified by O-linked N-acetylglucosamine. J Biol Chem. 1992;267:555–563. [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–7. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration. Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- Ross SA, Chen X, Hope HR, Sun S, McMahon EG, Broschat K, Gulve EA. Development and comparison of two 3T3-L1 adipocyte models of insulin resistance: increased glucose flux vs glucosamine treatment. Biochem Biophys Res Commun. 2000;273:1033–1041. doi: 10.1006/bbrc.2000.3082. [DOI] [PubMed] [Google Scholar]
- Roth C, Chan S, Offen WA, Hemsworth GR, Willems LI, King DT, Varghese V, Britton R, Vocadlo DJ, Davies GJ. Structural and functional insight into human O-GlcNAcase. Nat Chem Biol. 2017;13:610–612. doi: 10.1038/nchembio.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HB, Dietrich MO, Liu ZW, Zimmer MR, Li MD, Singh JP, Zhang K, Yin R, Wu J, Horvath TL, Yang X. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell. 2014;159:306–317. doi: 10.1016/j.cell.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, Zhao L, Bennett AM, Samuel VT, Wu J, Yates JR, Yang X. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab. 2012;16:226–237. doi: 10.1016/j.cmet.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu BR, Ko HW, Jou I, Noh JS, Gwag BJ. Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. J Neurobiol. 1999;39:536–546. [PubMed] [Google Scholar]
- Ryu IH, Do SI. Denitrosylation of S-nitrosylated OGT is triggered in LPS-stimulated innate immune response. Biochem Biophys Res Commun. 2011;408:52–57. doi: 10.1016/j.bbrc.2011.03.115. [DOI] [PubMed] [Google Scholar]
- Ryu IH, Lee KY, Do SI. Aβ-affected pathogenic induction of S-nitrosylation of OGT and identification of Cys-NO linkage triplet. Biochim Biophys Acta. 2016;1864:609–621. doi: 10.1016/j.bbapap.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Sacoman JL, Dagda RY, Burnham-Marusich AR, Dagda RK, Berninsone PM. Mitochondrial O-GlcNAc Transferase (mOGT) Regulates Mitochondrial Structure, Function, and Survival in HeLa Cells. J Biol Chem. 2017;292:4499–4518. doi: 10.1074/jbc.M116.726752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe K, Hart GW. O-GlcNAc transferase regulates mitotic chromatin dynamics. J Biol Chem. 2010;285:34460–34468. doi: 10.1074/jbc.M110.158170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi SD, Leon MJD, Rusinek H, Convit A. Hippocampal formation glucose metabolism and volume losses in MCI and AD. Neurobiology of …. 2001 doi: 10.1016/s0197-4580(01)00230-5. [DOI] [PubMed] [Google Scholar]
- Schubert D, Cole G, Saitoh T, Oltersdorf T. Amyloid beta protein precursor is a mitogen. Biochem Biophys Res Commun. 1989;162:83–88. doi: 10.1016/0006-291x(89)91965-7. [DOI] [PubMed] [Google Scholar]
- Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, Farhang-Fallah J, Dikkes P, Warot XM, Rio C, Corfas G, White MF. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci. 2003;23:7084–7092. doi: 10.1523/JNEUROSCI.23-18-07084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sean M, Smith1 AS, Jonathan1 Daniel, Declercq1 Ruben, Marcus2 Jacob, Toolan1 Dawn, Wang1 Xiaohai, Schachter1 Joel B, Cosden1 Mali, Pearson1 Michelle, Hess1 Fred, Selnick1 Harold, Salinas1 Cristian, Li1 Wenping, Duffy1 Joseph, McEachern3 Ernest, Vocadlo3 David, Renger1 John J, Eric1 Hostetler D, Forman1 Mark, Schoepp1 Darryle. EARLY CLINICAL RESULTS AND PRECLINICAL VALIDATION OF THE O-GLCNACASE (OGA) INHIBITOR MK-8719 AS A NOVEL THERAPEUTIC FOR THE TREATMENT OF TAUOPATHIES. Alzheimers and Dementia. 2016:261. [Google Scholar]
- Sekine O, Love DC, Rubenstein DS, Hanover JA. Blocking O-linked GlcNAc cycling in Drosophila insulin-producing cells perturbs glucose-insulin homeostasis. J Biol Chem. 2010;285:38684–38691. doi: 10.1074/jbc.M110.155192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih RH, Wang CY, Yang CM. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front Mol Neurosci. 2015;8:77. doi: 10.3389/fnmol.2015.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SH, Love DC, Hanover JA. Elevated O-GlcNAc-dependent signaling through inducible mOGT expression selectively triggers apoptosis. Amino Acids. 2011;40:885–893. doi: 10.1007/s00726-010-0719-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoghi-Jadid K, Small GW, Agdeppa ED. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. The American Journal of …. 2002 [PubMed] [Google Scholar]
- Simpson IA, Dwyer D, Malide D. The facilitative glucose transporter GLUT3: 20 years of distinction. American Journal of …. 2008 doi: 10.1152/ajpendo.90388.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Syrzycka M, Macauley MS, Rastgardani T, Komljenovic I, Vocadlo DJ, Brock HW, Honda BM. Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc) Proc Natl Acad Sci U S A. 2009;106:13427–13432. doi: 10.1073/pnas.0904638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorobogatko Y, Landicho A, Chalkley RJ, Kossenkov AV, Gallo G, Vosseller K. O-linked β-N-acetylglucosamine (O-GlcNAc) site thr-87 regulates synapsin I localization to synapses and size of the reserve pool of synaptic vesicles. J Biol Chem. 2014;289:3602–3612. doi: 10.1074/jbc.M113.512814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawson C, Lakshmanan T, Knapp S, Hart GW. A mitotic GlcNAcylation/phosphorylation signaling complex alters the posttranslational state of the cytoskeletal protein vimentin. Mol Biol Cell. 2008;19:4130–4140. doi: 10.1091/mbc.E07-11-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawson C, Zachara NE, Vosseller K, Cheung WD, Lane MD, Hart GW. Perturbations in O-linked beta-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J Biol Chem. 2005;280:32944–32956. doi: 10.1074/jbc.M503396200. [DOI] [PubMed] [Google Scholar]
- Smet-Nocca C, Broncel M, Wieruszeski JM, Tokarski C, Hanoulle X, Leroy A, Landrieu I, Rolando C, Lippens G, Hackenberger CP. Identification of O-GlcNAc sites within peptides of the Tau protein and their impact on phosphorylation. Mol Biosyst. 2011;7:1420–1429. doi: 10.1039/c0mb00337a. [DOI] [PubMed] [Google Scholar]
- Snow CM, Senior A, Gerace L. Monoclonal antibodies identify a group of nuclear pore complex glycoproteins. J Cell Biol. 1987;104:1143–1156. doi: 10.1083/jcb.104.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ. α-Synuclein in Lewy bodies. Nature. 1997 doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Sprung R, Nandi A, Chen Y, Kim SC, Barma D, Falck JR, Zhao Y. Tagging-via-substrate strategy for probing O-GlcNAc modified proteins. J Proteome Res. 2005;4:950–957. doi: 10.1021/pr050033j. [DOI] [PubMed] [Google Scholar]
- Srikanth B, Vaidya MM, Kalraiya RD. O-GlcNAcylation determines the solubility, filament organization, and stability of keratins 8 and 18. J Biol Chem. 2010;285:34062–34071. doi: 10.1074/jbc.M109.098996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr CM, D’Onofrio M, Park MK, Hanover JA. Primary sequence and heterologous expression of nuclear pore glycoprotein p62. J Cell Biol. 1990;110:1861–1871. doi: 10.1083/jcb.110.6.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr CM, Hanover JA. Glycosylation of nuclear pore protein p62. Reticulocyte lysate catalyzes O-linked N-acetylglucosamine addition in vitro. J Biol Chem. 1990;265:6868–6873. [PubMed] [Google Scholar]
- Su C, Schwarz TL. O-GlcNAc Transferase Is Essential for Sensory Neuron Survival and Maintenance. J Neurosci. 2017;37:2125–2136. doi: 10.1523/JNEUROSCI.3384-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sümegi M, Hunyadi-Gulyás E, Medzihradszky KF, Udvardy A. 26S proteasome subunits are O-linked N-acetylglucosamine-modified in Drosophila melanogaster. Biochem Biophys Res Commun. 2003;312:1284–1289. doi: 10.1016/j.bbrc.2003.11.074. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Nagatani S, Bucholtz DC, Ohkura S, Tsukamura H, Maeda K, Foster DL. Central action of insulin regulates pulsatile luteinizing hormone secretion in the diabetic sheep model. Biol Reprod. 2000;62:1256–1261. doi: 10.1095/biolreprod62.5.1256. [DOI] [PubMed] [Google Scholar]
- Taylor EW, Wang K, Nelson AR, Bredemann TM, Fraser KB, Clinton SM, Puckett R, Marchase RB, Chatham JC, McMahon LL. O-GlcNAcylation of AMPA receptor GluA2 is associated with a novel form of long-term depression at hippocampal synapses. J Neurosci. 2014;34:10–21. doi: 10.1523/JNEUROSCI.4761-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toleman C, Paterson AJ, Whisenhunt TR, Kudlow JE. Characterization of the histone acetyltransferase (HAT) domain of a bifunctional protein with activable O-GlcNAcase and HAT activities. J Biol Chem. 2004;279:53665–53673. doi: 10.1074/jbc.M410406200. [DOI] [PubMed] [Google Scholar]
- Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. 1984;259:3308–3317. [PubMed] [Google Scholar]
- Toth C, Brussee V, Martinez JA, McDonald D, Cunningham FA, Zochodne DW. Rescue and regeneration of injured peripheral nerve axons by intrathecal insulin. Neuroscience. 2006;139:429–449. doi: 10.1016/j.neuroscience.2005.11.065. [DOI] [PubMed] [Google Scholar]
- Trapannone R, Mariappa D, Ferenbach AT, van Aalten DM. Nucleocytoplasmic human O-GlcNAc transferase is sufficient for O-GlcNAcylation of mitochondrial proteins. Biochem J. 2016;473:1693–1702. doi: 10.1042/BCJ20160092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapannone R, Rafie K. O-GlcNAc transferase inhibitors: current tools and future challenges. Biochemical Society …. 2016 doi: 10.1042/BST20150189. [DOI] [PubMed] [Google Scholar]
- Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, Burlingame AL. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics. 2012;11:215–229. doi: 10.1074/mcp.O112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front Aging Neurosci. 2015;7:18. doi: 10.3389/fnagi.2015.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan K, Niranjan T, Selvan N, Teo CF, May M, Patel S, Weatherly B, Skinner C, Opitz J, Carey J, Viskochil D, Gecz J, Shaw M, Peng Y, Alexov E, Wang T, Schwartz C, Wells L. Identification and characterization of a missense mutation in the O-linked β-N-acetylglucosamine (O-GlcNAc) transferase gene that segregates with X-linked intellectual disability. J Biol Chem. 2017;292:8948–8963. doi: 10.1074/jbc.M116.771030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- van der Harg JM, van Heest JC, Bangel FN, Patiwael S, van Weering, JR, Scheper W. The UPR reduces glucose metabolism via IRE1 signaling. Biochim Biophys Acta. 2017;1864:655–665. doi: 10.1016/j.bbamcr.2017.01.009. [DOI] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, Jaarsma D, Gerritsen HC, Akhmanova A, Hoogenraad CC. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77:485–502. doi: 10.1016/j.neuron.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voll CL, Whishaw IQ, Auer RN. Postischemic insulin reduces spatial learning deficit following transient forebrain ischemia in rats. Stroke. 1989;20:646–651. doi: 10.1161/01.str.20.5.646. [DOI] [PubMed] [Google Scholar]
- Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 2002;99:5313–5318. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh-Wilson LC. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc Natl Acad Sci U S A. 2016;113:15120–15125. doi: 10.1073/pnas.1606899113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Hanover JA. Nutrient-driven O-GlcNAc cycling influences autophagic flux and neurodegenerative proteotoxicity. Autophagy. 2013;9:604–606. doi: 10.4161/auto.23459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Lazarus BD, Forsythe ME, Love DC, Krause MW, Hanover JA. O-GlcNAc cycling mutants modulate proteotoxicity in Caenorhabditis elegans models of human neurodegenerative diseases. Proc Natl Acad Sci U S A. 2012;109:17669–17674. doi: 10.1073/pnas.1205748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Udeshi ND, O’Malley M, Shabanowitz J. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer …. … & Cellular Proteomics. 2010a doi: 10.1074/mcp.M900268-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Udeshi ND, Slawson C, Compton PD, Sakabe K, Cheung WD, Shabanowitz J, Hunt DF, Hart GW. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci Signal. 2010b;3:ra2. doi: 10.1126/scisignal.2000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani WY, Chatham JC, Darley-Usmar V, McMahon LL, Zhang J. O-GlcNAcylation and neurodegeneration. Brain Res Bull. 2016a doi: 10.1016/j.brainresbull.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani WY, Chatham JC, Darley-Usmar V. O-GlcNAcylation and neurodegeneration. … Research Bulletin. 2016b doi: 10.1016/j.brainresbull.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner M, Khachaturian Z. The use of MRI and PET for clinical diagnosis of dementia and investigation of cognitive impairment: a consensus report. Alzheimer’s Association 2005 [Google Scholar]
- Wells L, Gao Y, Mahoney JA, Vosseller K, Chen C, Rosen A, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: further characterization of the nucleocytoplasmic beta-N-acetylglucosaminidase, O-GlcNAcase. J Biol Chem. 2002;277:1755–1761. doi: 10.1074/jbc.m109656200. [DOI] [PubMed] [Google Scholar]
- Wells L, Vosseller K, Hart GW. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell Mol Life Sci. 2003;60:222–228. doi: 10.1007/s000180300017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SA, Dias WB, Thiruneelakantapillai L, Lane MD, Hart GW. Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-Linked beta-N-acetylglucosamine in 3T3-L1 adipocytes. J Biol Chem. 2010;285:5204–5211. doi: 10.1074/jbc.M109.077818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SA, Hart GW. Proteomic approaches to analyze the dynamic relationships between nucleocytoplasmic protein glycosylation and phosphorylation. Circ Res. 2003;93:1047–1058. doi: 10.1161/01.RES.0000103190.20260.37. [DOI] [PubMed] [Google Scholar]
- Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur J Neurosci. 2011;33:1139–1151. doi: 10.1111/j.1460-9568.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- Winner B, Winkler J. Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2015;7:a021287. doi: 10.1101/cshperspect.a021287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak M, Rydzewski B, Baker SP, Raizada MK. The cellular and physiological actions of insulin in the central nervous system. Neurochem Int. 1993;22:1–10. doi: 10.1016/0197-0186(93)90062-a. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 2003;17:896–911. doi: 10.1101/gad.252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Yu RK. O-linked beta-N-acetylglucosaminylation in mouse embryonic neural precursor cells. J Neurosci Res. 2009;87:3535–3545. doi: 10.1002/jnr.22170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, Cho JW. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol. 2006;8:1074–1083. doi: 10.1038/ncb1470. [DOI] [PubMed] [Google Scholar]
- Yang WH, Park SY, Nam HW, Kim do H, Kang JG, Kang ES, Kim YS, Lee HC, Kim KS, Cho JW. NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc Natl Acad Sci U S A. 2008a;105:17345–17350. doi: 10.1073/pnas.0806198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, Kudlow JE, Michell RH, Olefsky JM, Field SJ, Evans RM. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature. 2008b;451:964–969. doi: 10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18:452–465. doi: 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhang F, Kudlow JE. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell. 2002;110:69–80. doi: 10.1016/s0092-8674(02)00810-3. [DOI] [PubMed] [Google Scholar]
- Yang YR, Song M, Lee H, Jeon Y, Choi EJ, Jang HJ, Moon HY, Byun HY, Kim EK, Kim DH, Lee MN, Koh A, Ghim J, Choi JH, Lee-Kwon W, Kim KT, Ryu SH, Suh PG. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell. 2012;11:439–448. doi: 10.1111/j.1474-9726.2012.00801.x. [DOI] [PubMed] [Google Scholar]
- Yao PJ, Coleman PD. Reduction of O-linked N-acetylglucosamine-modified assembly protein-3 in Alzheimer’s disease. J Neurosci. 1998a;18:2399–2411. doi: 10.1523/JNEUROSCI.18-07-02399.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao PJ, Coleman PD. Reduced O-glycosylated clathrin assembly protein AP180: implication for synaptic vesicle recycling dysfunction in Alzheimer’s disease. Neurosci Lett. 1998b;252:33–36. doi: 10.1016/s0304-3940(98)00547-3. [DOI] [PubMed] [Google Scholar]
- Yu Y, Zhang L, Li X, Run X, Liang Z, Li Y, Liu Y, Lee MH, Grundke-Iqbal I, Iqbal K, Vocadlo DJ, Liu F, Gong CX. Differential effects of an O-GlcNAcase inhibitor on tau phosphorylation. PLoS One. 2012;7:e35277. doi: 10.1371/journal.pone.0035277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Shan X, Jones BA, Zhao G, Woodward ML, Li X, Zhu Y, McEachern EJ, Silverman MA, Watson NV, Gong CX, Vocadlo DJ. Pharmacological inhibition of O-GlcNAcase (OGA) prevents cognitive decline and amyloid plaque formation in bigenic tau/APP mutant mice. Mol Neurodegener. 2014;9:42. doi: 10.1186/1750-1326-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Vocadlo DJ. O-GlcNAc modification and the tauopathies: insights from chemical biology. Curr Alzheimer Res. 2009;6:451–454. doi: 10.2174/156720509789207967. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Vocadlo DJ. Production of O-GlcNAc Modified Recombinant Tau in E. coli and Detection of Ser400 O-GlcNAc Tau In Vivo. Methods Mol Biol. 2017;1523:237–248. doi: 10.1007/978-1-4939-6598-4_13. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Yadav AK, Skorobogatko Y, Clark T, Vosseller K, Vocadlo DJ. Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody. Amino Acids. 2011;40:857–868. doi: 10.1007/s00726-010-0705-1. [DOI] [PubMed] [Google Scholar]
- Zachara NE, Cole RN, Hart GW, Gao Y. Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr Protoc Protein Sci. 2001 doi: 10.1002/0471140864.ps1208s25. Chapter 12:Unit 12.8. [DOI] [PubMed] [Google Scholar]
- Zachara NE, Hart GW, Cole RN, Gao Y. Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr Protoc Mol Biol. 2002 doi: 10.1002/0471142727.mb1706s57. Chapter 17:Unit 17.6. [DOI] [PubMed] [Google Scholar]
- Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell. 2003;115:715–725. doi: 10.1016/s0092-8674(03)00974-7. [DOI] [PubMed] [Google Scholar]
- Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, Alkon DL. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem. 1999;274:34893–34902. doi: 10.1074/jbc.274.49.34893. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Xiao J, Gong S, Clara JA, Ledoux MS. Neural expression of the transcription factor THAP1 during development in rat. Neuroscience. 2013;231:282–295. doi: 10.1016/j.neuroscience.2012.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Tao T, Zhang D, Liu X, Ke K, Shen A. NOS1AP O-GlcNAc Modification Involved in Neuron Apoptosis Induced by Excitotoxicity. Int J Mol Sci. 2015a;16:16560–16575. doi: 10.3390/ijms160716560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Liu TW, Cecioni S, Eskandari R, Zandberg WF, Vocadlo DJ. O-GlcNAc occurs cotranslationally to stabilize nascent polypeptide chains. Nat Chem Biol. 2015b;11:319–325. doi: 10.1038/nchembio.1774. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ. The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem. 2014;289:34472–34481. doi: 10.1074/jbc.R114.601351. [DOI] [PMC free article] [PubMed] [Google Scholar]