

Abstract
The deposition of misfolded β-sheet enriched amyloid protein is a shared feature of many neurodegenerative diseases. Recent studies demonstrated the existence of conformationally diverse strains as a common property for multiple amyloidogenic proteins including α-Synuclein (α-Syn). α-Syn is misfolded and aggregated in a group of neurodegenerative diseases collectively known as α-Synucleinopathies, which include Parkinson’s disease (PD), dementia with Lewy body, multiple system atrophy and also a subset of Alzheimer’s disease patients with concomitant PD-like Lewy bodies and neurites. While sharing the same pathological protein, different α-Synucleinopathies demonstrate distinct clinical and pathological phenotypes, which could result from the existence of diverse pathological α-Syn strains in patients. In this review, we summarized the characteristics of different α-Synucleinopathies and α-Syn strains generated with recombinant α-Syn monomers. We also make predictions of α-Syn strains that could potentially exist in patients based on the knowledge from other amyloid proteins and the clinical and pathological features of different α-Synucleinopathies.
Keywords: α-synuclein strains, protein aggregates, α-synucleinopathy, multiple system atrophy, Parkinson’s disease, dementia with lewy body
α-Synuclein
α-Synuclein (α-Syn) is a 140 amino acid protein discovered almost 30 years ago (Maroteaux and Scheller, 1991). In the central nervous system (CNS), α-Syn is mainly expressed by neurons and localized mostly in the presynapses. The N-terminus of α-Syn contains seven highly conserved hexameric motifs, which are predicted to form an amphipathic alpha-helix structure (Figure 1A). α-Syn monomers exist in equilibrium between a cytosolic, unfolded form and membrane bound, α-helical form (Bertoncini et al., 2005; Burre, 2015; Davidson et al., 1998; Pineda and Burre, 2017; Weinreb et al., 1996). However, there is also evidence indicating that native α-Syn forms folded helical tetramers (Bartels et al., 2011). The middle domain of α-Syn is known as the non-amyloid-component (NAC) domain (61–95), which was first identified in β-amyloid plaques in AD patients (Ueda et al., 1993). The NAC domain plays a key role in both the aggregation and cytotoxicity of α-Syn (Giasson et al., 2001; Luk et al., 2009). The C-terminus of α-Syn is enriched with charged residues and contains multiple phosphorylation sites. α-Syn has been shown to preferentially bind membranes with high curvature (Jensen et al., 2011), which probably explains its presynaptic localization, since synaptic vesicles are among the smallest biological membranes in the brain. The A30P mutation of α-Syn disrupts the interaction of α-Syn with membranes and its presynaptic localization indicating that the N-terminus of α-Syn is critical for membrane interaction (Fortin et al., 2004; Jensen et al., 1998).
Figure 1. α-Synuclein aggregations in different α-Synucleinopathies.
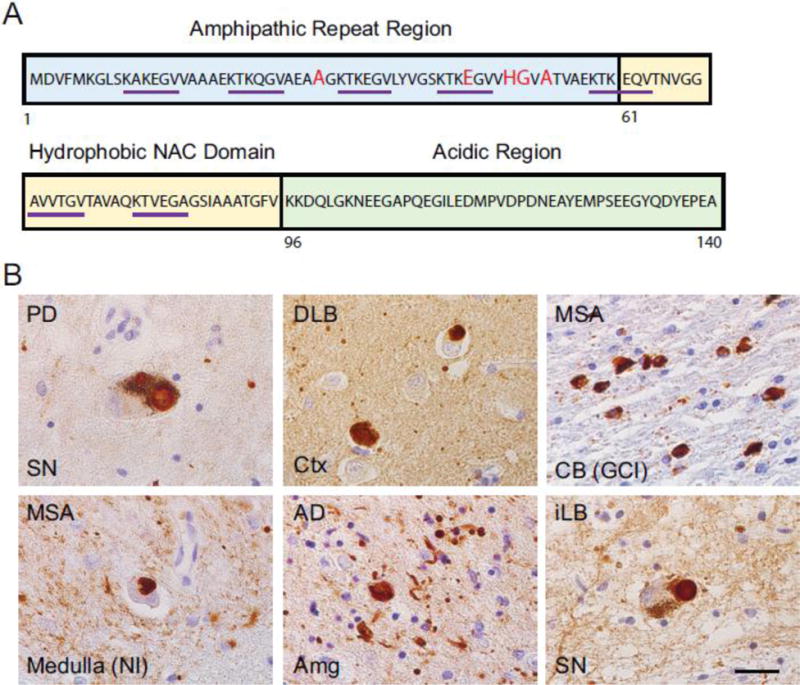
(A) Structural features of α-Synuclein. The N-terminal, NAC and C-terminal domain are highlighted in blue, yellow and green squares respectively. The five PD-related point mutations (A53T, A30P, E46K, H50Q, G51D) are marked in red. The seven hexameric motifs are underlined in purple. (B) α-Synuclein aggregations strained by anti-α-Synuclein antibody (303) in different α-Synucleinopathies. PD: Parkinson’s disease; SN: Substantia nigra; DLB: dementia with Lewy body; Ctx: Cortex; MSA: multiple system atrophy; CB: Cerebellum; GCI: Glial cytoplasmic inclusion; NI: Neuronal inclusion; AD: Alzheimer’s disease; Amg: Amygdala; iLB: Incidental LB disease. Scale bar: 25μm.
The physiological function of α-Syn is still largely unknown. Although α-Syn is localized mainly in the presynaptic terminals in axons, its association with synaptic vesicles is relatively weak and not essential for synapse development (Fortin et al., 2004; Kahle et al., 2000). α-Syn knockout mice only show very modest phenotype, but triple knockouts of α-, β- and γ-synucleins show smaller presynaptic boutons and die at about 1 year (Burre et al., 2010; Fernandez-Chacon et al., 2004; Greten-Harrison et al., 2010), indicating functional redundancy between different members of the synuclein family. Several studies suggested that α-Syn could inhibit neurotransmitter release. Defects in synaptic vesicle exocytosis have been observed in α-Syn transgenic mice and experiments using α-Syn knockout mice indicate that α-Syn deletion leads to increased dopamine release (Abeliovich et al., 2000; Yavich et al., 2006; Yavich et al., 2004). Similar phenomena has also been observed in primary neuron cultures overexpressing α-Syn. Interestingly, the A30P mutation could block the inhibitory effect of α-Syn on release, suggesting that the interaction of α-Syn with the membrane is critical for this effect. Furthermore, α-Syn has been shown to inhibit membrane fusion, suggesting a potential underlying mechanism for this inhibitory effect (DeWitt and Rhoades, 2013). On the other hand, it has been reported that knockout or knockdown of α-Syn lead to reduced levels of dopamine and “reserve” pool of synaptic vesicles (Abeliovich et al., 2000; Cabin et al., 2002; Murphy et al., 2000; Zharikov et al., 2015).
Previous studies have attempted to demonstrate the potential toxic effects of α-Syn in neurons. Even though viral mediated overexpression of α-Syn leads to dopaminergic neuron loss in vivo, transgenic mice overexpressing wild type α-Syn do not show obvious neurotoxicity (Matsuoka et al., 2001). Several studies suggested that α-Syn multimers can form pores on cell membranes, providing a potential mechanism for α-Syn mediated toxicity (Rochet et al., 2004; Tsigelny et al., 2007; Volles et al., 2001). While knocking out α-Syn in mice leads to very modest phenotypes, surprisingly, loss of α-Syn leads to increased resistance to MPTP toxicity, a drug that had been widely used in mice to mimic PD syndromes in animal models (Dauer et al., 2002). On the other hand, α-Syn knockout exacerbates the neurodegeneration cause by the loss of CSPa (Chandra et al., 2005). Thus, it appears that the neurotoxic effects of α-Syn are highly context-dependent.
α-Synucleinopathies
While normally a highly soluble protein, α-Syn forms β-sheet rich amyloid fibrils in a group of neurodegenerative diseases collectively known as α-synucleinopathies, which include Parkinson’s disease (PD) without and with dementia (PDD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA) (Figure 1B). In addition to these three major types of α-Synucleinopathies, about half of the Alzheimer’s disease (AD) patients and several rare disorders also have α-Syn pathology, such as various neuroaxonal dystrophies (Hamilton, 2000; Mikolaenko et al., 2005). Misfolded α-Syn has also been found in more than 20% of neurologically normal elderly individuals known as incidental Lewy body diseases (Markesbery et al., 2009). Different α-synucleinopathies have very distinct clinical and pathological presentations, which rises a very important question as how the same pathological protein lead to dramatically different diseases.
Parkinson’s disease
Parkinson’s Disease was first described by James Parkinson about 200 years ago. It is the most common neurodegenerative movement disorder worldwide and is by far the most common α-Synucleinopathies. Clinically, PD is characterized by three cardinal motor manifestations: bradykinesia, rigidity and rest tremor (Postuma et al., 2015). Other symptoms include posture instability and multiple nonmotor features such as problems with olfaction, insomnia, urinary symptoms and constipation. Cognitive dysfunction is also very common for PD patients and includes visual hallucinations, deficits in attention and executive dysfunction. PD patients were estimated to have fourfold higher chance to develop dementia compared with the general population (Aarsland et al., 2003; Levy et al., 2002). About 50% of PD patients develop cognitive problems and dementia within 10 years from diagnosis (Barker and Williams-Gray, 2016) and 20 years after disease onset, 83% of the patients were demented and 74% of them had hallucinations (Hely et al., 2008). Patients who develop dementia syndrome after more than one year after the original diagnosis of PD are defined as Parkinson’s Disease Dementia (PDD). Based on motor and non-motor symptoms, four PD subtypes have been defined: younger onset, tremor dominant, non-tremor dominant and rapid motor progression (Lewis et al., 2005).
Pathologically, the diagnostic criteria of PD include depigmentation of the substantia nigra and the existence of insoluble α-Syn aggregations in the form of Lewy bodies (LBs) and Lewy neurites (LNs) in neurons (Dickson et al., 2009). By detailed neuropathological analysis of a large number of sporadic PD cases, Braak and colleagues proposed a staging system for α-Syn pathology in PD patients (Braak et al., 2003; Del Tredici et al., 2002). According to the Braak staging system, α-Syn pathology is hypothesized to develop initially in the peripheral mucosa as well as enteric nervous system and then travel into the CNS, where α-Syn aggregates are first detected in brainstem structures at early stages of disease, and progress to limbic and neocortical regions in the later stages. α-Syn pathology in the cortical area is much more severe (up to 10-fold higher) in PDD than PD and the extent of LBs/LNs is believed to correlate with dementia (Compta et al., 2011; Lashley et al., 2008). PDD patients also have a higher Aβ plague and neurofibrillary tangle burden in the cortical area than PD patients (Compta et al., 2011; Irwin et al., 2012b; Jellinger and Attems, 2008; Kempster et al., 2010; Tsuboi et al., 2007) such that up to 50% of PDD patients could be assigned a diagnosis of PDD plus AD (Irwin et al., 2012b; Jellinger, 2007; Kotzbauer et al., 2012; Sabbagh et al., 2009). Importantly, AD pathologies in PDD patients correlated with shorter disease duration and shorter time to dementia (Jellinger et al., 2002; Sabbagh et al., 2009). However, PDD could be differentiated from AD by the presence of clinical symptoms such as sleep disturbance, hallucinations and depression (Lippa et al., 2007). Several pieces of evidence demonstrate a strong correlation between the burden of cortical α-Syn pathology and dementia: 1) In PDD patients, α-Syn pathology are almost exclusively found in the limbic or neocortical area (Apaydin et al., 2002; Compta et al., 2011; Irwin et al., 2012b). 2) Increasing cortical α-syn pathology burden has been found to correlate with diminishing cognitive performance (Braak et al., 2005; Kovari et al., 2003; Mattila et al., 2000). Detailed analyses of α-Syn pathology among above mentioned subtypes of PD illustrate that non-motor dominant subtype has more cortical α-Syn pathology than other subgroups as well as more amyloid-β in the cortical area than younger onset and tremor dominant group, which also confirmed the link between cognitive decline and LB deposition in the neocortex (Selikhova et al., 2009).
Mutations (A53T, A30P, E46K, G51D and H50Q) as well as duplication and triplication of SNCA, the gene coding for α-Syn, has been identified in familial PD patients (Appel-Cresswell et al., 2013; Kruger et al., 1998; Lesage et al., 2013; Polymeropoulos et al., 1997; Proukakis et al., 2013; Zarranz et al., 2004). Symptoms of patients with the A30P mutation are similar to sporadic PD with late disease onset and good response to levodopa therapy (Kruger et al., 2001), while A53T mutation carriers are usually characterized by early disease onset and rapid disease progression (Polymeropoulos et al., 1997; Schiesling et al., 2008). Some patients with E46K mutant have symptoms similar to DLB (Zarranz et al., 2004). Patients with H50Q substitution also develop cognitive impairments (Appel-Cresswell et al., 2013), while G51D mutation carriers are characterized by early disease onset, rapid disease progression and psychiatric symptoms without cognitive impairment during the early stage (Fujioka et al., 2014; Kiely et al., 2013). The symptoms of patients with SNCA duplication is very similar to sporadic PD (Nishioka et al., 2006), while carriers of SNCA triplications develop much more severe phenotype including rapid progression, reduce lifespan and early dementia (Fuchs et al., 2007; Golbe et al., 1996).
Dementia with Lewy Body
The second most common α-Synucleinopathy is DLB. While PD is characterized by the predominance of motor features, DLB is characterized by the predominance of dementia. About 25% of DLB patients demonstrate parkinsonian symptoms at the initial stage of the disease and 25% of DLB patients never develop parkinsonian symptoms (Kim et al., 2014). The cognitive feature and clinical syndrome of DLB is often indistinguishable from PDD. The diagnosis criteria to differentiate DLB and PDD is somewhat arbitrary. Patients who develop dementia before or within one year after the onset of motor symptoms are classified as DLB, while PDD is assigned to patients who develop dementia after one year of the initial diagnosis of PD. Many DLB patients have significant amounts of Aβ deposition in the cortical area and several studies claim that increased number of Aβ plaques in DLB patients could be used to differentiate DLB from PDD (Halliday et al., 2011; Jellinger and Attems, 2006). DLB patients with AD pathology also have a shorter disease duration and have a more rapid decline. Although significant amounts of Aβ plaques in the cortical area are present in many DLB patients, clinicopathological studies demonstrate that DLB is clinically closer to PDD than to AD (Kraybill et al., 2005). Finally, the initiation of α-Syn pathology in DLB is believed to be in the brainstem and eventually spreads to neocortical area, which is very different from AD since in most AD patients, α-Syn pathology is restricted to amygdala.
Multiple system atrophy
MSA is a sporadic orphan disease that is historically known as three separate diseases called oligopontocerebellar atrophy, striatonigral degeneration and Shy-Drager syndrome. The estimated mean incidence is 0.6 to 0.7 cases per 100,000 person-year (Bower et al., 1997). It is characterized by various combinations of autonomic failure, cerebellar ataxia and parkinsonism. MSA patients could be divided to two clinical subtypes: the parkinsonian subtype (MSA-P) with parkinsonism as the predominant feature and the cerebellar subtype (MSA-C) if cerebellar motor symptoms predominate. In most countries, the ratio of MSA-P versus MSA-C patients is 2:1 to 4:1 (Gilman et al., 2005; Kim et al., 2011; Kollensperger et al., 2010). However, MSA-C is more frequent in Japan (Watanabe et al., 2002). MSA is far more aggressive than PD and demonstrates much shorter disease duration than PD (6–9 years for MSA compared with ~12 years for PD) although the age of onset for both diseases is similar (~60 years) (Ben-Shlomo et al., 1997; Fanciulli and Wenning, 2015; McCann et al., 2014). The disease duration of MSA-P and MSA-C are similar. Motor symptoms that characterize MSA-P include rigidity, slowness of movement, nystagmus and a tendency to fall, while MSA-C patients are characterized by cerebellar features including a gait ataxia, action tremor and uncoordinated limb movements. No motor manifestations of MSA include orthostatic hypotension, respiratory disturbances. Unlike PD/PDD or DLB, dementia or visual hallucinations are not found in MSA patients (Gilman et al., 2008).
Pathologically, MSA is distinct from other α-synucleinopathies because it is characterized by the accumulation of pathological α-Syn in oligodendrocytes as glial cytoplasmic inclusions (GCIs) (Tu et al., 1998), while in all other α-synucleinopathies, α-Syn aggregates accumulate in neurons as LBs and LNs. α-Syn pathology in MSA patients distributes mainly in three functional systems: the olivopontocerebellar system, the striatonigral systems and the autonomic system (Kim et al., 2014). Since oligodendrocytes normally do not express or express very low level of α-Syn, the origin of α-Syn in oligodendrocytes still remains a puzzle. Two hypotheses have been put forth to explain the accumulation of GCIs in MSA patients: 1) induced expression of α-Syn in oligodendrocytes, however the evidence for significant expression of α-Syn in oligodendrocytes in MSA patients is controversial (Asi et al., 2014; Miller et al., 2005). 2) transmission of α-Syn from neuron to oligodendrocytes. Despite the ongoing debate about the source of α-Syn, in MSA patients α-Syn accumulates to particular high levels in oligodendrocytes. In addition to GCIs, α-Syn positive accumulation can also be found in the cytoplasm as well as nucleus of neurons (Ahmed et al., 2012). In most patients, neuronal inclusions (NIs) in MSA patients are much less prevalent than GCIs. However, a subgroup of patients with long disease duration and severe temporal atrophy have numerous NIs in limbic structures and the accumulation of NIs is more profound than GCIs in these patients (Yoshida, 2007). α-Syn pathology in both MSA-C and MSA-P patients has been graded between 0 to 3. In MSA-P patients, grade I pathology is restricted to substantia nigra and putamen with the involvement of external globus pallidus and caudate in grade II and III. For MSA-C, grade I is characterized by a mild loss of purkinje cells and cell loss in the substantia nigra and Grade II and III involves widespread pathology in the cerebellum, pons and inferior olives (Jellinger et al., 2005).
α-Syn transmission
The templated recruitment of normal protein by misfolded seeds was originally thought to be a unique feature of prion diseases, in which PrPSc, the conformationally altered protein, acts as a seed and corrupts the normal counterpart PrPC. However, recent surge of studies demonstrate that a similar mechanism is applicable to a large group of neurodegenerative disease related proteins including α-Syn, Aβ, tau, TDP-43, SOD1 and Huntingtin (Clavaguera et al., 2013; Clavaguera et al., 2009; Frost et al., 2009; Guo and Lee, 2011; Iba et al., 2013; Lasagna-Reeves et al., 2012; Luk et al., 2012a; Luk et al., 2012b; Luk et al., 2009; Masuda-Suzukake et al., 2013; Meyer-Luehmann et al., 2006; Munch et al., 2011; Nonaka et al., 2013; Ren et al., 2009; Stohr et al., 2012; Volpicelli-Daley et al., 2011). The spatiotemporal progression of α-Syn pathology in PD and DLB patients is highly stereotypical (Braak et al., 2003), suggesting the spreading of misfolded α-Syn among specific anatomic pathways. More direct evidence comes from the observation of α-Syn aggregation in the grafted fetal mesencephalic neurons in PD patients, which strongly suggests host-to-graft transmission of misfolded α-Syn (Kordower et al., 2008; Li et al., 2008). Consistent with this transmission model, ‘prion-like’ self-propagation of misfolded α-Syn has been demonstrated in cell and animal models using both fibrils assembled from recombinant proteins and aggregate-containing brain lysates obtained from either animals or diseased brain (Luk et al., 2012a; Luk et al., 2012b; Luk et al., 2009; Masuda-Suzukake et al., 2013; Paumier et al., 2015; Prusiner et al., 2015; Volpicelli-Daley et al., 2011; Woerman et al., 2015). α-Syn preformed fibrils (PFF) were able to induce α-Syn aggregation in both a HEK293 cell line stably overexpressing α-Syn and in primary neurons endogenously expressing α-Syn (Luk et al., 2009; Volpicelli-Daley et al., 2011). Brain extracts from symptomatic mutant α-Syn transgenic mice could facilitate the disease progression when inoculated into non-symptomatic young transgenic mice (Luk et al., 2012b). Injection of α-Syn PFF as well as brain lysates from DLB patients into WT mice not only induced α-Syn misfolding in the injection site but also lead to the spreading of α-Syn pathology to distant brain regions that are connected to the injection site. Importantly, the induced α-Syn pathology lead to dopamine neuron loss in the substantia nigra pars compacta in these mice, which is a key feature of PD (Luk et al., 2012b; Masuda-Suzukake et al., 2013). Brain lysates from MSA patients were also able to induce α-Syn aggregation in a transgenic mouse line expressing A53T mutant α-Syn (Prusiner et al., 2015; Watts et al., 2013). Moreover, intramuscular injection of α-Syn PFF could promote α-Syn aggregation in the central nervous system in α-Syn transgenic mice demonstrating that misfolded α-Syn could spread not only between different brain regions but also from peripheral nervous system to central nervous system (Sacino et al., 2014).
Strains of misfolded proteins
One of the most interesting finding for prions is that PrPSc are able to form aggregates with distinct pathological conformations giving rise to distinct histopathological lesion profiles with different brain region distribution and different clinical manifestation. These aggregates with distinct properties are named ‘strains’, which was used to describe PrPSc isolates that, when transmitted in the same host species, exhibit distinct prion-disease phenotypes (Aguzzi et al., 2007). More recently, similar conformational strains either generated with recombinant proteins or isolated from diseased brains have also been described for multiple amyloidogenic proteins including tau, Aβ, SOD1, huntingtin and α-Syn, indicating that the ability of these proteins to misfold into different strains is another shared property of neurodegenerative disease related proteins (Table 1).
Table 1.
Strains of non-prion protein aggregates
| Protein | Recourse | Characters of the strains | Reference |
|---|---|---|---|
| Aβ | Human brain lysate | Aβ fibrils seeded by brain lysates from two AD patients show different structure by nuclear magnetic resonance and electron microscopy. | (Lu et al., 2013) |
| Aβ | Human brain lysate | Brain homogenates from sporadic or Swedish AD versus Arctic AD patients induce Aβ pathology with different morphology after inoculation into APP transgenic mice. | (Watts et al., 2014) |
| Aβ | Human brain lysate | Aβ42 particle prepared from rapidly progressive Alzheimer’s disease shows expanded conformational heterogeneity compared with those from slowly progressive Alzheimer’s disease | (Cohen et al., 2015) |
| Aβ | Mouse brain lysate | β-amyloid containing brain extracts from two different APP transgenic mice (APP23 and APPPS1) could induce different β-amyloid deposits with characteristics of the original seeds. | (Heilbronner et al., 2013) |
| Aβ | Synthetic fibrils | Aβ fibrils generated in vertical dialysis tubes in an unstirred bath or in horizontal polypropylene tubes with gentle circular agitation show different molecular structures as revealed by electron microscopy and solid-state nuclear magnetic resonance measurements | (Petkova et al., 2005) |
| Tau | Human brain lysate | Pathological tau prepared from different tauopathies show different conformation as revealed by trypsin digestion and could induce tau pathologies resembling the corresponding human disease after injecting into tau transgenic mice. | (Boluda et al., 2015; Clavaguera et al., 2013; Taniguchi-Watanabe et al., 2016) |
| Tau | Human brain lysate | Misfolded four repeat (4R) tau isolated from tauopathy patient brains could only induce tau aggregation in cells expressing 4R tau, while misfolded three repeat (3R) tau could only induce tau pathology in cells expressing 3R tau. | (Woerman et al., 2016) |
| Tau | Cell lysate | After being transduced with tau fiber, cells expressing the tau repeat domain developed tau pathologies with distinct morphologies that could be propagated in cells and tau transgenic mice. | (Sanders et al., 2014) |
| Huntingtin | Synthetic fibrils | Huntingtin-exon 1 with expanded polyglutamines could form conformationally distinct aggregates under different temperatures. | (Nekooki-Machida et al., 2009) |
| α-Synuclein | Synthetic fibrils | α-Synuclein fibrils generated by repetitive seeded fibrillization could cross seed tau aggregation, while de novo generated α-Synuclein fibrils could only seed α-Synuclein aggregation in primary neurons and tau-transgenic mice. | (Guo et al., 2013) |
| α-Synuclein | Synthetic fibrils | α-Synuclein aggregates generate under physiological or low salt concentration have different conformation and also show different seeding ability and toxicity in cells and animals. | (Bousset et al., 2013; Peelaerts et al., 2015) |
| α-Synuclein | Synthetic fibrils | α-Syn fibers generated with or without lipopolysaccharide have different conformation and seeding ability | (Kim et al., 2016) |
| α-Synuclein | Human brain lysate | Brain lysates from MSA but not PDD or DLB brains induced α-Syn aggregates in cells and transgenic mice expressing A53T mutant α-Syn | (Prusiner et al., 2015; Woerman et al., 2015) |
Prion strains
Human prion diseases are epidemiologically and clinically diverse. They can occur as sporadic (sporadic Creutzfeldt-Jakob disease (sCJD)), infectious (iatrogenic CJD, variant CJD, Kuru) and hereditary forms [familial CJD (fCJD), fatal familial insomnia (FFI), Gerstmann-Sträussler-Scheinker syndrome (GSS)]. In sCJD, patients typically present initially with myoclonus and ataxia along with rapidly progressive dementia. Iatrogenic forms of CJD have distinct clinical presentations based on the patient’s means of exposure. Infection of the brain due to contamination of neurosurgical instruments or infected dura mater grafts results in a phenotype that is similar to sCJD whereas peripheral exposure from cadaveric human growth hormone results in progressive cerebellar ataxia and, later in disease progression, cognitive impairment (Rudge et al., 2015). Familial CJD is nearly indistinguishable from sCJD, presenting with rapid cognitive decline and myoclonus (Geschwind, 2015). Contrastingly, vCJD’s clinical presentation typically begins with psychiatric disorders, usually 6 months prior to the development of traditional cognitive dysfunction, involuntary movements and cerebellar dysfunction (Heath et al., 2011). Kuru presents, similarly to peripherally contracted iCJD, with fatal cerebellar ataxia along with athetoid movements, tremor and choreiform (Liberski et al., 2012). GSS typically presents with a slowly progressive ataxic disorder, as opposed to the rapidly progressive ataxia in CJD subfamilies, and proceeds into dementia in the late stages (Geschwind, 2015). FFI is distinct from other prion disorders in its clinical manifestations, presenting initially with a progressive insomnia, followed by dysautonomia and eventually motor and cognitive manifestations in the late stages of disease (Brown and Mastrianni, 2010). The mean disease durations of sCJD, fCJD, iCJD (5–6 months), vCJD (14 months), Kuru (12 months), FFI (18 months), and GSS (3–10 years) are as distinct as each disease’s clinical manifestation (Boelle et al., 2004; Gajdusek and Zigas, 1957; Gambetti et al., 2003; Johnson and Gibbs, 1998; Manix et al., 2015; Mead, 2006; Schenkein and Montagna, 2006).
The dramatic diversity of prion diseases have been partially attributed to the existence of various prion strains. Mammalian prion strains were first observed in goats (Pattison and Millson, 1961). Later, by infecting mice with five different strains of scrapie, Dickinson and colleagues found that all five strains could be reliably distinguished based on the degree of damage in different brain regions (Fraser and Dickinson, 1973). Since that time, many different prion strains have been identified in multiple species including human, based on their distinct structures, protein associations and histopathological lesion profiles.
The structural differences of prion strains has been revealed by the presence of diverse proteolytic fragments following proteinase K (PK) digestion, which degrades proteins based on the exposure of cleavage sites due to the three dimensional structure of the protein. Examples include PrPSc isolated from vCJD, and genetic form of TSE, FFI (Collinge et al., 1996; Telling et al., 1996). Moreover, nuclear magnetic resonance and electron paramagnetic resonance spectroscopy have also been used to explore the structure differences of prion strains (Tanaka et al., 2004). PrP can be glycosylated at two Thr residues, which results in three different glycosylation statuses: diglycosylated, monoglycosylated and unglycosylated forms. The ratios of these forms has been used as criteria to characterize the different strains (Collinge, 2005; Collinge et al., 1996). Even though biochemical and structural characteristics facilitate the identification and study of new strains, it should be pointed out that distinct prion strains could only be defined by bioassays that demonstrate the persistence of strain properties when passaged in new hosts. Moreover, the co-existence of different PrPSc types in patients has also been observed and could further contribute to disease diversity (Dickson and Brown, 1999; Polymenidou et al., 2005; Puoti et al., 1999; Yull et al., 2006).
Huntingtin strains
Huntington’s disease is caused by misfolding of the protein huntingtin with expanded polyglutamine repeats. Nekooki-Machida and colleagues have found that huntingtin amyloid fibrils formed at 4°C differ from those formed at 37°C (Nekooki-Machida et al., 2009). For example, when analyzed via circular dichroism spectroscopy and fourier transform infrared (FTIR) spectroscopy, these two strains are found to have differing loop/turn structures and β-sheet patterns. These two strains also show different thermal and physical stabilities, differential ability to be detected by the 1C2 antibody and differential resistance to SDS denaturation. More importantly, these two strains have different toxicities in mammalian cells. Similar huntingtin strains have also been identified in a transgenic mouse model of Huntington’s disease (Nekooki-Machida et al., 2009).
Tau strains
Tau is a microtubule binding protein that normally functions to stabilize microtubules (Avila et al., 2004; Lee et al., 2001). Tau is hyperphosphorylated and forms filamentous inclusions in a group of neurodegenerative diseases collectively known as tauopathies. Pathologically, tauopathies are often classified by the ratios of three repeat (3R) versus four repeat (4R) pathological tau and by the different cell type distribution of tau aggregates. AD is considered a secondary tauopathy with equal amount of 3R and 4R pathological tau (Braak and Del Tredici, 2011). PSP, CBD, and AGD are all considered 4R tauopathies due to the prevalence of 4R tau over 3R tau but all display different pathological hallmarks that are cell type specific. While PSP, CBD, and AGD all display coiled bodies in white matter, PSP is additionally characterized by globose tau inclusions and neuronal tangles in neurons and tufted astrocytes. (Esiri et al., 2004; Irwin et al., 2012a) Additionally, CBD and AGD both have pathological “ballooned neurons” in grey matter but have different distribution these “ballooned neurons” throughout the brain with CBD displaying ballooned neurons in limbic and neocortical structures while in AGD they are found in the amygdala. Furthermore, CBD features large astrocytic plaques in grey matter and astrocytic tau inclusions in white matter while AGD features spindle-shaped grains, pre-tangle tau inclusions, and neurofibrillary tangles in the grey matter (Esiri et al., 2004).
Different tauopathies also differ in symptomatic presentation and disease duration. The most common tauopathy, AD, is also the most common cause of dementia. The phenotypes of AD include impairment of memory, language, visuospatial function, executive function and praxis (Association and Association, 2000). PSP typically presents with pseudobulbar palsy, dementia, supranuclear ophthalmoplegia, axial dystonia and vertical supranuclear gaze palsy (Richardson et al., 1963). The phenotype of CBD differs greatly from that of other tauopathies with symptoms including frontal lobe behavioral changes, cortical sensory loss, non-fluent aphasia, dystonia, apraxia, dementia, and alien limb syndrome (Litvan et al., 2000). A relatively new tauopathy, AGD, accounts for approximately 5% of all dementia with symptoms including episodic memory loss, dementia, cognitive decline, behavioral abnormalities, personality changes (Ikeda et al., 2000). The disease durations of AD (3–9 years), PSP (6–9 years), CBD (<10 years), and AGD (1–15 years) are highly variable and often depend on how early in disease progression the tauopathy is diagnosed (Jellinger, 1998; Litvan et al., 2000; Querfurth and LaFerla, 2010).
Similar to prion, different tau strains has also been described (Table 1). Inoculation of brain extracts prepared from multiple different tauopathies into transgenic mice expressing wildtype human tau lead to the formation of distinct tau aggregates, which resemble the tau pathologies in the original diseased brains (Clavaguera et al., 2013). For example, injection of PSP brain homogenates results in the formation of astrocytic aggregates that resemble tufted astrocytes, the hallmark lesions of PSP, while CBD brain lysates induce the formation of astrocyte pathology reminiscent of astrocytic plaques. A more recent study using enriched pathological tau prepared from CBD and AD also showed that CBD-tau and AD-tau induce distinct tau pathologies after injection into transgenic mice expressing mutant tau, with CBD-tau inducing more oligodendrocyte pathology and AD-tau inducing predominantly neuronal pathology (Boluda et al., 2015). These results indicate the existence of different tau strains in diseased brains that contribute to the diverse pathological and clinical phenotypes of tauopathies. Recently, Woerman et al. showed that pathological tau isolated from Pick’s Disease (PiD-tau), which is composed mainly of 3R tau could induce tau aggregation in cells expressing the repeat domain of 3R tau, but not 4R tau, while CBD-tau and PSP-tau could only induce tau pathology in cells expressing the repeat domains of 4R tau (Woerman et al., 2016). These results further support the hypothesis that tau aggregates from different tauopathies represent different strains and that seeding reactions require isoform pairing between the seed and the substrate. In addition to different seeding behavior, conformational differences between pathological tau in different tauopathies as revealed by trypsin digestion have also been reported (Taniguchi-Watanabe et al., 2016). Tau strains, generated by the assembly of recombinant tau fibrils correlated with different patterns of tau pathology when induced in cells expressing the microtubule-binding domain of 4R tau (Sanders et al., 2014).
Amyloid β (Aβ) strains
Aβ, which is generated from amyloid precursor protein, is the primary misfolded protein in senile plaques of AD. Mutations in APP, the gene coding amyloid precursor protein, has been identified in familial AD. AD patients with APP mutations have earlier age of onsets than sporadic AD patients. Individuals with the arctic mutation of APP have a disease onset of 57 years and individuals with the Swedish mutation of APP have a disease onset of 55 years while the mean onset of sporadic AD is 73.1 years (Duara et al., 1993; Mullan et al., 1992). In addition to their earlier disease onset, carriers of Swedish and Arctic APP mutations may have a longer disease duration than sporadic AD, although direct comparison is problematic. Individuals that experience an earlier age of onset may be healthier during the most severe stages of the disease when compared to older patients with sporadic AD who may have additional comorbidities in sporadic AD (Ryan and Rossor, 2010).
Similarly, different strains have also been reported for amyloid β (Table 1), which can be identified by their unique conformations, morphology, toxicity, and transmission patterns. Aβ deposits prepared from sporadic or heritable (Arctic and Swedish) AD cases showed different resistance to denaturation indicating distinct conformations and they also induced different Aβ pathology after inoculation into a mutant APP transgenic mice (Watts et al., 2014). By amplifying Aβ 40 fibrils from two AD patients and analyzing them with nuclear magnetic resonance and electron microscopy, Lu and colleagues showed that Aβ 40 fibrils amplified from different AD patients have different structures and a single predominant Aβ 40 fibril structure exists for each patient (Lu et al., 2013). Brain extracts from two different APP transgenic mice (APP/PS1 and APP23) can induce β-amyloid deposition with different morphologies, conformations and Aβ40/Aβ42 ratios after injection into young APP transgenic mice (Heilbronner et al., 2013). Aβ 40 fibrils with different structures and toxicities have also been generated with recombinant proteins (Petkova et al., 2005).
α-Syn strains
Accumulating evidence has documented that recombinant α-Syn monomers can form synthetic α-Syn aggregates with distinct conformations and biological activities (Table 1). Through repetitive seeded fibrillization in vitro, whereby a small amount of preformed fibrils were included as seeds in the fibrillization reaction, Guo and colleagues generated α-Syn preformed fibrils (PFFs), which were defined as strain B, that showed different conformation compared with de novo generated α-Syn PFFs, which were defined as strain A (Figure 2). Strain B is less potent in seeding α-Syn aggregation than strain A but can very efficiently seed tau aggregation both in primary neurons and in tau transgenic mice (Guo et al., 2013). This finding not only demonstrates the existence of α-Syn strains but also illustrates the cross-seeding properties of misfolded α-Syn and tau. When taken together with the transmission hypothesis of α-Syn aggregates, every transmission event from one neuron to the other can be viewed as an independent seeded- fibrillization reaction. Therefore continuous transmission of α-Syn pathology in disease brains might lead to the generation of strain B like fibrils because of the repeated seeding process. For example, LBs in the substantia nigra of PDD patients could be different from LBs in neocortical areas because they are affected at different stages of the disease. Cortical LBs may be a consequence of many more rounds of seeding and propagation compared with those in substantia nigra. Consistent with this hypothesis, tau aggregates are more frequently detected in limbic and neocortical areas than in the midbrain in PDD or DLB cases with concomitant AD pathologies (Gearing et al., 1999; Gomez-Tortosa et al., 2000). Importantly, pathological α-Syn prepared from PDD cases with or without a secondary diagnosis of AD showed different proteolytic fragments after PK digestions, which likely indicates the existence of similar α-Syn strains in diseased brains (Guo et al., 2013).
Figure 2. Different α-Synuclein strains generated from recombinant proteins.
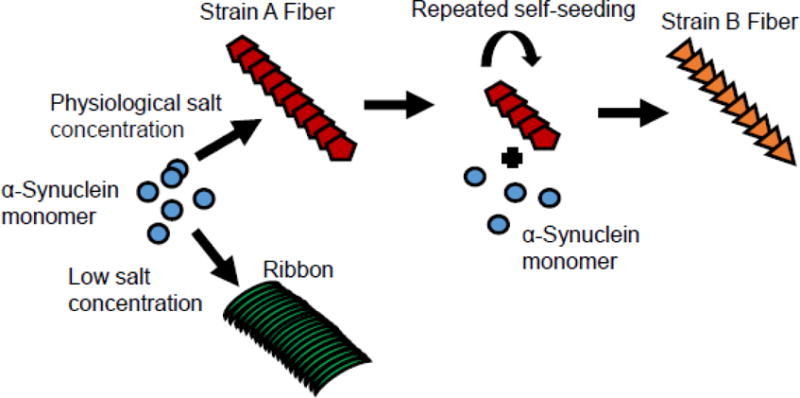
De novo generated α-Syn aggregates under physiological salt concentrations have a cylindrical aspect (Strain A), while those obtained under low salt concentrations are flat (Ribbons). Repeat seeding of Strain A fiber lead to the generation of Strain B fiber, which could cross seed tau aggregation in neurons.
Later on, Bousset and colleagues found that α-Syn aggregates generated under physiological salt concentrations have a cylindrical shape, which they referred to as fibrils, while those obtained under low salt concentrations are flat, which were termed ribbons (Figure 2). Multiple biophysical analysis such as FTIR spectra, X-ray fibre diffraction and solid-state nuclear magnetic resonance spectroscopy measurements illustrate that α-Syn fibrils and ribbons have different structures (Bousset et al., 2013; Peelaerts et al., 2015). Importantly, α-Syn fibrils and ribbons are able to imprint their intrinsic architecture onto soluble α-Syn monomer in seeded fibrillization reactions in vitro and also in cells. α-Syn fibrils are more toxic than ribbons to SH-SY5Y cells. Consistent with their higher toxicity, α-Syn fibrils also bind and permeabilize cell membranes to a greater extent than ribbons. In vivo studies showed that ribbons are more efficient in inducing α-Syn pathology in vivo and, in combination with virus mediated overexpression of α-Syn, could even lead to the development of oligodendrocyte pathology. However, α-Syn fibrils are also more toxic to dopaminergic neurons than ribbons, when combined with rAAV mediated α-Syn overexpression, and lead to motor deficits. α-Syn fibrils and ribbons are shown to maintain their conformation after in vivo passaging (Peelaerts et al., 2015). More recently, conformational differences have also been observed between α-Syn fibrils generated with or without lipopolysaccharide (Kim et al., 2016).
In addition to in vitro generated α-Syn aggregates, pathological α-Syn prepared from different α-Synucleinopathies has also been studied. In 2013, Masami Masuda-Suzukake and colleagues showed that the insoluble fraction from DLB brains could induce α-Syn aggregation in wild type mice (Masuda-Suzukake et al., 2013). Later on, it was found that brain lysate from MSA patients but not PD or DLB patients could facilitate α-Syn aggregation in a mouse model expressing A53T mutant α-Syn and in HEK293 cells overexpressing A53T mutant α-Syn-YPF fusion protein (Prusiner et al., 2015; Woerman et al., 2015). These results indicate potential differences between pathological α-Syn in MSA patients versus those in PDD and DLB patients. However, a more recent study claimed that pathological α-Syn prepared from MSA and incidental Lewy body disease patients showed similar seeding ability in a mouse model only expressing wild type human α-Syn (Bernis et al., 2015). Therefore, it is still unclear whether pathological α-Syn in MSA patients versus those in PDD and DLB patients represent different strains.
Potential α-Syn strains in diseased brains
The studies described above convincingly demonstrate that recombinant α-Syn monomers are able to form different α-Syn strains with distinct conformations and biological activities. One obvious next step is to identify and characterize α-Syn strains in α-Synucleinopathy patients. Currently, evidence supporting the existence of pathological α-Syn strains in human brains is still very limited and the properties of α-Syn strains are poorly characterized. More importantly, how α-Syn strains contribute to the diverse pathological and clinical presentations of α-Synucleinopathies remains largely unknown. Based on the knowledge from strains identified for other amyloid proteins and the characteristics of α-Synucleinopathies, here we make predictions of α-Syn strains that could potentially exist in patients. Characterizing the different α-Syn strains in α-Synucleinopathy patients is a priority for future studies.
GCI-α-Syn versus LB-α-Syn
Given the dramatically different cell type-specific distribution of GCIs in oligodendrocytes and LBs in neurons of disease brains and that GCI-α-Syn and LB-α-Syn have different ability to seed A53T mutant α-Syn (Prusiner et al., 2015; Woerman et al., 2015), pathological α-Syn in GCI and LB might be different α-Syn strains. Detailed analyses of GCI-α-Syn and LB-α-Syn purified from disease brains to illustrate their potential biochemical and more importantly seeding differences are critical to demonstrate that they are really different strains and to correlate their properties with the clinical behavior of patients.
It is possible that GCI-α-Syn and LB-α-Syn may preferentially seed oligodendrocytes and neurons respectively, which could contribute to their different cell type distribution in disease brains, as was observed for CBD-tau and AD-tau in tauopathies (Boluda et al., 2015; Clavaguera et al., 2013). Interestingly, after being injected into a mouse model expressing A53T mutant α-Syn, GCI-α-Syn induced α-Syn pathology in neurons but not oligodendrocytes (Watts et al., 2013). However, this surprising result could be due to the mutation, which has been reported to promote α-Syn aggregation, as well as the expression pattern of the transgene, which is mainly in neurons. It is also noteworthy that oligodendrocytes express very low levels α-Syn, therefore the lack of oligodendrocyte α-Syn aggregation in GCI-α-Syn injected mice likely resulted from the lack of substrate in these cells. It would be interesting to compare LB-α-Syn and GCI-α-Syn in the various MSA mouse models that express α-Syn in oligodendrocytes to see whether GCI-α-Syn could more efficiently induce oligodendrocyte pathology.
α-Syn aggregation with or without concomitant AD pathologies
The properties of strain B and more importantly the fact that pathological α-Syn from PDD cases with or without significant amount of tau deposition show different proteolytic fragments after PK digestions (Guo et al., 2013) suggests that pathological α-Syn in different patients or even in different brain regions of the same patient with or without concomitant AD pathology may represent different strains. α-Syn aggregates in regions with tau pathology may have ‘strain B’-like properties and can cross-seed tau aggregation. Similarly, there might also be pathological α-Syn strains that could induce or facilitate Aβ deposition given the fact that Aβ is also commonly observed in the cortical area of PDD and DLB patients. The different α-Syn strains that can cross-seed other amyloid proteins could be responsible for the co-deposition of tau and Aβ in PDD and DLB patients.
However, it is technically challenging to demonstrate the cross-seeding of α-Syn with tau or Aβ using disease brain derived material. First, theoretically, such α-Syn strains should exist in brain regions with concomitant tau or Aβ deposition, therefore any cross-seeding effect of the pathological α-Syn prepared from these brain tissues could potentially be attributed to the contaminated pathological tau or Aβ in the brain lysates. An efficient way to generate highly purified pathological α-Syn without the contamination of tau or Aβ pathology, such as IP purification, will be critical to demonstrate the existence and to analyze the properties of these strains. Second, the amount of pathological α-Syn that can be recovered from disease brains is very low as indicated by the work from Prusiner’s group and Hasegawa’s group (Masuda-Suzukake et al., 2013; Prusiner et al., 2015; Woerman et al., 2015). Therefore, an efficient way to amplify pathological α-Syn with recombinant α-Syn monomer, similar to the method used to amplify Aβ fibrils derived from AD brains (Lu et al., 2013), will greatly facilitate the analysis of potential α-Syn strains. On the bright side, Guo and colleagues have generated a strain selective antibody that could preferentially recognize strain B over strain A α-Syn fibrils (Guo et al., 2013), which could be a very powerful tool to identify potential α-Syn strains in vivo and correlate the distribution of different strains with specific clinical behavior.
α-Syn aggregation in ‘pure’ PD and DLB patients
Since DLB patients develop cortical and limbic α-Syn pathology much faster than PD patients, who show a slow progression of α-Syn pathology in the limbic area with a long interval between the onset of motor symptoms and dementia, it is possible that pathological α-Syn in DLB patients might represent a different strain that is more potent in seeding cortical neurons than the α-Syn strain in PD patients. One pitfall of this hypothesis is that DLB patients usually have significant amount AD pathology in cortical areas, therefore an obvious alternative hypothesis is that AD pathology facilitates the transmission of α-Syn pathology into cortical areas in DLB patients. However, ‘pure’ DLB (without AD neuropathology) also shows accelerated progression of α-Syn pathology in the limbic area compared to PD patients (Irwin et al., 2013). Therefore, either there are other unknown pathological changes in DLB patients that promote the development of LBs/LNs in cortical areas or the aggregated α-Syn is intrinsically different between DLB and PD patients.
Therapeutic implications of α-Syn strains
If distinct α-Syn strains truly exist in α-Synucleinopathy patients, developing therapies targeting α-Syn, such as immunotherapies, will assuredly become more complex and challenging. Antibodies that efficiently recognize one strain of α-Syn aggregation may not necessary recognize another strain. Similarly, small molecules that are designed to reverse or block the amplification of aggregated α-Syn may need to be tested for different stains to determine their clinical application. Regardless, the recent explosion of knowledge on α-Syn strains will lead to a deeper understanding of the development and progression of α-Synucleinopathies and will facilitate the design of new therapies. Moreover, detailed analyses of α-Syn strains in α-Synucleinopathy patients could facilitate the development of a more mechanism based classification systems of α-Synucleinopathies, which would help to refine the design of clinical trials and guide the treatment of α-Synucleinopathies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarsland D, et al. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol. 2003;60:387–92. doi: 10.1001/archneur.60.3.387. [DOI] [PubMed] [Google Scholar]
- Abeliovich A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–52. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Aguzzi A, et al. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–61. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, et al. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol. 2012;38:4–24. doi: 10.1111/j.1365-2990.2011.01234.x. [DOI] [PubMed] [Google Scholar]
- Apaydin H, et al. Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch Neurol. 2002;59:102–12. doi: 10.1001/archneur.59.1.102. [DOI] [PubMed] [Google Scholar]
- Appel-Cresswell S, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord. 2013;28:811–3. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- Asi YT, et al. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia. 2014;62:964–70. doi: 10.1002/glia.22653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Association, A. P., Association, A. P. DSM-IV-TR: Diagnostic and statistical manual of mental disorders, text revision. Washington, DC: American Psychiatric Association; 2000. p. 75. [Google Scholar]
- Avila J, et al. Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004;84:361–84. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- Barker RA, Williams-Gray CH. Review: The spectrum of clinical features seen with alpha synuclein pathology. Neuropathol Appl Neurobiol. 2016;42:6–19. doi: 10.1111/nan.12303. [DOI] [PubMed] [Google Scholar]
- Bartels T, et al. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–10. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shlomo Y, et al. Survival of patients with pathologically proven multiple system atrophy: a meta-analysis. Neurology. 1997;48:384–93. doi: 10.1212/wnl.48.2.384. [DOI] [PubMed] [Google Scholar]
- Bernis ME, et al. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun. 2015;3:75. doi: 10.1186/s40478-015-0254-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoncini CW, et al. Familial mutants of alpha-synuclein with increased neurotoxicity have a destabilized conformation. J Biol Chem. 2005;280:30649–52. doi: 10.1074/jbc.C500288200. [DOI] [PubMed] [Google Scholar]
- Boelle PY, et al. Epidemiological evidence of higher susceptibility to vCJD in the young. BMC Infect Dis. 2004;4:26. doi: 10.1186/1471-2334-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boluda S, et al. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129:221–37. doi: 10.1007/s00401-014-1373-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousset L, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower JH, et al. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology. 1997;49:1284–8. doi: 10.1212/wnl.49.5.1284. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011;121:171–81. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- Braak H, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Braak H, et al. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology. 2005;64:1404–10. doi: 10.1212/01.WNL.0000158422.41380.82. [DOI] [PubMed] [Google Scholar]
- Brown K, Mastrianni JA. The prion diseases. J Geriatr Psychiatry Neurol. 2010;23:277–98. doi: 10.1177/0891988710383576. [DOI] [PubMed] [Google Scholar]
- Burre J. The Synaptic Function of alpha-Synuclein. J Parkinsons Dis. 2015;5:699–713. doi: 10.3233/JPD-150642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J, et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–7. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabin DE, et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci. 2002;22:8797–807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, et al. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–96. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–40. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ML, et al. Rapidly progressive Alzheimer’s disease features distinct structures of amyloid-beta. Brain. 2015;138:1009–22. doi: 10.1093/brain/awv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J. Molecular neurology of prion disease. J Neurol Neurosurg Psychiatry. 2005;76:906–19. doi: 10.1136/jnnp.2004.048660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, et al. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–90. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- Compta Y, et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain. 2011;134:1493–505. doi: 10.1093/brain/awr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A. 2002;99:14524–9. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, et al. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Del Tredici K, et al. Where does parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol. 2002;61:413–26. doi: 10.1093/jnen/61.5.413. [DOI] [PubMed] [Google Scholar]
- DeWitt DC, Rhoades E. alpha-Synuclein can inhibit SNARE-mediated vesicle fusion through direct interactions with lipid bilayers. Biochemistry. 2013;52:2385–7. doi: 10.1021/bi4002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009;8:1150–7. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Brown P. Multiple prion types in the same brain: is a molecular diagnosis of CJD possible? Neurology. 1999;53:1903–4. doi: 10.1212/wnl.53.9.1903. [DOI] [PubMed] [Google Scholar]
- Duara R, et al. A comparison of familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1377–84. doi: 10.1212/wnl.43.7.1377. [DOI] [PubMed] [Google Scholar]
- Esiri MM, et al. The Neuropathology of Dementia. Cambridge University Press; 2004. [Google Scholar]
- Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med. 2015;372:1375–6. doi: 10.1056/NEJMc1501657. [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon R, et al. The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron. 2004;42:237–51. doi: 10.1016/s0896-6273(04)00190-4. [DOI] [PubMed] [Google Scholar]
- Fortin DL, et al. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24:6715–23. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser H, Dickinson AG. Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. J Comp Pathol. 1973;83:29–40. doi: 10.1016/0021-9975(73)90024-8. [DOI] [PubMed] [Google Scholar]
- Frost B, et al. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–52. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs J, et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68:916–22. doi: 10.1212/01.wnl.0000254458.17630.c5. [DOI] [PubMed] [Google Scholar]
- Fujioka S, et al. Update on novel familial forms of Parkinson’s disease and multiple system atrophy. Parkinsonism Relat Disord. 2014;20(Suppl 1):S29–34. doi: 10.1016/S1353-8020(13)70010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native population. N Engl J Med. 1957;257:974–8. doi: 10.1056/NEJM195711142572005. [DOI] [PubMed] [Google Scholar]
- Gambetti P, et al. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–39. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- Gearing M, et al. Neurofibrillary pathology in Alzheimer disease with Lewy bodies: two subgroups. Arch Neurol. 1999;56:203–8. doi: 10.1001/archneur.56.2.203. [DOI] [PubMed] [Google Scholar]
- Geschwind MD. Prion Diseases. Continuum (Minneap Minn) 2015;21:1612–38. doi: 10.1212/CON.0000000000000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, et al. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–6. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- Gilman S, et al. The North American Multiple System Atrophy Study Group. J Neural Transm (Vienna) 2005;112:1687–94. doi: 10.1007/s00702-005-0381-6. [DOI] [PubMed] [Google Scholar]
- Gilman S, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbe LI, et al. Clinical genetic analysis of Parkinson’s disease in the Contursi kindred. Ann Neurol. 1996;40:767–75. doi: 10.1002/ana.410400513. [DOI] [PubMed] [Google Scholar]
- Gomez-Tortosa E, et al. Clinical and neuropathological correlates of dementia with Lewy bodies. Ann N Y Acad Sci. 2000;920:9–15. doi: 10.1111/j.1749-6632.2000.tb06899.x. [DOI] [PubMed] [Google Scholar]
- Greten-Harrison B, et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–8. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–17. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–31. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday GM, et al. Striatal beta-amyloid in dementia with Lewy bodies but not Parkinson’s disease. J Neural Transm (Vienna) 2011;118:713–9. doi: 10.1007/s00702-011-0641-6. [DOI] [PubMed] [Google Scholar]
- Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–84. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath CA, et al. Diagnosing variant Creutzfeldt-Jakob disease: a retrospective analysis of the first 150 cases in the UK. J Neurol Neurosurg Psychiatry. 2011;82:646–51. doi: 10.1136/jnnp.2010.232264. [DOI] [PubMed] [Google Scholar]
- Heilbronner G, et al. Seeded strain-like transmission of beta-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013;14:1017–22. doi: 10.1038/embor.2013.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hely MA, et al. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord. 2008;23:837–44. doi: 10.1002/mds.21956. [DOI] [PubMed] [Google Scholar]
- Iba M, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33:1024–37. doi: 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, et al. Clinical aspects of argyrophilic grain disease. Clin Neuropathol. 2000;19:278–84. [PubMed] [Google Scholar]
- Irwin DJ, et al. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain. 2012a;135:807–18. doi: 10.1093/brain/aws013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, et al. Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci. 2013;14:626–36. doi: 10.1038/nrn3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012b;72:587–98. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Dementia with grains (argyrophilic grain disease) Brain Pathol. 1998;8:377–86. doi: 10.1111/j.1750-3639.1998.tb00161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Morphological substrates of parkinsonism with and without dementia: a retrospective clinico-pathological study. J Neural Transm Suppl. 2007:91–104. doi: 10.1007/978-3-211-73574-9_12. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Attems J. Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol. 2006;112:253–60. doi: 10.1007/s00401-006-0088-2. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol. 2008;115:427–36. doi: 10.1007/s00401-008-0347-5. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, et al. Grading of neuropathology in multiple system atrophy: proposal for a novel scale. Mov Disord. 2005;20(Suppl 12):S29–36. doi: 10.1002/mds.20537. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, et al. Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J Neural Transm (Vienna) 2002;109:329–39. doi: 10.1007/s007020200027. [DOI] [PubMed] [Google Scholar]
- Jensen MB, et al. Membrane curvature sensing by amphipathic helices: a single liposome study using alpha-synuclein and annexin B12. J Biol Chem. 2011;286:42603–14. doi: 10.1074/jbc.M111.271130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PH, et al. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273:26292–4. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- Johnson RT, Gibbs CJ., Jr Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994–2004. doi: 10.1056/NEJM199812313392707. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–73. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempster PA, et al. Relationships between age and late progression of Parkinson’s disease: a clinico-pathological study. Brain. 2010;133:1755–62. doi: 10.1093/brain/awq059. [DOI] [PubMed] [Google Scholar]
- Kiely AP, et al. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013;125:753–69. doi: 10.1007/s00401-013-1096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, et al. Exposure to bacterial endotoxin generates a distinct strain of alpha-synuclein fibril. Sci Rep. 2016;6:30891. doi: 10.1038/srep30891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, et al. Survival of Korean patients with multiple system atrophy. Mov Disord. 2011;26:909–12. doi: 10.1002/mds.23580. [DOI] [PubMed] [Google Scholar]
- Kim WS, et al. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res Ther. 2014;6:73. doi: 10.1186/s13195-014-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollensperger M, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord. 2010;25:2604–12. doi: 10.1002/mds.23192. [DOI] [PubMed] [Google Scholar]
- Kordower JH, et al. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–6. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Kotzbauer PT, et al. Pathologic accumulation of alpha-synuclein and Abeta in Parkinson disease patients with dementia. Arch Neurol. 2012;69:1326–31. doi: 10.1001/archneurol.2012.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovari E, et al. Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol. 2003;106:83–8. doi: 10.1007/s00401-003-0705-2. [DOI] [PubMed] [Google Scholar]
- Kraybill ML, et al. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology. 2005;64:2069–73. doi: 10.1212/01.WNL.0000165987.89198.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R, et al. Familial parkinsonism with synuclein pathology: clinical and PET studies of A30P mutation carriers. Neurology. 2001;56:1355–62. doi: 10.1212/wnl.56.10.1355. [DOI] [PubMed] [Google Scholar]
- Kruger R, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–8. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. doi: 10.1038/srep00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashley T, et al. Cortical alpha-synuclein load is associated with amyloid-beta plaque burden in a subset of Parkinson’s disease patients. Acta Neuropathol. 2008;115:417–25. doi: 10.1007/s00401-007-0336-0. [DOI] [PubMed] [Google Scholar]
- Lee VM, et al. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lesage S, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. 2013;73:459–71. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- Levy G, et al. Combined effect of age and severity on the risk of dementia in Parkinson’s disease. Ann Neurol. 2002;51:722–9. doi: 10.1002/ana.10219. [DOI] [PubMed] [Google Scholar]
- Lewis SJ, et al. Heterogeneity of Parkinson’s disease in the early clinical stages using a data driven approach. J Neurol Neurosurg Psychiatry. 2005;76:343–8. doi: 10.1136/jnnp.2003.033530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–3. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- Liberski PP, et al. Kuru: the first prion disease. Adv Exp Med Biol. 2012;724:143–53. doi: 10.1007/978-1-4614-0653-2_12. [DOI] [PubMed] [Google Scholar]
- Lippa CF, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68:812–9. doi: 10.1212/01.wnl.0000256715.13907.d3. [DOI] [PubMed] [Google Scholar]
- Litvan I, et al. Phenotypes and prognosis: clinicopathologic studies of corticobasal degeneration. Adv Neurol. 2000;82:183–96. [PubMed] [Google Scholar]
- Lu JX, et al. Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell. 2013;154:1257–68. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012a;338:949–53. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, et al. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012b;209:975–86. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A. 2009;106:20051–6. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manix M, et al. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39:E2. doi: 10.3171/2015.8.FOCUS15328. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, et al. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol. 2009;68:816–22. doi: 10.1097/NEN.0b013e3181ac10a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res. 1991;11:335–43. doi: 10.1016/0169-328x(91)90043-w. [DOI] [PubMed] [Google Scholar]
- Masuda-Suzukake M, et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain. 2013;136:1128–38. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, et al. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–9. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- Mattila PM, et al. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 2000;100:285–90. doi: 10.1007/s004019900168. [DOI] [PubMed] [Google Scholar]
- McCann H, et al. alpha-Synucleinopathy phenotypes. Parkinsonism Relat Disord. 20 Suppl. 2014;1:S62–7. doi: 10.1016/S1353-8020(13)70017-8. [DOI] [PubMed] [Google Scholar]
- Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14:273–81. doi: 10.1038/sj.ejhg.5201544. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–4. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Mikolaenko I, et al. Alpha-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: evidence from the Baltimore Longitudinal Study of Aging (BLSA) J Neuropathol Exp Neurol. 2005;64:156–62. doi: 10.1093/jnen/64.2.156. [DOI] [PubMed] [Google Scholar]
- Miller DW, et al. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J Neural Transm (Vienna) 2005;112:1613–24. doi: 10.1007/s00702-005-0378-1. [DOI] [PubMed] [Google Scholar]
- Mullan M, et al. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–7. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Munch C, et al. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A. 2011;108:3548–53. doi: 10.1073/pnas.1017275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, et al. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–20. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekooki-Machida Y, et al. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci U S A. 2009;106:9679–84. doi: 10.1073/pnas.0812083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, et al. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson’s disease. Ann Neurol. 2006;59:298–309. doi: 10.1002/ana.20753. [DOI] [PubMed] [Google Scholar]
- Nonaka T, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4:124–34. doi: 10.1016/j.celrep.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Pattison IH, Millson GC. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J Comp Pathol. 1961;71:101–9. doi: 10.1016/s0368-1742(61)80013-1. [DOI] [PubMed] [Google Scholar]
- Paumier KL, et al. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis. 2015;82:185–99. doi: 10.1016/j.nbd.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peelaerts W, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–4. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- Petkova AT, et al. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- Pineda A, Burre J. Modulating membrane binding of alpha-synuclein as a therapeutic strategy. Proc Natl Acad Sci U S A. 2017;114:1223–1225. doi: 10.1073/pnas.1620159114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, et al. Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol. 2005;4:805–14. doi: 10.1016/S1474-4422(05)70225-8. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Postuma RB, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30:1591–601. doi: 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- Proukakis C, et al. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–4. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A. 2015;112:E5308–17. doi: 10.1073/pnas.1514475112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puoti G, et al. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology. 1999;53:2173–6. doi: 10.1212/wnl.53.9.2173. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Ren PH, et al. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–25. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JC, et al. Supranuclear Ophthalmoplegia, Pseudobulbar Palsy, Nuchal Dystonia and Dementia. A Clinical Report on Eight Cases of “Heterogenous System Degeneration”. Trans Am Neurol Assoc. 1963;88:25–9. [PubMed] [Google Scholar]
- Rochet JC, et al. Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 2004;23:23–34. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- Rudge P, et al. Iatrogenic CJD due to pituitary-derived growth hormone with genetically determined incubation times of up to 40 years. Brain. 2015;138:3386–99. doi: 10.1093/brain/awv235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan NS, Rossor MN. Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark Med. 2010;4:99–112. doi: 10.2217/bmm.09.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbagh MN, et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord. 2009;23:295–7. doi: 10.1097/WAD.0b013e31819c5ef4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, et al. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A. 2014;111:10732–7. doi: 10.1073/pnas.1321785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–88. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkein J, Montagna P. Self-management of fatal familial insomnia. Part 2: case report. MedGenMed. 2006;8:66. [PMC free article] [PubMed] [Google Scholar]
- Schiesling C, et al. Review: Familial Parkinson’s disease—genetics, clinical phenotype and neuropathology in relation to the common sporadic form of the disease. Neuropathol Appl Neurobiol. 2008;34:255–71. doi: 10.1111/j.1365-2990.2008.00952.x. [DOI] [PubMed] [Google Scholar]
- Selikhova M, et al. A clinico-pathological study of subtypes in Parkinson’s disease. Brain. 2009;132:2947–57. doi: 10.1093/brain/awp234. [DOI] [PubMed] [Google Scholar]
- Stohr J, et al. Purified and synthetic Alzheimer’s amyloid beta (Abeta) prions. Proc Natl Acad Sci U S A. 2012;109:11025–30. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, et al. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–8. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- Taniguchi-Watanabe S, et al. Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 2016;131:267–80. doi: 10.1007/s00401-015-1503-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–82. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- Tsigelny IF, et al. Dynamics of alpha-synuclein aggregation and inhibition of pore-like oligomer development by beta-synuclein. FEBS J. 2007;274:1862–77. doi: 10.1111/j.1742-4658.2007.05733.x. [DOI] [PubMed] [Google Scholar]
- Tsuboi Y, et al. Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat Disord. 2007;13(Suppl 3):S221–4. doi: 10.1016/S1353-8020(08)70005-1. [DOI] [PubMed] [Google Scholar]
- Tu PH, et al. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol. 1998;44:415–22. doi: 10.1002/ana.410440324. [DOI] [PubMed] [Google Scholar]
- Ueda K, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–6. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, et al. Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry. 2001;40:7812–9. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain. 2002;125:1070–83. doi: 10.1093/brain/awf117. [DOI] [PubMed] [Google Scholar]
- Watts JC, et al. Serial propagation of distinct strains of Abeta prions from Alzheimer’s disease patients. Proc Natl Acad Sci U S A. 2014;111:10323–8. doi: 10.1073/pnas.1408900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. 2013;110:19555–60. doi: 10.1073/pnas.1318268110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb PH, et al. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–15. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- Woerman AL, et al. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci U S A. 2016;113:E8187–E8196. doi: 10.1073/pnas.1616344113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerman AL, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A. 2015;112:E4949–58. doi: 10.1073/pnas.1513426112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavich L, et al. Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in alpha-synuclein knockout and A30P transgenic mice. J Neurochem. 2006;99:724–32. doi: 10.1111/j.1471-4159.2006.04098.x. [DOI] [PubMed] [Google Scholar]
- Yavich L, et al. Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24:11165–70. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M. Multiple system atrophy: alpha-synuclein and neuronal degeneration. Neuropathology. 2007;27:484–93. doi: 10.1111/j.1440-1789.2007.00841.x. [DOI] [PubMed] [Google Scholar]
- Yull HM, et al. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol. 2006;168:151–7. doi: 10.2353/ajpath.2006.050766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zharikov AD, et al. shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J Clin Invest. 2015;125:2721–35. doi: 10.1172/JCI64502. [DOI] [PMC free article] [PubMed] [Google Scholar]