

Abstract
Objective:
To expand the clinical phenotype associated with STXBP1 gene mutations and to understand the effect of STXBP1 mutations in the pathogenesis of focal cortical dysplasia (FCD).
Methods:
Patients with STXBP1 mutations were identified in various ways: as part of a retrospective cohort study of epileptic encephalopathy; through clinical referrals of individuals (10,619) with developmental delay (DD) for chromosomal microarray; and from a collection of 5,205 individuals with autism spectrum disorder (ASD) examined by whole-genome sequencing.
Results:
Seven patients with heterozygous de novo mutations affecting the coding region of STXBP1 were newly identified. Three cases had radiologic evidence suggestive of FCD. One male patient with early infantile epileptic encephalopathy, DD, and ASD achieved complete seizure remission following resection of dysplastic brain tissue. Examination of excised brain tissue identified mosaicism for STXBP1, providing evidence for a somatic mechanism. Cell-type expression analysis suggested neuron-specific expression. A comprehensive analysis of the published data revealed that 3.1% of severe epilepsy cases carry a pathogenic de novo mutation within STXBP1. By contrast, ASD was rarely associated with mutations in this gene in our large cohorts.
Conclusions:
STXBP1 mutations are an important cause of epilepsy and are also rarely associated with ASD. In a case with histologically proven FCD, an STXBP1 somatic mutation was identified, suggesting a role in its etiology. Removing such tissue may be curative for STXBP1-related epilepsy.
Patients with STXBP1 gene mutations live with 3 key phenotypic abnormalities: intellectual disability (ID), epilepsy, and movement disorders.1–4 Epilepsy is often drug resistant and most commonly presents as severe early infantile epileptic encephalopathy (EIEE; Ohtahara syndrome) or West syndrome.4–6 However, STXBP1 mutations have been identified in patients with ID without epilepsy or with autism spectrum disorder (ASD).2,4,7 This broad STXBP1 encephalopathy phenotype is perhaps unsurprising, as STXBP1 is involved in the synaptic release of neurotransmitters, with heterozygous mutations resulting in a reduction of both STXBP1's protein product, syntaxin-binding protein 1, and syntaxin-1.8 Moreover, homozygous deletions in knockout models are characterized by widespread brain apoptosis.9 Although there is evidence for a role in neurite growth,10 the clinical pathophysiology remains unclear and sometimes spontaneously remitting epilepsy suggests developmental mechanisms beyond neurotransmission.
Radiologic evidence of gross brain pathology with heterozygous STXBP1 mutations is variable.4,11 When present, it suggests cerebral atrophy, abnormal myelination, or hypogenesis of the corpus callosum.12 STXBP1 germline mutation was reported only once in focal cortical dysplasia (FCD) but never a somatic mutation.13 Molecular genetic studies of FCD to date has focused on MTOR pathway mutations.14 We present a case series with STXBP1 encephalopathy including a case with a radiologic diagnosis of FCD that went on to have successful epilepsy surgery.
Our aim was twofold: first, to investigate the histopathology and molecular biology of the excised brain tissue from an STXBP1-positive FCD patient; and second, to characterize the phenotypic spectrum of STXBP1 through our case series and meta-analysis.
METHODS
Patients.
Patient 1 was part of a retrospective cohort study of patients with epileptic encephalopathy recruited using a standard protocol and with patient consent at The Hospital for Sick Children in Toronto between January 2012 and June 2014.15 All patients underwent a series of investigations as part of their clinical workup, including chromosomal microarray and targeted next-generation sequencing using epilepsy panels for between 35 and 70 known epilepsy genes as previously described.15 Additional patients with STXBP1 deletions were identified through Toronto's Hospital for Sick Children, having been referred for clinical microarray and/or exome sequencing from a variety of clinical services in connection with concerns regarding epilepsy and/or developmental delay (DD) (10,619 microarrays).16 In addition, an ASD cohort, forming part of the MSSNG project (mss.ng), was searched for STXBP1 mutations. This project aims to sequence a minimum of 10,000 families and obtain detailed phenotypes. These families have been recruited from across Canada and/or are in the Autism Genetic Resource Exchange.17,18 Molecular and computational methods have been previously described,19,20 and lollipops software21 was used to map mutations.
For meta-analysis, we have identified 162 cases with STXBP1 mutations (single nucleotide variants [SNVs] or copy number variants [CNVs]) from numerous independent scientific reports (tables e-1 at http://links.lww.com/NXG/A14, and e-2 at http://links.lww.com/NXG/A15). For meta-analysis, we have used PubMed to review all of the described STXBP1 scientific reports in the literature that were published up to September 2016. A total of 162 patients with heterozygous mutations of STXBP1 have been described so far across 35 studies.
Standard protocol approvals, registrations, and patient consents.
All patients described in this report have provided a written consent from the individuals involved or their substitute decision makers, and the study was approved by The Hospital for Sick Children Research Ethics Board, Toronto, Canada.
DNA extraction from resected brain tissue.
A portion of stock brain tissue (from patient 1) kept on dry ice was removed, added directly to cell lysis solution, and then homogenized using a sterile probe and an Omni tissue homogenizer. The homogenized tissue was digested overnight with Proteinase K at 55°C. This was repeated for the remaining visible tissue fragments for a further 5 hours at 55°C to ensure complete digestion. The tissue digest was subsequently spun down, and the supernatant was removed for extraction. High-salt Gentra protein precipitation solution was next added, and the sample was vortexed for 20 seconds and then put on ice for 5 minutes for complete precipitation. The sample was spun at 2,000g for 10 minutes, and all the supernatant was removed down to the protein pellet, leaving a thin layer above the pellet to prevent protein contamination. The supernatant was then transferred into isopropanol and inverted gently to mix 50 times to precipitate the DNA, which is then washed with 70% ethanol. This was then centrifuged and the ethanol was removed. Tris-EDTA buffer was next added to the dried DNA pellet for rehydration. After heating the DNA to 65°C for 1 hour, it was maintained on a rotator at room temperature overnight. DNA was then heated at 55°C for 1 hour and subsequently quantitated with a fluorometer using ds dye Hoescht.
Immunohistochemistry.
For immunohistochemical studies on the resected brain tissue slice, hematoxylin and eosin staining was applied following a standard protocol, Olig2 staining against oligodendroglia-like cells and antineuronal nuclear antigen NeuN (Cat# MAB377, Millipore, Temeuza, CA) against neurons with a dilution ratio of 1:50 were used following established protocols.22
Droplet digital PCR.
We applied droplet digital PCR (ddPCR) technology to validate CNVs and to quantify the exact copy number. Copy number estimation of STXBP1 was performed using the QX200 ddPCR system (Bio-Rad Laboratories, Inc., Hercules, CA) using the TaqMan copy number probe Hs00269332_cn (Life Technologies, Carlsbad, CA). Prior to the copy number experiment, 250 ng of genomic DNA was digested with 4U of BtgI in a 10-μL reaction buffer (New England Biolabs, Ipswich, MA), 1 hour × 37°C incubation, and no enzyme denaturation. The 20-μL copy number reaction mix consisted of 10 μL of 2× ddPCR supermix for probes (Bio-Rad Laboratories), 1 μL of the copy number target assay (labeled with FAM), 1μL of the copy number reference assay (RNAseP, Life Technologies, part 4403326, labeled with VIC), 6 μL of water, and 2 μL of 25 ng/μL digested genomic DNA. The copy number assay was validated by temperature gradient to ensure optimal separation of target and RNAseP-containing droplets. Cycling conditions for the reaction were 95°C for 10 minutes, followed by 45 cycles of 94°C for 30 seconds and 60°C for 1 minute, 98°C for 10 minutes and finally a 4°C hold on a Life Technologies Veriti thermal cycler. Data were analyzed with QuantaSoft v1.4 (Bio-Rad Laboratories). Two reference DNA samples (NA10843 and HuRef) plus 3 nontemplate controls were included with the study samples.
RESULTS
We have identified 7 new patients with heterozygous de novo mutations affecting the coding region of STXBP1. Patient 1 was diagnosed with infantile spasms and global DD at age 3.5 months (table). Further evaluation at age 5 years identified social and communication impairments and behavioral features consistent with a diagnosis of ASD. At this time, he continued to present with DD, and his frequent focal dyscognitive seizures (paroxysmal onset of altered awareness, gazing around with an open mouth, and ending with a cough with or without emesis), with secondary generalization, were resistant to pharmacologic anticonvulsant treatments. EEG demonstrated bilateral frontotemporal spikes and waves, and MRI suggested FCD in the left medial temporal lobe, specifically affecting the superior temporal, hippocampal, and parahippocampa1 gyri (figure 1A). Genetic evaluation using clinical microarray and a commercial 70-gene next-generation sequencing epilepsy panel (GeneDx, Gaithersburg, MD; table e-3 at http://links.lww.com/NXG/A14) identified a heterozygous partial deletion of exons 3–4 in STXBP1. Examination of parental genotypes confirmed the de novo nature of this deletion. No other clinically relevant genetic abnormality was identified on sequencing or microarray. In view of the intractable nature of his seizures, patient 1 subsequently underwent lesionectomy of the left anterior temporal horn at the age of 6 years, with complete remission of seizures achieved following resection and continuing as of the last follow-up 2 years later (both Engel and International League Against Epilepsy outcome Class 1). His ID and ASD remain symptomatically unchanged after surgery.
Table.
Clinical characteristics of patients with de novo STXBP1 mutations
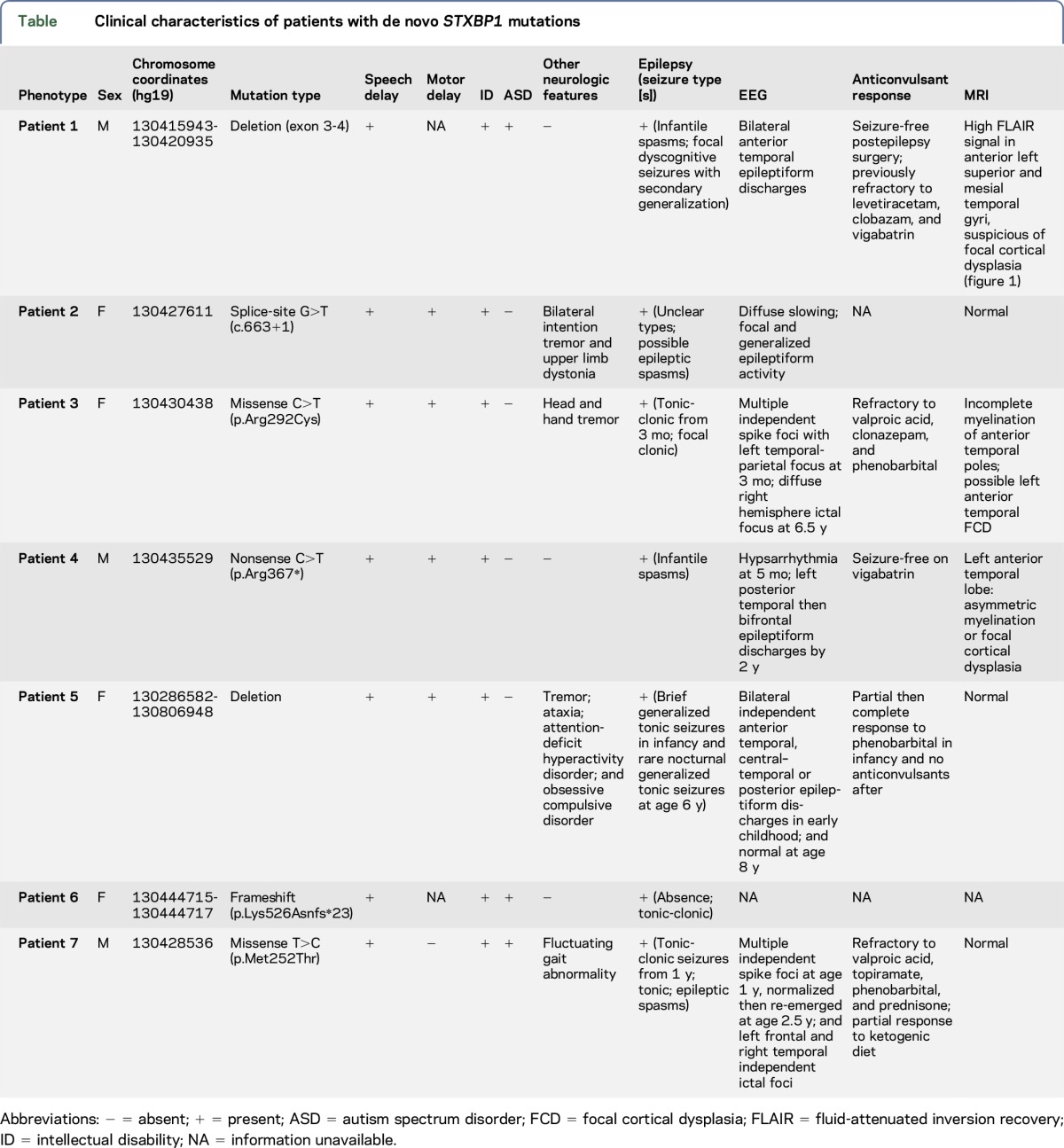
Figure 1. MRI of patient 1 with fluid-attenuated inversion recovery signal.

(A) Axial MRI of patient 1 demonstrating diffuse cortical/subcortical high fluid-attenuated inversion recovery (FLAIR) signal confined to the anterior left superior temporal gyrus and mesial temporal structures (white boxes). (B–C) Subtle increased FLAIR signal in the anterior temporal lobes of patients 2 and 3, respectively, suggestive of possible cortical dysplasia.
Resected dysplastic tissue and marginal nondysplastic tissue were histologically examined. While normal histology was evident in most of the imaging-suggested dysplastic tissue, the superior temporal gyrus showed heterotopic neurons in the white matter, blurring the gray–white matter junction, and in the deeper temporal white matter, there were nodular heterotopias comprising dysplastic tissue representing collections of cells with small round nuclei and clear cytoplasm, possibly immature neurons (figure 2, A–D). Immunohistochemistry labeling with both NeuN and Olig2 was negative in the latter. These findings were most consistent with FCD-type Ib.23 To further evaluate the molecular basis of patient 1 with epilepsy and ID, we undertook ddPCR on resected left lateral temporal cortical tissue and marginally resected normal brain tissue, hypothesizing that dysplastic tissue may be characterized by mosaicism for STXBP1. As predicted, copy number quantification was consistent with haploinsufficiency in marginal tissue but mosaicism in dysplastic tissue. The quantification (below 1 copy; figure 2E) was confirmed on 7 independent replications, consistent with cells mosaic for heterozygous and homozygous STXBP1 mutations in the dysplastic tissue. To characterize the cellular impact of mosaicism, we analyzed STXBP1 transcriptome profiles from 466 single cells24 for 6 major brain cells—astrocytes, microglia, neurons, endothelial, oligodendrocytes, and oligodendrocyte precursor cells. STXBP1 encodes syntaxin-binding protein that plays a role in the release of neurotransmitter through the regulation of syntaxin. Our analysis confirmed the principally neuronal expression of STXBP1 (figure 2F) compared with other primary brain cell types. This neuron-specific expression pattern and the known pathway of syntaxin imply a functional relevance of this gene into the etiology of epileptogenicity.
Figure 2. Histopathology of surgically excised dysplastic brain tissue (patient 1).

Circled areas of the left temporal lobe juxtacortical demonstrate linear neuronal heterotopia on (A) hematoxylin and eosin (H&E) and (B) NeuN stains, highlighting neurons; superior temporal gyrus nodular heterotopia are seen in boxes with (C) NeuN and (D) H&E stains, highlighting likely immature neurons; (E) Copy number variant mosaicism quantification using (ddPCR) within focal cortical dysplasia (red) and normal (black) tissue. The standard error bar shows the inferred copy number of approximately 20,000 droplets generated from each tissue type by the ddPCR; (F) single expression profile of the STXBP1 gene in primary brain cell types. RNA-seq reads were obtained (GEO GSE67835) for STXBP1 gene expression from 6 primary brain cell types (astrocytes, endothelial, microglia, neurons, oligodendrocytes, and oligodendrocyte progenitor cells [OPCs]). The bar showing lower 25th and upper 75th percentiles of the data, and the dots represent the outliers. Each color represents each cell types.
We identified 4 additional cases with heterozygous STXBP1 mutations (patients 2, 3, and 4) or deletion (patient 5) through clinics and 10,619 samples referred for chromosomal microarray at The Hospital for Sick Children. These patients were identified with a de novo mutation or deletion effecting one or more exons of STXBP1 (table). Two (patients 2 and 5) of our reported de novo mutations are novel and 2 (patients 3 and 4) were found to be recurrent mutations reported in previous literature. All presented with speech delay, global DD/ID, and epilepsy. Other more variable clinical features included motor delay and upper limb tremor (patients 2 and 3). Patients 3 and 4 also had MRI evidence of possible FCD (figure 1, B and C). The 7-year-old female (patient 3) presented at 3 months of age with multiple seizure types (including myoclonic, atonic, tonic–clonic, and focal dyscognitive seizures) and DD, with MRI demonstrating asymmetric increased T2 signal in the white matter of the anterior temporal poles, consistent with either delayed myelination or possibly left anterior temporal FCD. Although interictal EEG was abnormal bilaterally, ictal EEG demonstrated seizures arising from the contralateral right hemisphere. The 4-year-old male (patient 4) was similarly suspected to have delayed myelination or FCD at the left anterior temporal pole based on the increased T2 signal in the white matter. His interictal EEGs did not demonstrate epileptiform activity.
In view of patient 1's diagnosis of ASD and the indication in the literature of several patients with “autistic features” or ASD diagnoses, we next investigated our ASD cohort for the evidence of STXBP1 mutations. Of the currently sequenced individuals (5,205), 2 were identified with novel de novo variants overlapping STXBP1 (table; patients 6 and 7). A female individual (patient 6) has a de novo frameshift mutation truncating the protein's domain 2. Patient 7 is a male individual with a de novo missense mutation and primarily diagnosed with ASD with severe epilepsy. Both individuals were diagnosed with ASD using the ADI-R in conjunction with the ADOS. Both are nonverbal, with severe ASD in association with ID. Many of these individuals have also been investigated for the presence of CNVs by microarray.17
We subsequently reviewed all of the described STXBP1 reports in the literature (up to September 2016). There have been a total of 162 patients with heterozygous mutations of STXBP1 described so far across 35 studies (table e-1). For each mutation, we have converted the mutations into hg19 track and inferred the corresponding protein changes. The mutation spectrum comprises different variants spread across all 3 domains of the gene, with 21 recurrent mutations in unrelated individuals occurring in domains 2 and 3 (figure 3A). Mutations include 121 SNVs and 15 indels (of size <100 bp), and a total of 26 cases were reported to have a CNV affecting STXBP1. Sex was available for 126 cases, and we observed a higher proportion of females reported to carry an STXBP1 mutation compared with males, 57.1% (72) and 42.8% (54), respectively (figure 3B). Ascertainment for these 162 cases included 80% with a form of epilepsy syndrome (described as EIEE, Ohtahara syndrome, West syndrome, and Dravet syndrome) and 20% with one or more of the following: ASD, DD, movement disorder, nonsyndromic ID, and atypical Rett syndrome (figure 3C). Information pertaining to comorbidity for these 162 cases is not available.
Figure 3. Mapping of mutations within the STXBP1 gene.

(A) Mutations mapped within the 3 protein domains (cyan— domain 1, green—domain 2, and red—domain 3a/b) of the STXBP1 gene; (B) percentage of male and female cases reported to carry a pathogenic mutation for the STXBP1 gene. (C) Clinical spectrum associated with de novo STXBP1 mutations reported for early infantile epileptic encephalopathy (EIEE), focal cortical dysplasia (FCD), Ohtahara syndrome (OS), West syndrome (WS), Dravet syndrome, infantile spasms (ISs), neonatal refractory seizures, autism spectrum disorder, developmental delay, nonsyndromic intellectual disabilities (IDs), and atypical Rett syndrome.
A total of 140 variants (of 162) were reported to be de novo including case reports. To determine the rate of de novo variants, we excluded all case reports and retained only those studies that analyzed a cohort (with at least 20 samples); all 19 studies were ascertained by way of a primary diagnosis if epilepsy. From these 19 studies (table e-1), a total of 2,569 cases were analyzed, among whom 80 were reported to have a variant within STXBP1. More than 92.5% (74 of 80) carried a de novo variant, with the remainder of unknown inheritance. We can compute, therefore, a rate of 3.1% for de novo mutations within STXBP1 for cases with epilepsy. We realize that this de novo rate is high due to the selection of samples that are clinically well defined for epilepsy-related conditions.
DISCUSSION
Through a review of over 18,393 cases of ASD and DD, with molecular and/or cytogenetic analysis and meta-analysis of the literature, we report the prevalence of STXBP1 gene mutations in epilepsy as well as present an interesting finding of somatic STXBP1 mutation associated with FCD. Our detailed molecular analysis of excised brain tissue from patient 1 has revealed the observation of the postzygotic STXBP1 mutation in excised focally dysplastic tissue, with sustained remission of the epilepsy following resective surgery, which highlights the clinical importance of this finding. Only one other patient has been described in the literature with a similar pattern.13 This other patient, with a germline heterozygous deletion, presented clinically with ID and epilepsy. Despite having a normal MRI, she underwent surgical management of her seizures with near-complete remission achieved. FCD was demonstrated in resected tissue within the temporal lobe, although detailed molecular biology of the resected tissue, as we have newly done here, is not reported.
Patient 1's exemplary outcome following surgery is important and supports the relevance of a previous study with the observation of a good outcome.13 Treatment-resistant epilepsy significantly decreases the quality of life and increases the risk of sudden unexpected death in an epilepsy patient.25 Our observation suggests that presurgical evaluations should be aggressively pursued in patients with STXBP1 when there is evidence of dominant focality and not limited out of concern for the presence of a germline mutation. Although only about 50% of cases described so far with heterozygous deletions of STXBP1 have MRI evidence of brain pathology, milder type II and type I classified cortical dysplasias may be undiagnosed. Certainly, the limitation of current imaging sensitivity, even 3T MRI, is evidenced by a previous study13 and the abnormal myelination vs FCD differential reported in 2 of our patients who did not have surgery.
Although more extensive studies are required to fully elucidate the genetics of FCD, the case described in this study provides a new lead. It remains possible that the FCD was an incidental finding in our reported case, but it does raise the possibility of a causal relationship between neuronal expression of mosaic STXBP1 mutations and FCD. This association is supported by the homozygous STXBP1 deletion being only identified in the dysplastic portions of the resected brain, and at low levels, perhaps just in neurons. Recent studies reported somatic mutations in MTOR in excised dysplastic brain tissue in 12 of 77 patients with epilepsy.26,27 The implication is that somatic mutations may arise in brain tissue during development by an as-yet undetermined mechanism.28 As has been presented and discussed at length in a previous study, even low levels of mosaicism in MTOR pathway genes may result in significant clinical pathology.14 The MTOR gene was not sequenced in the 70-gene panel of patient 1 (table e-2), and there was insufficient dysplastic tissue to do this separately. With regard to potential MTOR mutations in our case, even if present, we would not be able to tell whether those were in the same subclonal population as the dysplasia-specific STXBP1 mosaicism. While somatic mutations have received much attention in cancer and certain nonbrain disorders, a possible etiopathologic connection with neurodevelopmental disorders, such as epilepsy and ASD, requires further elucidation. Our own study provides some impetus to further explore this possibility.
From a clinical genetics perspective, our study has calculated that 3.1% of clinically identified epilepsy cases harbor a rare/de novo mutation within STXBP1. Furthermore, our wider sampling of separate very large, “classic autism” ASD and DD cohorts allows us to conclude that STXBP1 seems to be associated only with severe ASD and DD in the presence of epilepsy. We have shown that the epilepsy observed with STXBP1 encephalopathy may be controlled through surgery by identifying dysplastic tissue, which is associated with further mosaic changes in STXBP1 limited to dysplasia. These rare observations help define this disorder's phenotype and introduce interesting genotypic information underlying FCD.
ACKNOWLEDGMENT
The authors thank The Centre for Applied Genomics (TCAG), which is funded by Genome Canada and the Ontario Genomics Institute, the Canada Foundation for Innovation (CFI), and the Ontario Research Fund of the Government of Ontario. The project was also supported by funds from the University of Toronto McLaughlin Centre and Genome Canada. M.U. holds a Banting postdoctoral fellowship from the Canadian Institutes of Health Research (CIHR).
GLOSSARY
- ASD
autism spectrum disorder
- CNV
copy number variant
- DD
developmental delay
- ddPCR
droplet digital PCR
- EIEE
early infantile epileptic encephalopathy
- FCD
focal cortical dysplasia
- ID
intellectual disability
- SNV
single nucleotide variant
AUTHOR CONTRIBUTIONS
M.U. and C.B. contributed to the conception and design of the study. A.C., L.B., S.L., G.P., R.K.C.Y., M.F., and A.S. helped analyze data. D.J.S, J.D, C.D.H., C.H., and L.A.T. contributed to sample recruitment, clinical data acquisition, and analysis. M.W.-S., C.R.M, B.M., S.W.S, M.U., and C.B. contributed to drafting the text and preparing the figures.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
M. Uddin has received travel funding from a nonprofit entity; was a Banting postdoctoral fellow from the Canadian Institutes of Health Research (CIHR); and has applied for a US patent for classifying mutation for autism and neurodevelopmental disorders. M. Woodbury-Smith is supported by a Clinical Investigatorship Award from the Canadian Institutes of Health Research's (CIHR) Institute of Genetics and has served on the editorial board of the Journal of Autism and Developmental Disorders. A. Chan has received research support from the Government of Ontario/Ontario Graduate Scholarship and the Government of Canada/Frederick Banting and Charles Best Scholarship. L. Brunga, S. Lamoureux, G. Pellecchia, R. Yuen, M. Faheem, D.J. Stavropoulos, and J. Drake report no disclosures. C. Hahn has served on the scientific advisory boards of SAGE Therapeutics and Marinus Pharmaceuticals; has served on the editorial boards of the Journal of Clinical Neurophysiology and PLoS One; is the second vice president of the American Clinical Neurophysiology Society and the President of the Canadian Society of Child Neurology; and has received research support from The Hospital for Sick Children Foundation and the American Epilepsy Society and Epilepsy Foundation of America. C. Hawkins has served on the scientific advisory board of the Pediatric Brain Tumor Consortium (nonprofit entity); has served on the editorial boards of Acta Neuropathologica Communications and the Journal of Neuropathology and Experimental Neurology; holds a patent for BOC modulation and uses thereof for treating, preventing and/or diagnosing cancer; and has received research support from Bayer, Grants4Targets, the Canadian Institutes of Health Research, Medical Sciences Proof of Principle (MsC POP) Program, Genome Canada, the Canadian Cancer Society Research Institute, DIPG collaborative, and Curing Kids Cancer. A. Shlien has received research support from the Terry Fox Foundation. C.R. Marshall has received travel funding from Affymetrix and Life Technologies and has served on the editorial boards of Genes, Genomes, Genetics and the Journal of Neurodevelopmental Disorders. L.A. Turner has received research support from Shire and CIHR. B. Minassian holds patents for diagnostic testing of the following genes: EPM2A, EPM2B, MECP2, and VMA21; has received research support from NIH; and has received license fee payments/royalty payments from patents for diagnostic testing of the following genes: EPM2A, EPM2B, MECP2, and VMA21. S.W. Scherer holds the GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children; has served on the scientific advisory board of Population Diagnostics; has served on the editorial boards of Genomic Medicine, Genes, Genomes, Genetics, the Journal of Personalized Medicine, the Open Genomics Journal, the Hugo Journal, Genome Medicine, the Journal of Neurodevelopmental Disorders, Autism Research, PathoGenetics, Comparative and Functional Genomics, BMC Medical Genomics, and Cytogenetics and Genome Research; and has received research support from Genome Canada/Ontario Genomics Institute, Canadian Institutes of Health Research, Canadian Institute for Advanced Research, McLaughlin Centre, Canada Foundation for Innovation, government of Ontario, and NIH. C. Boelman reports no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Gburek-Augustat J, Beck-Woedl S, Tzschach A, Bauer P, Schoening M, Riess A. Epilepsy is not a mandatory feature of STXBP1 associated ataxia-tremor-retardation syndrome. Eur J Paediatr Neurol 2016;20:661–665. [DOI] [PubMed] [Google Scholar]
- 2.Hamdan FF, Piton A, Gauthier J, et al. . De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol 2009;65:748–753. [DOI] [PubMed] [Google Scholar]
- 3.Mignot C, Moutard ML, Trouillard O, et al. . STXBP1-related encephalopathy presenting as infantile spasms and generalized tremor in three patients. Epilepsia 2011;52:1820–1827. [DOI] [PubMed] [Google Scholar]
- 4.Stamberger H, Nikanorova M, Willemsen MH, et al. . STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology 2016;86:954–962. [DOI] [PubMed] [Google Scholar]
- 5.Otsuka M, Oguni H, Liang JS, et al. . STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome–result of Japanese cohort study. Epilepsia 2010;51:2449–2452. [DOI] [PubMed] [Google Scholar]
- 6.Saitsu H, Kato M, Mizuguchi T, et al. . De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008;40:782–788. [DOI] [PubMed] [Google Scholar]
- 7.Milh M, Villeneuve N, Chouchane M, et al. . Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia 2011;52:1828–1834. [DOI] [PubMed] [Google Scholar]
- 8.Patzke C, Han Y, Covy J, et al. . Analysis of conditional heterozygous STXBP1 mutations in human neurons. J Clin Invest 2015;125:3560–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grone BP, Marchese M, Hamling KR, et al. . Epilepsy, behavioral abnormalities, and physiological comorbidities in syntaxin-binding protein 1 (STXBP1) mutant zebrafish. PLoS One 2016;11:e0151148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamashita S, Chiyonobu T, Yoshida M, et al. . Mislocalization of syntaxin-1 and impaired neurite growth observed in a human iPSC model for STXBP1-related epileptic encephalopathy. Epilepsia 2016;57:e81–e86. [DOI] [PubMed] [Google Scholar]
- 11.Di Meglio C, Lesca G, Villeneuve N, et al. . Epileptic patients with de novo STXBP1 mutations: key clinical features based on 24 cases. Epilepsia 2015;56:1931–1940. [DOI] [PubMed] [Google Scholar]
- 12.Barcia G, Chemaly N, Gobin S, et al. . Early epileptic encephalopathies associated with STXBP1 mutations: could we better delineate the phenotype? Eur J Med Genet 2014;57:15–20. [DOI] [PubMed] [Google Scholar]
- 13.Weckhuysen S, Holmgren P, Hendrickx R, et al. . Reduction of seizure frequency after epilepsy surgery in a patient with STXBP1 encephalopathy and clinical description of six novel mutation carriers. Epilepsia 2013;54:e74–e80. [DOI] [PubMed] [Google Scholar]
- 14.Mirzaa GM, Campbell CD, Solovieff N, et al. . Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol 2016;73:836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mercimek-Mahmutoglu S, Patel J, Cordeiro D, et al. . Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia 2015;56:707–716. [DOI] [PubMed] [Google Scholar]
- 16.Uddin M, Pellecchia G, Thiruvahindrapuram B, et al. . Indexing effects of copy number variation on genes involved in developmental delay. Scientific Rep 2016;6:28663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinto D, Delaby E, Merico D, et al. . Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 2014;94:677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uddin M, Tammimies K, Pellecchia G, et al. . Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat Genet 2014;46:742–747. [DOI] [PubMed] [Google Scholar]
- 19.Yuen RK, Merico D, Cao H, et al. . Genome-wide characteristics of de novo mutations in autism. NPJ Genomic Med 2016;1:160271–1602710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuen RK, Thiruvahindrapuram B, Merico D, et al. . Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med 2015;21:185–191. [DOI] [PubMed] [Google Scholar]
- 21.Jay JJ, Brouwer C. Lollipops in the clinic: information dense mutation plots for precision medicine. PLoS One 2016;11:e0160519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krzisch M, Temprana SG, Mongiat LA, et al. . Pre-existing astrocytes form functional perisynaptic processes on neurons generated in the adult hippocampus. Brain Struct Funct 2015;220:2027–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blumcke I, Thom M, Aronica E, et al. . The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011;52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darmanis S, Sloan SA, Zhang Y, et al. . A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci USA 2015;112:7285–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall-Patch L, Gayatri N, Ferrie C, Morrall MC. Clinical reasoning and investigations for resective surgery for medically refractory epilepsies in children. Epilepsy Behav 2012;23:442–446. [DOI] [PubMed] [Google Scholar]
- 26.Lim JS, Kim WI, Kang HC, et al. . Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015;21:395–400. [DOI] [PubMed] [Google Scholar]
- 27.Lim ET, Uddin M, De Rubeis S, et al. . Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci 2017;20:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science 2013;341:1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]