

Summary
Nasu-Hakola disease (NHD) is a rare autosomal recessive leukoencephalopathy caused by a loss-of-function mutation of either TYROBP (DAP12) or TREM2 expressed in microglia. A rare variant of the TREM2 gene encoding p.Arg47His causes a 3-fold increase in the risk for late-onset Alzheimer's disease (LOAD). A recent study demonstrated that a rare coding variant p.Ser209Phe in the ABI family member 3 (ABI3) gene, a regulator of actin cytoskeleton organization, confers risk of developing of LOAD, although the pattern of ABI3 expression in AD and NHD brains with relevance to microglial pathology remains to be characterized. We investigated the cell type-specific expression of ABI3 in the brains derived from four non-neurological controls (NC), ten AD and five NHD cases by immunohistochemistry. We identified an intense ABI3 immunoreactivity chiefly on a subset of microglia with ramified or amoeboid morphology located in the grey matter and the white matter of the frontal cortex and the hippocampus of NC, AD, and NHD cases. The immunolabeled area of ABI3-positive microglia was not significantly different among NC, AD, and NHD cases due to great variability from case to case. The clusters of ABI3-immunoreactive microglia were found exclusively in AD brains and they were associated with amyloid plaques. Although these observations do not actively support the view that ABI3-immunoreactive microglia play a central role in the development of leukoencephalopathy in NHD brains and the neurodegeneration in AD brains, the intense expression of ABI3 on microglia might regulate their migration under conditions of health and disease in the central nervous system (CNS).
Keywords: ABI3, Alzheimer's disease, leukoencephalopathy, microglia, Nasu-Hakola disease
1. Introduction
Nasu-Hakola disease (NHD), also designated polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL; OMIM 221770), is a rare autosomal recessive disorder, characterized by progressive presenile dementia and formation of multifocal bone cysts, caused by genetic mutations of either TYROBP(DAP12) or TREM2 (1). Clinically, the patients with NHD show recurrent bone fractures during the third decade of life, and a frontal lobe syndrome during the fourth decade of life, and progressive dementia and death until the fifth decade of life (2). Pathologically, the brains of NHD patients exhibit extensive demyelination designated leukoencephalopathy, astrogliosis, accumulation of axonal spheroids, and remarkable activation of microglia predominantly in the white matter of frontal and temporal lobes and the basal ganglia (3). Although NHD patients are clustered in Japan and Finland, approximately 200 NHD cases are presently reported worldwide. TREM2 and DAP12 constitute a receptor/adaptor signaling complex expressed exclusively on osteoclasts, dendritic cells, macrophages, and microglia (4). It is generally supposed that a complete loss of function of the TREM2-DAP12 signaling pathway in microglia induces leukoencephalopathy in NHD. Thus, NHD is a primary disease of microglia termed “microgliopathy” (5), although the mechanisms how functionally aberrant microglia evoke leukoencephalopathy remain unknown.
Alzheimer's disease (AD) is characterized by the hallmark pathology comprised of widespread amyloid-β (Aβ) deposition, neurofibrillary tangle (NFT) formation, extensive neurodegeneration, and profound activation of microglia in the brain (6). The complex interaction between multiple genetic and environmental factors affecting various molecular pathways plays a key role in AD, although the precise molecular mechanism underlying AD remains largely unknown.
Microglia, resident myeloid cells in the central nervous system (CNS), play a pivotal role in maintenance of brain homeostasis, along with progression of neurodegenerative diseases (7). Microglia originate from erythromyeloid progenitors in the yolk sac and populate the CNS during early embryonic development (8). Microglia actively survey the surrounding microenvironment with dynamic processes. A battery of the genes, such as TREM2, BIN1, ABCA7, PICALM, CASS4, HLA-DRB1, MS4A6A, ZCWPW1, INPP5D and CD33, whose polymorphic variants modulate the risk of late-onset AD (LOAD), are expressed abundantly in microglia (9). The most notable example is a significant association between an approximately 3-fold increase in the risk for LOAD and a rare variant of the TREM2 gene encoding p.Arg47His located in the ligand binding domain of TREM2 (10). These observations suggest that a partial loss of function of the TREM2-DAP12 signaling pathway in microglia causes amyloid pathology, tau pathology, and neurodegeneration in AD.
Recently, by genotyping using a whole-exome microarray followed by de novo genotyping and analysis of imputed genotypes, a three-stage case-control study of 85,133 subjects identified rare coding missense variants associated with LOAD (11). They are composed of a protective variant of p.Pro522Arg in PLCG2, a risk variant of p.Ser209Phe in ABI3, and a risk variant of p.Arg62His in TREM2. Among them, the Phe209 allele of ABI3 showed consistent evidence for increasing the LOAD risk with a minor allele frequency (MAF) of 0.011 in cases and 0.008 in controls. However, the pattern of expression of ABI3 in the brains of AD, along with NHD with relevance to microglial pathology remains unknown. In the present study, for the first time, we attempt to clarify the cell type-specific expression of ABI3 in the brains of AD and NHD by immunohistochemistry.
2. Materials and Methods
2.1. Human brain tissues
The brain autopsies were performed at the National Center Hospital, National Center of Neurology and Psychiatry (NCNP), Japan, Kohnodai Hospital, National Center for Global Health and Medicine (NCGM), Japan, and affiliated hospitals of Research Resource Network (RRN), Japan. The comprehensive examination by established neuropathologists (YS and TI) validated the pathological diagnosis. In all cases, written informed consent was obtained. The Ethics Committee of the NCNP for the Human Brain Research, the Ethics Committee of the NCGM on the Research Use of Human Samples, and the Human Research Ethics Committee (HREC) of the Meiji Pharmaceutical University (MPU) approved the present study.
For immunohistochemical studies, serial sections of the frontal cortex and the hippocampus were prepared from four subjects who died of non-neurological causes (NC), composed of a 63-year-old man who died of prostate cancer and acute myocardial infarction (NC1), a 67-year-old man who died of dissecting aortic aneurysm (NC2), a 57-year-old man who died of alcoholic liver cirrhosis (NC3), and a 61-year-old man who died of rheumatoid arthritis with interstitial pneumonia (NC4), ten AD patients, composed of a 68-year-old woman (AD1), a 70-year-old woman (AD2), a 68-year-old woman (AD3), a 56-year-old man (AD4), a 59-year-old man (AD5), an 81-year-old man (AD6), a 68-year-old woman (AD7), an 80-year-old man (AD8), a 72 year-old man (AD9), and a 77-year-old woman (AD11), and five NHD patients, composed of a 42-year-old man (NHD1), a 48-year-old woman (NHD2), a 44-year-old man (NHD3), a 32-year-old woman (NHD4), and a 38-year-old man (NHD5). The homozygous mutation of a single base deletion of 141G (c.141delG) in exon 3 of DAP12 was identified in NHD1, NHD2, and NHD5, while the genetic analysis was not performed in NHD3 or NHD4. All AD cases were satisfied with the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria for diagnosis of definite AD (12). They were categorized into the stage C of amyloid deposition and the stage VI of neurofibrillary degeneration, following the Braak's staging (13).
2.2. Immunohistochemistry
After deparaffination, tissue sections were heated in 10 mM citrate sodium buffer, pH 6.0 by autoclave at 110°C for 15 min in a temperature-controlled pressure chamber (Biocare Medical, Pacheco, CA, USA). They were treated at room temperature (RT) for 15 min with 3% hydrogen peroxide-containing methanol to block the endogenous peroxidase activity. They were then incubated with phosphate-buffered saline (PBS) containing 10% normal goat serum at RT for 15 min to block non-specific staining, followed by incubation in a moist chamber at 4°C overnight with rabbit polyclonal anti-ABI3 antibody (HPA017345; Atlas Antibodies, Bromma, Sweden) or rabbit polyclonal anti-Iba1 antibody (Wako Pure Chemical, Tokyo, Japan) for a marker specific for microglia. The specificity of anti-ABI3 antibody was validated by western blot analysis of recombinant human ABI3 protein expressed in HEK293 cells, which were transfected with the pcDNA4/HisMax TOPO vector (Thermo Fisher Scientific, Carlsbad, CA, USA) containing the full-length ABI3 sequence. After washing with PBS, tissue sections were incubated at RT for 30 min with horseradish peroxidase (HRP)-conjugated secondary antibody (Nichirei, Tokyo, Japan), followed by incubation with diaminobenzidine tetrahydrochloride (DAB) substrate (Vector, Burlingame, CA, USA), and DAB enhancing solution (Vector) for detection of ABI3 immunolabeling. They were processed for a counterstain with hematoxylin. Negative controls underwent all the steps except for exposure to primary antibody. In limited experiments, double immunolabeling was performed using rabbit anti-ABI3 antibody and mouse monoclonal antibodies against glial fibrillary acidic protein (GFAP) (GA5; Nichirei, Tokyo, Japan), neuronal nuclei antigen (NeuN) (1B7; Abcam, Cambridge, UK), 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) (11-5B; Sigma, St. Louis, MO, USA), gp91phox (Abcam), or amyloid-β peptide (12B2; Immunobiological Laboratories, Gunma, Japan), followed by incubation with HRP-conjugated or alkaline phosphatase-conjugated anti-rabbit or anti-mouse secondary antibody and exposure to DAB substrate and Warp Red chromogen (Biocare Medical).
2.3. Quantification of ABI3 immunoreactivity
To quantify immunolabeled areas, the images derived from three fields of the white matter or the grey matter per each section were captured at a 200× magnification on the Olympus BX51 universal microscope. They were then processed for quantification by using ImageJ software (National Institute of Health, Bethesda, MD, USA). The ABI3-immunolabeled area was calibrated by the Iba1-immunolabeled area. The differences in the ABI3/Iba1 ratio among NC, AD, and NHD subjects were evaluated statistically by one-way analysis of variance (ANOVA) followed by post-hoc Tukey's test.
2.4. Quantitative RT-PCR analysis
To investigate the effects of inflammatory mediators on ABI3 expression, v-myc-immortalized human microglial cells named HMO6 (14), incubated in 10% fetal bovine serum (FBS)-containing Dulbecco's Modified Eagle's Medium (DMEM), were exposed for 24 hours to 1 µg/mL lipopolysaccharide (LPS; Sigma), recombinant human IFNγ, IL-4, IL-13 or TGFβ1 (50 ng/mL each; Peprotech, London, UK), followed by extraction of total cellular RNA. For quantitative RT-PCR (qPCR) analysis, cDNA was amplified by PCR on LightCycler 96 (Roche Diagnostics, Tokyo, Japan) with SYBR Green I and a primer set composed of 5′catgcatatggagaaggtggcc3′ and 5′tgcctgttcttgacagctgcgt3′ for detection of a 200 bp product of the ABI3 gene (NM_016428.2). The expression levels of ABI3 were standardized against the levels of glyceraldehyde-3-phosphate dehydrogenase (G3PDH) detected in the corresponding cDNA samples. All the assays were performed in triplicate.
3. Results
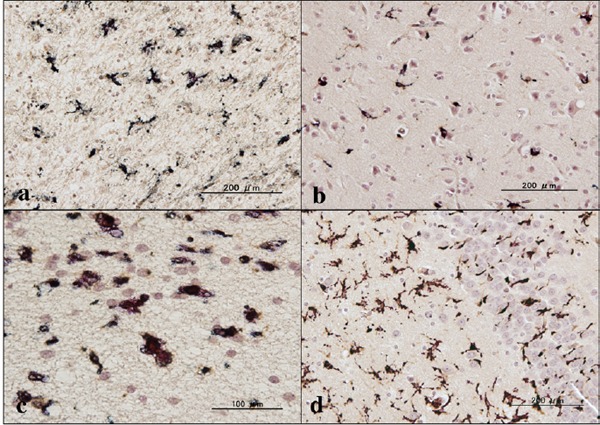
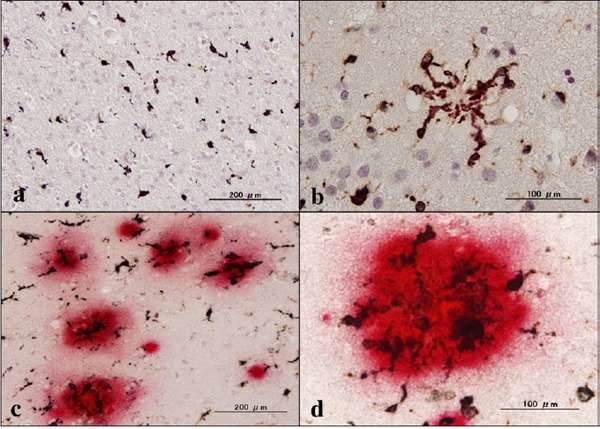
First of all, we validated the specificity of anti-ABI3 antibody HPA017345 by western blot analysis of Xpress-tagged recombinant human ABI3 protein expressed in HEK293 cells (Figure 1, panels a, b). Then, by immunohistochemistry using HPA017345, we identified an intense expression of ABI3 immunoreactivity chiefly on a subset of microglia with ramified or amoeboid morphology located in the grey matter and the white matter of the frontal cortex and the hippocampus derived from NC, AD, and NHD subjects (Figures 2–4, panels a–d). ABI3 immunoreactivity was found to be located in the cytoplasm. We occasionally found inconsistent staining of capillary walls, and perivascular and intravascular macrophages/monocytes. In AD brains, some of ABI3-immunoreactive microglia formed the clusters and they were often closely associated with amyloid deposition (Figure 3, panels c, d). In contrast, the clusters of ABI3-expressing microglia were almost undetectable in both NC and NHD brains. By double immunolabeling, the expression pattern of ABI3 overlapped with that of gp91phox, a marker of microglia (15) (Figure 5, panel a), whereas GFAP-positive astrocytes, CNPase-positive oligodendrocytes, and NeuN-positive neurons did not express ABI3 (Figure 5, panels b–d), suggesting that astrocytes, oligodendrocytes and neurons do not express discernible levels of ABI3 in the human brain. By quantitative analysis, the immunolabeled area of ABI3-positive microglia was not significantly different among NC, AD, and NHD cases in the grey matter (p = 0.8689) and the white matter (p = 0.8237) due to great variability in the area of ABI3-immunolabeled microglia from case to case (Figure 6). These observations do not actively support the view that ABI3-immunoreactive microglia play a central role in the development of leukoencephalopathy in NHD brains and the neurodegeneration in AD brains. In HMO6 cells, a culture model of human microglia, the treatment with TGFβ1 but not with LPS, IFNγ, IL-4 or IL-13 significantly elevated ABI3 mRNA expression levels (p = 0.0001) (Figure 7).
Figure 1.
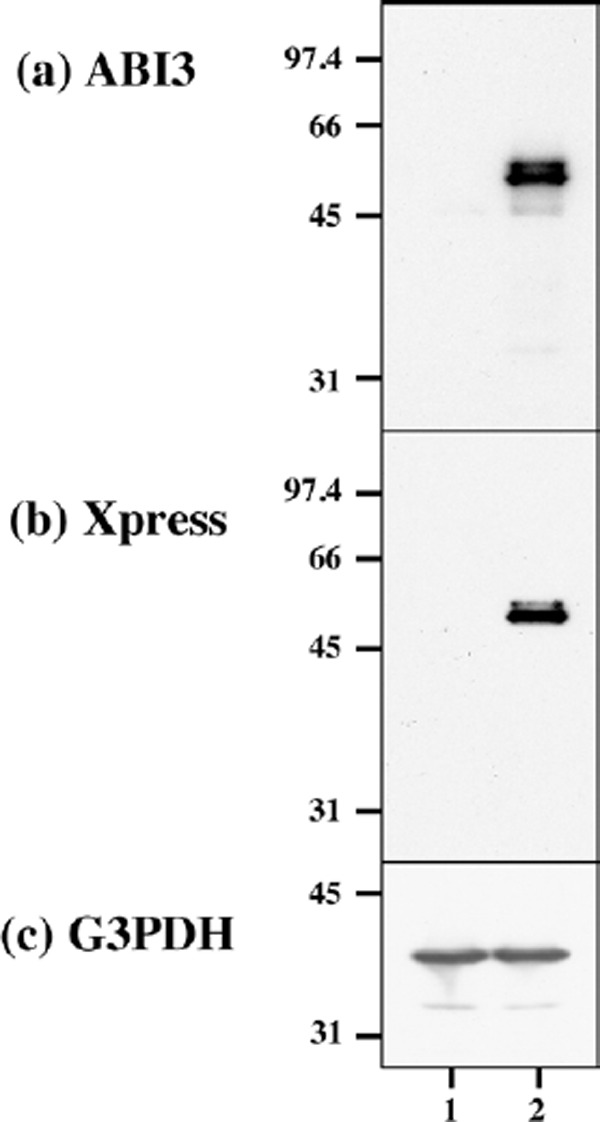
The specificity of ABI3 antibody. Western blot of non-transfected HEK293 cells (lane 1) and the cells transfected with the vector containing the full-length ABI3 sequence (lane 2). (a) ABI3, (b) Xpress tag, and (c) G3PDH as a loading control.
Figure 2.
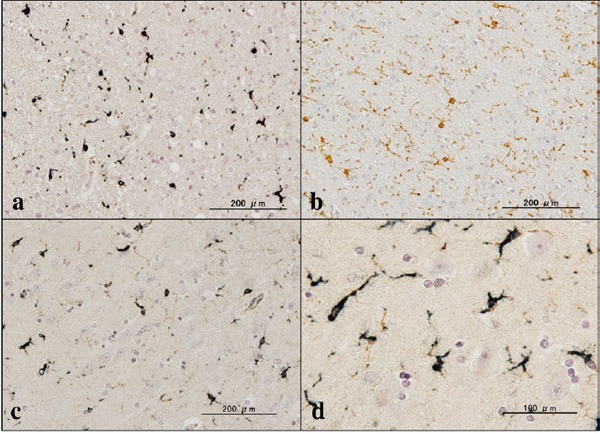
Expression of ABI3 in NC brains. (a) the frontal lobe white matter, ABI3, (b) the same area of (a), Iba1, (c) the CA1 region of the hippocampus, ABI3, and (d) the same area of (c), ABI3.
Figure 4.
Expression of ABI3 in NHD brains. (a) the frontal lobe white matter, ABI3, (b) the frontal cortex, ABI3, (c) the frontal lobe white matter, ABI3, and (d) the granular cell layer of dentate gyrus of the hippocampus, ABI3.
Figure 3.
Expression of ABI3 in AD brains. (a) the frontal lobe white matter, ABI3, (b) the granular cell layer of the dentate gyrus of the hippocampus, ABI3, (c) the frontal cortex, ABI3 (dark brown), amyloid-β (red), and (d) the same area of (c), ABI3 (dark brown), amyloid-β (red).
Figure 5.
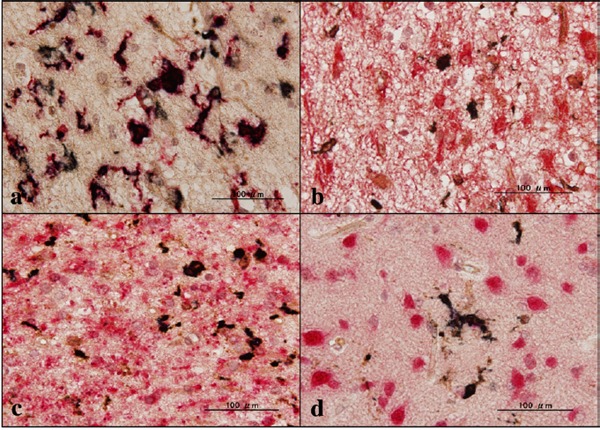
Expression of ABI3 in AD brains. (a) the periventricular white matter of the the hippocampus, ABI3 (dark brown), gp91phox (red), (b) the periventricular white matter of the hippocampus, ABI3 (dark brown), GFAP (red), (c) the temporal lobe white matter, ABI3 (dark brown), CNPase (red), and (d) the frontal cortex, ABI3 (dark brown), NeuN (red)
Figure 6.
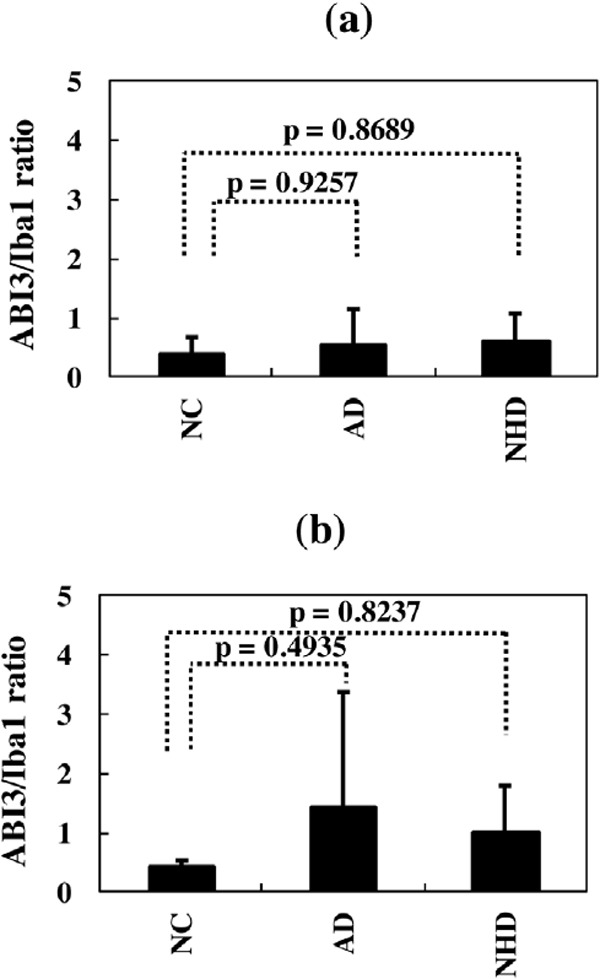
Quantitative analysis of ABI3 expression on microglia in NC, AD, and NHD brains. (a) The ABI3-immunolabeled area/Iba1-immunolabelled area ratio, the grey matter of the frontal cortex, and (b) The ABI3-immunolabeled area/Iba1-immunolabeled area ratio, the white matter of the frontal cortex.
Figure 7.
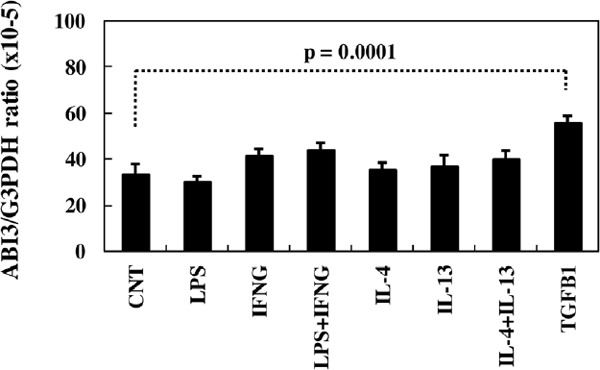
Quantitative RT-PCR analysis of ABI3 expression in HMO6 microglia in culture. HMO6 cells were exposed for 24 hours to 1 µg/ml lipopolysaccharide (LPS), recombinant human IFNγ, IL-4, IL-13 or TGFβ1 (50 ng/mL each), followed by extraction of total cellular RNA that is processed for qRT-PCR. The expression levels of ABI3 were standardized against the levels of G3PDH.
4. Discussion
A recent study identified p.Ser209Phe in the ABI3 gene as a risk variant for LOAD, although the expression of ABI3 in the human brain tissues remains to be determined (11). NHD is regarded as microgliopathy due to the complete loss of function of the TREM2-DAP12 signaling pathway in microglia, while AD is characterized by the partial loss of function of this pathway. Therefore, we attempted to investigate the pattern of ABI3 expression with relevance to microglial pathology in both AD and NHD brains. As a result, we identified an intense ABI3 immunoreactivity almost exclusively on a subset of microglia in NC, AD, and NHD brains.
ABI3 belongs to the Abelson-interactor (ABI) family of proteins, including ABI1, ABI2 and ABI3, which serve as a class of cytoplasmic molecular adaptors. Both ABI1 and ABI2 were originally identified as a binding partner for the c-ABL kinase, a non-receptor tyrosine kinase, whose activation induces cell growth, transformation, and cytoskeletal organization. ABI3 was initially cloned as a new molecule including Src-homology 3 (SH3) domain (NESH) (16). Members of ABI family encode a protein containing an amino-terminal homeodomain homologous region (HHR), a proline-rich (PR) region, and a carboxy-terminal SH3 domain. They constitute the WAVE regulatory complex (WRC) that regulates actin cytoskeleton organization (17,18). WRC is a macromolecular complex, composed of five-subunit proteins that include ABI (ABI1, ABI2 or ABI3), WAVE (WAVE1, WAVE2 or WAVE3), Nap1, PIR121/Sra1, and HSPC300 (19). WRC regulates Arp2/3-mediated actin filament nucleation and actin network assembly in response to the Rac GTPase. WRC containing ABI3 is functionally distinct from that containing ABI1 (17,18). Both ABI1 and ABI2 but not ABI3 promote the c-ABL-mediated phosphorylation of WAVE2, since ABI3 does not directly interact with c-ABL (17,18). WAVE2 phosphorylation by c-ABL activates WRC, leading to formation of lamellipodial membrane protrusions. A linker region between the PR region and the SH3 domain of ABI1 is crucial for its interaction with c-ABL (18).
At present, the precise biological function of ABI3 in microglia remains unknown. ABI3 expression is often lost in invasive cancer cells (20). This is attributable to transcriptional silencing by methylation of specific CpG sites located within the ABI3 promoter (21). Forced expression of ABI3 in v-src-transformed NIH3T3 (SRD) cells, thyroid cancer cells, or colon cancer cells inhibits cell growth, invasiveness and migration, and enhances cellular senescence in vitro and reduces the tumor growth and metastasis in vivo, indicating that ABI3 acts as a tumor suppressor gene (20,22). In contrast, treatment of SRD cells overexpressing ABI3 with a tyrosine kinase inhibitor imatinib mesylate markedly promotes formation of invadopodia, actin-rich ventral membrane protrusions with extracellular matrix-degrading activity, and enhances the metastatic potential (18,23). Microglia, along with macrophages, are capable of producing podosomes, a matrix-degrading ventral cell surface structure similar to invadopodia (24). These observations suggest that ABI3 expressed on microglia in NC, AD, and NHD brains might positively regulate their motility and migration capacity via formation of podosomes, particularly when the activity of c-ABL tyrosine kinase is suppressed. In contrast, ABI3 mRNA expression is upregulated in TREM2-deficient microglia that exhibit a reduction in chemotaxis and migration (25).
Forced expression of ABI3 suppresses proliferation of thyroid and colon cancer cells (22). ABI3 expression in these cells increases the percentage of cells located in G0/G1 phase by upregulating the cell cycle inhibitor p21WAF1 and downregulating ERK phosphorylation and E2F1 expression. Therefore, the possibility exists that an intense expression of ABI3 on microglia in NC, AD, and NHD brains might negatively regulate cell proliferation. The SH3 domain of ABI3 interacts with p21-activated kinase (PAK), whose activation positively regulates cell motility. The SRD cells overexpressing ABI3 show a significant decrease in PAK2 phosphorylation at Thr402 (20). Forced expression of ABI3 in thyroid cancer cells substantially reduced the phosphorylation of AKT at both Thr308 and Ser473 and GSK3β at Ser9 (26). ABI3 is phosphorylated at Ser342 by signaling through the PI3K/AKT pathway. Non-phosphorylated form of ABI3 is preferentially incorporated in WRC, while the phosphorylated form represents an inactive protein not included in the complex. ABI3 serves as a downstream mediator of the PI3K/AKT pathway that disrupts WRC via phosphorylation of ABI3 (26).
Microglia adopt two distinct activation phenotypes, composed of a proinflammatory and neurotoxic "classical" activation (M1) phenotype by exposure to LPS and IFNγ and an anti-inflammatory and neuroprotective “alternative” activation (M2) phenotype following treatment with IL-4 and IL-13 (27). We found upregulation of ABI3 mRNA expression in HMO6 microglial cells by exposure to TGFβ1 but not by LPS, IFNγ, IL4 or IL-13. TGFβ1 plays a pivotal role in development and maintenance of microglia both in vitro and in vivo (28). Microglia is lost in the CNS-specific TGFβ1 deficient mice that exhibit progressive motor paralysis and death by 23–25 weeks of age, indicating that TGFβ1 acts as a major differentiation and survival factor for microglia (28). TGFβ1-treated microglia show a quiescent phenotype that resembles M0 cells (29). Importantly, TGFβ1 promotes microglial clearance of amyloid-β (30). We identified an intense ABI3 immunoreactivity on the microglia associated with amyloid plaques in AD, possibly related to their migration onto amyloid plaques.
In conclusions, we found that ABI3 is expressed almost exclusively on a subset of microglia in NC, AD, and NHD brains. The pattern of ABI3 expression was similar among the cases, although the clusters of ABI3-immunoreactive microglia were found predominantly in AD brains, associated with amyloid plaques. The biological implications of an intense expression of ABI3 on microglia in AD and NHD brains remain to be clarified.
Acknowledgements
The authors thank Drs. Kenji Jinnai, Nobutaka Arai, Kiyotaka Nakamagoe, Nobutaka Motohashi, and Saburo Yagishita for providing us brain samples. This work was supported by grants from the Research on Intractable Diseases, entitled “Clinicopathological and genetic studies of Nasu-Hakola disease” (H21-Nanchi-Ippan-201; H22-Nanchi-Ippan-136), the Ministry of Health, Labour and Welfare of Japan, and grants from the JSPS KAKENHI (C25430054 and 16K07043) and the Dementia Drug Development Research Center (DRC) project (S1511016), the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
References
- 1. Klünemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE, Peltonen L, Paloneva J. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. 2005; 64:1502-1507. [DOI] [PubMed] [Google Scholar]
- 2. Bianchin MM, Capella HM, Chaves DL, Steindel M, Grisard EC, Ganev GG, da Silva Júnior JP, Neto Evaldo S, Poffo MA, Walz R, Carlotti Júnior CG, Sakamoto AC. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy - PLOSL): A dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol. 2004; 24:1-24. [DOI] [PubMed] [Google Scholar]
- 3. Satoh J, Tabunoki H, Ishida T, Yagishita S, Jinnai K, Futamura N, Kobayashi M, Toyoshima I, Yoshioka T, Enomoto K, Arai N, Arima K. Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology. 2011; 31:363-375. [DOI] [PubMed] [Google Scholar]
- 4. Xing J, Titus AR, Humphrey MB. The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: A molecular genetics perspective. Res Rep Biochem. 2015; 5:89-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Satoh J. Possible role of microgliopathy in the pathogenesis of Nasu-Hakola disease. Clin Exp Neuroimmunol. 2013; 4 Suppl 1:17-26. [Google Scholar]
- 6. Sarlus H, Heneka MT. Microglia in Alzheimer's disease. J Clin Invest. 2017; 127:3240-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017; 35:441-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ginhoux F, Prinz M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb Perspect Biol. 2015; 7:a020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yeh FL, Hansen DV, Sheng M. TREM2, microglia, and neurodegenerative diseases. Trends Mol Med. 2017; 23:512-533. [DOI] [PubMed] [Google Scholar]
- 10. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013; 368:107-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sims R, van der Lee SJ, Naj AC, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer's disease. Nat Genet. 2017; 49:1373-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991; 41:479-486. [DOI] [PubMed] [Google Scholar]
- 13. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006; 112:389-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagai A, Nakagawa E, Hatori K, Choi HB, McLarnon JG, Lee MA, Kim SU. Generation and characterization of immortalized human microglial cell lines: Expression of cytokines and chemokines. Neurobiol Dis. 2001; 8:1057-1068. [DOI] [PubMed] [Google Scholar]
- 15. Satoh JI, Kino Y, Yanaizu M, Tosaki Y, Sakai K, Ishida T, Saito Y. Expression of gp91phox and p22phox, catalytic subunits of NADPH oxidase, on microglia in Nasu-Hakola disease brains. Intractable Rare Dis Res. 2016; 5:275-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miyazaki K, Matsuda S, Ichigotani Y, Takenouchi Y, Hayashi K, Fukuda Y, Nimura Y, Hamaguchi M. Isolation and characterization of a novel human gene (NESH) which encodes a putative signaling molecule similar to e3B1 protein. Biochim Biophys Acta. 2000; 1493:237-241. [DOI] [PubMed] [Google Scholar]
- 17. Hirao N, Sato S, Gotoh T, Maruoka M, Suzuki J, Matsuda S, Shishido T, Tani K. NESH (Abi-3) is present in the Abi/WAVE complex but does not promote c-Abl-mediated phosphorylation. FEBS Lett. 2006; 580:6464-6470. [DOI] [PubMed] [Google Scholar]
- 18. Sekino S, Kashiwagi Y, Kanazawa H, Takada K, Baba T, Sato S, Inoue H, Kojima M, Tani K. The NESH/Abi-3-based WAVE2 complex is functionally distinct from the Abi-1-based WAVE2 complex. Cell Commun Signal. 2015; 13:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen B, Brinkmann K, Chen Z, Pak CW, Liao Y, Shi S, Henry L, Grishin NV, Bogdan S, Rosen MK. The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell. 2014; 156:195-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ichigotani Y, Yokozaki S, Fukuda Y, Hamaguchi M, Matsuda S. Forced expression of NESH suppresses motility and metastatic dissemination of malignant cells. Cancer Res. 2002; 62:2215-2219. [PubMed] [Google Scholar]
- 21. Moraes L, Galrão AL, Rubií I, Cerutti JM. Transcriptional regulation of the potential tumor suppressor ABI3 gene in thyroid carcinomas: Interplay between methylation and NKX2-1 availability. Oncotarget. 2016; 7:25960-25970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Latini FR, Hemerly JP, Freitas BC, Oler G, Riggins GJ, Cerutti JM. ABI3 ectopic expression reduces in vitro and in vivo cell growth properties while inducing senescence. BMC Cancer. 2011; 11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsuda S, Ichigotani Y, Okumura N, Yoshida H, Kajiya Y, Kitagishi Y, Shirafuji N. NESH protein expression switches to the adverse effect of imatinib mesylate. Mol Oncol. 2008; 2:16-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vincent C, Siddiqui TA, Schlichter LC. Podosomes in migrating microglia: Components and matrix degradation. J Neuroinflammation. 2012; 9:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mazaheri F, Snaidero N, Kleinberger G, et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017; 18:1186-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moraes L, Zanchin NIT, Cerutti JM. ABI3, a component of the WAVE2 complex, is potentially regulated by PI3K/AKT pathway. Oncotarget. 2017; 8:67769-67781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016; 53:1181-1194. [DOI] [PubMed] [Google Scholar]
- 28. Butovsky O, Jedrychowski MP, Moore CS, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014; 17:131-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abutbul S, Shapiro J, Szaingurten-Solodkin I, Levy N, Carmy Y, Baron R, Jung S, Monsonego A. TGF-β signaling through SMAD2/3 induces the quiescent microglial phenotype within the CNS environment. Glia. 2012; 60:1160-1171. [DOI] [PubMed] [Google Scholar]
- 30. Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat Med. 2001; 7:612-618. [DOI] [PubMed] [Google Scholar]