

Abstract
Primary ubiquinone (UQ) deficiency is an important subset of mitochondrial disease that is caused by mutations in UQ biosynthesis genes. To guide therapeutic efforts we sought to estimate the number of individuals who are born with pathogenic variants likely to cause this disorder. We used the NCBI ClinVar database and literature reviews to identify pathogenic genetic variants that have been shown to cause primary UQ deficiency, and used the gnomAD database of full genome or exome sequences to estimate the frequency of both homozygous and compound heterozygotes within seven genetically-defined populations. We used known population sizes to estimate the number of afflicted individuals in these populations and in the mixed population of the USA. We then performed the same analysis on predicted pathogenic loss-of-function and missense variants that we identified in gnomAD. When including only known pathogenic variants, our analysis predicts 1,665 affected individuals worldwide and 192 in the USA. Adding predicted pathogenic variants, our estimate grows to 123,789 worldwide and 1,462 in the USA. This analysis predicts that there are many undiagnosed cases of primary UQ deficiency, and that a large proportion of these will be in developing regions of the world.
Introduction
Mitochondrial disease is a complex and heterogeneous collection of disorders that can result in death or prolonged disability. The prevalence of these disorders could be as high as 1 in 4,300 individuals, which makes it one of the most common forms of inherited illness1,2. There is no general cure for these disorders but the most widespread treatments are vitamin and nutritional supplementation, most commonly with L-carnitine, creatine and ubiquinone (UQ; a.k.a. Coenzyme Q), despite the fact that there is little evidence supporting their effectiveness3,4.
UQ is a redox-active lipid-like molecule that plays a number of critical roles in biological membranes. Its best characterized role is as a key electron carrier of the mitochondrial electron transport chain5. UQ is also a co-factor in a number of other enzymatic processes as well as a potential membrane antioxidant. The rationale for generalized UQ supplementation in mitochondrial disease is thus the hope that it might support mitochondrial function, and that its antioxidant function could ameliorate any increase in oxidative stress. Furthermore, UQ deficiency secondary to other mitochondrial defects is observed in a substantial subset of mitochondrial disease patients6.
There is, however, one patient population that could directly benefit from effective UQ supplementation: individuals suffering from primary UQ deficiency due to mutations in genes required for UQ biosynthesis. Although such patients have been much discussed7–9, we are not aware of any formal attempt to estimate the prevalence of primary UQ deficiency. At this point, approximately 70 patients have been described in the published literature, and it has been informally estimated that their prevalence may be less than 1 in 100,0008. Despite clear genetic evidence that UQ deficiency is the primary cause in these patients, UQ supplementation has not met with consistent success, possibly due to poor bioavailability of the highly lipophilic UQ molecule10,11. A better understanding of the possible prevalence of this disorder would help guide decisions regarding investigations into novel UQ formulations or potential drugs which could modulate the UQ biosynthesis pathway.
UQ is composed of a redox-active benzoquinone ring with a lipid tail consisting of a species-specific number of isoprenoid sub-units (ten in humans). Although UQ biosynthesis has been most extensively studied in yeast, human homologues of the critical genes have been identified7–9. Thirteen yeast genes are required for UQ biosynthesis (COQ1 – COQ11, YAH1, ARH1). In brief, COQ1 (or the human homologues PDSS1 and PDSS2 acting as a hetero-tetramer) assembles an isoprenoid tail from precursors produced by the mevalonate pathway. COQ2 joins this isoprenoid tail to a tyrosine-derived benzoquinone ring precursor, and COQ3, COQ5, COQ6 and COQ7 are responsible for various methylation and hydroxylation reactions affecting the benzoquinone ring. COQ8 appears to play a regulatory role by modulating phosphorylation of COQ3, COQ5 and COQ7. COQ8 has two human homologues, COQ8A (also known as ADCK3 or CABC1) and COQ8B (ADCK4), both of which can independently result in UQ deficiency12,13. The roles of COQ4 and COQ9 are not well defined, although COQ4 appears to play a role in the assembly of COQ2 – COQ7 into a complex and COQ9 is required for COQ7 function. ARH1 (human homologue FDX1L) and YAH1 (FDXR) transfer electrons to COQ6, while also participating in other pathways. There are two modification steps of the UQ benzoquinone ring that have yet to be assigned an enzyme.
To date, pathogenic variants in nine of these proteins (PDSS1, PDSS2, COQ2, COQ4, COQ6, COQ7, COQ8A, COQ8B and COQ9) have been shown to cause UQ deficiency in human patients7,9. We sought to leverage the recent availability of exome or genome sequences of very large numbers of individuals in order to estimate the frequency of known pathogenic variants in these genes. We used the NCBI ClinVar database14 and conducted a literature search to identify variants in the known UQ biosynthesis genes that result in illness and UQ deficiency. The gnomAD exome and genome database15, with sequences for almost 138,632 individuals divided into seven genetically-distinct populations, was used to estimate the frequencies of these variants. Using these frequencies, we estimated the birth prevalence of individuals homozygous or compound heterozygous for known or predicted pathogenic genetic variants for primary UQ deficiency (assuming Hardy-Weinberg equilibria) on a population-by-population basis and used known population sizes and distributions to estimate the actual numbers of afflicted individuals due to each variant world-wide, as well as in a population with the particular size and mix of the USA. Importantly, the calculation of the number of afflicted individuals on a per-variant, per-population, basis eliminates a potential confounding factor when working with large numbers of variants present at very low frequencies – namely, that many individual variants may be too rare to result in any homozygous or compound heterozygous individuals, and the traditional method of summing these frequencies could yield frequencies high enough to artificially suggest that individuals are affected.
It is likely that many pathogenic variants simply have not been clinically documented at this relatively early stage in our awareness of primary UQ deficiency. To account for this, we also estimated the number of individuals who would be homozygous or compound heterozygous for variants observed in gnomAD but that have not yet been observed in the clinic, focusing on predicted loss-of-function (LoF) or pathogenic missense mutations.
There are many challenges to making estimates of this nature. For example, it is not possible to conclusively determine the pathogenicity of missense variants based on sequence information alone. We attempt to address this by conservatively included only those variants independently predicted to be pathogenic by two separate bioinformatic algorithms (see Methods). There is also extreme variability in severity of primary UQ deficiency, ranging from neonatal lethality (with mouse studies suggesting that embryonic lethality is a possible outcome for null alleles for some genes16–19) to mild disease that becomes apparent only in later decades of life. This makes accurate predictions of disease prevalence based on allelic frequencies extrapolated from public databases of genomic variants challenging, which is why our results are best interpreted as birth prevalence of individuals homozygous or compound heterozygous for variants likely to cause disease. Actual disease prevalence would be expected to diverge from these estimates. We discuss these issues in greater detail below.
We found that the carrier frequencies for most previously identified pathogenic variants were low (averaging 1/6,420 for the populations in which they were present), and given known population sizes we estimated they would result in a total of 1,016 individuals worldwide due to homozygosity and an additional 649 due to compound heterozygosity, with a total of 192 in the USA. The addition of all predicted loss-of-function and pathogenic missense variants results in a predicted total of 123,789 individuals worldwide and 1,462 in the USA.
Methods
Identification of known pathogenic variants
We identified pathogenic variants of UQ biosynthesis genes (PDSS1, PDSS2, COQ2 – COQ7, COQ8A/ADCK3, COQ8B/ADCK4 and COQ9) using the NCBI ClinVar database and via PubMed literature searches. ClinVar is a public archive (https://www.ncbi.nlm.nih.gov/clinvar/) describing human genetic variants and their relationship to human health14. Variants are extracted from the peer-reviewed literature or directly reported by CLIA certified or ISO 1589 accredited clinical testing laboratories. Variant pathogenicity is reported by the submitter according to the ordinal scale recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (“pathogenic”, “likely pathogenic”, “uncertain significance”, “likely benign” or “benign”)20. Note that ClinVar results cannot be used to directly estimate birth prevalence and the database does not include fields for incidence frequency. Each ClinVar entry describes a unique variant, and may be derived from multiple submissions.
We queried ClinVar (search conducted on 2017-03) for each gene (e.g., ‘COQ2[gene]’) and identified pathogenic variants using the following inclusion criteria:
(i) At least one submission describes the variant as “pathogenic” or “likely pathogenic”.
(ii) No submitter assigns a significance as “benign” or “likely benign”.
(iii) Variant only affects one gene (i.e., no multi-gene deletions or duplications).
Complete records for all variants meeting our inclusion criteria were manually reviewed, including confirming that the record matches the description in any cited studies. To ensure as complete as possible a record of known variants, we also conducted a systematic literature search via pubmed (search conducted on 2017-01), where we reviewed all clinical studies in the search results for each gene name.
COQ2 transcript start
It was recently shown that the canonical COQ2 transcript, used by most previous studies, erroneously includes a 150 base N-terminal region that is only rarely, if ever, transcribed in humans21. Thus, any variant involving this region is unlikely to be pathogenic. For ease of comparability with previous studies we have retained the canonical numbering, but we have not included any variant affecting this region. For this reason, we did not consider variants such as p.Ala17Argfs (ClinVar allele ID 237155).
Estimation of variant birth prevalence
To determine the birth prevalence of these variants we used the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/), an updated version of the previously released dataset from the Exome Aggregation Consortium (ExAC)15. The gnomAD release includes exome or genome sequences from a total of 138,632 individuals without severe pediatric diseases. The database assigns ancestry by a principal components analysis based upon a subset of samples of known ancestry. Most sequences clustered into one of seven geographic or endogamous groups (African, Ashkenazi Jews, East Asian, Finnish, non-Finnish Europeans, Latin Americans, and South American), with the remainder (3,234) considered to be ‘other’. Hence, almost all samples included in this dataset have an ancestry that is well-defined on a genetic basis. This dataset has undergone extensive quality control measures to remove poor-quality sequences, related individuals and to flag variants of questionable reliability15, and assessments of variant pathogenicity are provided via the SIFT and PolyPhen2 tools.
When querying gnomAD for each of our genes of interest, variants affecting protein-coding regions were considered equivalent to known pathogenic variants if they resulted in the same change to protein structure (i.e., the same amino acid conversion, a stop codon introduced in the same location, or a frameshift resulting in the same residue changes). For variants affecting splice sites, only variants that exactly matched the nucleotide changes of the pathogenic variants were included. We only considered variants that had passed gnomAD random forest filters.
We acquired estimates of population sizes from various sources (Table S1). The population estimates summed to 6 billion, accounting for 80% of total global population. Estimates of population sizes within the USA summed to 309 million (vs. a total population of approx. 319 million). To estimate the number of affected individuals, we used the individual frequencies for each variant in a population (i.e., not on summed frequencies), and estimates for each variant for a population were rounded down to the nearest whole number.
Rates of compound heterozygosity were determined using data tables of missense and loss-of-function variants for each gene obtained from the gnomAD browser. An R script (available upon request) was written to systematically strip out undesirable variants (e.g., those affecting non-canonical transcripts) and make the multiple comparisons required. We calculated the predicted frequency of compound heterozygotes within each population in which it was possible (i.e., some variants were not observed in the same population, making compound heterozygosity for that variant impossible in that population). When determining rates of compound heterozygosity for the group of predicted loss-of-function (LoF) and pathogenic missense variants, we included known pathogenic/predicted LoF variant pairs in our calculations.
Pearson’s chi-square test of goodness of fit was calculated in Excel 14.0.7180.5002 (Microsoft, USA), and other calculations were performed in R. 95 percent confidence intervals were calculated with the exact binomial test.
Identification of predicted pathogenic variants
To identify predicted pathogenic variants in the gnomAD database, we first excluded variants that did not pass quality-control filters and those in non-canonical transcripts (as defined by gnomAD, the canonical transcript is the longest consensus coding sequence translation with no stop codons). To identify LoF variants, we extracted those annotated as “stop gained”, “frameshift”, “splice donor” or “splice acceptor” and excluded variants which gnomAD had flagged as low-confidence LoF. To identify the missense variants that were most likely to be pathogenic, we extracted only those variants for which gnomAD reported an assessment of “probably damaging” and “deleterious” by PolyPhen2 and SIFT respectively. To reduce the risk of obtaining false positives, we excluded variants with high minor allele (MAF) frequencies. Although a MAF cut-off of 0.5% has been suggested22, we chose a more conservative approach, instead using the highest observed MAF in the list of “known” pathogenic variants as a threshold: thus variants with a global MAF greater than 0.019% or a MAF for any population greater than 0.31% were excluded.
Data Availability
The datasets analyzed during the current study are available at www.ncbi.nlm.nih.gov/clinvar/ and http://gnomad.broadinstitute.org/.
Results
Through ClinVar, we identified 552 reported genetic variants affecting UQ biosynthesis genes (for complete listing, see File S1). Of these, 143 were deletions or duplications affecting multiple genes (all 17 variants reported for COQ3 and COQ5 fell into this category), and were not considered further because the pathogenicity of these variants could potentially be related to the activity of multiple genes. Of the remainder, 315 were excluded because submitters did not assess them as pathogenic (in only one case, ClinVar variation 3645, COQ8A p.Phe331=, were there both pathogenic and benign interpretations – in this case, a MAF of 1.57% supports the benign interpretation). Thirteen of those remaining were subsequently excluded because close inspection of the records and cited works revealed a number of problems, including single-copy variants not consistent with the typically recessive nature of UQ deficiencies (4 records), duplicate records (4), risk factors mis-categorized as causative pathogenic variants (2), an incomplete ClinVar entry (1), a multi-variant haplotype not testable in gnomAD (1), reliance on a secondary, unreferenced, source (1), and one variant present in the untranscribed N-terminal region of COQ2 (see Methods).
Of the remaining 80 records, the majority (49) had been extracted from the peer-reviewed literature, 22 were from the genetic testing company GeneDx (MD, USA), with the remainder from 6 other testing labs (see Table S2 for detailed information). GeneDx and five other testing labs provided detailed assertion criteria for the determination of variant pathogenicity, all adhering to established standards.
To account for the possibility that not all known pathogenic variants are included in ClinVar, we carried out an independent review of the literature, identifying 18 additional pathogenic variants: 2 affecting COQ2, 3 COQ4, 1 COQ6, 9 COQ8A, and 2 COQ8B (see Table S2 for literature references).
In total, we identified 97 pathogenic variants. Of these, 57 resulted in a single residue substitution, 21 in frameshifts, 10 premature stop codons, 7 variants altering splice-site donor or acceptor regions in ways predicted to be pathogenic, and 3 single-residue indels (see Table S2 for a complete listing of all identified known pathogenic variants). COQ8A was most frequently affected, with 40 variants.
To better understand the birth prevalence of these variants we queried the gnomAD exome and genome database. We found 441 carriers, with 49 of 97 pathogenic variants present (Table 1). No variants were present in homozygous form, all missense variants were predicted to be damaging by PolyPhen2, SIFT, or both, and all premature stop, frameshift or splice site-disrupting variants were predicted to be high-confidence loss-of-function. All of these findings are fully consistent with the reported pathogenic nature of these variants. Global allele frequencies ranged from 4.1 × 10−6 to 1.7 × 10−4, yielding a combined frequency of 1.76 × 10−3, implying that 1/321,368 individuals will be homozygous for pathogenic variants at birth.
Table 1.
Known pathogenic variants from ClinVar or literature review that are represented in gnomAD sequence database.
| Gene | Consequence | Source1 | Clinical Sign.2 | Predicted effect on protein function | Carriers | Allele Freq. | |||
|---|---|---|---|---|---|---|---|---|---|
| Lit. | Lab | PolyPhen2 | SIFT | LoF | |||||
| PDSS1 | p.Asp308Glu | 1 | 0 | P | prob. damaging | deleterious | 1 | 4.07E-06 | |
| PDSS2 | p.His92Pro | 0 | 1 | LP | prob. damaging | deleterious | 11 | 3.97E-05 | |
| p.Gln322* | 1 | 0 | P | N/A | N/A | high-conf. | 1 | 4.07E-06 | |
| p.Ser382Leu | 1 | 0 | P | prob. damaging | deleterious | 7 | 2.84E-05 | ||
| COQ2 | p.Ser146Asn | 1 | 0 | P | prob. damaging | deleterious | 4 | 1.67E-05 | |
| p.Arg197His | 1 | 0 | P | prob. damaging | deleterious | 2 | 8.14E-06 | ||
| p.Asn228Ser | 1 | 0 | P | benign | deleterious | 32 | 1.16E-04 | ||
| p.Leu234Argfs*14 | 1& | 0 | N/A | N/A | N/A | high-conf. | 1 | 4.19E-06 | |
| p.Tyr297Cys | 1 | 0 | P | prob. damaging | deleterious | 1 | 4.11E-06 | ||
| p.Thr317Met | 0 | 1 | P | prob. damaging | deleterious | 5 | 2.72E-05 | ||
| p.Gly390Ala | 1& | 0 | N/A | prob. damaging | deleterious | 6 | 2.45E-05 | ||
| p.Asn401Ilefs*15 | 1 | 0 | P | N/A | N/A | high-conf. | 3 | 1.23E-05 | |
| COQ4 | p.Pro64Ser | 1 | 0 | P | prob. damaging | deleterious | 2 | 9.07E-06 | |
| p.Asp68His | 1 | 0 | P | prob. damaging | deleterious | 10 | 4.22E-05 | ||
| p.Pro119Leu | 0 | 1 | LP | prob. damaging | deleterious | 3 | 1.22E-05 | ||
| p.Arg145Gly | 1 | 0 | P | prob. damaging | tolerated | 1 | 4.75E-06 | ||
| p.Arg240Cys | 2 | 1 | P | prob. damaging | deleterious | 47 | 1.72E-04 | ||
| COQ6 | p.Pro261Leu | 1& | 0 | N/A | prob. damaging | deleterious | 19 | 6.86E-05 | |
| p.Asn380Ser | 0 | 1 | LP | prob. damaging | deleterious | 6 | 2.16E-05 | ||
| COQ8A | c.589–3 C > G | 1& | 0 | N/A | N/A | N/A | conf.3 | 1 | 3.23E-05 |
| p.Arg213Trp | 1 | 0 | P | prob. damaging | damaging | 2 | 6.46E-05 | ||
| p.Arg213Glnfs*71 | 0 | 1 | P | N/A | N/A | high-conf. | 1 | 4.07E-06 | |
| p.Arg271Cys | 2 | 1 | LP;US | prob. damaging | deleterious | 29 | 1.08E-04 | ||
| p.Leu277Pro | 0 | 1 | LP | pos. damaging | deleterious | 1 | 4.49E-06 | ||
| p.Arg299Trp | 1& | 0 | N/A | prob. damaging | deleterious | 8 | 2.89E-05 | ||
| p.Ala304Thr | 1& | 0 | N/A | prob. damaging | deleterious | 4 | 1.63E-05 | ||
| p.Ala304Val | 0 | 1 | LP | prob. damaging | deleterious | 11 | 3.97E-05 | ||
| p.Arg348Ter | 0 | 1 | P | N/A | N/A | high-conf. | 10 | 4.04E-05 | |
| p.Leu379* | 1 | 0 | N/A | N/A | N/A | high-conf. | 1 | 4.06E-06 | |
| p.Arg410* | 0 | 1 | N/A | N/A | N/A | high-conf. | 5 | 1.82E-05 | |
| p.Thr445Argfs*52 | 0 | 1 | P | N/A | N/A | high-conf. | 1 | 4.07E-06 | |
| p.Thr511Met | 0 | 1 | LP | pos. damaging | deleterious | 13 | 4.69E-05 | ||
| p.Gly549Ser | 1 | 0 | P | prob. damaging | deleterious | 35 | 1.27E-04 | ||
| p.Glu551Lys | 1 | 0 | P | prob. damaging | deleterious | 5 | 1.81E-05 | ||
| p.Met555Ile | 1 | 0 | LP | pos. damaging | deleterious | 41 | 1.49E-04 | ||
| p.Glu568* | 0 | 1 | P | N/A | N/A | high-conf. | 1 | 4.07E-06 | |
| p.Ser582Glufs*148 | 0 | 1 | P | N/A | N/A | high-conf. | 4 | 1.63E-05 | |
| p.Thr584del | 1 | 1 | P | N/A | N/A | N/A4 | 30 | 1.22E-04 | |
| p.Pro602Arg | 1& | 0 | N/A | prob. damaging | deleterious | 9 | 3.25E-05 | ||
| p.Pro602Gln | 0 | 1 | P | prob. damaging | deleterious | 10 | 4.06E-05 | ||
| p.Ser616Leufs*114 | 0 | 1 | P | N/A | N/A | high-conf. | 16 | 5.77E-05 | |
| COQ8B | p.Phe215Leufs*14 | 1 | 0 | N/A | N/A | N/A | high-conf. | 10 | 3.67E-05 |
| p.Asp286Gly | 1 | 0 | P | pos. damaging | deleterious | 4 | 1.45E-05 | ||
| p.Arg320Trp | 1 | 0 | P | prob. damaging | deleterious | 5 | 2.03E-05 | ||
| p.His400Glnfs*11 | 1 | 0 | P | N/A | N/A | high-conf. | 1 | 4.06E-06 | |
| p.Glu447Glyfs*10 | 1& | 0 | N/A | N/A | N/A | high-conf. | 6 | 2.48E-05 | |
| p.Glu483* | 1 | 0 | P | N/A | N/A | high-conf. | 11 | 4.83E-05 | |
| COQ9 | c.521 + 1delG | 1 | 1 | P | N/A | N/A | high-conf. | 1 | 4.08E-06 |
| p.Arg244* | 1 | 0 | P | N/A | N/A | high-conf. | 3 | 1.22E-05 | |
| Sum: | 441 | 1.76E-03 | |||||||
See Table S2 for more information on all known pathogenic variants.
1Number of reports in the peer-reviewed literature (&: sources not included in ClinVar) or directly submitted by testing labs.
2Clinical significance, as stated in ClinVar record: Pathogenic, Likely Pathogenic, Uncertain Significance.
3Acceptor splice site mutation c.589–3 C > G demonstrated by in vitro model to result in non-functional product p.Leu197Valfs*20.
4In-frame deletion; mutant ADCK3 failed to rescue growth in ADCK3-null yeast.
Through casual observation it was apparent that several of the known pathogenic variants were not distributed evenly within the different populations. For example, the COQ8A p.Met555Ile variant was observed in 39 European or Finnish individuals, but in no other population, and the COQ8B p.Glu483* variant was observed in 10 individuals from South Asia but only in 1 European, despite the almost 4-fold greater number of European alleles genotyped. Indeed, the six variants with the greatest numbers of carriers had frequencies that were distributed unevenly between populations (Pearson’s chi-squared 24.3 to 537.5, p < 0.001) (Figure S1 - statistically significant differences were rarer among the variants with lower allele counts, potentially due to the decreased statistical power inherent in a lower sample size). Because of this unevenness, subsequent analysis was conducted on a population-by-population basis.
Each pathogenic variant was observed in an average of 1.9 populations (not counting ‘Other’), with an average allele frequency of 1.56 × 10–4 (Table 2). Combined estimates of Hardy-Weinberg homozygosity for all variants for each of the 7 populations averaged 1/5,492,983, ranging from 1/12,021,014 (Latin Americans) to 1/60,113 (Ashkenazi Jews). Predicted homozygous frequency for individual variants averaged 1/5.4 M, with the variant found at the greatest frequency being COQ4 p.Arg240Cys (with a 1/162 carrier frequency among Ashkenazi Jews which would result in the birth of homozygotes at a frequency of 1/104,733). With an estimated worldwide population of 10 M, this would imply 95 afflicted Ashkenazi Jews susceptible to UQ deficiency due to homozygosity for this one variant alone. Considering all variants across all populations, we can predict 1,016 homozygous-at-birth individuals globally, or 122 in the USA (Table 2).
Table 2.
Population breakdown and predicted prevalence of afflicted individuals for known pathogenic variants present in the gnomAD database.
| Gene | Consequence | Popn1 | Allele Frequency | Predicted Homozygous (with 95% CIs)2 | USA4 | ||
|---|---|---|---|---|---|---|---|
| Actual | Freq. | Frequency | Global3 | ||||
| PDSS1 | p.Asp308Glu | Ash | 1/9,834 | 1.02E-04 | 1.03E-8 (6.63E-12 – 3.21E-7) | <1 (<1–3) | <1 (<1–1) |
| PDSS2 | p.His92Pro | Afr | 2/23,986 | 8.34E-05 | 6.95E-9 (1.02E-10–9.07E-8) | 8 (<1–107) | <1 (<1–3) |
| Eur | 9/126,452 | 7.12E-05 | 5.07E-9 (1.06E-9–1.83E-8) | 3 (<1–13) | 1 (<1–3) | ||
| p.Gln322* | Eur | 1/111,562 | 8.96E-06 | 8.03E-11 (5.15E-14–2.49E-9) | <1 (<1 – 1) | <1 (<1 –<1) | |
| p.Ser382Leu | EAs | 1/17,240 | 5.80E-05 | 3.36E-9 (2.16E-12 – 1.04E-7) | 5 (<1–168) | <1 (<1 –<1) | |
| Eur | 5/111,682 | 4.48E-05 | 2.0E-9 (2.11E-10–1.09E-8) | 1 (<1–7) | <1 (<1–2) | ||
| Lat | 1/33,582 | 2.98E-05 | 8.87E-10 (5.68E-13–2.75E-8) | <1 (<1–17) | <1 (<1–1) | ||
| COQ2 | p.Ser146Asn | Eur | 4/107,234 | 3.73E-05 | 1.39E-9 (1.03E-10–9.12E-9) | 1 (<1–6) | <1 (<1–1) |
| p.Arg197His | EAs | 1/17,222 | 5.81E-05 | 3.37E-9 (2.16E-12–1.05E-7) | 5 (<1–168) | <1 (<1 –<1) | |
| Eur | 1/111,452 | 8.97E-06 | 8.05E-11 (5.16E-14–2.50E-9) | <1 (<1–1) | <1 (<1 –<1) | ||
| p.Asn228Ser | Eur | 30/126,254 | 2.38E-04 | 5.65E-8 (2.57E-8–1.15E-7) | 41 (18–84) | 11 (5–22) | |
| Afr | 1/23,990 | 4.17E-05 | 1.74E-9 (1.11E-12–5.39E-8) | 2 (<1–63) | <1 (<1–2) | ||
| Lat | 1/34,214 | 2.92E-05 | 8.54E-10 (5.48E-13–2.65E-8) | <1 (<1–16) | <1 (<1–1) | ||
| p.Leu234Argfs*14 | Eur | 1/108,122 | 9.25E-06 | 8.55E-11 (5.48E-14–2.66E-9) | <1 (<1–1) | <1 (<1 –<1) | |
| p.Tyr297Cys | SAs | 1/30,338 | 3.30E-05 | 1.09E-9 (6.96E-13–3.37E-8) | 1 (<1–61) | <1 (<1 –<1) | |
| p.Thr317Met | EAs | 1/11,902 | 8.40E-05 | 7.06E-9 (4.52E-12–2.19E-7) | 11 (<1–353) | <1 (<1–1) | |
| Afr | 1/16,630 | 6.01E-05 | 3.62E-9 (2.32E-12–1.12E-7) | 4 (<1–133) | <1 (<1–4) | ||
| Eur | 3/73,824 | 4.06E-05 | 1.65E-9 (7.02E-11–1.41E-8) | 1 (<1–10) | <1 (<1–2) | ||
| p.Gly390Ala | Lat | 5/33,178 | 1.51E-04 | 2.27E-8 (2.39E-9–1.24E-7) | 14 (1–78) | 1 (<1–6) | |
| Eur | 1/111,216 | 8.99E-06 | 8.08E-11 (5.18E-14–2.51E-9) | <1 (<1–1) | <1 (<1 –<1) | ||
| p.Asn401Ilefs*15 | Eur | 3/110,906 | 2.70E-05 | 7.32E-10 (3.11E-11–6.25E-9) | <1 (<1–4) | <1 (<1–1) | |
| COQ4 | p.Pro64Ser | Eur | 2/97,392 | 2.05E-05 | 4.22E-10 (6.18E-12–5.50E-9) | <1 (<1–4) | <1 (<1–1) |
| p.Asp68His | Lat | 3/28,866 | 1.04E-04 | 1.08E-8 (4.59E-10–9.22E-8) | 6 (<1–58) | <1 (<1–5) | |
| Eur | 6/107,660 | 5.57E-05 | 3.11E-9 (4.18E-10–1.47E-8) | 2 (<1–10) | <1 (<1–2) | ||
| Oth | 1/5,584 | 1.79E-04 | — | — | — | ||
| p.Pro119Leu | Afr | 3/15,302 | 1.96E-04 | 3.84E-8 (1.63E-9–3.28E-7) | 45 (1–389) | 1 (<1–13) | |
| p.Arg145Gly | SAs | 1/26,964 | 3.71E-05 | 1.38E-9 (8.82E-13–4.27E-8) | 2 (<1–77) | <1 (<1 –<1) | |
| p.Arg240Cys | Ash | 31/10,032 | 3.09E-03 | 9.55E-6 (4.41E-6–1.92E-5) | 95 (44–192) | 48 (22–98) | |
| Eur | 9/124,924 | 7.20E-05 | 5.19E-9 (1.09E-9–1.87E-8) | 3 (<1–13) | 1 (<1–3) | ||
| Lat | 2/33,836 | 5.91E-05 | 3.49E-9 (5.12E-11–4.56E-8) | 2 (<1–28) | <1 (<1–2) | ||
| Oth | 5/6,362 | 7.86E-04 | — | — | — | ||
| COQ6 | p.Pro261Leu | Afr | 3/24,026 | 1.25E-04 | 1.56E-8 (6.63E-10–1.33E-7) | 18 (<1–157) | <1 (<1–5) |
| EAs | 2/18,870 | 1.06E-04 | 1.12E-8 (1.65E-10–1.47E-7) | 18 (<1–236) | <1 (<1 –<1) | ||
| Eur | 13/126,674 | 1.03E-04 | 1.05E-8 (2.99E-9–3.08E-8) | 7 (2–22) | 2 (<1–6) | ||
| SAs | 1/30,782 | 3.25E-05 | 1.06E-9 (6.76E-13–3.28E-8) | 1 (<1–59) | <1 (<1 –<1) | ||
| p.Asn380Ser | Afr | 5/24,036 | 2.08E-04 | 4.33E-8 (4.56E-9–2.36E-7) | 51 (5–279) | 1 (<1–9) | |
| EAs | 1/18,870 | 5.30E-05 | 2.81E-9 (1.80E-12–8.72E-8) | 4 (<1–140) | <1 (<1 –<1) | ||
| COQ8A | c.589–3 C > G | Eur | 1/15,000 | 6.67E-05 | 4.44E-9 (2.85E-12–1.38E-7) | 3 (<1–101) | <1 (<1–27) |
| p.Arg213Trp | Eur | 2/14,998 | 1.33E-04 | 1.78E-8 (2.61E-10–2.32E-7) | 13 (<1–169) | 3 (<1–45) | |
| p.Arg213Glnfs*71 | Eur | 1/111,444 | 8.97E-06 | 8.05E-11 (5.16E-14–2.50E-9) | <1 (<1–1) | <1 (<1 –<1) | |
| p.Arg271Cys | Fin | 20/25,144 | 7.95E-04 | 6.33E-7 (2.36E-7–1.51E-6) | 3 (1–8) | <1 (<1 –<1) | |
| Eur | 7/122,222 | 5.73E-05 | 3.28E-9 (5.30E-10–1.39E-8) | 2 (<1–10) | <1 (<1–2) | ||
| Oth | 2/6,304 | 3.17E-04 | — | — | — | ||
| p.Leu277Pro | Eur | 1/99,064 | 1.01E-05 | 1.02E-10 (6.53E-14–3.16E-9) | <1 (<1–2) | <1 (<1 –<1) | |
| p.Arg299Trp | Lat | 3/34,414 | 8.72E-05 | 7.60E-9 (3.23E-10–6.49E-8) | 4 (<1–41) | <1 (<1–3) | |
| Eur | 5/126,472 | 3.95E-05 | 1.56E-9 (1.65E-10–8.51E-9) | 1 (<1–6) | <1 (<1–1) | ||
| p.Ala304Thr | Ash | 1/9,836 | 1.02E-04 | 1.03E-8 (6.63E-12–3.21E-7) | <1 (<1–3) | <1 (<1–1) | |
| Afr | 1/15,270 | 6.55E-05 | 4.29E-9 (2.75E-12–1.33E-7) | 5 (<1–157) | <1 (<1–5) | ||
| SAs | 1/30,782 | 3.25E-05 | 1.06E-9 (6.76E-13–3.28E-8) | 1 (<1–59) | <1 (<1 –<1) | ||
| Eur | 1/111,486 | 8.97E-06 | 8.05E-11 (5.16E-14–2.50E-9) | <1 (<1–1) | <1 (<1 –<1) | ||
| p.Ala304Val | Eur | 10/126,438 | 7.91E-05 | 6.26E-9 (1.44E-9–2.12E-8) | 4 (1–15) | 1 (<1–4) | |
| Oth | 1/6,456 | 1.55E-04 | — | — | — | ||
| p.Arg348* | Lat | 3/31,814 | 9.43E-05 | 8.89E-9 (3.78E-10–7.59E-8) | 5 (<1–48) | <1 (<1–4) | |
| Eur | 7/110,946 | 6.31E-05 | 3.98E-9 (6.44E-10–1.69E-8) | 2 (<1–12) | <1 (<1–3) | ||
| p.Leu379* | Eur | 1/111,658 | 8.96E-06 | 8.02E-11 (5.14E-14–2.49E-9) | <1 (<1–1) | <1 (<1 –<1) | |
| p.Arg410* | EAs | 2/18,820 | 1.06E-04 | 1.13E-8 (1.66E-10–1.47E-7) | 18 (<1–237) | <1 (<1 –<1) | |
| SAs | 1/30,750 | 3.25E-05 | 1.06E-9 (6.78E-13–3.28E-8) | 1 (<1–59) | <1 (<1 –<1) | ||
| Eur | 2/125,096 | 1.60E-05 | 2.56E-10 (3.75E-12–3.34E-9) | <1 (<1–2) | <1 (<1 –<1) | ||
| p.Thr445Argfs*52 | Lat | 1/33,562 | 2.98E-05 | 8.88E-10 (5.69E-13–2.76E-8) | <1 (<1–17) | <1 (<1–1) | |
| p.Thr511Met | Lat | 5/34,420 | 1.45E-04 | 2.11E-8 (2.22E-9–1.15E-7) | 13 (1–72) | 1 (<1–6) | |
| Ash | 1/10,150 | 9.85E-05 | 9.71E-9 (6.22E-12–3.01E-7) | <1 (<1–3) | <1 (<1–1) | ||
| Eur | 7/126,688 | 5.53E-05 | 3.05E-9 (4.94E-10–1.30E-8) | 2 (<1–9) | <1 (<1–2) | ||
| p.Gly549Ser | Eur | 30/126,484 | 2.37E-04 | 5.63E-8 (2.56E-8–1.15E-7) | 41 (18–83) | 11 (5–22) | |
| Fin | 2/25,596 | 7.81E-05 | 6.11E-9 (8.95E-11–7.97E-8) | <1 (<1 –<1) | <1 (<1 –<1) | ||
| Afr | 1/23,988 | 4.17E-05 | 1.74E-9 (1.11E-12–5.39E-8) | 2 (<1–63) | <1 (<1–2) | ||
| Lat | 1/34,410 | 2.91E-05 | 8.45E-10 (5.41E-13–2.62E-8) | <1 (<1–16) | <1 (<1–1) | ||
| Oth | 1/6,456 | 1.55E-04 | — | — | — | ||
| p.Glu551Lys | Eur | 4/126,382 | 3.17E-05 | 1.0E-9 (7.44E-11–6.57E-9) | <1 (<1–4) | <1 (<1–1) | |
| Oth | 1/6,454 | 1.55E-04 | — | — | — | ||
| p.Met555Ile | Fin | 18/24,584 | 7.32E-04 | 5.36E-7 (1.88E-7–1.34E-6) | 2 (1–7) | <1 (<1 –<1) | |
| Eur | 21/126,250 | 1.66E-04 | 2.77E-8 (1.06E-8–6.46E-8) | 20 (7–47) | 5 (2–12) | ||
| Oth | 2/6,446 | 3.10E-04 | — | — | — | ||
| p.Glu568* | Eur | 1/111,654 | 8.96E-06 | 8.02E-11 (5.14E-14–2.49E-9) | <1 (<1–1) | <1 (<1 –<1) | |
| p.Ser582Glufs*148 | Lat | 1/33,580 | 2.98E-05 | 8.87E-10 (5.68E-13–2.75E-8) | <1 (<1–17) | <1 (<1–1) | |
| Eur | 3/111,712 | 2.69E-05 | 7.21E-10 (3.07E-11–6.16E-9) | <1 (<1–4) | <1 (<1–1) | ||
| p.Thr584del | Ash | 26/9,848 | 2.64E-03 | 6.97E-6 (2.98E-6–1.49E-5) | 69 (29–149) | 35 (15–76) | |
| Eur | 4/111,702 | 3.58E-05 | 1.28E-9 (9.52E-11–8.41E-9) | <1 (<1–6) | <1 (<1–1) | ||
| p.Pro602Arg | Ash | 3/10,148 | 2.96E-04 | 8.74E-8 (3.72E-9–7.46E-7) | <1 (<1–7) | <1 (<1–3) | |
| Lat | 2/34,420 | 5.81E-05 | 3.38E-9 (4.95E-11–4.41E-8) | 2 (<1–27) | <1 (<1–2) | ||
| SAs | 1/30,782 | 3.25E-05 | 1.06E-9 (6.76E-13–3.28E-8) | 1 (<1–59) | <1 (<1 –<1) | ||
| Eur | 2/126,676 | 1.58E-05 | 2.49E-10 (3.66E-12–3.25E-9) | <1 (<1–2) | <1 (<1 –<1) | ||
| Oth | 1/6,464 | 1.55E-04 | — | — | — | ||
| p.Pro602Gln | SAs | 2/30,782 | 6.50E-05 | 4.22E-9 (6.19E-11–5.51E-8) | 7 (<1–100) | <1 (<1 –<1) | |
| Eur | 7/111,706 | 6.27E-05 | 3.93E-9 (6.35E-10–1.67E-8) | 2 (<1–12) | <1 (<1–3) | ||
| Oth | 1/5,486 | 1.82E-04 | — | — | — | ||
| p.Ser616Leufs*114 | EAs | 6/18,866 | 3.18E-04 | 1.01E-7 (1.36E-8–4.79E-7) | 163 (21–772) | <1 (<1–2) | |
| SAs | 4/30,782 | 1.30E-04 | 1.69E-8 (1.25E-9–1.11E-7) | 30 (2–201) | <1 (<1 –<1) | ||
| Afr | 1/24,020 | 4.16E-05 | 1.73E-9 (1.11E-12–5.38E-8) | 2 (<1–63) | <1 (<1–2) | ||
| Eur | 5/126,684 | 3.95E-05 | 1.56E-9 (1.64E-10–8.48E-9) | 1 (<1–6) | <1 (<1–1) | ||
| COQ8B | p.Phe215Leufs*14 | Eur | 1/107,204 | 9.33E-06 | 8.70E-11 (5.58E-14–2.70E-9) | <1 (<1–1) | <1 (<1 –<1) |
| p.Phe215Leufs*14 | Eur | 7/122,270 | 5.73E-05 | 3.28E-9 (5.30E-10–1.39E-8) | 2 (<1–10) | <1 (<1–2) | |
| EAs | 1/18,840 | 5.31E-05 | 2.82E-9 (1.81E-12–8.74E-8) | 4 (<1–140) | <1 (<1 –<1) | ||
| Lat | 1/34,350 | 2.91E-05 | 8.48E-10 (5.43E-13–2.63E-8) | <1 (<1–16) | <1 (<1–1) | ||
| Oth | 1/6,400 | 1.56E-04 | — | — | — | ||
| p.Asp286Gly | Fin | 4/24,036 | 1.66E-04 | 2.77E-8 (2.06E-9–1.82E-7) | <1 (<1 –<1) | <1 (<1 –<1) | |
| p.Arg320Trp | Afr | 2/15,262 | 1.31E-04 | 1.72E-8 (2.52E-10–2.24E-7) | 20 (<1–265) | <1 (<1–9) | |
| SAs | 2/30,780 | 6.50E-05 | 4.22E-9 (6.19E-11–5.51E-8) | 7 (<1–100) | <1 (<1 –<1) | ||
| Eur | 1/111,380 | 8.98E-06 | 8.06E-11 (5.17E-14–2.50E-9) | <1 (<1–1) | <1 (<1 –<1) | ||
| p.His400Glnfs*11 | Eur | 1/111,710 | 8.95E-06 | 8.01E-11 (5.14E-14–2.49E-9) | <1 (<1–1) | <1 (<1 –<1) | |
| p.Glu447Glyfs*10 | Eur | 6/108,260 | 5.54E-05 | 3.07E-9 (4.14E-10–1.46E-8) | 2 (<1–10) | <1 (<1–2) | |
| p.Glu483* | SAs | 10/29,404 | 3.40E-04 | 1.16E-7 (2.66E-8–3.91E-7) | 210 (48–712) | <1 (<1–1) | |
| Eur | 1/101,248 | 9.88E-06 | 9.75E-11 (6.25E-14–3.03E-9) | <1 (<1–2) | <1 (<1 –<1) | ||
| COQ9 | c.521 + 1delG | Eur | 1/111,218 | 8.99E-06 | 8.08E-11 (5.18E-14–2.51E-9) | <1 (<1–1) | <1 (<1 –<1) |
| p.Arg244* | Fin | 2/22,296 | 8.97E-05 | 8.05E-9 (1.18E-10–1.05E-7) | <1 (<1 –<1) | <1 (<1 –<1) | |
| SAs | 1/30,782 | 3.25E-05 | 1.06E-9 (6.76E-13–3.28E-8) | 1 (<1–59) | <1 (<1 –<1) | ||
| Totals: | 1,016 (200–6956) | 122 (49–444) | |||||
1 Ashkenazi Jewish, African/African American, European (non-Finnish), East Asian, South Asian, Latin American (admixed American), Finnish, Other.
2Predicted homozygosity assuming Hardy-Weinberg equilibrium.
3Predicted number of homozygotes in population size equivalent to that of the given ethnicity globally.
4Predicted number of homozygotes in a population size equivalent to that the given ethnicity in the USA.
The presence of multiple variants within the same populations is consistent with the numerous reports of compound heterozygosity in patients with primary UQ deficiency7. When estimating birth prevalence of compound heterozygosity, pathogenic variants in COQ8A again exhibited a greater prevalence relative to other genes. In fact, the birth prevalence of compound heterozygotes for COQ8A among Ashkenazi Jews (1/725,578), Finns (1/1.4 M) or non-Finnish Europeans (1/1.6 M) alone were all individually greater than the combined prevalence of all other genes (1/17.1 M) (Table 3, full variant-by-variant breakdown in Table S3). We can estimate that 649 individuals worldwide are born as compound heterozygous for pathogenic genetic variants causing UQ deficiency, with 70 in the USA-like population.
Table 3.
The predicted occurrence of compound heterozygotes of known pathogenic variants.
| Gene | Popn | Unique Genotypes | Summed Frequency1 | Afflicted Worldwide | Afflicted USA |
|---|---|---|---|---|---|
| PDSS2 | Eur | 3 | 4.23E-09 | 2 | 0 |
| All | 3 | 5.28E-10 | 2 | 0 | |
| COQ2 | Afr | 1 | 2.51E-09 | 2 | 0 |
| EAs | 1 | 4.88E-09 | 7 | 0 | |
| Eur | 21 | 3.81E-08 | 21 | 3 | |
| Lat | 1 | 4.40E-09 | 2 | 0 | |
| All | 22 | 6.24E-09 | 32 | 3 | |
| COQ4 | Eur | 3 | 6.64E-09 | 3 | 0 |
| Lat | 1 | 6.14E-09 | 3 | 0 | |
| Oth | 1 | 1.41E-07 | — | — | |
| All | 3 | 1.92E-08 | 6 | 0 | |
| COQ6 | Afr | 1 | 2.60E-08 | 30 | 1 |
| EAs | 1 | 5.62E-09 | 9 | 0 | |
| All | 1 | 3.95E-09 | 39 | 1 | |
| COQ8A | Afr | 3 | 7.19E-09 | 8 | 0 |
| Ash | 6 | 1.38E-06 | 11 | 6 | |
| EAs | 1 | 3.38E-08 | 54 | 0 | |
| Eur | 210 | 6.20E-07 | 357 | 60 | |
| Fin | 3 | 7.02E-07 | 3 | 0 | |
| Lat | 21 | 9.03E-08 | 47 | 0 | |
| SAs | 10 | 3.06E-08 | 48 | 0 | |
| Oth | 21 | 8.58E-07 | — | — | |
| All | 216 | 4.65E-07 | 528 | 66 | |
| COQ8B | Eur | 15 | 7.87E-09 | 2 | 0 |
| SAs | 1 | 2.21E-08 | 40 | 0 | |
| All | 15 | 3.75E-09 | 42 | 0 | |
| Totals: | 259 | 4.99E-07 | 649 | 70 | |
See Table S3 for full variant-by-variant breakdown with confidence intervals.
1Subtotals (“All”) represent the average across all 8 populations, and the Total is the sum of those subtotals.
Premature stop codons, frameshifts or the disruption of canonical splice sites (LoF) or critical protein residues (via missense mutations) are all expected to result in significant impairments to protein function. Although we can expect an unknown proportion of these predicted pathogenic variants to result in embryonic lethality, those that do allow survival to birth are likely to result in clinically significant illness. We therefore determined the birth prevalence of all predicted pathogenic variants in UQ biosynthesis genes, as described in Methods. Across all UQ biosynthesis genes there were a total of 782 predicted pathogenic variants (including all known pathogenic variants), and 618 possible compound heterozygote combinations (summarized in Table 4, complete variant list in Table S4 and Table S5). The two genes with the highest frequency of predicted pathogenic variants (combining homozygotes and compound heterozygotes) were COQ8A and COQ8B, with cross-population average incidences of 1/193,621 and 1/198,391, resulting in a predicted 27,321 and 44,727 afflicted individuals worldwide, respectively, and 391 and 398 afflicted individuals in the USA. The gene with the lowest frequency was COQ3 (1/57 M), with only 146 predicted affected individuals worldwide, and none predicted in the USA. The population with the greatest total frequency of pathogenic variants was that of East Asia (1/20,170), with a predicted 79,423 afflicted individuals worldwide. The variant with the greatest prevalence in any population was COQ4 p.Arg240Cys in the Ashkenazi Jewish population, with a MAF of 0.0001719 (Table S4).
Table 4.
Predicted prevalence of homozygous and compound heterozygous afflicted individuals for all known and predicted pathogenic variants.
| Gene | Popn | Homozygous | Compound Heterozygous | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Unique Genotypes | Summed Frequency1 | Afflicted Worldwide | Afflicted USA | Unique Genotypes | Summed Frequency1 | Afflicted Worldwide | Afflicted USA | ||
| PDSS1 | Afr | 10 | 6.68E-08 | 77 | <1 | 45 | 2.69E-07 | 297 | <1 |
| Ash | 2 | 2.06E-08 | <1 | <1 | 1 | 1.03E-08 | <1 | <1 | |
| EAs | 8 | 7.81E-07 | 1,256 | 4 | 28 | 8.60E-07 | 1,372 | 2 | |
| Eur | 33 | 2.46E-08 | 12 | 2 | 528 | 1.70E-07 | 27 | 1 | |
| Fin | 3 | 2.21E-08 | <1 | <1 | 3 | 1.41E-08 | <1 | <1 | |
| Lat | 11 | 1.53E-06 | 963 | 84 | 55 | 1.06E-06 | 643 | 50 | |
| SAs | 7 | 4.44E-08 | 76 | <1 | 21 | 8.13E-08 | 132 | <1 | |
| Oth | 3 | 9.98E-08 | — | - | 3 | 9.98E-08 | — | — | |
| All | 62 | 3.23E-07 | 2,384 | 90 | 674 | 3.21E-07 | 2,471 | 53 | |
| PDSS2 | Afr | 12 | 5.14E-07 | 607 | 18 | 66 | 9.00E-07 | 1,038 | 17 |
| Ash | 1 | 2.03E-06 | 20 | 10 | 0 | — | — | — | |
| EAs | 13 | 5.03E-06 | 8,100 | 29 | 78 | 2.22E-06 | 3,532 | 3 | |
| Eur | 37 | 2.14E-08 | 9 | 1 | 666 | 2.15E-07 | 27 | <1 | |
| Lat | 6 | 7.94E-09 | 2 | <1 | 15 | 1.76E-08 | 5 | <1 | |
| SAs | 12 | 1.74E-07 | 309 | <1 | 66 | 4.20E-07 | 721 | <1 | |
| Oth | 5 | 2.47E-07 | — | — | 10 | 4.16E-07 | — | — | |
| All | 68 | 1.00E-06 | 9,047 | 58 | 879 | 5.23E-07 | 5,323 | 20 | |
| COQ2 | Afr | 10 | 1.55E-07 | 180 | 2 | 45 | 5.53E-07 | 628 | 6 |
| Ash | 1 | 1.70E-06 | 17 | 8 | 0 | — | — | — | |
| EAs | 5 | 4.01E-07 | 644 | 2 | 10 | 1.99E-07 | 315 | <1 | |
| Eur | 31 | 7.51E-08 | 48 | 11 | 465 | 2.99E-07 | 106 | 12 | |
| Fin | 2 | 2.24E-08 | <1 | <1 | 1 | 8.18E-09 | <1 | <1 | |
| Lat | 7 | 4.53E-08 | 26 | 1 | 21 | 9.31E-08 | 47 | <1 | |
| SAs | 10 | 1.86E-08 | 29 | 0 | 45 | 7.24E-08 | 119 | <1 | |
| Oth | 3 | 1.10E-06 | — | — | 3 | 3.80E-07 | — | — | |
| All | 53 | 4.40E-07 | 944 | 24 | 573 | 2.00E-07 | 1,215 | 18 | |
| COQ3 | Afr | 2 | 4.51E-08 | 53 | 1 | 1 | 8.67E-09 | 10 | <1 |
| EAs | 3 | 2.10E-08 | 33 | <<1 | 3 | 1.72E-08 | 27 | <1 | |
| Eur | 16 | 7.30E-09 | 3 | <1 | 120 | 2.62E-08 | 1 | <1 | |
| Lat | 1 | 3.55E-09 | 2 | <1 | 0 | — | — | — | |
| SAs | 4 | 4.75E-09 | 6 | <1 | 6 | 7.09E-09 | 11 | <1 | |
| All | 23 | 1.02E-08 | 97 | 1 | 130 | 7.40E-09 | 49 | <1 | |
| COQ4 | Afr | 19 | 2.22E-07 | 257 | 2 | 171 | 1.60E-06 | 1,811 | 5 |
| Ash | 1 | 9.55E-06 | 95 | 48 | 0 | — | — | — | |
| EAs | 11 | 2.61E-06 | 4,195 | 13 | 55 | 2.60E-06 | 4,169 | 7 | |
| Eur | 40 | 5.10E-08 | 29 | 4 | 780 | 4.90E-07 | 129 | 8 | |
| Fin | 5 | 1.11E-06 | 5 | <1 | 10 | 2.01E-07 | <1 | <1 | |
| Lat | 17 | 8.40E-08 | 44 | 2 | 136 | 4.04E-07 | 187 | 2 | |
| SAs | 17 | 3.19E-08 | 45 | <1 | 136 | 2.30E-07 | 334 | <1 | |
| Oth | 9 | 1.87E-06 | — | — | 36 | 3.61E-06 | — | — | |
| All | 81 | 1.94E-06 | 4,670 | 69 | 1,236 | 1.14E-06 | 6,630 | 22 | |
| COQ5 | Afr | 4 | 1.71E-08 | 20 | <1 | 6 | 2.56E-08 | 30 | <1 |
| EAs | 5 | 5.11E-07 | 822 | 2 | 10 | 3.92E-07 | 627 | 1 | |
| Eur | 11 | 1.32E-08 | 6 | <1 | 55 | 4.02E-08 | 11 | <1 | |
| Fin | 1 | 2.01E-09 | <1 | <1 | 0 | — | — | — | |
| Lat | 2 | 1.77E-09 | <1 | <1 | 1 | 8.87E-10 | <1 | <1 | |
| SAs | 1 | 1.06E-09 | 1 | <1 | 0 | — | — | — | |
| All | 23 | 6.83E-08 | 849 | 2 | 72 | 5.74E-08 | 668 | 1 | |
| COQ6 | Afr | 20 | 2.32E-07 | 270 | 2 | 190 | 1.79E-06 | 2,045 | 6 |
| Ash | 3 | 3.09E-08 | <1 | <1 | 3 | 3.09E-08 | <1 | <1 | |
| EAs | 18 | 1.75E-07 | 274 | <1 | 153 | 1.08E-06 | 1,642 | <1 | |
| Eur | 58 | 1.26E-07 | 80 | 17 | 1,653 | 1.06E-06 | 385 | 48 | |
| Fin | 4 | 7.03E-09 | <1 | <1 | 6 | 1.05E-08 | <1 | <1 | |
| Lat | 18 | 1.53E-06 | 962 | 83 | 153 | 1.66E-06 | 984 | 67 | |
| SAs | 21 | 3.06E-07 | 540 | <1 | 210 | 6.91E-07 | 1,076 | <1 | |
| Oth | 9 | 1.52E-06 | — | — | 36 | 3.40E-06 | — | — | |
| All | 110 | 4.91E-07 | 2,126 | 102 | 2,284 | 1.22E-06 | 6,132 | 121 | |
| COQ7 | Afr | 2 | 1.38E-08 | 16 | <1 | 1 | 6.92E-09 | 8 | <1 |
| EAs | 2 | 6.96E-09 | 10 | <1 | 1 | 3.48E-09 | 5 | <1 | |
| Eur | 10 | 8.85E-09 | 5 | <1 | 45 | 1.90E-08 | 3 | <1 | |
| Fin | 2 | 4.46E-07 | 2 | <1 | 1 | 5.14E-08 | <1 | <1 | |
| Lat | 1 | 8.87E-10 | <1 | <1 | 0 | — | — | — | |
| SAs | 5 | 3.84E-08 | 66 | <1 | 10 | 4.99E-08 | 85 | <1 | |
| Oth | 2 | 5.72E-08 | — | — | 1 | 2.82E-08 | — | — | |
| All | 18 | 7.15E-08 | 99 | <1 | 58 | 1.99E-08 | 101 | <1 | |
| ADCK3 | Afr | 23 | 3.16E-07 | 369 | 7 | 253 | 2.39E-06 | 2,736 | 19 |
| Ash | 4 | 7.08E-06 | 69 | 35 | 6 | 1.38E-06 | 11 | 6 | |
| EAs | 17 | 2.47E-06 | 3,970 | 12 | 136 | 6.50E-06 | 10,392 | 15 | |
| Eur | 97 | 1.99E-07 | 120 | 23 | 4,656 | 3.81E-06 | 1,437 | 154 | |
| Fin | 12 | 1.77E-06 | 7 | <1 | 66 | 3.95E-06 | 11 | <1 | |
| Lat | 26 | 8.76E-07 | 541 | 45 | 325 | 2.41E-06 | 1,361 | 72 | |
| SAs | 34 | 1.23E-06 | 2,209 | 3 | 561 | 2.47E-06 | 4,088 | <1 | |
| Oth | 14 | 6.57E-07 | — | — | 91 | 3.82E-06 | — | — | |
| All | 149 | 1.82E-06 | 7,285 | 125 | 5,635 | 3.34E-06 | 20,036 | 266 | |
| ADCK4 | Afr | 34 | 8.77E-07 | 1,031 | 26 | 561 | 6.97E-06 | 8,043 | 109 |
| Ash | 6 | 3.37E-06 | 32 | 16 | 15 | 1.74E-06 | 11 | 3 | |
| EAs | 24 | 3.70E-06 | 5,945 | 18 | 276 | 1.63E-05 | 26,171 | 44 | |
| Eur | 83 | 1.34E-07 | 79 | 14 | 3,403 | 2.20E-06 | 725 | 74 | |
| Fin | 11 | 1.54E-07 | <1 | <1 | 55 | 4.68E-07 | <1 | <1 | |
| Lat | 30 | 5.79E-07 | 352 | 27 | 435 | 2.43E-06 | 1,349 | 67 | |
| SAs | 17 | 1.51E-07 | 262 | <1 | 136 | 4.54E-07 | 727 | <1 | |
| Oth | 6 | 2.38E-07 | — | — | 15 | 5.40E-07 | — | — | |
| All | 135 | 1.15E-06 | 7,701 | 101 | 4,449 | 3.89E-06 | 37,026 | 297 | |
| COQ9 | Afr | 9 | 8.83E-07 | 1,045 | 33 | 36 | 1.19E-06 | 1,401 | 38 |
| EAs | 10 | 1.71E-06 | 2,751 | 9 | 45 | 1.98E-06 | 3,171 | 4 | |
| Eur | 34 | 1.82E-08 | 7 | 1 | 561 | 1.59E-07 | 18 | <1 | |
| Fin | 5 | 1.62E-07 | <1 | <1 | 10 | 1.74E-07 | <1 | <1 | |
| Lat | 15 | 1.06E-07 | 59 | 3 | 105 | 4.28E-07 | 221 | 4 | |
| SAs | 11 | 4.34E-08 | 70 | <1 | 55 | 1.31E-07 | 193 | <1 | |
| Oth | 3 | 1.55E-07 | — | — | 3 | 1.34E-07 | — | — | |
| All | 60 | 3.85E-07 | 3,932 | 46 | 768 | 5.25E-07 | 5,004 | 46 | |
| Totals: | 782 | 7.70E-06 | 39,134 | 618 | 16,758 | 1.12E-05 | 84,655 | 844 | |
See Tables S4 and S5 for full variant-by-variant breakdowns with confidence intervals.
1Frequencies for each population are the sum of all homozygous or compound heterozygote frequencies for each variant in that population; Subtotals for all populations are the average of the 8 populations considered, and Total is the sum of those subtotals.
Considering the occurrence of both homozygotes and compound heterozygotes averaged across all populations, our results predict a global birth prevalence of 1/52,092. However, not all the populations considered are of equal size, and the predicted number of afflicted individuals worldwide was 41,555 due to homozygosity and 85,581 due to compound heterozygosity, for a total of 123,789 (1/48,495). In the USA, our analysis predicts 1,462 afflicted individuals (1/211,917).
Discussion
Overall, our results predict a worldwide total of 123,789 individuals suffering from primary UQ deficiency, and 1,462 in a population with a composition similar to the USA. Of these, 1,665 and 192 respectively are due to variants that are known to be pathogenic, with the remainder due to predicted LoF and pathogenic missense variants (summarized in Fig. 1A and B). However, the extent to which known pathogenic variants contributed to the total varied between populations. The addition of predicted LoF variants has less impact for Western populations (Ashkenazi Jews, Finnish and non-Finish Europeans: blue in Fig. 1), with inclusion of predicted pathogenic variants resulting in an average 3.5-fold increase in the number of afflicted individuals, relative to known pathogenic variants only (Fig. 1c). In contrast, in populations from non-Western, developing regions (South and East Asians, Latin Americans and Africans: red in Fig. 1), the addition of predicted LoF variants resulted in an average 122-fold increase in the number of afflicted individuals (Fig. 1c). The increased likelihood of pathogenic variants to have been identified in Western populations is consistent with the reality of their relatively higher clinical coverage compared to non-Western populations, where the expense of clinical sequencing has limited the genetic characterization of patients suffering from mitochondrial disease. Our results imply that primary UQ deficiency is substantially under-diagnosed in Latin American, African and Asian populations.
Figure 1.
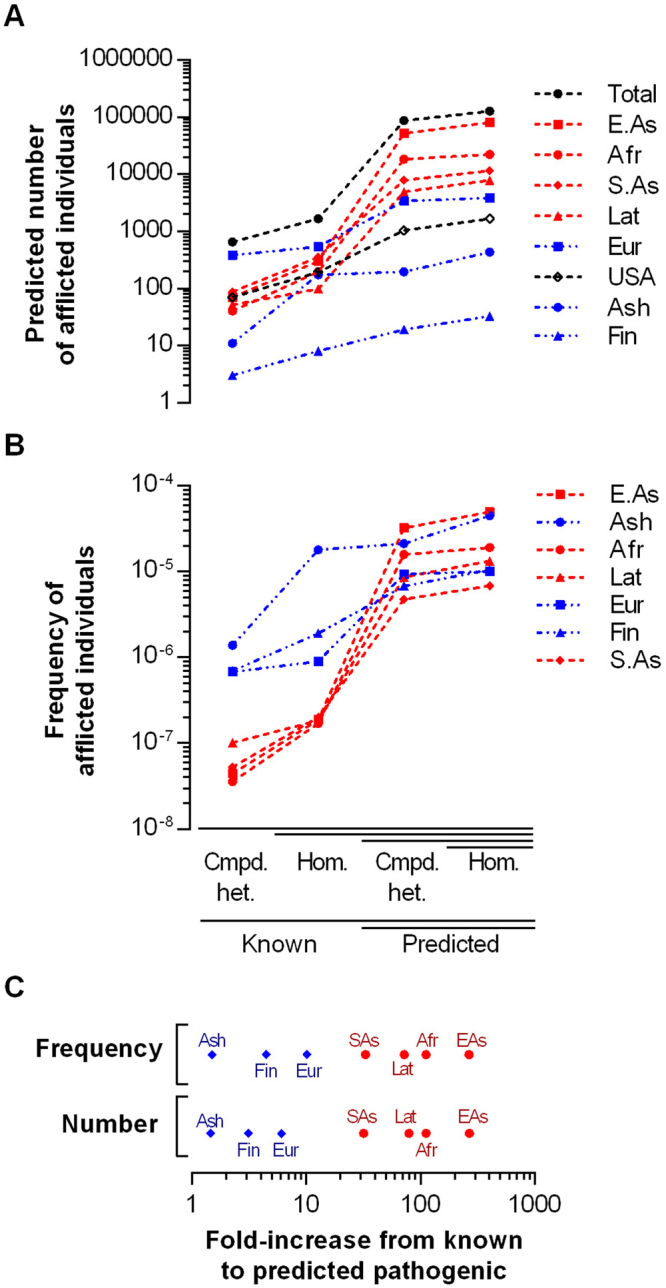
Prevalence of primary UQ deficiency based on known and predicted pathogenic variants. (A) Predicted number of afflicted individuals due to compound heterozygosity or homozygosity of known or predicted pathogenic variants, as denoted on x-axis, for each population. (B) Frequency of afflicted individuals within each analyzed population. (C) Contribution of known or predicted pathogenic variants to the frequency or number of afflicted individuals within each population. The fold-difference between known pathogenic variants only, and the total of known and predicted pathogenic variants, is shown on the x-axis.
There are several factors that could induce error in our predictions. For example, LoF variants may be so harmful that a homozygous individual is not viable in the first place. That this is possible is supported by the embryonic lethality of the complete genetic ablation of PDSS2, COQ2, COQ3, COQ6 and COQ7 in mice16–19, with COQ4 exhibiting pre-weaning lethality17. In contrast, COQ8A23 and COQ924 –null mice have been reported as viable. Indeed, among patients with pathogenic variants in the UQ biosynthesis genes likely to be necessary for life (PDSS1 – COQ7), very few are homozygous or compound heterozygous for severe variants expected to result in significant LoF. Among the severe variants (nonsense, frameshift, splice site affecting) for these genes described in the literature we reviewed, only COQ2 p.Asn401Ilefs*15 was present in homozygous form, resulting in multi-organ failure and death in an infant patient25, and there was only one patient compound heterozygous for LoF variants (COQ6 p.Trp447* and p.Gln461fs*47826). In all three variants the region affected was close to the C-terminus (closer than any other known pathogenic variant for these genes), implying that these patients may have retained some partially functional protein, and that other severe variants may have resulted in complete LoF and embryonic lethality.
It is therefore likely that some of the LoF variants that contribute to our final totals may not actually contribute to disease rates due to embryonic or pre-natal lethality. Variant severity is not easy to predict – for example, COQ9R239X mice that express a partial protein have a much more severe phenotype than COQ9Q95X mice with no measurable protein expression, presumably due to the destabilization of a multiprotein UQ biosynthesis complex by the truncated protein27. However, homozygous or compound heterozygous severe variants in PDSS1 through COQ7 account for only 6,142 out of 123,789 predicted individuals worldwide, and 219 out 1,462 in the US. This suggests that our predictions are not greatly inflated by the inclusion of embryonically lethal allelic combinations.
Our predictions may also suffer from the opposite problem - missense variants identified as damaging by SIFT or PolyPhen2 may, in fact, not have deleterious physiological effects. We attempted to address this by requiring our “predicted pathogenic” variants to be rated as highly likely to be deleterious by both PolyPhen2 and SIFT, but such prediction algorithms are clearly not infallible. For example, COQ4 p.Arg145Gly was rated as “tolerated” by SIFT, yet was reported in homozygous form in a neonate who died 4 h after birth, and it also failed to rescue Δcoq4 yeast28. It is therefore reasonable to expect a certain proportion of predicted-pathogenic missense variants to result in asymptomatic individuals. Interestingly, missense variants seem to be responsible for a lesser proportion of COQ8A and COQ8B-deficient individuals, with patients homozygous or compound heterozygous for LoF variants being relatively common29–32. Given that COQ8A alone can rescue COQ8-null yeast33, and COQ8A patients with truncating nonsense mutations shown to result in nonsense-mediated decay remained viable in their mid-20’s32, it is likely that these genes may be relatively insensitive to some borderline-pathogenic missense variants. This has the potential to greatly impact our predictions, with homozygous or compound heterozygous variants in COQ8A and COQ9B accounting for 46,654 out of 123,789 predicted affected individuals worldwide, and 559 out of 1,462 in the USA.
COQ8A and COQ8B are also noteworthy in that most of the known patients have relatively well-defined, gene-specific, pathologies. Specifically, symptoms of ataxia (often associated with cerebellar atrophy or other neurological abnormalities) are found with 26 of the 29 known pathogenic variants of COQ8A, and all 13 of the published COQ8B pathogenic variants exhibited nephrotic syndrome (citations provided in Table S2). It would therefore be tempting to claim that our predicted patients would exhibit similar clinical conditions, with, for example, all predicted COQ8B patients suffering from nephrotic syndrome34. However, it is likely (and our results support) that only a subset of primary UQ deficiency patients have been identified at this point, and they may be non-representative of the actual patient population. Of note, many of the known pathogenic variants were identified in studies where clinicians screened cohorts of patients with specific subsets of well-defined symptoms. For example, our knowledge of COQ8B variants largely comes from two studies in which large numbers of patients with nephrotic syndrome were subjected to sequencing of either whole exomes or multi-gene panels designed for nephrotic syndrome29,35. A similar issue can be raised for the ataxic nature of COQ8A variants. For example, two studies described how, after identifying pathogenic COQ8A variants in ataxic patients, they proceeded to sequence COQ8A in other ataxic patients, identifying additional novel pathogenic variants30,32. Additional pathogenic variants were found in later studies in which COQ8A, alone or in combination with other UQ biosynthesis genes, was specifically sequenced in ataxic patients31,36. We hypothesize that future COQ8A or COQ8B patients identified via less targeted methodologies may present with more diverse clinical phenotypes, as is characteristic of other UQ biosynthesis genes such as COQ2 or COQ4.
There are also several factors that could increase the number of afflicted individuals beyond our estimates. For example, we conservatively assumed that primary UQ deficiency is always recessive; however, haploinsufficiency of COQ4 has been shown to cause clinically significant primary UQ deficiency37. Also, violations of Hardy-Weinberg equilibrium (e.g., consanguinity or populations with a large degree of endogamy) could increase the likelihood of an individual being born with two pathogenic variants. It is also noteworthy that 6 of the 29 missense variants known to be pathogenic would not have met our criteria for inclusion as “predicted” pathogenic variants, since they were not assigned the highest level of confidence for pathogenicity by both SIFT and PolyPhen2 (Table 1). This supports the conservative nature of our selection criteria.
Furthermore, there are several reasons why truly pathogenic variants may not appear on our list of known variants. Some variants may have been identified in clinics without being formally described in the literature. For example, COQ2 p.Met128Val and p.Arg387* have been cited as pathogenic in the secondary literature38, but without a formal research citation they would not have met our inclusion criteria as known pathogenic variants. Furthermore, although the latter variant was included as a predicted pathogenic variant, the former was assessed as ‘benign’ and ‘tolerated’ by Polyphen and SIFT respectively, excluding it from our list of predicted pathogenic variants. In addition, many predicted pathogenic variants were more common in non-western populations, meaning that they are less likely to have been identified in the existing clinical reports, which have focussed on western populations. Additionally, our list of known pathogenic variants may not have included variants detected as part of recent large-scale studies39–41, and the fact that some UQ biosynthesis genes were found to be associated with disease earlier than others (e.g., COQ2 was first found in 200642, vs. COQ8B in 201329 and COQ7 in 201543) could have delayed the introduction of some genes into widely used genetic screening panels44, meaning that more patients were screened for some genes compared to others. Finally, after the literature review phase of our analysis was concluded, novel pathogenic variants have continued to be described in the clinical literature (e.g., COQ445, COQ646, COQ747, ADCK448,49), indicating that many remain to be reported.
Several aspects of our results point towards their general reliability. For example, there have been no reports of pathogenic variants in COQ3 or COQ5, which is consistent with our prediction of few individuals with primary UQ deficiency due to pathogenic variants in these genes (less than 2,000 individuals worldwide, and only 4 in the USA). Conversely, more patients with defects in COQ8A and COQ8B have been described than for any other UQ biosynthesis gene8, which corresponds to our finding that pathogenic variants in these genes make the greatest contribution to the number of individuals worldwide predicted to suffer from primary UQ deficiency, together accounting for more than half of the predicted 127,136 patients worldwide.
In conclusion, we have made the first estimates of the worldwide and within-population birth prevalence of individuals who are homozygous or compound heterozygous for pathogenic variants causing primary UQ deficiency by combining a decades-worth of clinical genetics with the recently available large-scale full exome/genome sequencing. Our calculations suggest a minimum of 1,665 afflicted individuals worldwide or 192 in the USA (using only variants clinically shown to be pathogenic), up to a maximum of 123,789 worldwide or 1,462 in the USA (with all variants predicted to be pathogenic). Notably, the gap between predictions made using “known” vs. “predicted” pathogenic variants appears smallest in populations expected to have the greatest access to the modern methodologies of clinical genetics. This implies that healthcare providers have already made substantial headway in identifying individuals suffering from this disorder. However, it remains likely that the bulk of patients worldwide suffering from primary UQ deficiency have yet to be recognized.
Electronic supplementary material
Acknowledgements
Research in SH laboratory is funded by grants from the Canadian Institutes of Health Research: MOP-114891, MOP-123295 and MOP-97869, as well as by McGill University. S.H. is Campbell Chair of Developmental Biology.
Author Contributions
B.G.H. and S.H. devised the study; B.G.H. and P.M.H. conducted the analysis; B.G.H. and S.H. wrote the manuscript. All authors read and approved the final manuscript.
Competing Interests
Funding to Dr. Hekimi was provided by the Canadian Institutes of Health Research.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-17564-y.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 2.Gorman GS, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Annals of Neurology. 2015;77:753–759. doi: 10.1002/ana.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kerr DS. Review of Clinical Trials for Mitochondrial Disorders: 1997–2012. Neurotherapeutics. 2013;10:307–319. doi: 10.1007/s13311-013-0176-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfeffer, G., Majamaa, K., Turnbull, D. M., Thorburn, D. & Chinnery, P. F. Treatment for mitochondrial disorders. Cochrane Database of Systematic Reviews (2012). [DOI] [PMC free article] [PubMed]
- 5.Wang Y, Hekimi S. Understanding Ubiquinone. Trends in Cell Biology. 2016;26:367–378. doi: 10.1016/j.tcb.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Yubero D, et al. Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion. 2016;30:51–58. doi: 10.1016/j.mito.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Hekimi S. Molecular genetics of ubiquinone biosynthesis in animals. Critical Reviews in Biochemistry and Molecular Biology. 2013;48:69–88. doi: 10.3109/10409238.2012.741564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desbats MA, Lunardi G, Doimo M, Trevisson E, Salviati L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. Journal of inherited metabolic disease. 2015;38:145–156. doi: 10.1007/s10545-014-9749-9. [DOI] [PubMed] [Google Scholar]
- 9.Acosta MJ, et al. Coenzyme Q biosynthesis in health and disease. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2016;1857:1079–1085. doi: 10.1016/j.bbabio.2016.03.036. [DOI] [PubMed] [Google Scholar]
- 10.López LC, et al. Treatment of CoQ10 Deficient Fibroblasts with Ubiquinone, CoQ Analogs, and Vitamin C: Time- and Compound-Dependent Effects. PLoS ONE. 2010;5:e11897. doi: 10.1371/journal.pone.0011897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Oxer D, Hekimi S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat Commun. 2015;6:6393. doi: 10.1038/ncomms7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashraf S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. The Journal of Clinical Investigation. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barca E, et al. Cerebellar ataxia and severe muscle CoQ10 deficiency in a patient with a novel mutation in ADCK3. Clinical Genetics. 2016;90:156–160. doi: 10.1111/cge.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landrum MJ, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng M, et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. Plos Genetics. 2008;4:14. doi: 10.1371/journal.pgen.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koscielny G, et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data (IMPC data Release 5.0) Nucleic Acids Res. 2014;42:D802–D809. doi: 10.1093/nar/gkt977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lapointe J, Wang Y, Bigras E, Hekimi S. The submitochondrial distribution of ubiquinone affects respiration in long-lived Mclk1 +/− mice. The Journal of Cell Biology. 2012;199:215–224. doi: 10.1083/jcb.201203090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levavasseur F, et al. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J Biol Chem. 2001;276:46160–46164. doi: 10.1074/jbc.M108980200. [DOI] [PubMed] [Google Scholar]
- 20.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desbats MA, et al. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Human Molecular Genetics. 2016;25:4256–4265. doi: 10.1093/hmg/ddw257. [DOI] [PubMed] [Google Scholar]
- 22.Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stefely JA, et al. Cerebellar Ataxia and Coenzyme Q Deficiency Through Loss of Unorthodox Kinase Activity. Molecular Cell. 2016;63:608–620. doi: 10.1016/j.molcel.2016.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.García-Corzo L, et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Human Molecular Genetics. 2013;22:1233–1248. doi: 10.1093/hmg/dds530. [DOI] [PubMed] [Google Scholar]
- 25.Mollet J, et al. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. Journal of Clinical Investigation. 2007;117:765–772. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heeringa SF, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. The Journal of Clinical Investigation. 2011;121:2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luna Sánchez M, et al. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Molecular Medicine. 2015;7:670–687. doi: 10.15252/emmm.201404632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brea-Calvo G, et al. COQ4 Mutations Cause a Broad Spectrum of Mitochondrial Disorders Associated with CoQ(10) Deficiency. American Journal of Human Genetics. 2015;96:309–317. doi: 10.1016/j.ajhg.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashraf S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ(10) biosynthesis disruption. The Journal of Clinical Investigation. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lagier-Tourenne C, et al. ADCK3, an Ancestral Kinase, Is Mutated in a Form of Recessive Ataxia Associated with Coenzyme Q(10) Deficiency. American Journal of Human Genetics. 2008;82:661–672. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mignot C, et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet Journal of Rare Diseases. 2013;8:173–173. doi: 10.1186/1750-1172-8-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerards M, et al. Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion. 2010;10:510–515. doi: 10.1016/j.mito.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 33.Xie LX, et al. Expression of the Human Atypical Kinase ADCK3 Rescues Coenzyme Q Biosynthesis and Phosphorylation of Coq Polypeptides in Yeast coq8 Mutants. Biochimica et biophysica acta. 2011;1811:348–360. doi: 10.1016/j.bbalip.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadowski CE, et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. Journal of the American Society of Nephrology: JASN. 2015;26:1279–1289. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korkmaz E, et al. ADCK4-Associated Glomerulopathy Causes Adolescence-Onset FSGS. Journal of the American Society of Nephrology: JASN. 2016;27:63–68. doi: 10.1681/ASN.2014121240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horvath R, et al. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. Journal of Neurology, Neurosurgery & Psychiatry. 2012;83:174–178. doi: 10.1136/jnnp-2011-301258. [DOI] [PubMed] [Google Scholar]
- 37.Salviati L, et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. Journal of Medical Genetics. 2012;49:187–191. doi: 10.1136/jmedgenet-2011-100394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salviati, L., Trevisson, E., Doimo, M. & Navas, P. Primary Coenzyme Q10 Deficiency. GeneReviews® [Internet] https://www.ncbi.nlm.nih.gov/books/NBK410087/. (2017).
- 39.Legati A, et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2016;1857:1326–1335. doi: 10.1016/j.bbabio.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 40.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. Jama. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neveling K, et al. A Post-Hoc Comparison of the Utility of Sanger Sequencing and Exome Sequencing for the Diagnosis of Heterogeneous Diseases. Human mutation. 2013;34:1721–1726. doi: 10.1002/humu.22450. [DOI] [PubMed] [Google Scholar]
- 42.Quinzii C, et al. A Mutation in Para-Hydroxybenzoate-Polyprenyl Transferase (COQ2) Causes Primary Coenzyme Q(10) Deficiency. American Journal of Human Genetics. 2006;78:345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freyer C, et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4–dihydroxybensoic acid. Journal of Medical Genetics. 2015;52:779–783. doi: 10.1136/jmedgenet-2015-102986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parikh S, et al. Practice patterns of mitochondrial disease physicians in North America. Part 1: Diagnostic and clinical challenges. Mitochondrion. 2014;14:26–33. doi: 10.1016/j.mito.2013.07.116. [DOI] [PubMed] [Google Scholar]
- 45.Sondheimer N, et al. Novel recessive mutations in COQ4 cause severe infantile cardiomyopathy and encephalopathy associated with CoQ(10) deficiency. Molecular Genetics and Metabolism Reports. 2017;12:23–27. doi: 10.1016/j.ymgmr.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park, E. et al. COQ6 Mutations in Children With Steroid-Resistant Focal Segmental Glomerulosclerosis and Sensorineural Hearing Loss. American Journal of Kidney Diseases (2017). [DOI] [PubMed]
- 47.Wang, Y. et al. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. Journal of cellular and molecular medicine, n/a-n/a (2017). [DOI] [PMC free article] [PubMed]
- 48.Park, E. et al. Focal segmental glomerulosclerosis and medullary nephrocalcinosis in children with ADCK4 mutations. Pediatric Nephrology, 1–8 (2017). [DOI] [PubMed]
- 49.Lolin K, et al. Early-onset of ADCK4 glomerulopathy with renal failure: a case report. BMC Medical Genetics. 2017;18:28. doi: 10.1186/s12881-017-0392-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed during the current study are available at www.ncbi.nlm.nih.gov/clinvar/ and http://gnomad.broadinstitute.org/.