

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is a prevalent, incurable myopathy, linked to hypomethylation of D4Z4 repeats on chromosome 4q causing expression of the DUX4 transcription factor. However, DUX4 is difficult to detect in FSHD muscle biopsies and it is debatable how robust changes in DUX4 target gene expression are as an FSHD biomarker. PAX7 is a master regulator of myogenesis that rescues DUX4-mediated apoptosis. Here, we show that suppression of PAX7 target genes is a hallmark of FSHD, and that it is as major a signature of FSHD muscle as DUX4 target gene expression. This is shown using meta-analysis of over six FSHD muscle biopsy gene expression studies, and validated by RNA-sequencing on FSHD patient-derived myoblasts. DUX4 also inhibits PAX7 from activating its transcriptional target genes and vice versa. Furthermore, PAX7 target gene repression can explain oxidative stress sensitivity and epigenetic changes in FSHD. Thus, PAX7 target gene repression is a hallmark of FSHD that should be considered in the investigation of FSHD pathology and therapy.
Facioscapulohumeral muscular dystrophy is a myopathy linked to ectopic expression of the DUX4 transcription factor. The authors show that the suppression of targets genes of the myogenesis regulator PAX7 is a signature of FSHD, and might explain oxidative stress sensitivity and epigenetic changes.
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is a prevalent inherited skeletal myopathy (12/100,000)1 that typically presents during the second decade of life in males and third decade in females. FSHD is characterised by an asymmetric, descending skeletal muscle atrophy affecting specific muscle groups, including the orbicularis occuli/orbis, the scapular fixator muscles, the biceps and latterly, the tibialis anterior and certain other lower limb muscles2. Curiously, muscles such as the quadriceps, diaphragm and deltoids are spared2, 3. FSHD is also linked to retinal telangiectasia and sensorineural hearing loss, pointing to more systemic mechanisms4, 5.
Genetically, FSHD is associated with hypomethylation of the D4Z4 macrosatellite repeat at chromosome 4q35, coupled with a permissive 4qA haplotype containing a polyadenylation signal in the pLAM region distal to the final D4Z4 repeat. Hypomethylation is caused either by D4Z4 repeat length truncation to between 1–10 D4Z4 units in FSHD1 (MIM 158900) (~ 95% of cases)6 or by mutation in epigenetic modifiers in FSHD2 (MIM 158901), particularly SMCHD1 7, but with DNMT3B also recently identified in rare cases8. Each D4Z4 unit contains an open reading frame for a retrogene termed double homeobox 4 (DUX4) (MIM 606009). D4Z4 hypomethylation permits transcription of DUX4 from the final D4Z4 repeat, with DUX4 message stabilised by addition of a polyadenylation tail by the signal in the flanking DNA on a permissive haplotype, and translated. The consensus is that such DUX4 expression underlies pathology9 and DUX4 target gene expression has been suggested as the major molecular signature in FSHD10. However, detection of DUX4 is difficult in FSHD muscle biopsies and a consistent molecular biomarker of FSHD muscle based on DUX4 expression has yet to be fully validated.
DNA binding, and so target gene selection, is defined by the homeodomains of DUX4, which display significant amino-acid sequence homology to the homeodomains of the transcription factors PAX3 and PAX711. Given this homology, it has been hypothesised that competitive inhibition of PAX3 and/or PAX7 by DUX4 may also contribute to FSHD pathology. PAX3 and PAX7 are critical to development in the early embryo12, most notably playing essential, non-redundant roles, in the generation of the skeletal myogenic lineage13, and derivatives of the dorsal neural ectoderm12. PAX3 and PAX7 are also highly homologous, with 86% sequence similarity14 and are well conserved between mouse and man. In adult, Pax7 is expressed in quiescent, activated and proliferating muscle satellite cells15, while the Pax3 locus is active in some satellite cells in certain muscles, such as the diaphragm, and may be transiently expressed during activation13, 16–18. Both genes promote cell survival and proliferation, while preventing precocious differentiation15, 19. In man, PAX7 is also detected in satellite cells20, 21 but little is known about PAX3 in postnatal skeletal muscle. However, mutations in PAX3 in man are associated with Waardenburg syndrome22, a condition whose characteristics include high frequency, sensorineural hearing loss, a symptom also reported in FSHD.
In line with a competitive inhibition model, it has been demonstrated that cytotoxicity caused by expression of DUX4 or its murine ortholog mDUX is rescued by over-expression of Pax3 or Pax7 in murine myoblasts11, 23. We have recently shown in a transgenic mouse model of FSHD that DUX4 levels rise during the early phases of muscle regeneration in activated and proliferating satellite cells, a phase when Pax7 is operating24. It has also been shown that FSHD patient-derived cells express high levels of DUX4 during a muscle progenitor phase characterised by high levels of PAX3 and PAX7 during in vitro differentiation of embryonic stem (ES) cells, providing another time point in FSHD myogenic differentiation where competitive inhibition may occur25. A more recent study of myogenic differentiation of ES cells and induced pluripotent stem (iPS) cells from FSHD patients showed that although PAX7 and DUX4 mRNA are found in a myogenic progenitor phase, the two proteins are not present in the same cell26. Interestingly, the frequency of co-expression of PAX7 and DUX4 in FSHD cells was significantly lower than expected, indicating that some non-competitive inhibitory interaction between the two proteins may ensure such mutual exclusivity26. PAX7 also drives long term epigenetic changes associated with de-repression of gene expression, including DNA demethylation27. Hence, inhibition of PAX7 by DUX4 in FSHD during a muscle progenitor stage may result in a global suppression of PAX7 transcriptional targets that persists in terminally differentiated FSHD muscle. Such repression may represent a novel biomarker of FSHD muscle, and define a new set of therapeutic target genes. Examination of PAX7 target gene expression in FSHD patient-derived samples is lacking, however, and so the translational implications of this mechanism are unresolved.
Here, we adopt a meta-analysis approach across six independent FSHD muscle biopsy studies to analyse several FSHD molecular biomarkers based on DUX4 target gene expression, and to compare them to a novel biomarker based on Pax7 target gene repression. We find that only the new biomarker based on Pax7 target gene repression is consistently able to discriminate FSHD from control skeletal muscle biopsies in every FSHD biopsy data set, while the discriminatory power of DUX4 target gene expression is limited to just two studies10, 28. The discriminatory power of Pax7 target gene repression is verified via RNA-seq of immortalised human myoblasts isolated from FSHD patients and matched controls. We also show that the Pax7 target genes repressed in FSHD are enriched for factors that suppress the HIF1α-mediated hypoxic response, indicating over-activation of HIF1α in FSHD as a putative pathomechanism driving oxidative stress sensitivity, as we previously suggested29. Lastly, co-expression of DUX4 and Pax7 results in repression of their respective target genes, confirming mutual target gene inhibition between these two transcription factors. Overall, our findings demonstrate that Pax7 target gene repression is at least as strong a signature in FSHD skeletal muscle as DUX4 target gene over-expression. Thus, Pax7 target genes should be considered in the investigation of pathology and design of therapies in FSHD.
Results
DUX4 target gene expression as an FSHD biomarker
Transcriptomic biomarkers of FSHD muscle are important both for understanding molecular processes perturbed in pathology and for monitoring therapeutic responses in clinical trials3. In recent years, a number of transcriptomic biomarkers for identifying FSHD muscle biopsies have been proposed, notably the biopsy-derived 15 gene signature described by Rahimov et al.3, and the 114 gene signature derived from human myoblasts over-expressing DUX4, described by Yao et al.10. Outside of a small number of samples, however, these biomarkers have received little validation.
We identified six independent transcriptomic studies profiling FSHD muscle biopsies alongside suitable controls (five microarray studies totalling 82 FSHD and 82 controls and one RNA-seq study with 15 FSHD and 8 control samples)3, 10, 28, 30–32 and assessed the discriminatory capacity of the Rahimov et al.3 and Yao et al.10 biomarkers via meta-analysis (Fig. 1). Meta-analysis over five independent data sets revealed that neither biomarker could reliably discriminate FSHD from control samples. Interestingly however, both signatures were able to discriminate between FSHD and control muscle biopsies on one microarray data set independent of their discovery data sets, namely that described by Tasca et al.28. Unlike the other studies, this data set profiled affected FSHD muscle as confirmed by magnetic resonance imaging (MRI), indicating that an MRI guided muscle biopsy approach may be optimal for identifying tissue displaying molecular changes associated with FSHD.
Fig. 1.

Reported FSHD biomarkers do not validate on most published FSHD muscle biopsy gene expression data sets. a Box plot confirms that the Yao et al.10 DUX4 114 target gene signature validates as a biomarker on the RNA-seq FSHD muscle biopsy data set published by Yao et al.10. The box represents the interquartile range (IQR), with the median indicated by a line. Whiskers denote min (1.5*IQR, max (observed value)). ‘‘o’’ represents data points greater than 1.5 IQR from the median, n = 15 FSHD and n = 8 control muscle biopsies. b A forest plot displays the results of meta-analysis of the discriminatory power of the Yao et al.10 DUX4 target gene signature across five published microarray FSHD muscle biopsy data sets (in total n = 82 FSHD and n = 82 control muscle biopsies). The differential scores (FSHD score minus control score) alongside 95% confidence intervals are provided. c A box plot confirms that the Rahimov et al.3 FSHD 15 gene biomarker validates as a biomarker on the microarray FSHD muscle biopsy discovery data set published by Rahimov et al.3. The box represents the interquartile range (IQR), with the median indicated by a line. Whiskers denote min (1.5*IQR, max (observed value)). ‘‘o’’ represents data points greater than 1.5 IQR from the median, n = 26 FSHD and n = 24 control muscle biopsies. d A forest plot displays the results of meta-analysis of the discriminatory power of the Rahimov et al.3 FSHD biomarker across four published microarray and one RNA-seq FSHD muscle biopsy data sets (in total n = 71 FSHD and n = 66 control muscle biopsies). The differential scores (FSHD score minus control score) alongside 95% confidence intervals are provided. Neither biomarker is able to discriminate FSHD from control samples on meta-analysis. However, both are able to discriminate on the Tasca et al.28 MRI-guided muscle biopsy microarray data set. For single studies a two-tailed Wilcoxon U-test was performed to assess significance, while a Fisher’s combined test was employed for overall assessment: either the p-value is given, or an asterisk denotes p < 0.05
The DUX4-based biomarker of Yao et al.10 was derived from RNA-seq data of two human myoblast samples over-expressing DUX4, compared to two qualitatively different controls, one of which was contaminated and contained reads from a DUX4-expressing sample33. Hence it is unclear how representative the Yao et al.10 biomarker is of DUX4 expression. Unfortunately, the only data set profiling FSHD muscle biopsies by RNA-seq was also produced by Yao et al.10 and our other validation data sets were profiled by microarray. Therefore, it is also unclear whether failure of these genes to discriminate FSHD status is due to DUX4 target genes not being a biomarker, or due to differences in the technology platforms used, namely microarray vs. RNA-seq.
To address these issues, we derived two further DUX4 over-expression signatures from two independent human myoblast DUX4 over-expression studies. The first signature was derived from a recent RNA-seq data set profiling human myoblasts over-expressing DUX4 for 8 h via a doxycycline inducible promotor against suitable controls, as described by Choi et al.34. We identified 212 transcripts that were significantly upregulated in DUX4-expressing samples (FDR < 0.05, log FC > 2, Supplementary Data 1). The Choi et al.34 DUX4 target gene signature was significantly elevated in FSHD samples from the Yao et al.10 RNA-seq muscle biopsy data set. However, we found no significant difference in Choi et al.34 DUX4 target levels between FSHD and control samples in any of the five microarray data sets or on meta-analysis (Fig. 2a–b).
Fig. 2.

Novel DUX4 target gene signatures only discriminate some FSHD muscle biopsy gene expression data sets from controls. a A box plot demonstrates that the Choi et al.34 RNA-seq-based 212 DUX4 target gene signature validates as a biomarker on the RNA-seq FSHD muscle biopsy data set published by Yao et al.10. The box represents the interquartile range (IQR), with the median indicated by a line. Whiskers denote min (1.5*IQR, max (observed value)). ‘‘o’’ represents data points greater than 1.5 IQR from the median, n = 15 FSHD and n = 8 control muscle biopsies. b A forest plot displays the results of meta-analysis of the discriminatory power of the Choi et al.34 DUX4 target gene signature across five published microarray FSHD muscle biopsy data sets (in total n = 82 FSHD and n = 82 control muscle biopsies). The differential scores (FSHD score minus control score) alongside 95% confidence intervals are provided. The Choi et al.34 DUX4 target gene signature is not a significant biomarker on any microarray data set nor on meta-analysis. Note: in the Choi et al.34 study, target genes were analysed after 8 h of DUX4 induction. c A box plot demonstrates that the Geng et al.35 microarray based 165 DUX4 target gene signature validates as a biomarker on the RNA-seq FSHD muscle biopsy data set published by Yao et al.10. The box represents the interquartile range (IQR), with the median indicated by a line. Whiskers denote min (1.5*IQR, max (observed value)). ‘‘o’’ represents data points greater than 1.5 IQR from the median, n = 15 FSHD and n = 8 control muscle biopsies. d A forest plot displays the results of meta-analysis of the discriminatory power of the Geng et al.35 DUX4 target gene signature across five published microarray FSHD muscle biopsy data sets (in total n = 82 FSHD and n = 82 control muscle biopsies). The differential scores (FSHD score minus control score) alongside 95% confidence intervals are provided. The Geng et al.35 DUX4 target gene signature is not a significant biomarker on any individual microarray data set, however, it is significant on meta-analysis. For single studies a two-tailed Wilcoxon U-test was performed to assess significance, while a Fisher’s combined test was employed for overall assessment: either the p-value is given, or an asterisk denotes p < 0.05
A second DUX4 signature was derived from a microarray data set published by Geng et al.35, profiling DUX4 lentivirus transduced human myoblasts after 24 h against suitable controls35. We identified 165 transcripts that were significantly upregulated in DUX4-expressing samples (FDR < 0.05, log FC > 2, Supplementary Data 2). The Geng et al.35 DUX4 target gene signature was significantly upregulated on the FSHD muscle biopsy samples from the Yao et al.10 RNA-seq data set. Moreover, across the five microarray FSHD studies, although no individual study showed statistically significant elevation of DUX4 target gene expression, all studies showed a positive trend towards this. On meta-analysis, we identified a small, but significant, upregulation of DUX4 targets in FSHD samples (Fisher’s combined test p = 0.045, Fig. 2c–d).
As DUX4 suppresses MyoD11, 24 we also evaluated expression of MyoD target genes identified by de la Serna et al.36 in the five FSHD microarray studies. Although no individual data set was significant, there was a trend towards repression of MyoD target genes across microarray studies of FSHD muscle biopsies, but this was also not significant on meta-analysis (Supplementary Fig. 1)
Our results indicate that DUX4 target gene expression is a weak, but significant, biomarker of FSHD status but points to a lack of compatibility between RNA-seq and microarray studies for evaluating DUX4 transcriptional target genes. Despite overlap between the three DUX4 signatures (28 genes shared between Geng et al.35 and Choi et al.34, 45 between Yao et al.10 and Geng et al.35 and 29 between Choi et al.34 and Yao et al.10), we see that RNA-seq-derived DUX4 upregulated target genes are only capable of discriminating FSHD biopsies from controls, if the biopsies were also profiled by RNA-seq. In contrast, microarray-based DUX4 upregulated target genes show discriminatory power on both microarray (on meta-analysis) and RNA-seq data sets (Figs. 1 and 2). This is likely due to differences in coverage and dynamic range across the two technologies and is a consideration when evaluating DUX4 transcriptional target genes on FSHD samples.
PAX7 target gene repression hallmarks FSHD skeletal muscle
We next considered repression of PAX7 target genes as a biomarker of FSHD muscle. To derive a set of PAX7 target genes, we assayed primary murine satellite cell-derived myoblasts over-expressing Pax7, a dominant negative Pax7 fusion protein, comprising the DNA binding domains of Pax7 and the engrailed repressor domain (Pax7-ERD)15 and a control retroviral construct. PAX7 is highly conserved between mouse and man with 97% amino-acid sequence homology13, hence a conserved set of target genes is expected. We assayed gene expression via microarray to facilitate compatibility with the publically available FSHD muscle biopsy data sets. Microarray data were pre-processed and normalised as described in Methods. Hierarchical clustering and principal component analysis confirmed reproducibility of the transcriptional landscapes induced by the PAX7 constructs and close clustering of replicates.
Unlike DUX4, which is a potent transcriptional activator24, 29, PAX7 likely modulates transcription in more complex ways12. Hence, to derive a biomarker of Pax7 expression in murine satellite cells, rather than focus only on the most strongly induced target genes, we performed differential expression analysis to derive a set of 311 upregulated target genes (defined as induced by PAX7 over-expression and suppressed by PAX7-ERD) and a set of 290 downregulated target genes (defined as suppressed by PAX7 over-expression and induced by PAX7-ERD) (Supplementary Data 3). A biomarker of PAX7 was then defined from consideration of the ratio of mean upregulated to downregulated target gene expression in a given sample (see Methods). To validate this ratio as a biomarker of PAX7, we evaluated it on an independent microarray dataset describing Pax7 retroviral over-expression in primary murine satellite cells alongside control retrovirus in triplicate14. Our PAX7 biomarker demonstrated significantly higher values on samples over-expressing Pax7 in this independent data set, so confirming its validity (Supplementary Fig. 2).
To evaluate the functional relevance of PAX7 target genes to FSHD pathology, we performed gene set enrichment analysis (GSEA) of activated and repressed target genes of PAX7 separately against the Molecular Signatures database (MSigDB)37, 38. The top 100 enriched gene sets for the PAX7 targets are presented in Supplementary Data 4 and 5. To refine our search we identified gene sets which displayed inverse enrichments across activated and repressed targets (Fig. 3a). PAX7 is associated with repression of a hypoxia gene set. Importantly of 48 hypoxia gene sets in MSigDB37, 38, only one was identified, describing genes upregulated or downregulated by over-expression of an active form of HIF1α39. We have previously implicated over-activation of HIF1α as a putative mechanism for oxidative stress sensitivity in FSHD29. Another gene set repressed by PAX7 is EZH2 target genes (Fig. 3a). EZH2 is a critical component of the polycomb repressor complex 2 (PRC2), which has been associated with perturbed methylation in FSHD40, 41. Thus PAX7 target genes are enriched for pathways previously identified in FSHD molecular pathology.
Fig. 3.

PAX7 target gene signature is a robust hallmark of FSHD skeletal muscle. a Gene set enrichment analysis was performed separately for activated and repressed targets of PAX7 against the Molecular Signatures database (MSigDB)37, 38. Gene sets in MSigDB are frequently partitioned into ‘‘genes upregulated in phenotype’’ and ‘‘genes downregulated in phenotype’’. If such opposing gene sets were inversely significantly enriched across activated and repressed PAX7 targets (as assessed by Fisher’s exact test), they were considered robust. A bar plot displays the −log10 (enrichment p-value) for PAX7 robust gene sets. A horizontal green line denotes p = 0.05. UP and DOWN on the x-axis refers to the partition of labeled gene sets into genes positively and negatively associated with the gene set respectively. PAX7 targets are enriched for repression of a HYPOXIA gene set, which specifically describes genes upregulated and downregulated following over-expression of a constitutively active form of HIF1α. Another gene set affected by PAX7 repression is EZH2 target genes. b A box plot demonstrates that the PAX7 target gene signature derived from 311 upregulated target genes and 290 downregulated target genes, validates as a biomarker on the RNA-seq FSHD muscle biopsy data set published by Yao et al.10. The box represents the interquartile range (IQR), with the median indicated by a line. Whiskers denote min (1.5*IQR, max (observed value)). ‘‘o’’ represents data points greater than 1.5 IQR from the median, n = 15 FSHD and n = 8 control muscle biopsies. The two-tailed Wilcoxon U-test p-value is given. c A forest plot displays the results of meta-analysis of the discriminatory power of the PAX7 target gene signature across five published microarray FSHD muscle biopsy data sets (in total n = 82 FSHD and n = 82 control muscle biopsies). The differential scores (FSHD score minus control score) alongside 95% confidence intervals are provided. Our PAX7 target gene signature is a significant biomarker on every individual FSHD muscle biopsy data set, and is strongly significant on meta-analysis. For single studies, a two-tailed Wilcoxon U-test was performed to assess significance, while a Fisher’s combined test was employed for overall assessment: *denotes p < 0.05, **denotes p < 0.01 and ***denotes p < 0.001
We next evaluated our PAX7 biomarker on the six FSHD muscle biopsy data sets. We found that the levels of PAX7 target genes were significantly repressed in FSHD muscle biopsy samples profiled by RNA-seq (Fig. 3b). Importantly, PAX7 target gene repression was also found to be a significant biomarker of FSHD status in each of the five microarray FSHD muscle biopsy studies independently, leading to a highly significant repression of PAX7 target genes on meta-analysis (Fisher’s combined test p = 3.5 × 10−9, Fig. 3c).
To confirm that PAX7 target gene repression is specific to FSHD and not attributable to general muscle wasting, we identified four published microarray studies profiling Duchenne muscular dystrophy (DMD) muscle biopsies alongside matched controls31, 42, 43. Meta-analysis revealed no consistent significant difference in PAX7 target gene expression between DMD muscle and controls (Supplementary Fig. 3).
The PAX7 and DUX4 biomarkers were derived differently, so it is necessary to consider how the results would change if this were not the case. Firstly, if repressed DUX4 targets are combined with the induced targets to construct a biomarker of DUX4 analogous to our derivation of the PAX7 biomarker, DUX4 target genes are lost as a discriminator of FSHD status. This indicates that only robustly upregulated DUX4 targets have discriminatory power in FSHD and that suppressed targets introduce noise that masks this signal. Conversely, if we separately consider the upregulated or downregulated target genes of PAX7, rather than together as a biomarker of PAX7 (analogous to the DUX4 biomarker), we see that upregulated target genes are significantly repressed in FSHD microarray samples on meta-analysis, while downregulated target genes are significantly over-expressed, confirming that both activated and suppressed PAX7 target genes are perturbed in FSHD. However, the upregulated or downregulated target genes alone are unable to discriminate between FSHD and control muscle biopsies on the RNA-seq data set (Supplementary Fig. 4).
This cross comparison demonstrates that the biomarker construction which maximises DUX4 target gene discriminatory power in FSHD focuses only on upregulated targets, while that which maximises PAX7 target gene discriminatory power utilises both upregulated and downregulated target genes.
To confirm that our PAX7 biomarker does not discriminate FSHD status from control by chance, we performed a resampling procedure, selecting 1000 random gene sets of equivalent size to both the upregulated and downregulated PAX7 target gene sets. For each randomisation, we defined a biomarker analogously to the PAX7 biomarker using the ratio of upregulated to downregulated target genes and evaluated its capacity to discriminate FSHD and control muscle biopsies on meta-analysis across the five FSHD microarray muscle biopsy data sets. None of the random biomarkers were able to significantly discriminate FSHD from control samples on meta-analysis (Supplementary Fig. 5), hence the probability that PAX7 target gene repression in FSHD is attributed to chance is less than 1/1000.
PAX7 or DUX4 target genes form equivalent FSHD biomarkers on RNA-seq data
To compare the discriminatory capacity of our PAX7 target gene biomarker against the various DUX4 target gene biomarkers, we employed a receiver operating characteristic (ROC) approach. Given the data compatibility issues explored above, we considered the microarray and RNA-seq muscle biopsy data separately. An impact of this is that analysis over the RNA-seq data is underpowered relative to the microarray data due to large differences in sample size (RNA-seq data: 8 controls, 15 FSHD; pooled microarray data: 82 controls, 82 FSHD), and such a discrepancy should be taken into account on data interpretation.
Considering the microarray data, the only significant biomarkers on meta-analysis were our PAX7 target gene repression signature and the robustly upregulated DUX4 targets described by Geng et al.35 while Choi et al.34 and Yao et al.10 failed to reach significance. Both biomarkers were computed for each sample on each microarray study independently and z-normalised within each study to ensure identically distributed predictors, studies were then pooled for ROC analysis. We found that PAX7 target gene repression significantly outperformed DUX4 target expression as a classifier of FSHD status (Pax7 AUC = 0.81, DUX4 AUC = 0.59, DeLong’s test: p < 5.7 × 10−6, Fig. 4a), indicating that in microarray data, PAX7 target gene repression is a more robust signature of FSHD skeletal muscle than DUX4 target gene expression.
Fig. 4.
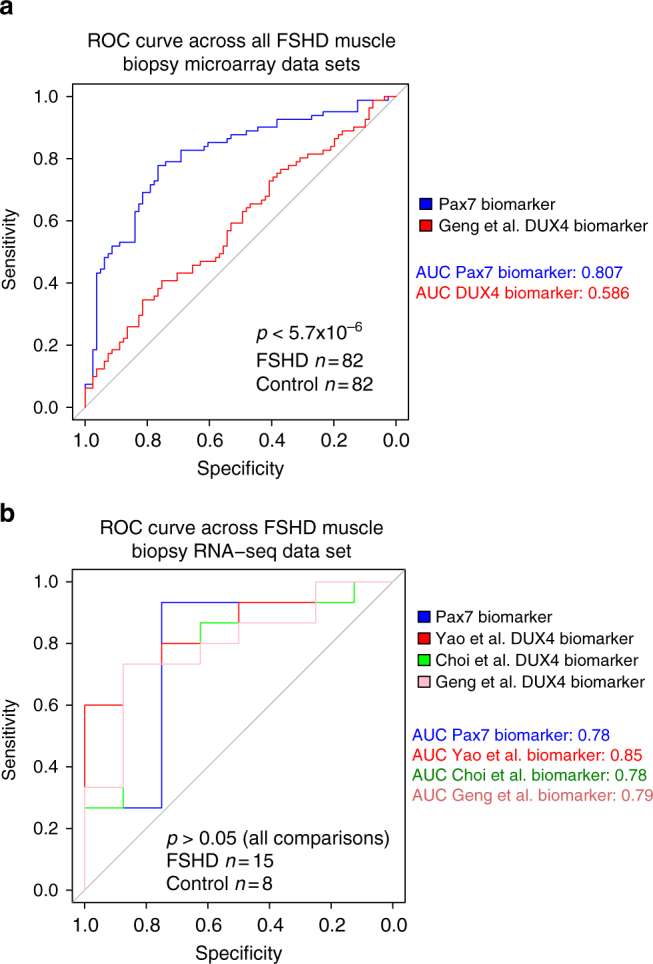
PAX7 target gene repression is an equivalent FSHD biomarker to DUX4 target gene expression. a A ROC curve compares the discriminatory power of our PAX7 biomarker with the DUX4 target gene signature that we derived from Geng et al.35, across five microarray data sets (in total n = 82 FSHD and n = 82 control muscle biopsies). De-Long’s test p-value is given and demonstrates that the PAX7 biomarker is a significantly better discriminator of FSHD status. b A ROC curve compares the discriminatory power of our PAX7 biomarker with the DUX4 target gene signatures that we derived from Geng et al.35 and Choi et al.34 and that described in Yao et al.10 across the RNA-seq data set of n = 15 FSHD and n = 8 control muscle biopsies. De-Long’s test reveals no significant differences in the discriminatory power of these four biomarkers
Considering the smaller RNA-seq data set, all three DUX4 upregulated target gene signatures (Geng et al.35, Choi et al.34 and Yao et al.10), as well as our PAX7 target gene repression signature, proved to be significant biomarkers of FSHD status. Though the Yao et al.10 signature demonstrated a slightly higher AUC on the RNA-seq samples, than the Pax7 biomarker (Yao et al.10 AUC = 0.85, Geng et al.35 AUC = 0.79, Choi et al.34 AUC = 0.78, PAX7 AUC = 0.78, Fig. 4b), DeLong’s test revealed no significant differences between the discriminatory power of any of the biomarkers on this data set. Thus PAX7 target gene repression is at least as major a signature of FSHD skeletal muscle as DUX4 target gene expression.
PAX7 or DUX4 target genes are equivalent biomarkers on FSHD myoblasts
As muscle biopsies do not represent a pure myogenic population of cells, we next investigated our PAX7 or DUX4 target gene biomarkers in FSHD patient-derived myoblast cell lines. We performed RNA-sequencing on immortalised human myoblasts isolated from three independent FSHD patients alongside matched controls44, 45. One of the FSHD patients considered is mosaic for the FSHD genotype and multiple clones are available45, of which we considered five. These clones are isogenic with exception of the D4Z4 region, which is truncated to three repeats in clones 54–12, 54-2 and 54-A5 (an FSHD genotype), whereas clones 54-6 and 54-A10 have 13 repeats (a ‘‘healthy’’ number). The two further FSHD patient cell lines considered were the 12Abic and 16Abic immortalised myoblasts obtained from the UMMS Wellstone centre for FSHD, Worcester, USA, alongside sibling-matched controls44. RNA-sequencing was performed in triplicate on confluent myoblasts for each cell line, giving a total of 15 FSHD samples (three patients corresponding to five lines in triplicate) and 12 control samples (three individuals corresponding to four lines in triplicate).
Our PAX7 biomarker, alongside the DUX4 target gene signatures (Yao et al.10, Choi et al.34 and Geng et al.35), were computed for each sample and scores for each patient and matched control were z-normalised and pooled for analysis. We found that our PAX7 biomarker, the Yao et al.10 and Geng et al.35 DUX4 target gene signatures were all significant discriminators of FSHD status (Wilcoxon signed rank test p < 0.05, Fig. 5a–d), in line with our results from the muscle biopsy RNA-seq data, but the Choi et al.34 DUX4 target gene signature was not (Fig. 5a). ROC curve analysis demonstrated that there was no significant difference between the discriminatory power of our PAX7 biomarker and the DUX4 biomarkers on FSHD patient cell lines (De-Long’s test p > 0.05, Fig. 5e). This provides further evidence that PAX7 target gene repression is at least as major a signature as DUX4 target gene expression in FSHD skeletal muscle.
Fig. 5.

PAX7 and DUX4 target gene biomarkers validate on FSHD myoblast gene expression data sets. Box plots show the a Choi et al.34, b Yao et al.10 and c Geng et al.35 DUX4 target gene signatures and d our PAX7 biomarker, computed for five FSHD patient lines and four matched control lines corresponding to three patients and controls RNA-sequenced in triplicate (in total n = 15 FSHD and n = 12 Control). In line with previous findings, PAX7 repression and DUX4 expression as assessed by the Yao et al.10 and Geng et al.35 DUX4 target signatures are significant biomarkers of FSHD status. The Choi et al.34 DUX4 target gene biomarker is not a significant discriminator of FSHD status, where target genes were identified after 8 h of DUX4 induction. The box represents the interquartile range (IQR), with the median indicated by a line. Whiskers denote min (1.5*IQR, max (observed value)). ‘‘o’’ represents data points greater than 1.5 IQR from the median. The two-tailed Wilcoxon U-test p-value is given. e A ROC curve compares the discriminatory power of our PAX7 biomarker with the Geng et al.35, Choi et al.34 and Yao et al.10 DUX4 target gene signatures across the RNA-seq data set of FSHD and control immortalised myoblasts. De-Long’s test reveals no significant differences in the discriminatory power of these three DUX4 and one PAX7 biomarkers
PAX7 and DUX4 mutually inhibit target gene activation
Having established that PAX7 target genes are repressed in FSHD skeletal muscle, we next investigated if DUX4 could lead to repression of PAX7 transcriptional target genes, as suggested by the homology between their homeodomains11. A reporter construct assay was performed on human embryonic kidney (HEK-293) or NIH-3T3 cells transfected with plasmids encoding DUX4, Pax7, both DUX4 and Pax7 together, dominant negative DUX4 (DUX4-ERD)29 or PAX7 (Pax7-ERD)15, and GFP as a control. To measure the ability of PAX7 to activate its transcriptional targets, we measured activity of the p34 reporter construct, consisting of concatermerised PAX3/7-binding sites driving thymidine kinase-nlacZ 15, along with endogenous expression of the PAX7 target gene SELP 14. To quantify DUX4 activity, we used DUX4 reporter constructs RFPL4B-luc or ZSCAN4-luc controlling a luciferase reporter gene46 and also measured endogenous expression of the DUX4 target genes ZSCAN4, RFPL4B, MBD3L2 and TRIM48.
PAX7 robustly elevated p34 reporter activity, as expected. Interestingly, DUX4 also increased p34 reporter activity, but to a smaller extent than PAX7. Strikingly both DUX4 and PAX7 proteins together resulted in no change of p34 reporter activity, with the level unaltered from that achieved by transfection of control GFP-encoding plasmid (Fig. 6a). Reverse transcription quantitative PCR (RT-qPCR) of PAX7 target gene SELP also confirmed its activation by PAX7, but when both DUX4 and PAX7 were present together, transcription of SELP dropped to control levels (Fig. 6b). Thus, while DUX4 can bind and activate the PAX7 reporter gene, when DUX4 is present with PAX7, the effect is to repress activation of PAX7 transcriptional target genes. Levels of PAX7 target gene activation achieved with DUX4 and PAX7 co-expression are lower than achieved by either PAX7 or DUX4 alone, implying that the mechanism by which DUX4 represses PAX7 transcriptional target gene activation may encompass mechanisms in addition to competitive inhibition of DNA-binding.
Fig. 6.

Co-expression of PAX7 and DUX4 causes suppression of the transcriptional activity of both proteins. HEK-293 or NIH-3T3 cells were transfected with plasmids encoding DUX4, Pax7, DUX4 and Pax7, or dominant negative-version DUX4-ERD, or Pax7-ERD. a The ability of PAX7 to activate its transcriptional target genes was measured via the p34 plasmid driving lacZ, co-transfected with the constructs listed above, together with a control RSV luciferase plasmid as a transfection normaliser, into NIH-3T3 cells. Reporter gene intensities were measured using a Glomax-Multi + plate reader and normalised to RSV luciferase. PAX7 activated the p34 PAX7 reporter construct, but both PAX7 and DUX4 together suppressed activity of this PAX7 reporter compared to PAX7 alone. b RT-qPCR for PAX7 transcriptional target gene SELP confirms that co-expression of Pax7 and DUX4 suppresses the activation of this endogenous PAX7 transcriptional target compared to PAX7 alone, in HEK-293. The ability of DUX4 to activate its transcriptional target genes was measured via two separate DUX4 reporter constructs: c ZSCAN4-luc and d RFPL4B-luc, both controlling a luciferase reporter gene, co-transfected with DUX4 and/or Pax7 constructs, or DUX4-ERD or GFP, together with a RSV lacZ as a transfection normaliser, into HEK-293. Reporter gene intensities were measured using a Glomax-Multi + plate reader and normalised to β-galactosidase activity. While DUX4 activated both DUX4 reporters, both PAX7 and DUX4 together, suppressed activity of these DUX4 reporters compared to DUX4 alone. RT-qPCR for DUX4 endogenous transcriptional target genes. e TRIM48, f ZSCAN4, g RFPL4B and h MBD3L2 confirms that the presence of both PAX7 and DUX4 together suppresses activation of all these endogenous DUX4 transcriptional target genes compared to the levels achieved by DUX4 alone in HEK-293 cells. Boxes represents the interquartile range (IQR), with the median indicated by a line. Whiskers denote min (1.5*IQR, max (observed value)). For bar graphs, error bars denote standard error of the mean, n = 3 or 4 for each cell line, ANOVA revealed significant intensity differences, post hoc unpaired two tailed t-tests were employed to assess significant pairwise differences: *denotes p < 0.05, and n.s. denotes non-significance of pairwise t-tests
Performing reciprocal experiments using DUX4 reporters, we found that while DUX4 activated both the RFPL4B-luc or ZSCAN4-luc reporters, PAX7 did not activate either DUX4-reporter (Fig. 6c, d). As observed with the PAX7 reporter assay, expression of both PAX7 and DUX4 proteins together, significantly reduced DUX4 reporter activity compared to DUX4 alone, and back to control levels with the RFPL4B-luc reporter (Fig. 6d). These findings were confirmed via RT-qPCR of endogenous DUX4 transcriptional target genes, which showed that DUX4 induced expression of ZSCAN4, RFPL4B, MBD3L2 and TRIM48, while both DUX4 and PAX7 together failed to increase transcript levels of these DUX4 target genes to the same level as DUX4 alone, and in 3 out of 4 cases, not above control levels (Fig. 6e–h).
Therefore, inhibition between DUX4 and PAX7 proteins is reciprocal, whereby co-expression of PAX7 and DUX4 acts to suppress the respective transcriptional target genes of both DUX4 and PAX7.
Discussion
Here, we demonstrate that PAX7 target gene repression is a hallmark of FSHD skeletal muscle. Moreover, it is a superior FSHD biomarker to DUX4 target gene expression across microarray data sets, and at least as major a signature of FSHD skeletal muscle when considering RNA-seq data.
DUX4 is currently the leading candidate gene in FSHD molecular pathology9. Recently, it has been suggested that DUX4 target gene expression is also the major molecular signature in FSHD skeletal muscle43. This has led to an acceleration of investigations into DUX4 target genes in FSHD, which are pro-apoptotic and suppress myogenic progression11, 24, 29, 47–49. In addition to these DUX4 focused studies, others have compared FSHD and control skeletal muscle in an hypothesis free manner. Using this approach, we and others, have revealed FSHD molecular mechanisms, which may not be direct consequences of DUX4 target gene expression, such as HIF1α target gene activation29, 50 and epigenetic alterations in PCR2 genes41. Such mechanisms are an important line of investigation into FSHD pathology and therapeutics, but may not be fully understood by investigation of DUX4 target genes alone.
The idea that DUX4 and PAX7 protein may mutually inhibit the activation of their respective transcriptional target genes was originally proposed by Bosnakovski et al.11 and was based on sequence similarity between the homeodomains of the two transcription factors. This model provides an alternative mechanism by which DUX4 may drive molecular changes in FSHD. However, besides the demonstration that Pax7 can mitigate DUX4 driven apoptosis in murine myoblasts11, 23, the role of PAX7 in human FSHD pathology has been little investigated. Recently, it has been demonstrated by several groups, including our own, that DUX4 and PAX7 mRNA are co-expressed in muscle progenitor cells from FSHD patients25 and during regeneration in an FSHD mouse model24, providing opportunity for the proteins to interact. A recent study investigating myogenic differentiation of ES and iPS cells derived from FSHD and control patients, reported that both PAX7 and DUX4 mRNA expression could be detected both during a myogenic precursor phase and in myotubes26. However, immunofluorescence suggested that PAX7 and DUX4 protein were not localised to the same cells. Although embryonic muscle is usually unaffected in FSHD9, it is interesting that the authors could not find cells containing both PAX7 and DUX4. The statistically significant low frequency of co-expression indicates that PAX7 and DUX4 protein may interact to cause mutual exclusivity26, resulting in repression of PAX7 transcriptional target genes in FSHD muscle, as we report.
Here, we show that when PAX7 and DUX4 are present in the same cell, the effect is the mutual inhibition of the respective transcriptional target genes of both proteins. Moreover, suppression of PAX7 transcriptional target genes when DUX4 and PAX7 are co-expressed is greater than would be expected under a competitive inhibition model. There are several possible mechanisms by which DUX4 and PAX7 proteins may interact to cause a suppression of PAX7 target genes in FSHD in addition to competitive inhibition. One is a direct protein–protein interaction, such as dimerization, which could disrupt target gene activation. Since PAX3 and PAX7 bind DNA as cooperative dimers14, 51 via motifs in the homeobox domain52, the homeobox domains of DUX4 may interfere with such dimerization to disrupt DNA-binding.
DUX4-mediated suppression of PAX7 transcriptional target genes, coupled with the report that PAX7 target genes can induce long-term epigenetic changes that persist in differentiated muscle27, may explain our finding that PAX7 target gene repression is a feature of FSHD skeletal muscle. Such repression may directly contribute to FSHD molecular pathology. Indeed, we show here that PAX7 target gene repression is associated with over-expression of EZH2 target genes, an important pathway in epigenetic modification in skeletal muscle. EZH2 is a well-known epigenetic transcriptional repressor and component of PRC2 complex, which plays critical roles in development and cancer53. It has been shown that EZH2 is enriched at the D4Z4 region in healthy control primary myocytes, but not in FSHD patient-derived cells, contributing to epigenetic de-repression of DUX4 in FSHD41. Our data suggests that DUX4 inhibition of PAX7, resulting in perturbation of EZH2 targets, may also contribute to epigenetic de-repression of DUX4 in FSHD54.
We further show that PAX7 target gene repression is associated with over-expression of HIF1α-mediated hypoxic response genes. Oxidative stress sensitivity is a well-known phenotype of FSHD myoblasts54, 55, however, the precise mechanism is unclear. Studies focusing on DUX4 target gene expression have implicated perturbation of the glutathione redox pathway11, while studies investigating FSHD patient-derived tissue in an unbiased manner have implicated HIF1α transcriptional dysregulation29, 50. Our results suggest that this latter mechanism could be attributed to DUX4-mediated inhibition of PAX7 target genes.
Mechanistically, HIF1α plays a complex role in cellular response to hypoxia and can be either pro- and anti-apoptotic, depending on oxygen levels, cell type and experimental set-up56. In normoxia, HIF1α undergoes proteasomal degradation, following oxygen-dependent ubiquitination by the Von Hippel Lindau tumour suppressor57. Hypoxia thus results in nuclear accumulation of HIF1α, where it dimerizes with HIF1β to form a transcription factor. HIF1α upregulation, following transient, mild levels of hypoxia is typically associated with cell survival and proliferation56. During sustained hypoxia, however, HIF1α induces apoptosis via a p53-dependent mechanism58. In FSHD myoblasts, DUX4 inhibition of PAX7, resulting in HIF1α upregulation, may mimic a sustained hypoxic response, resulting in increased p53-mediated apoptosis.
To conclude, our findings indicate that DUX4 likely operates in two ways to cause FSHD. Firstly, by driving the expression of a cohort of transcriptional target genes, many of which are pro-apoptotic, DUX4 can cause muscle pathology and cell death59. Secondly, by perturbing the ability of PAX7 to activate/maintain its transcriptional target genes, or by interfering with epigenetic changes elicited by PAX7, DUX4 represses PAX7 transcriptional target genes, likely causing pathology by mechanisms such as HIF1α over-activation and aberrant epigenetic changes. Thus, we propose that the model of FSHD molecular pathogenesis should be updated to incorporate PAX7 target gene repression as an important molecular mechanism and to refine current therapeutic strategies.
Methods
Published gene expression data
Publicly available gene expression data used in this study fell into two distinct classes: data sets describing over-expression of Pax7 and DUX4, and data sets describing FSHD and control muscle biopsies. These ten independent studies encompassed 222 samples: four studies10, 14, 33–35, totalling 35 samples described Pax7 and DUX4 over-expression. Six studies3, 10, 28, 30–32, totalling 187 samples described FSHD and matched control muscle biopsies. Study references, public database accession numbers, experimental design descriptions, gene expression assay used and sample counts for each study are provided in Supplementary Data 6. For the RNA-seq FSHD muscle biopsy study published by Yao et al.10, we removed control sample C6 from our analysis as it was derived from tibialis anterior, whereas all other samples in that study were isolated from quadriceps.
Publicly available microarray studies were obtained log normalised, from the GEO database. Quantile normalisation was subsequently performed across all samples within each study. To enable comparison across data sets and evaluation of the Pax7 and DUX4 scores, probes in each microarray data set and sequences in the RNA-seq data were matched to unique gene identifiers (EntrezGene for human and Ensemble for mouse). Probes or sequences mapping to the same gene identifier were averaged.
Microarray of Pax7 constructs
Procedures were carried out under the Animals (Scientific Procedures) Act 1986, as approved by King’s College London Ethical Review Process committee. Satellite cells were obtained from myofibres isolated from the extensor digitorum longus (EDL) muscles of three adult (6–8-week-old) male C57BL/10 mice. Mice were killed by cervical dislocation and the EDL muscles removed intact and incubated in 0.2% Collagenase Type 1 in Dulbecco's modified Eagle medium (DMEM)-GlutaMAX (Invitrogen) at 37 °C for 90 min. Myofibres were then dissociated in DMEM-GlutaMAX under a stereo dissecting microscope using a series of heat-polished glass Pasteur pipettes and rinsed in changes of DMEM-GlutaMAX60. Intact myofibres were plated on serum-reduced Matrigel (BD) coated plates in proliferation medium (DMEM-GlutaMAX with 30% fetal bovine serum (PAA), 10% horse serum (Gibco), 1% chick embryo extract (ICN Flow), and 1% penicillin/streptomycin (Sigma)) at 37 °C and 5% CO2 60. Expanded satellite cell-derived myoblasts were transduced with retroviral constructs encoding Pax7 and dominant negative Pax7-ERD 15 or control retroviruses using 4 μg/ml Polybrene for 48 h. PAX7 is highly conserved, with 97% amino-acid sequence similarity between mouse and human. RNA was isolated using RNeasy-mini columns (Qiagen), processed using WT Gene Expression (Ambion) and GeneChip v4 Whole Transcript Sense Target Labelling Kit as per the manufacturers’ instructions, and hybridised to Mouse Gene Chip 1.0 ST arrays using the GCS3000 microarray system (Affymetrix). Array data were normalised using robust multichip average (RMA), MvA transformation, and processed and statistically analysed using Partek Genomics Suite software (Partek Inc.). Principal component analysis on the 1000 most variable probes confirmed validity of biological replicates.
Differential expression analysis
Differential gene expression analysis was performed within two DUX4 over-expression studies17, 35 and our Pax7 construct over-expression study to derive target gene sets. For microarray studies an empirical Bayes approach was employed utilising the limma package in R61, while for RNA-seq data the DESeq2 package62 was used to assess fold change. For DUX4 studies, to replicate the approach used in previous analyses10, genes, which showed log FC > 2 and FDR < 0.05 were considered significantly upregulated by DUX4. DUX4 target genes from the Choi et al.34 RNA-seq data and Geng et al.35 microarray data are displayed in Supplementary Data 1 and 2. For our Pax7 microarray study a slightly different approach was employed to take advantage of the data from the dominant negative Pax7-ERD construct. Genes that showed both a log FC > 1 and p < 0.05 under Pax7 transduction and a log FC < 1 and p < 0.05 under Pax7-ERD transduction, were considered induced by Pax7. Genes that showed both a log FC < 1 and p < 0.05 under Pax7 transduction and a log FC > 1 and p < 0.05 under Pax7-ERD transduction, were considered repressed by Pax7: listed in Supplementary Data 3.
Derivation and validation of Pax7 and DUX4 target genes
Three DUX4 target gene scores were defined corresponding to DUX4 activated targets derived from the Yao et al.10, Choi et al.34 and Geng et al.35 DUX4 over-expression. For each study, a single sample DUX4 target gene score was defined as the average expression of the DUX4 activated targets in a given sample.
Our Pax7 target gene score was defined from our Pax7 construct microarray derived induced and repressed targets (described above). A single sample Pax7 score was then defined as the t-statistic comparing the expression of induced and repressed Pax7 targets within the sample, analogous to an approach we previously employed63, 64. We validated our Pax7 score on a published data set14, demonstrating that the score was significantly elevated in Pax7-expressing samples (p < 5 × 10−4, Supplementary Fig. 2).
Scores were z-normalised within each study, to reduce study-dependent effects, and evaluation of score differences between FSHD and control samples in a single study was performed via a Wilcoxon U-test.
Meta-analysis of target gene scores
Meta-analysis to assess the discriminatory powers of each of the target gene scores across the five microarray FSHD and control muscle biopsy data sets, was performed using a random effects model to combine statistics across studies. The p-values denoting the significance of the scores on meta-analysis were derived from a Fisher’s combined test.
ROC curve analysis was performed using the pROC package in R65.
GSEA of the target gene scores
GSEA was performed using a Fisher’s exact test to compare Pax7 target gene sets against the gene sets defined by the Molecular Signatures Database37, 38. Computations were performed via software downloaded from the Molecular Signatures database (http://www.broadinstitute.org/msigdb).
Human myoblast cell culture
Five immortalised human myoblast cell lines from a mosaic FSHD1 patient 54-6, 54-A10 (control, 13 D4Z4 repeats) and 54-12, 54-A5, 54-2 (FSHD, 3 D4Z4 repeats)45, were kind gifts from Dr. Vincent Mouly (Center for Research In Myology, UMRS 974 UPMC-INSERM, FRE 3617 CNRS, Paris, France) and Professor Silvère der Maarel (Leiden University Medic’al Center, Leiden, The Netherlands). Four immortalised human myoblast lines from two FSHD1 patients and two sibling matched controls: 12Ubic, 12Abic, 16Ubic and 16Abic, were obtained from Professor Charles Emerson (UMMS Wellstone centre for FSHD, Worcester, MA, USA)44. Immortalised human myoblasts were cultured in Skeletal Muscle Cell Growth Medium (Promocel) supplemented with 20% fetal bovine serum, 50 μg/ml Fetuin (bovine), 10 ng/ml epidermal growth factor (recombinant human), 1 ng/ml basic fibroblast growth factor (recombinant human), 10 μg/ml insulin (recombinant human), 0.4 μg/ml dexamethasone and 50 μg/ml gentamycin, at 37 °C under 5% CO2. RNA was harvested from proliferating myoblasts as they reached confluency for each line in triplicate using RNeasy kit (Qiagen) according to the manufacturer’s recommendation, including an additional DNA removal step using RNase-Free DNase Set (Qiagen).
RNA seq and alignment
RNA-seq libraries were prepared using the Agilent sureselect stranded RNAseq protocol, which allows polyA selection but was modified to work with ribodepletion. Prepared libraries were sequenced on an Illumina HiSeq2500. The raw reads were trimmed using trim-galore, utilising cutadapt66 (v0.4.0) to remove the Illumina Sequencing Adapter (AGATCGGAAGAGC) at the 3′-end. Additionally, 12 bases were also trimmed from the 5′-end of the reads since they showed a biased distribution of bases. The reads were mapped to the human transcriptome using the human genome sequence GRCh38 and v82 gene annotations downloaded from Ensembl. The mapping was performed using tophat67 (v2.1.0) and bowtie68 (v1.1.0), enabling the fr-firststrand option of tophat to restrict mapping to the sense strand of the transcript. Reads were assigned to genes using the featureCounts program69 (v1.5.0), counting fragments and ignoring multi-mapping reads. Gene assignment was also restricted so to the sense strand of the transcript. The resulting matrix of read counts was then loaded into R and normalised using DESeq262 before computation of Pax7 and DUX4 target gene scores.
Reporter gene assays
HEK-293 or NIH-3T3 cells (12,500 cells/well) were transfected with pMSCV-IRES-eGFP plasmids encoding DUX4 and/or Pax7, or dominant negative DUX4-ERD, or dominant negative Pax7-ERD 15, 29, together with reporter plasmids encoding either DUX4-responsive promoters driving luciferase expression (pZSCAN4-luc35 or RFPL4B-luc) or a Pax3/7-responsive element driving β-galactosidase (p34 plasmid15) using Lipofectamin LTX (Thermofisher). A pRSV vector encoding either β-galactosidase or luciferase as appropriate was co-transfected as an internal control for normalisation of transfection efficiency. Total amount of DNA transfected per reaction was 1.5 mg, in equal ratios for reporters and plasmids encoding genes of interest, and 250 ng/reaction of the internal normaliser vector. Cells were harvested 24 h after transfection, and assayed using the Dual-light Reporter system (Thermofisher) according to the manufacturer’s instructions. 3-4 independent transfections were performed in technical triplicate. Reporter gene activity was measured using Glomax-Multi + plate reader (Promega) and normalised for transfection efficiency. N = 3/4 for each cell line, analysis of variance (ANOVA) revealed significant intensity differences, post hoc unpaired two-tailed t-tests were employed to assess significant pairwise differences.
Reverse transcription quantitative PCR
HEK-293 (150 000 cells/well) were transfected with plasmids encoding DUX4 and/or Pax7, or dominant negative DUX4-ERD, or dominant negative Pax7-ERD 15, 29. RNA was isolated after 24 h and reverse-transcribed using the Reverse Transcription Kit with genomic DNA wipeout (Qiagen); RT-qPCR was performed on a Viia7 qPCR system (Life Technologies) with MESA Blue qPCR MasterMix Plus and ROX reference dye (Eurogentec). Primers used were as follows:
RPLPO (FWD: 5′-TCTACAACCCTGAAGTGCTTGAT-3′, REV: 5′-CAATCTGCAGACAGACACTGG-3′)
ZSCAN4 (FWD: 5′-TGGAAATCAAGTGGCAAAAA-3′, REV: 5′-CTGCATGTGGACGTGGAC-3′)
TRIM48 (FWD: 5′-TGAATGTGGAAACCACCAGA-3′, REV: 5′-GTTGAGCCTGTCCCTCAGTC-3′)
RFPL4B (FWD: 5′-GAGACGTAGGCTTCGGATCTT-3′, REV: 5′-GGCTGAATTCAAGTGGGTCT-3′)
MBD3L2 (FWD: 5′-GCGTTCACCTCTTTTCCAAG-3′, REV: 5′-GCCATGTGGATTTCTCGTTT-3′)
SELP (FWD: 5′-CGCCTGCCTCCAGACCATCTTC-3′, REV: 5′-CTATTCACATTCCAGAAACTCACCACAGC-3′)
Data availability
Microarray and RNA-seq data are available from the GEO data base (https://www.ncbi.nlm.nih.gov/geo/), accession numbers GSE77478 (microarray) and GSE102812 (RNA-seq).
Electronic supplementary material
Description of Additional Supplementary Files
Acknowledgements
We are grateful to colleagues who generously shared cells lines, including Dr. Vincent Mouly (Center for Research In Myology, UMRS 974 UPMC-INSERM, FRE 3617 CNRS, Paris, France), Professor Silvère der Maarel (Leiden University Medical Center, Leiden, The Netherlands) and Professor Charles Emerson (UMMS Wellstone centre for FSHD, Worcester, MA, USA). We heartily thank Dr. Stephen Tapscott (Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N. Seattle, WA 98109-1024) for providing the DUX4 reporter constructs. We are grateful to Efthymios Fidanis and Dr. Alka Saxena of the Genomics Research Platform, Biomedical Research Centre at Guy’s and St Thomas’ Trust and Kings College London for Library preparation and Sequencing: supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London. C.R.S.B. was supported by CoMPLEX Ph.D studentship and the British Heart Foundation (SP/08/004) and then the FSH Society (FSHS-82016-03). S.S. is supported by the Royal Society, the EPSRC and the National Natural Science Foundation of China. R.B.W. received funding from Muscular Dystrophy UK (RA3/762) and The Wellcome Trust (085137/Z/08/Z). M.P. is funded by Muscular Dystrophy UK (RA3/3052/1). H.H. was supported by the British Heart Foundation (PG/13/1930059). We are particularly thankful to the FSH Society Shack Family and Friends research grant (FSHS-82013-06), The King's Health Partners Research and Development Challenge Fund (R151006) and Association Francaise contre les Myopathies (17865). The Zammit laboratory is additionally supported by the Medical Research Council (MR/P023215/1), together with BIODESIGN (262948-2) from EU FP7.
Author contributions
The overall study was conceived by C.R.S.B. and P.S.Z. PAX7 microarray data was generated during a project conceived and directed by P.S.Z. and F.R. Statistical analysis was performed by C.R.S.B. Experiments were performed by C.R.S.B., M.P. and R.B.W. RNA-Seq data was pre-processed and mapped by C.R.S.B. and H.H. and analysed by C.R.S.B. The manuscript was mainly written by C.R.S.B. with input from P.S.Z. and contributions from M.P. and S.S. and proof-read by all authors.
Competing interests
The authors declare no competing financial interests.
Footnotes
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-017-01200-4.
Change history: A correction to this article has been published and is linked from the HTML version of this paper.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Christopher R. S. Banerji, Email: christopher.banerji15@imperial.ac.uk
Peter S. Zammit, Email: peter.zammit@kcl.ac.uk
References
- 1.Deenen JC, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056–1059. doi: 10.1212/WNL.0000000000000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orrell RW. Facioscapulohumeral dystrophy and scapuloperoneal syndromes. Handb. Clin. Neurol. 2011;101:167–180. doi: 10.1016/B978-0-08-045031-5.00013-X. [DOI] [PubMed] [Google Scholar]
- 3.Rahimov F, et al. Transcriptional profiling in facioscapulohumeral muscular dystrophy to identify candidate biomarkers. Proc. Natl Acad. Sci. USA. 2012;109:16234–16239. doi: 10.1073/pnas.1209508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fitzsimons RB, Gurwin EB, Bird AC. Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy. A general association with genetic and therapeutic implications. Brain. 1987;110:631–648. doi: 10.1093/brain/110.3.631. [DOI] [PubMed] [Google Scholar]
- 5.Lutz KL, Holte L, Kliethermes SA, Stephan C, Mathews KD. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology. 2013;81:1374–1377. doi: 10.1212/WNL.0b013e3182a84140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemmers RJ, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemmers RJ, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Boogaard ML, et al. Mutations in DNMT3B Modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am. J. Hum. Genet. 2016;98:1020–1029. doi: 10.1016/j.ajhg.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tawil R, van der Maarel SM, Tapscott SJ. Facioscapulohumeral dystrophy: the path to consensus on pathophysiology. Skelet. Muscle. 2014;4:12. doi: 10.1186/2044-5040-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao Z, et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 2014;23:5342–5352. doi: 10.1093/hmg/ddu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bosnakovski D, et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008;27:2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buckingham M. Skeletal muscle progenitor cells and the role of Pax genes. C. R. Biol. 2007;330:530–533. doi: 10.1016/j.crvi.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Relaix F, et al. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol. 2006;172:91–102. doi: 10.1083/jcb.200508044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soleimani VD, et al. Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev. Cell. 2012;22:1208–1220. doi: 10.1016/j.devcel.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zammit PS, et al. Pax7 and myogenic progression in skeletal muscle satellite cells. J. Cell Sci. 2006;119:1824–1832. doi: 10.1242/jcs.02908. [DOI] [PubMed] [Google Scholar]
- 16.Boutet SC, Biressi S, Iori K, Natu V, Rando TA. Taf1 regulates Pax3 protein by monoubiquitination in skeletal muscle progenitors. Mol. Cell. 2010;40:749–761. doi: 10.1016/j.molcel.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Montarras D, et al. Direct isolation of satellite cells for skeletal muscle regeneration. Science. 2005;309:2064–2067. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- 18.Calhabeu F, Hayashi S, Morgan JE, Relaix F, Zammit PS. Alveolar rhabdomyosarcoma-associated proteins PAX3/FOXO1A and PAX7/FOXO1A suppress the transcriptional activity of MyoD-target genes in muscle stem cells. Oncogene. 2013;32:651–662. doi: 10.1038/onc.2012.73. [DOI] [PubMed] [Google Scholar]
- 19.Relaix F, Rocancourt D, Mansouri A, Buckingham M. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435:948–953. doi: 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- 20.Reimann J, et al. Pax7 distribution in human skeletal muscle biopsies and myogenic tissue cultures. Cell Tissue Res. 2004;315:233–242. doi: 10.1007/s00441-003-0833-y. [DOI] [PubMed] [Google Scholar]
- 21.Marg A, et al. Human satellite cells have regenerative capacity and are genetically manipulable. J. Clin. Invest. 2014;124:4257–4265. doi: 10.1172/JCI63992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldwin CT, et al. Mutations in PAX3 associated with Waardenburg syndrome type I. Hum. Mutat. 1994;3:205–211. doi: 10.1002/humu.1380030306. [DOI] [PubMed] [Google Scholar]
- 23.Bosnakovski D, Daughters RS, Xu Z, Slack JM, Kyba M. Biphasic myopathic phenotype of mouse DUX, an ORF within conserved FSHD-related repeats. PLoS ONE. 2009;4:e7003. doi: 10.1371/journal.pone.0007003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knopp P, et al. DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis. J. Cell Sci. 2016;129:3816–3831. doi: 10.1242/jcs.180372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caron L, et al. A human pluripotent stem cell model of facioscapulohumeral muscular dystrophy-affected skeletal muscles. Stem Cells Transl. Med. 2016;5:1145–1161. doi: 10.5966/sctm.2015-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haynes P, Kernan K, Zhou SL, Miller DG. Expression patterns of FSHD-causing DUX4 and myogenic transcription factors PAX3 and PAX7 are spatially distinct in differentiating human stem cell cultures. Skelet. Muscle. 2017;7:13. doi: 10.1186/s13395-017-0130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carrio E, et al. Muscle cell identity requires Pax7-mediated lineage-specific DNA demethylation. BMC Biol. 2016;14:30. doi: 10.1186/s12915-016-0250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tasca G, et al. Different molecular signatures in magnetic resonance imaging-staged facioscapulohumeral muscular dystrophy muscles. PLoS ONE. 2012;7:e38779. doi: 10.1371/journal.pone.0038779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banerji CR, et al. beta-Catenin is central to DUX4-driven network rewiring in facioscapulohumeral muscular dystrophy. J. R. Soc. Interface. 2015;12:20140797. doi: 10.1098/rsif.2014.0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osborne RJ, Welle S, Venance SL, Thornton CA, Tawil R. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology. 2007;68:569–577. doi: 10.1212/01.wnl.0000251269.31442.d9. [DOI] [PubMed] [Google Scholar]
- 31.Bakay M, et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain. 2006;129:996–1013. doi: 10.1093/brain/awl023. [DOI] [PubMed] [Google Scholar]
- 32.Dixit M, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl Acad. Sci. USA. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young JM, et al. DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genet. 2013;9:e1003947. doi: 10.1371/journal.pgen.1003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi SH, et al. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016;44:5161–5173. doi: 10.1093/nar/gkw141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geng LN, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell. 2012;22:38–51. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de la Serna IL, et al. MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol. Cell. Biol. 2005;25:3997–4009. doi: 10.1128/MCB.25.10.3997-4009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liberzon A, et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manalo DJ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 40.Balog J, et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics. 2015;10:1133–1142. doi: 10.1080/15592294.2015.1113798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cabianca DS, et al. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell. 2012;149:819–831. doi: 10.1016/j.cell.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haslett JN, et al. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc. Natl Acad. Sci. USA. 2002;99:15000–15005. doi: 10.1073/pnas.192571199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pescatori M, et al. Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007;21:1210–1226. doi: 10.1096/fj.06-7285com. [DOI] [PubMed] [Google Scholar]
- 44.Homma S, et al. A unique library of myogenic cells from facioscapulohumeral muscular dystrophy subjects and unaffected relatives: family, disease and cell function. Eur. J. Hum. Genet. 2012;20:404–410. doi: 10.1038/ejhg.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krom YD, et al. Generation of isogenic D4Z4 contracted and noncontracted immortal muscle cell clones from a mosaic patient: a cellular model for FSHD. Am. J. Pathol. 2012;181:1387–1401. doi: 10.1016/j.ajpath.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferreboeuf M, et al. DUX4 and DUX4 downstream target genes are expressed in fetal FSHD muscles. Hum. Mol. Genet. 2014;23:171–181. doi: 10.1093/hmg/ddt409. [DOI] [PubMed] [Google Scholar]
- 47.Rickard AM, Petek LM, Miller DG. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 2015;24:5901–5914. doi: 10.1093/hmg/ddv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, et al. DNA-binding sequence specificity of DUX4. Skelet. Muscle. 2016;6:8. doi: 10.1186/s13395-016-0080-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jagannathan S, et al. Model systems of DUX4 expression recapitulate the transcriptional profile of FSHD cells. Hum. Mol. Genet. 2016;25:4419–4431. doi: 10.1093/hmg/ddw271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsumagari K, et al. Gene expression during normal and FSHD myogenesis. BMC Med. Genomics. 2011;4:67. doi: 10.1186/1755-8794-4-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schafer BW, Czerny T, Bernasconi M, Genini M, Busslinger M. Molecular cloning and characterization of a human PAX-7 cDNA expressed in normal and neoplastic myocytes. Nucleic Acids Res. 1994;22:4574–4582. doi: 10.1093/nar/22.22.4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Birrane G, Soni A, Ladias JA. Structural basis for DNA recognition by the human PAX3 homeodomain. Biochemistry. 2009;48:1148–1155. doi: 10.1021/bi802052y. [DOI] [PubMed] [Google Scholar]
- 53.Sellers WR, Loda M. The EZH2 polycomb transcriptional repressor—a marker or mover of metastatic prostate cancer? Cancer Cell. 2002;2:349–350. doi: 10.1016/S1535-6108(02)00187-3. [DOI] [PubMed] [Google Scholar]
- 54.Barro M, et al. Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. J. Cell. Mol. Med. 2010;14:275–289. doi: 10.1111/j.1582-4934.2008.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turki A, et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic. Biol. Med. 2012;53:1068–1079. doi: 10.1016/j.freeradbiomed.2012.06.041. [DOI] [PubMed] [Google Scholar]
- 56.Piret JP, Mottet D, Raes M, Michiels C. Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem. Pharmacol. 2002;64:889–892. doi: 10.1016/S0006-2952(02)01155-3. [DOI] [PubMed] [Google Scholar]
- 57.Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004;57:1009–1014. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmid T, Zhou J, Brune B. HIF-1 and p53: communication of transcription factors under hypoxia. J. Cell. Mol. Med. 2004;8:423–431. doi: 10.1111/j.1582-4934.2004.tb00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wallace LM, et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol. 2011;69:540–552. doi: 10.1002/ana.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moyle LA, Zammit PS. Isolation, culture and immunostaining of skeletal muscle fibres to study myogenic progression in satellite cells. Methods Mol. Biol. 2014;1210:63–78. doi: 10.1007/978-1-4939-1435-7_6. [DOI] [PubMed] [Google Scholar]
- 61.Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article 3, (2004). [DOI] [PubMed]
- 62.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banerji CR, Severini S, Caldas C, Teschendorff AE. Intra-tumour signalling entropy determines clinical outcome in breast and lung cancer. PLoS Comput. Biol. 2015;11:e1004115. doi: 10.1371/journal.pcbi.1004115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banerji CR, et al. Cellular network entropy as the energy potential in Waddington’s differentiation landscape. Sci. Rep. 2013;3:3039. doi: 10.1038/srep03039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robin X, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011;17:10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 67.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Microarray and RNA-seq data are available from the GEO data base (https://www.ncbi.nlm.nih.gov/geo/), accession numbers GSE77478 (microarray) and GSE102812 (RNA-seq).