

Abstract
Autism Spectrum Disorders (ASD) are heterogeneous neurodevelopmental disorders with a complex genetic architecture. They are characterized by impaired social communication, stereotyped behaviors and restricted interests and are frequently associated with comorbidities such as intellectual disability, epilepsy and severe sleep disorders. Hyperserotonemia and low melatonin levels are among the most replicated endophenotypes reported in ASD, but their genetic causes remain largely unknown. Based on the biochemical profile of 717 individuals including 213 children with ASD, 128 unaffected siblings and 376 parents and other relatives, we estimated the heritability of whole-blood serotonin, platelet N-acetylserotonin (NAS) and plasma melatonin levels, as well as the two enzymes arylalkylamine N-acetyltransferase (AANAT) and acetylserotonin O-methyltransferase (ASMT) activities measured in platelets. Overall, heritability was higher for NAS (0.72 ± 0.091) and ASMT (0.59 ± 0.097) compared with serotonin (0.31 ± 0.078), AANAT (0.34 ± 0.077) and melatonin (0.22 ± 0.071). Bivariate analyses showed high phenotypic and genetic correlations between traits of the second step of the metabolic pathway (NAS, ASMT and melatonin) indicating the contribution of shared genetic factors. A better knowledge of the heritability of the melatonin synthesis variability constitutes an important step to identify the factors that perturb this pathway in individuals with ASD.
Introduction
Autism Spectrum Disorders (ASD) are complex neurodevelopmental disorders characterized by deficits in social communication/interaction as well as restricted interests and repetitive patterns of behaviors. Although ASD are highly heritable disorders1–3, they are multifactorial and clinically heterogeneous and the ASD-risk genes remain largely unknown. Large-scale genetic studies revealed a complex genetic architecture, involving combined effects of multiple low risk common variants, rare and de novo deleterious mutations4. Endophenotypes are measurable markers with potentially reduced genetic heterogeneity compared to the disease itself and they can be used to overcome the genetic and phenotypic heterogeneity. They facilitate the stratification of patients into more homogeneous subgroups5, increase the power of quantitative genetic analyses to detect variants, genes or pathways associated with the disease of interest and they reveal underlying biological mechanisms.
Hyperserotonemia is one of the most replicated endophenotypes in ASD6–10. A recent meta-analysis including 739 patients and 868 controls estimated an elevated whole-blood serotonin in 28.3% of the patients with ASD compared to 5% of controls11. Low melatonin levels in urine, plasma and pineal gland have also been described in ASD individuals10,12–15. Melatonin derives from serotonin, which is successively converted into N-acetylserotonin (NAS) and melatonin by the enzymes arylalkylamine N-acetyltransferase (AANAT, EC: 2.3.1.87) and acetylserotonin O-methyltransferase (ASMT, EC: 2.1.1.4). Melatonin biosynthesis essentially occurs in the pineal gland, following a marked circadian rhythm with a maximal secretion at night. A global disruption of this pathway was observed in patients compared to relatives and controls, including hyperserotonemia, deficits in AANAT and ASMT platelet activity, increased platelet NAS and melatonin deficit10,15. These alterations were also observed to a lesser extent, in the relatives of patients with ASD, compared to controls10,15, but familial correlations and heritability have never been assessed for both steps of the melatonin synthesis pathway in families with ASD.
Serotonin is involved in a wide range of central processes, such as brain development, emotion, learning, memory or cognitive functions16,17. NAS is an agonist of melatonin receptors and activates TrkB in mice, a receptor of the brain-derived neurotrophic factor (BDNF), in a selective and circadian manner18,19. Melatonin is a pleiotropic neuroendocrine molecule essential for synchronizing circadian and seasonal rhythms, as well as sleep/wake cycles, but also displays antioxidant, neuroprotective, or immunomodulatory effects20–22. Biochemical alterations of this pathway could thus be related to the core symptoms of autism or the comorbidities such as cognitive problems, epilepsy, sleep and gastrointestinal disorders observed in ASD17,23–25.
In order to identify shared underlying genetic factors between an endophenotype and a disease, the endophenotype must be heritable with either high broad sense heritability (H2), defined as the ratio of total genetic variance (additive, dominance and epistasis) to phenotypic variance, or high narrow sense heritability (h2), reflecting only the additive part (the average effect of the alleles on the trait) of the genetic contribution to the phenotypic variance (Supplementary Note). Several studies in humans and in animal models have demonstrated relatively high heritability for serotonin26–28 and melatonin29–32. In humans, serotonin narrow sense heritability estimates range from 0.2 to 0.5126,27,33 and urinary melatonin narrow sense heritability estimate was 0.53 in families with acute intermittent porphyria32. To our knowledge, NAS, AANAT and ASMT heritability have never been investigated. In this study, we estimated the narrow sense heritability of all five quantitative traits (whole blood serotonin, platelet AANAT and ASMT activities, platelet NAS and morning plasma melatonin) in 717 individuals including 213 children with ASD, 364 parents, 128 unaffected siblings and 12 other relatives (Supplementary Fig. S1)10,15. We also evaluated the correlations of genetic and environmental factors between pairs of traits. The assessment of additive factors contribution to the inter-individual phenotypic variability should provide crucial information for further quantitative genetic investigations of this pathway in families with ASD.
Results
Biochemical ascertainment of the melatonin synthesis pathway
The study sample included 717 participants (182 patients with ASD, 364 parents, 128 unaffected siblings, 31 affected siblings and 12 other relatives) with a mean age of 31.3 years and 59.3% males (425/717). As frequently observed in ASD34,35, the sex ratio for the patients in our cohort was one female for 4.5 males (39 females and 174 males) (Table 1), whereas the number of males and females was similar in the unaffected siblings (M/F = 62/66 = 0.94). As previously reported for this cohort10,15, there were significant biochemical differences between patients and their relatives (parents or unaffected siblings) for each biochemical parameter: hyperserotonemia, increased platelet NAS, and deficit in platelet ASMT, platelet AANAT and plasma melatonin (Table 1).
Table 1.
Characteristics of the studied population including 185 families with ASD investigated for the melatonin synthesis pathway biochemical traits.
| All (415 males, 290 females) | Affected (A) (174 males, 39 females) | Unaffected (U) (62 males, 66 females) | Parents (P) (179 males, 185 females) | Wilcoxon rank-sum test | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | M (SD) | n | M (SD) | n | M (SD) | n | M (SD) | PA/U | PA/P | PU/P | |
| Age | 705 | 31.1 (18.6) | 213 | 14.4 (9.1) | 128 | 14.8 (8.0) | 364 | 46.5 (9.5) | 0.33 | ||
| Serotonin | 621 | 491.1 (319.7) | 189 | 645.8 (407.6) | 111 | 433.5 (252.2) | 321 | 419.9 (241.2) | 9.4 × 10 −6 | 4.4 × 10 −10 | 0.63 |
| AANAT | 486 | 3.9 (0.9) | 155 | 3.6 (0.9) | 72 | 4.0 (0.8) | 259 | 4.0 (0.8) | 1.1 × 10 −3 | 2.8 × 10 −6 | 0.87 |
| NAS | 348 | 36.9 (15.0) | 107 | 44.3 (15.9) | 56 | 35.0 (13.3) | 185 | 33.2 (13.3) | 6.4 × 10 −5 | 3.2 × 10 −10 | 0.41 |
| ASMT | 381 | 1.1 (0.7) | 118 | 0.8 (0.6) | 56 | 1.2 (0.7) | 207 | 1.2 (0.8) | 1.6 × 10 −4 | 2.3 × 10 −10 | 0.38 |
| Melatonin | 521 | 0.12 (0.08) | 157 | 0.10 (0.071) | 100 | 0.14 (0.08) | 264 | 0.14 (0.08) | 3.6 × 10 −8 | 2.4 × 10 −11 | 0.70 |
Affected include probands with ASD and affected siblings. Unaffected include unaffected siblings. The 12 other relatives (affected parents, grandparents, and uncles) are not included in this table. Significant p-values after Bonferroni correction are indicated in bold: 16 tests were performed including 15 critical tests (age was not relevant) p-values < 3.33 × 10−3 (0.05/15) were considered as significant. Blood serotonin (nM); platelet AANAT and ASMT activities (pmol/109 platelets/30 min); platelet NAS (nmol/109 platelets); melatonin (nM); age (years); M, Mean; SD, Standard Deviation; n, numbers of subjects; PA/U, p-value of the test comparing affected children to unaffected children; PA/P, p-value of the test comparing affected children to the parents; PU/P, p-value of the test comparing unaffected children to the parents.
Regarding the distribution of the biochemical traits within pedigrees, different categories of families could be determined (Fig. 1a). We observed that for some families, affected and/or unaffected children displayed values within the range defined by their corresponding parental values, while in other families, children biochemical values were outside this range. In most cases, children with ASD had the most extreme values compared to unaffected children. To quantify the difference between parents and affected or unaffected children, we used quartet families including one affected child and one unaffected child (Fig. 1b). For each trait, the distance between children and average parental values were compared. We observed that affected children values were significantly more distant (P < 0.005) from their corresponding average parental values than unaffected children. We therefore performed detailed familial correlation and heritability analyses of each step of the melatonin synthesis pathway.
Figure 1.

Biochemical values of molecules of the melatonin synthesis pathway. (a) Families studied in this work are divided into those with only ASD children, those with only unaffected children, and quartet families. Quartet families are then separated between those with 1) both children values within parental values, 2) both children values higher or lower than parental values, one children value within parental values and the other 3) ASD or 4) unaffected children value higher or lower, and 5) both children values outside the range of parental values but on opposite directions. Grey areas indicate pathological ranges based on previous studies in control populations (95th percentile of the controls for serotonin and NAS, 5th percentile for melatonin)10. All families are ordered according to the highest (serotonin and NAS) or lowest (AANAT, ASMT and melatonin) value observed for this family in the trait studied. (b) Quartet families selected are those for which the sibling and the two parents of the ASD proband are unaffected. For each trait monitored, the average of the two parental values was computed, and the distance from this average was measured for the two children. Distances of the ASD and the unaffected children are compared based on a Wilcoxon sign test and significance assessed after Bonferroni correction (6 tests were performed, p-values < 8.3 × 10−3 (0.05/6) were considered as significant).
Heritability of the melatonin synthesis variability
We first calculated the correlation coefficients for parent-offspring (P-O), sibling-sibling (S-S) and father-mother (F-M) pairs (Table 2). When all individuals of the families were taken into account (affected and unaffected), the P-O correlation coefficients were significantly different from 0 for all traits after Bonferroni correction for multiple testing, except for melatonin (uncorrected p-value = 0.027), and the correlation was particularly high for NAS compared with other parameters (NAS: 0.38; ASMT: 0.28; AANAT: 0.23; serotonin: 0.15, and melatonin: 0.13) (Table 2). No S-S correlations remained significant after Bonferroni correction. Interestingly, F-M correlations were significant for AANAT and melatonin.
Table 2.
Familial correlations of the biochemical traits in the studied cohort.
| Data | Group | Parent-offspring correlation (P-O) | Sibling-sibling correlation (S-S) | Father-mother correlation (F-M) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N pairs | Correlation (SE) | P-value | N pairs | Correlation (SE) | P-value | *N pairs | Correlation (SE) | P-value | ||
| Serotonin | bAll | 606 | 0.15 (0.043) | 0.0005 | 187 | 0.13 (0.080) | 0.11 | 165 | 0.058 (0.078) | 0.46 |
| aASD | 382 | 0.12 (0.054) | 0.029 | 30 | 0.094 (0.21) | 0.66 | 162 | 0.066 (0.079) | 0.41 | |
| aUnaffected | 216 | 0.20 (0.070) | 0.0063 | 35 | 0.18 (0.32) | 0.59 | 84 | 0.094 (0.11) | 0.40 | |
| AANAT | bAll | 460 | 0.23 (0.057) | 0.0001 | 138 | 0.26 (0.11) | 0.022 | 133 | 0.34 (0.077) | 0.0001 |
| bASD | 314 | 0.22 (0.064) | 0.0007 | 25 | 0.19 (0.21) | 0.38 | 133 | 0.34 (0.077) | <0.0001 | |
| cUnaffected | 140 | 0.36 (0.094) | 0.0006 | 26 | 0.099 (0.22) | 0.65 | 54 | 0.59 (0.091) | <0.0001 | |
| NAS | aAll | 328 | 0.38 (0.056) | <0.0001 | 101 | 0.21 (0.12) | 0.11 | 96 | 0.13 (0.10) | 0.20 |
| aASD | 214 | 0.40 (0.063) | <0.0001 | 12 | 0.42 (0.25) | 0.13 | 95 | 0.16 (0.10) | 0.13 | |
| cUnaffected | 102 | 0.32 (0.11) | 0.0077 | 20 | 0.63 (0.20) | 0.014 | 38 | 0.080 (0.17) | 0.64 | |
| ASMT | aAll | 354 | 0.28 (0.053) | <0.0001 | 104 | 0.23 (0.12) | 0.071 | 107 | −0.089 (0.097) | 0.37 |
| aASD | 240 | 0.24 (0.061) | 0.0001 | 13 | 0.59 (0.19) | 0.013 | 107 | −0.082 (0.097) | 0.4019 | |
| aUnaffected | 108 | 0.32 (0.090) | 0.0012 | 21 | 0.51 (0.23) | 0.058 | 40 | −0.21 (0.16) | 0.20 | |
| Melatonin | aAll | 518 | 0.13 (0.057) | 0.027 | 173 | 0.27 (0.088) | 0.0035 | 136 | 0.30 (0.078) | 0.0003 |
| bASD | 318 | 0.055 (0.072) | 0.45 | 28 | 0.23 (0.21) | 0.29 | 132 | 0.31 (0.079) | 0.0003 | |
| aUnaffected | 192 | 0.21 (0.090) | 0.025 | 35 | 0.62 (0.16) | 0.0023 | 73 | 0.20 (0.11) | 0.10 | |
Significant p-values after Bonferroni correction are indicated in bold: 45 tests were performed, p-values < 1.11 × 10−3 (0.05/45) were considered as significant. N pairs, number of relative pairs included in the analysis; SE, Standard Error; aage was included in the model as a covariate; bage and sex were included in the model as covariates; cno covariates were included in the model. *Number of father-mother pairs (F-M): parents included in the analyses using all family members and in stratified analyses as described in Supplementary Table S4.
We then estimated the heritability of each trait. When all individuals were taken into account, the narrow sense estimates of heritability (reflecting additive effects) were all significant (Bonferroni corrected p-value < 0.05; Fig. 2 and Supplementary Table S1). The heritability estimates ranged from 0.22 for melatonin to 0.72 for NAS (serotonin: 0.31; AANAT: 0.34; NAS: 0.72; ASMT: 0.59; melatonin: 0.22) providing support for a significant genetic contribution to the melatonin synthesis variability. Proportions of variance due to all covariates included were relatively low, ranging from 0.029 to 0.072 (Supplementary Table S2). When the sample was stratified by ASD status, all heritability estimates tended to be higher for unaffected children than for children with ASD (Fig. 2 and Supplementary Table S1). For example, both serotonin and melatonin heritability estimates were not significant for children with ASD, after Bonferroni correction for multiple testing.
Figure 2.
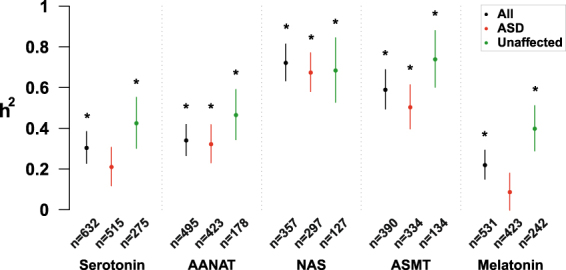
Narrow sense heritability estimates (h2) of the biochemical traits associated with the melatonin synthesis pathway. Heritability was calculated using the variance component analysis with maximum likelihood estimation method. When the effect of the covariates (age, sex) was significant (P < 0.1), they were included in the model. The significance of heritability estimates is assessed using a loglikelihood ratio test that compares the polygenic model to a sporadic model in which the additive genetic effect is constrained to zero. n, number of participants; dots, h2 estimates; error bars, standard errors. *Bonferroni corrected P-value < 0.05 (15 tests were performed, p-values < 3.3 × 10−3 (0.05/15) were considered as significant).
To investigate the relationship between each pair of traits and evaluate the proportion of heritability of one trait that is attributable to the other trait, we calculated bivariate trait correlation coefficients for ASD and unaffected children separately (Fig. 3a). In both groups, we observed phenotypic correlations ρP > 0.4 for ASMT-melatonin, ρP < −0.8 for NAS-ASMT and a ρP < −0.2 for NAS-melatonin. These results confirmed a high correlation between the three parameters of the last step of the melatonin biosynthesis pathway (NAS, ASMT and melatonin), as previously reported15. When only children with ASD were included, these phenotypic correlations were significantly different from 0 (NAS-ASMT: ρP = −0.84; ASMT-melatonin: ρP = 0.57; NAS-melatonin: ρP = −0.42) (Fig. 3a). For unaffected children, only ASMT-melatonin phenotypic correlation remained significant after Bonferroni correction (ASMT-melatonin: ρP = 0.42). Regarding genetic correlations, two negative genetic correlations were significantly different from 0 in children with ASD (NAS-ASMT: ρG = −0.96; NAS-melatonin: ρG = −1.0) and in unaffected children, only one genetic correlation was significant (NAS-ASMT: ρG = −0.90) (Fig. 3a). These genetic correlations were not significantly different from 1 or −1. There were also significant common environmental influences for NAS-ASMT (ρE = −0.70) and for ASMT-melatonin (ρE = 0.48) in children with ASD (Fig. 3a). In conclusion, these results showed an overlap of additive genetic effects as well as shared environmental factors influencing phenotypic variability in families with ASD, particularly for NAS, ASMT and melatonin (Fig. 3b).
Figure 3.

Heritability analysis of the melatonin synthesis pathway in families with ASD. (a) Bivariate heritability analyses. Pairwise phenotypic correlations (ρP), additive genetic correlations (ρG) and shared environmental correlations (ρE). Correlations calculated in patients with ASD are on the upper diagonal; correlations calculated in unaffected siblings are on the lower diagonal. P-values test the significance of the difference of ρP, ρG and ρE from zero. The diamond shape used for the phenotypic correlation between ASMT and NAS in unaffected individuals means that the p-value could not be tested according to SOLAR. *Significant p-values after Bonferroni correction: 60 tests were performed, p-values < 8.33 × 10−4 (0.05/60) were considered as significant. (b) Overview of heritability analyses results for the biochemical traits associated with the melatonin synthesis pathway in patients with ASD and in unaffected individuals. Circle sizes represent heritability estimates. Circle colors represent the proportion of patients or unaffected siblings with biochemical values >95th percentile of controls in purple and <5th percentile of controls in green, as previously published13,19. Grey curved lines represent environmental and genetic correlations with nominal P-values < 0.05 and their thickness is proportional to correlation values. *Significant correlations after Bonferroni correction.
Discussion
Very few heritability estimates for serotonin and melatonin are available for humans in the literature (Table 3) and this study is the first to characterize the heritability of the melatonin synthesis variability in a large sample of families with ASD. We first calculated familial correlations of the five traits (serotonin, AANAT, NAS, ASMT and melatonin). Using all family members, we observed significant P-O correlations except for melatonin, but no S-S correlations remained significant after correction for multiple testing, probably due to the relatively small number of S-S pairs compared to P-O pairs. Unexpectedly, we also observed significant correlations between fathers and mothers for AANAT and melatonin. AANAT activity and melatonin production display marked circadian variations in the pineal gland15,36. These processes are closely related to light-dark cycle37–39 and their regulation might depend on environmental factors, such as sleep-wake cycle and artificial light exposure. Thus, synchronization could be observed between parents that share daily habits and life rhythms.
Table 3.
Review of the literature on serotonin and melatonin heritability estimates in humans.
| Trait | Study | Heritability estimates and main findings | N | Subjects | Tissue | Method |
|---|---|---|---|---|---|---|
| Serotonin | Abney et al.27 | h2 = 0.51 (SE: 0.11) H2 = 1.0 (SE: 0.25) | 567 | Hutterite families, large pedigrees | Blood | Maximum-likelihood with variance component model. Best-fitting model: ADE58 |
| Coutinho et al.59 | H2 = 0.64 | 327 | Trio families with ASD | Platelet | Maximum-likelihood with variance component model. Polygenic model (QTDT60) | |
| Shin et al.26 | h2 = 0.331 (95% CI: 0.168, 0.426) | 866 MZ pairs 878 DZ pairs | TwinsUK cohort | Blood | Maximum-likelihood with variance component model. ACE model (OpenMx61) | |
| Rhee et al.33 | h2 ∼ 0.2 | 2,076 | Framingham Heart Study Offspring cohort families | Blood | Maximum-likelihood with variance component model. Additive genetic model (SOLAR54) | |
| This study | h2 = 0.31 (SE: 0.08) | 632 | PARIS cohort families (all individuals) | Blood | Maximum-likelihood with variance component model. Additive genetic model (SOLAR54) | |
| h2 = 0.21 (SE: 0.09) | 515 | PARIS cohort families (parents and affected children) | ||||
| h2 = 0.43 (SE: 0.13) | 275 | PARIS cohort families (parents and unaffected children) | ||||
| Melatonin | Wetterberg et al.32 | h2 = 0.53 | 107 | Swedish families with acute intermittent porphyria | Urine | Morton and MacLean mixed model for complex segregation analysis62,63 |
| Hallam et al.31 | rMZ = 0.928 rDZ = 0.867 | 6 MZ pairs 11 DZ pairs | Australian Twin Registry | Plasma | Monozygotic and dizygotic twin correlations | |
| This study | h2 = 0.22 (SE: 0.07) | 531 | PARIS cohort families (all individuals) | Plasma | Maximum-likelihood with variance component model. Additive genetic model (SOLAR54) | |
| h2 = 0.09 (SE: 0.09) | 423 | PARIS cohort families (parents and affected children) | ||||
| h2 = 0.40 (SE: 0.11) | 242 | PARIS cohort families (parents and unaffected children) |
N, number of subjects; h2, narrow sense heritability; H2, broad sense heritability; SE, standard error; MZ, monozygotic twins; DZ, dizygotic twins; A, additive; D, dominant; E, unique environment; C, common environment; rMZ, monozygotic twin correlation; rDZ, dizygotic twin correlation.
We then estimated the heritability of the five traits. In the literature, the first estimate of blood serotonin heritability was obtained by Abney et al.27 in an inbred founder population of Hutterite families. They found respectively 0.51 and 1.0 for narrow and broad sense heritability, suggesting a very strong genetic influence with both additive and dominance components. Our estimation of serotonin narrow sense heritability was lower (0.31) than the one reported by Abney et al., but very similar to the one obtained in a large cohort of 866 monozygotic and 878 dizygotic unaffected twin pairs (0.33; Table 3)26. For melatonin, our estimation of the heritability (0.22) ranged between two previous estimates31,32. Wetterberg et al. (1983) estimated melatonin heritability to 0.53 in 107 families with acute intermittent porphyria32. In contrast, a twin study measuring late night plasma melatonin in nine monozygotic and 11 dizygotic twin pairs, reported a high monozygotic correlation (r MZ = 0.928) and a high dizygotic correlation (r DZ = 0.867)31. Using these correlations and the Falconer’s formula (H2 = 2(rMZ - rDZ)), the heritability for melatonin would be 2(0.928-0.867) = 0.12.
All the five traits studied here are not expected to be independent since they belong to the same metabolic pathway. Using a bivariate approach, we estimated pairwise phenotypic, genetic and environmental correlations in samples including only children with ASD or only unaffected children. In both groups, phenotypic correlations appeared to be particularly high for the last step of melatonin biosynthesis pathway (NAS, ASMT and melatonin) (Fig. 3b). Therefore, considering these traits as dependent might be useful for further association studies exploring variants associated with these phenotypes. Interestingly, although ASMT and NAS were phenotypically correlated with melatonin, their heritability differed, particularly in children with ASD (high for ASMT and NAS, low for melatonin). In children with ASD, this discrepancy could be explained for ASMT and melatonin by the fact that phenotypic correlation between these traits was mainly attributable to shared environment while genetic factors seemed to be more divergent (Fig. 3a). However, for NAS and melatonin, genetic correlation was high and environmental correlation was not significant. These results suggest that even if there are probably common genetic mechanisms in the regulation of these three traits, environmental factors such as light exposure or drugs interacting with cytochromes, might act differentially on NAS and melatonin levels.
There are several limitations of this study such as the absence of data on circadian rhythm. However, previous studies showed that the deficit in melatonin in ASD was also observed in blood and urine samples collected during the night12,13,40. Another limitation is the absence of analyses in the pineal gland. Finally, our estimations for unaffected children might not reflect the heritability the melatonin synthesis variability in the general population. Nevertheless, for serotonin and melatonin, our heritability estimates are very similar from those obtained in previous studies (Table 3)26,27,32,33.
In summary, our results revealed that the resemblance between unaffected children and parents seems to be mostly due to additive genetic effects, while patients resemblance to their parents, which was lower, probably includes additional factors such as de novo events, dominant effects, or non-shared environmental influences related to ASD condition. Another interesting finding is that NAS displayed the highest heritability estimate within the melatonin synthesis pathway. In contrast to serotonin and melatonin, NAS received much less interest from the scientific and clinical community10,41. This is unfortunate, especially since NAS could have its own biological functions, such as TrkB activation, immunomodulation or analgesia19,41–44. We thus propose to use NAS, in addition to serotonin and melatonin, as a suitable endophenotype for further quantitative genetic studies in ASD. Further studies using larger study samples and molecular genetic analyses such as genome-wide association studies are now required to identify the variants involved in melatonin synthesis variability in the general population and in patients suffering from sleep disorders and circadian rhythms abnormalities.
Subjects and Methods
Ethics statement
The local Institutional Review Boards (IRB) at the Institut Pasteur in Paris (France) approved the study. Methods were performed in accordance with the relevant guidelines and regulations. Written informed consents were obtained from all participants. For the patients who were unable to consent for themselves, a parent or legal guardian consented to the study on their behalf.
Subjects and clinical evaluations
Clinical evaluations of patients with ASD, their relatives and control subjects have been detailed previously10. All the subjects were recruited into the Paris Autism Research International Sib-pair (PARIS) study. The ASD diagnosis was based on clinical expert assessment including the Autism Diagnostic Interview – Revised (ADI-R)45 and the Autism Diagnostic Observation Schedule (ADOS)46. Intellectual Quotient (IQ) was measured using an age-appropriate Wechsler scale. For the most severe and/or non-verbal patients, the Raven’s Standard Progressive Matrices and the Peabody Picture Vocabulary test were used.
Biochemical measurements
Blood samples were collected in the morning, between 8:30 and 10:30 as described previously10,14. Subjects were asked to avoid food with high content of tryptophan and/or serotonin two days before blood sampling and individuals receiving exogenous melatonin were not included in this study. Melatonin was measured in plasma using a radio-immunoassay (RK-MEL, Bühlmann, Switzerland) according to the manufacturer’s instructions. Whole-blood serotonin (5-hydroxytryptamine, 5-HT) was measured by high-performance liquid chromatography47. NAS, as well as enzyme activities of AANAT and ASMT were determined in platelets by radio-enzymology48. Patients who were receiving melatonin for treatment of sleep disorders were not included in the analyses. Only pedigrees with both parents and at least one child were included in the following analyses. The overlap between the biochemical data for serotonin, AANAT, NAS, ASMT and melatonin is illustrated in Supplementary Fig. S2. A detailed study of the differences between groups has been published elsewhere10,15.
Statistical methods
Data management and graphs were performed using JMP Pro 12 (SAS, USA) and R software49. Because some of the studied traits were not normally distributed, nonparametric statistical tests (Wilcoxon rank-sum test) were preferred to compare groups of individuals. For familial correlation and heritability analyses, serotonin, ASMT and melatonin values were logarithmically transformed in order to conform more closely to a normal distribution (Supplementary Fig. S3). Since melatonin and serotonin are known to be age and sex dependent50–53, they were included in the models using residuals from regression models obtained with the program Sequential Oligogenic Linkage Analysis Routines (SOLAR) 7.2.5 (Southern Foundation for Biomedical Research, San Antonio, TX, USA)54,55, when their effect as covariates (age, sex) were significant at the threshold of P < 0.1. Individuals at more than three standard deviations (SD) from the mean were considered as outliers and were removed. Depending on the biochemical trait and the subgroup considered, the number of outliers was always very low, ranging from 0 to 7 (Supplementary Table S3). A total of 717 individuals (182 ASD patients and 535 relatives) were included in this study. There were 185 families, including from three to nine relatives (Supplementary Table S4): 68 trios, 87 quartets, 22 quintets, four sextets, one septet, one nonet and two extended pedigrees. For three families, biochemical measurements could be obtained for the parents and unaffected children, but not for the ASD probands and these families were included in the analyses. For each analysis, uncorrected p-values are shown, but due to multiple testing, the significance was assessed after Bonferroni correction.
Estimation of familial correlation and heritability
Parent-offspring (P-O), sibling-sibling (S-S) and father-mother (F-M) correlations were measured for the five biochemical traits using the Family Correlations (FCOR) module of the Statistical Analysis for Genetic Epidemiology (S.A.G.E.) 6.3 software package (Human Genetic Analysis Resource, Case Western Reserve University, Cleveland, OH, USA)56. For each relative pair types available in the sample pedigrees, FCOR calculates sibling-sibling (S-S), parent-offspring (P-O) and father-mother (F-M) familial correlations with their asymptotic standard errors. S.A.G.E then tests the difference of the correlation coefficients from 0 using Fisher’s z-transformation.
Narrow sense heritability was estimated using the variance component-based program SOLAR 7.2.5 with a maximum likelihood estimation method and polygenic models. When the effect of the covariates (age, sex) was significant (P < 0.1), relative proportion of variance explained by known covariates was estimated and they were included in the model.
SOLAR can extend the variance component model to a bivariate analysis that maximizes the model for two dependent traits57. The trait pairs included all pairwise combinations and the same covariates (age and sex) significant at the threshold of P < 0.1 were used. Three parameters were estimated for each pair of traits: the additive genetic correlation (ρG), the shared environmental correlation (ρE) and the total phenotypic correlation (ρP) (Supplementary Note).
Familial correlations and univariate heritability analyses were first performed on all individuals, including parents and their affected children as well as their unaffected children (Supplementary Fig. S4). Secondly, stratified analyses were conducted on parents with their affected children and on parents with their unaffected children (Supplementary Fig. S4).
Electronic supplementary material
Acknowledgements
The authors gratefully acknowledge the families who accepted to participate in this study. The Clinical Investigation Centers of Robert-Debré and Henri Mondor Hospitals obtained and processed blood samples, the Hematology departments from both hospitals (Dr MF Hurtaud and Professor M Imbert) performed platelet counts. We also thank Audrey Grant at Institut Pasteur for her advices on heritability analyses. This work was supported by the Institut Pasteur, CNRS, INSERM, AP-HP, University Paris Diderot, University Paris Descartes, the Fondation pour la Recherche Médicale (FRM: FDT20160435693), the Cognacq-Jay foundation, the Bettencourt-Schueller foundation, the Orange foundation, the FondaMental foundation, the Conny-Maeva foundation, the ANR (SynDivAutism) and the Labex GenMed and BioPsy.
Author Contributions
M.B. and T.B. designed the study. R.D., M.L., C.P., and J.C. contributed to data acquisition. M.B., T.R., C.S.L., G.M., G.H., C.P., E.M. and T.B. participated to data analysis and interpretation. M.B. and T.B. drafted the manuscript. All authors contributed to the critical revision and approved the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-18016-3.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sandin S, et al. The familial risk of autism. JAMA. 2014;311:1770–1777. doi: 10.1001/jama.2014.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tick B, Bolton P, Happé F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: a meta-analysis of twin studies. J. Child Psychol. Psychiatry. 2016;57:585–595. doi: 10.1111/jcpp.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaugler T, et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vijayakumar NT, Judy MV. Autism spectrum disorders: Integration of the genome, transcriptome and the environment. J. Neurol. Sci. 2016;364:167–176. doi: 10.1016/j.jns.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 5.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 6.Schain RJ, Freedman DX. Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. J. Pediatr. 1961;58:315–320. doi: 10.1016/S0022-3476(61)80261-8. [DOI] [PubMed] [Google Scholar]
- 7.Launay JM, et al. Serotonin metabolism and other biochemical parameters in infantile autism. A controlled study of 22 autistic children. Neuropsychobiology. 1988;20:1–11. doi: 10.1159/000118465. [DOI] [PubMed] [Google Scholar]
- 8.Leboyer M, et al. Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biol. Psychiatry. 1999;45:158–163. doi: 10.1016/S0006-3223(97)00532-5. [DOI] [PubMed] [Google Scholar]
- 9.Mulder EJ, et al. Platelet serotonin levels in pervasive developmental disorders and mental retardation: diagnostic group differences, within-group distribution, and behavioral correlates. J. Am. Acad. Child Adolesc. Psychiatry. 2004;43:491–499. doi: 10.1097/00004583-200404000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Pagan C, et al. The serotonin-N-acetylserotonin-melatonin pathway as a biomarker for autism spectrum disorders. Transl. Psychiatry. 2014;4:e479. doi: 10.1038/tp.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabriele S, Sacco R, Persico AM. Blood serotonin levels in autism spectrum disorder: A systematic review and meta-analysis. Eur. Neuropsychopharmacol. 2014;24:919–929. doi: 10.1016/j.euroneuro.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Kulman G, et al. Evidence of pineal endocrine hypofunction in autistic children. Neuro Endocrinol. Lett. 2000;21:31–34. [PubMed] [Google Scholar]
- 13.Tordjman S, et al. Day and nighttime excretion of 6-sulphatoxymelatonin in adolescents and young adults with autistic disorder. Psychoneuroendocrinology. 2012;37:1990–1997. doi: 10.1016/j.psyneuen.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 14.Melke J, et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol. Psychiatry. 2007;13:90–98. doi: 10.1038/sj.mp.4002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pagan C, et al. Disruption of melatonin synthesis is associated with impaired 14-3-3 and miR-451 levels in patients with autism spectrum disorders. Sci. Rep. 2017;7:2096. doi: 10.1038/s41598-017-02152-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sundström E, et al. Neurochemical differentiation of human bulbospinal monoaminergic neurons during the first trimester. Brain Res. Dev. Brain Res. 1993;75:1–12. doi: 10.1016/0165-3806(93)90059-J. [DOI] [PubMed] [Google Scholar]
- 17.Whitaker-Azmitia PM. Serotonin and brain development: role in human developmental diseases. Brain Res. Bull. 2001;56:479–485. doi: 10.1016/S0361-9230(01)00615-3. [DOI] [PubMed] [Google Scholar]
- 18.Nonno R, et al. Ligand efficacy and potency at recombinant human MT2 melatonin receptors: evidence for agonist activity of some mt1-antagonists. Br. J. Pharmacol. 1999;127:1288–1294. doi: 10.1038/sj.bjp.0702658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jang S-W, et al. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. USA. 2010;107:3876–3881. doi: 10.1073/pnas.0912531107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiter RJ. The melatonin rhythm: both a clock and a calendar. Experientia. 1993;49:654–664. doi: 10.1007/BF01923947. [DOI] [PubMed] [Google Scholar]
- 21.Reiter RJ, Tan D-X, Fuentes-Broto L. Melatonin: a multitasking molecule. Prog. Brain Res. 2010;181:127–151. doi: 10.1016/S0079-6123(08)81008-4. [DOI] [PubMed] [Google Scholar]
- 22.Goldman BD. The circadian timing system and reproduction in mammals. Steroids. 1999;64:679–685. doi: 10.1016/S0039-128X(99)00052-5. [DOI] [PubMed] [Google Scholar]
- 23.Claustrat B, Brun J, Chazot G. The basic physiology and pathophysiology of melatonin. Sleep Med. Rev. 2005;9:11–24. doi: 10.1016/j.smrv.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Ibrahim SH, Voigt RG, Katusic SK, Weaver AL, Barbaresi WJ. Incidence of gastrointestinal symptoms in children with autism: a population-based study. Pediatrics. 2009;124:680–686. doi: 10.1542/peds.2008-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malow BA, et al. Sleep Difficulties and Medications in Children With Autism Spectrum Disorders: A Registry Study. Pediatrics. 2016;137(Suppl 2):S98–S104. doi: 10.1542/peds.2015-2851H. [DOI] [PubMed] [Google Scholar]
- 26.Shin S-Y, et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014;46:543–550. doi: 10.1038/ng.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abney M, McPeek MS, Ober C. Broad and Narrow Heritabilities of Quantitative Traits in a Founder Population. Am. J. Hum. Genet. 2001;68:1302–1307. doi: 10.1086/320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye, R. et al. Evaluation of heritable determinants of blood and brain serotonin homeostasis using recombinant inbred mice. Genes Brain Behav. 13, 247–260, 10.1111/gbb.12092 (2013). [DOI] [PMC free article] [PubMed]
- 29.Zarazaga LA, Malpaux B, Bodin L, Chemineau P. The large variability in melatonin blood levels in ewes is under strong genetic influence. Am. J. Physiol. 1998;274:E607–610. doi: 10.1152/ajpendo.1998.274.4.E607. [DOI] [PubMed] [Google Scholar]
- 30.Allain D, et al. Genetic variability of the pattern of night melatonin blood levels in relation to coat changes development in rabbits. Genet. Sel. Evol. GSE. 2004;36:207–216. doi: 10.1186/1297-9686-36-2-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hallam KT, et al. The heritability of melatonin secretion and sensitivity to bright nocturnal light in twins. Psychoneuroendocrinology. 2006;31:867–875. doi: 10.1016/j.psyneuen.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 32.Wetterberg L, Iselius L, Lindsten J. Genetic regulation of melatonin excretion in urine. A preliminary report. Clin. Genet. 1983;24:399–402. doi: 10.1111/j.1399-0004.1983.tb00093.x. [DOI] [PubMed] [Google Scholar]
- 33.Rhee EP, et al. A Genome-wide Association Study of the Human Metabolome in a Community-Based Cohort. Cell Metab. 2013;18:130–143. doi: 10.1016/j.cmet.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miles JH, et al. Essential versus complex autism: definition of fundamental prognostic subtypes. Am. J. Med. Genet. A. 2005;135:171–180. doi: 10.1002/ajmg.a.30590. [DOI] [PubMed] [Google Scholar]
- 35.Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 2013;26:146–153. doi: 10.1097/WCO.0b013e32835ee548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simonneaux V, Ribelayga C. Generation of the melatonin endocrine message in mammals: a review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol. Rev. 2003;55:325–395. doi: 10.1124/pr.55.2.2. [DOI] [PubMed] [Google Scholar]
- 37.Ackermann K, Stehle JH. Melatonin synthesis in the human pineal gland: advantages, implications, and difficulties. Chronobiol. Int. 2006;23:369–379. doi: 10.1080/07420520500464379. [DOI] [PubMed] [Google Scholar]
- 38.Maronde E, Stehle JH. The mammalian pineal gland: known facts, unknown facets. Trends Endocrinol. Metab. TEM. 2007;18:142–149. doi: 10.1016/j.tem.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Brainard GC, et al. Sensitivity of the human circadian system to short-wavelength (420-nm) light. J. Biol. Rhythms. 2008;23:379–386. doi: 10.1177/0748730408323089. [DOI] [PubMed] [Google Scholar]
- 40.Nir I, et al. Brief report: circadian melatonin, thyroid-stimulating hormone, prolactin, and cortisol levels in serum of young adults with autism. J. Autism Dev. Disord. 1995;25:641–654. doi: 10.1007/BF02178193. [DOI] [PubMed] [Google Scholar]
- 41.Carter MD, Wade Calcutt M, Malow BA, Rose KL, Hachey DL. Quantitation of melatonin and n-acetylserotonin in human plasma by nanoflow LC-MS/MS and electrospray LC-MS/MS: Melatonin and N-acetylserotonin quantitation. J. Mass Spectrom. 2012;47:277–285. doi: 10.1002/jms.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perianayagam MC. Immune-Modulating Effects of Melatonin, N-Acetylserotonin, and N-Acetyldopamine. Ann. N. Y. Acad. Sci. 2005;1053:386–393. doi: 10.1196/annals.1344.033. [DOI] [PubMed] [Google Scholar]
- 43.Psarakis S, Brown GM, Grota LJ. Analgesia induced by N-acetylserotonin in the central nervous system. Life Sci. 1988;42:1109–1116. doi: 10.1016/0024-3205(88)90567-X. [DOI] [PubMed] [Google Scholar]
- 44.Sompol P, et al. N-acetylserotonin promotes hippocampal neuroprogenitor cell proliferation in sleep-deprived mice. Proc. Natl. Acad. Sci. USA. 2011;108:8844–8849. doi: 10.1073/pnas.1105114108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 46.Lord C, et al. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 2000;30:205–223. doi: 10.1023/A:1005592401947. [DOI] [PubMed] [Google Scholar]
- 47.Kema IP, et al. High performance liquid chromatographic profiling of tryptophan and related indoles in body fluids and tissues of carcinoid patients. Clin. Chim. Acta Int. J. Clin. Chem. 1993;221:143–158. doi: 10.1016/0009-8981(93)90029-4. [DOI] [PubMed] [Google Scholar]
- 48.Launay JM, Geoffroy C, Costa JL, Alouf JE. Purified -SH-activated toxins (streptolysin O, alveolysin): new tools for determination of platelet enzyme activities. Thromb. Res. 1984;33:189–196. doi: 10.1016/0049-3848(84)90179-8. [DOI] [PubMed] [Google Scholar]
- 49.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/ (2014).
- 50.Cain SW, et al. Sex Differences in Phase Angle of Entrainment and Melatonin Amplitude in Humans. J. Biol. Rhythms. 2010;25:288–296. doi: 10.1177/0748730410374943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ortiz J, Artigas F, Gelpí E. Serotonergic status in human blood. Life Sci. 1988;43:983–990. doi: 10.1016/0024-3205(88)90543-7. [DOI] [PubMed] [Google Scholar]
- 52.Ritvo E, et al. Maturational changes in blood serotonin levels and platelet counts. Biochem. Med. 1971;5:90–96. doi: 10.1016/0006-2944(71)90077-9. [DOI] [PubMed] [Google Scholar]
- 53.Waldhauser F, Ehrhart B, Förster E. Clinical aspects of the melatonin action: impact of development, aging, and puberty, involvement of melatonin in psychiatric disease and importance of neuroimmunoendocrine interactions. Experientia. 1993;49:671–681. doi: 10.1007/BF01923949. [DOI] [PubMed] [Google Scholar]
- 54.Blangero, J. & Almasy, L. Solar: sequential oligogenic linkage analysis routines. Popul. Genet. Lab. Tech. Rep. 6 (1996).
- 55.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.S.A.G. E. Statistical Analysis for Genetic Epidemiology, Release 6.3. Human Genetic Analysis Resource, Case Western Reserve University, Cleveland, OH, USA. (2012).
- 57.Almasy L, Dyer TD, Blangero J. Bivariate quantitative trait linkage analysis: pleiotropy versus co-incident linkages. Genet. Epidemiol. 1997;14:953–958. doi: 10.1002/(SICI)1098-2272(1997)14:6<953::AID-GEPI65>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 58.Abney M, McPeek MS, Ober C. Estimation of Variance Components of Quantitative Traits in Inbred Populations. Am. J. Hum. Genet. 2000;66:629–650. doi: 10.1086/302759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coutinho AM, et al. Evidence for epistasis between SLC6A4 and ITGB3 in autism etiology and in the determination of platelet serotonin levels. Hum. Genet. 2007;121:243–256. doi: 10.1007/s00439-006-0301-3. [DOI] [PubMed] [Google Scholar]
- 60.Abecasis GR, Cardon LR, Cookson WO. A general test of association for quantitative traits in nuclear families. Am. J. Hum. Genet. 2000;66:279–292. doi: 10.1086/302698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boker S, et al. OpenMx: An Open Source Extended Structural Equation Modeling Framework. Psychometrika. 2011;76:306–317. doi: 10.1007/s11336-010-9200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morton NE, MacLean CJ. Analysis of family resemblance. 3. Complex segregation of quantitative traits. Am. J. Hum. Genet. 1974;26:489–503. [PMC free article] [PubMed] [Google Scholar]
- 63.Lalouel JM, Morton NE. Complex segregation analysis with pointers. Hum. Hered. 1981;31:312–321. doi: 10.1159/000153231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.