

Abstract
Purpose
Checkpoint molecules like programmed death-1 (PD-1) and T-cell immunoglobulin mucin-3 (TIM-3) are negative immune regulators that may be upregulated in the setting of glioblastoma multiforme. Combined PD-1 blockade and stereotactic radiosurgery (SRS) have been shown to improve antitumor immunity and produce long-term survivors in a murine glioma model. However, tumor-infiltrating lymphocytes (TIL) can express multiple checkpoints, and expression of ≥2 checkpoints corresponds to a more exhausted T-cell phenotype. We investigate TIM-3 expression in a glioma model and the antitumor efficacy of TIM-3 blockade alone and in combination with anti-PD-1 and SRS.
Experimental Design
C57BL/6 mice were implanted with murine glioma cell line GL261-luc2 and randomized into 8 treatment arms: (i) control, (ii) SRS, (iii) anti-PD-1 antibody, (iv) anti-TIM-3 antibody, (v) anti-PD-1 + SRS, (vi) anti-TIM-3 + SRS, (vii) anti-PD-1 + anti-TIM-3, and (viii) anti-PD-1 + anti-TIM-3 + SRS. Survival and immune activation were assessed.
Results
Dual therapy with anti-TIM-3 antibody + SRS or anti-TIM-3 + anti-PD-1 improved survival compared with anti- TIM-3 antibody alone. Triple therapy resulted in 100% overall survival (P < 0.05), a significant improvement compared with other arms. Long-term survivors demonstrated increased immune cell infiltration and activity and immune memory. Finally, positive staining for TIM-3 was detected in 7 of 8 human GBM samples.
Conclusions
This is the first preclinical investigation on the effects of dual PD-1 and TIM-3 blockade with radiation. We also demonstrate the presence of TIM-3 in human glioblastoma multiforme and provide preclinical evidence for a novel treatment combination that can potentially result in long-term glioma survival and constitutes a novel immunotherapeutic strategy for the treatment of glioblastoma multiforme.
Introduction
Glioblastoma multiforme is the most common primary malignancy of the central nervous system (CNS) and is associated with a 14.6-month median survival with standard-of-care surgery, chemotherapy, and radiation (1, 2). Glioblastoma multiforme pathogenesis is characterized by tissue invasion, angiogenesis, local tissue hypoxia and necrosis, and evasion of the innate and adaptive antitumor immune response. Tumor-associated local and systemic immunosuppression has garnered significant interest, as recent studies have shown that glioblastoma multiforme induces tumor-infiltrating lymphocyte (TIL) anergy, recruit immunosuppressive regulatory T cells (Treg), and activate immune checkpoints (3–8).
Checkpoint molecules, such as cytotoxic T lymphocyte–associated protein 4 (CTLA-4) and programmed death-1 (PD-1), are critical negative regulators of the immune system that protect the body from inappropriate immune activation. Several solid tumors, including glioblastoma multiforme, are protected from immunologic pressure by constitutive activity of immune checkpoint pathways (8). On the basis of these data, clinical development of antibodies that prevent checkpoint:ligand binding has proven to be a major advancement in cancer immunotherapy. Ipilimumab (anti-CTLA-4) was approved for metastatic melanoma in 2011, and approval of nivolumab (anti-PD-1) followed in 2014. Combination checkpoint blockade has the potential to dramatically improve response rates, albeit with an increased incidence of immune-related adverse events (9). Taken together, these data illustrate the potential effectiveness and feasibility of combination checkpoint blockade while highlighting the need to identify new targets and combination strategies.
T-cell immunoglobulin mucin-3 (TIM-3) is a negative regulator of lymphocyte function and survival that, like PD-1, is a marker of CD4 and CD8 T-cell exhaustion (10). PD-1 and TIM-3–coexpressing lymphocytes have been identified in colon adenocarcinoma, breast adenocarcinoma, and melanoma and represent a more severely impaired TIL population (compared with PD-1+ or TIM-3+ only) as measured by inflammatory cytokine production and proliferation capacity (11, 12). At present, dual checkpoint expression on TILs has not yet been described. However, clinical studies have demonstrated TIM-3 expression to be significantly elevated on both circulating blood lymphocytes and TILs in glioma patients. This expression was found to be positively correlated with glioma grade and negatively correlated with Karnofsky performance status score (13, 14). Using our glioma model, we hypothesized that dual blockade of PD-1 and TIM-3 would result in a more robust antiglioma immune response and improved survival compared with either antibody alone. In addition, in light of the synergistic potential of stereotactic radiosurgery (SRS) as demonstrated by Zeng and colleagues (3), it was further hypothesized that the addition of SRS would enhance the efficacy of dual checkpoint blockade against murine gliomas.
Materials and Methods
Mice and cell lines
Six- to 8-week-old C57BL/6J wild-type female mice were maintained at the Johns Hopkins University Animal Facility. All animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee. Orthotopic gliomas were established using GL261-Luc cells grown in DMEM (Life Technologies) + 10% FBS (Sigma-Aldrich) + 1% penicillin–streptomycin (Life Technologies) with the addition of 100 μg/mL G418 (Corning) selection media at 37°C, as described previously (3). GL261-Luc cells (130,000) in a volume of 1 μL were stereotactically injected into the left striatum as defined by the following coordinates: 2 mm posterior to the coronal suture, 2 mm lateral to the sagittal suture, and 3mm deep to the cortical surface. Mice were randomly segregated and assigned to treatment arms, and presence of tumor was monitored by bioluminescent IVIS imaging (PerkinElmer) on posttumor implantation day 7, 14, 21, 28, and 42. Survival experiments were repeated in triplicate with 6 to 10 mice in each control or treatment arm. Animals were euthanized according to humane endpoints, including CNS disturbances, hunched posture, lethargy, weight loss, and inability to ambulate.
Therapeutic antibodies
Hamster mAbs against murine PD-1 were purified from hybridoma (G4) as described previously (15). Individual treatment dose was 200 μg per animal. Anti-murine TIM-3 antibodies were purchased from Bio X Cell and stored at −80°C in 1 mg/mL aliquots. Anti-TIM-3 clone RMT3-23 is a non-depleting, blocking antibody (16). Individual treatment dose was 250 μg/animal.
Stereotactic radiation
A Small Animal Radiation Research Platform (SARRP) was used to irradiate tumor-bearing animals in in vivo experiments as described previously (3, 17, 51–52). CT imaging was used for each animal to localize the burr hole from tumor implantation. A 3-mm beam centered on the burr hole and underlying tumor was used to administer a total of 10 Gy radiation per animal at a rate of 1.9 Gy/minute. Dosimetric data were as described previously by Deng and colleagues (Supplementary Fig. S1; ref. 18).
Immune cell isolation
To analyze peripheral lymphocytes, blood (150 μL, retro-orbital), lungs, livers, lymph nodes (brachial and inguinal), and spleens were harvested from naïve mice after transcardial perfusion with PBS. Solid organs were mechanically homogenized in RPMI medium + 10% FBS + 1% penicillin–streptomycin and filtered through a 100-μm mesh cell strainer (BD Falcon). Lymph nodes were washed and resuspended in PBS. Red blood cells were lysed from lung and spleen samples and washed with PBS. Livers were resuspended in 5 mL 70% Percoll (GE Healthcare). Percoll gradients were prepared by layering cells below 7 mL of 40% Percoll (44%/70%) and centrifuged at 2,000 rpm for 20 minutes without brake at room temperature. Lymphocyte bands at the gradient interface were collected and washed with PBS.
To isolate brain-infiltrating lymphocytes (BIL), blood (150 μL) and brains were harvested after transcardial perfusion with PBS on postimplantation day 21. Blood samples were centrifuged at 1,250 rpm for 4 minutes, resuspended in ACK lysing buffer (Life Technologies), and then washed with PBS. Brains were mechanically homogenized, filtered, resuspended in 5 mL 80% Percoll, layered below 7 mL of 40% Percoll (40%/80%), and centrifuged at 2,000 rpm for 20 minutes at room temperature. Cell layer at the 40%/80% interface was collected and washed with PBS. For cytokine analysis, cells were stimulated in RPMI + Cell Stimulation Cocktail plus protein transport inhibitors (eBioscience) at 37°C for 4 hours and then washed with PBS.
To isolate microglia, macrophages, and dendritic cells (DC), brains were mechanically homogenized in 70% Percoll and laid beneath 30% Percoll (30%/70% gradient) and then centrifuged and processed in the same manner as BILs.
Flow cytometry and immunophenotyping
For analysis of surface markers, lymphocytes were stained for CD3, CD4, CD8, PD-1, and TIM-3 (Supplementary Table S1 for antibody clones and dilutions), fixed in 1:3 fixation/permeabilization concentrate:diluent mixture (eBioscience) for 30 minutes, and stained for FoxP3 in permeabilization buffer. For cytokine analysis, cells were stained for CD3, CD4, and CD8, fixed as described above, and then stained for IFNγ, IL2, IL17A, and TNFα. For analysis of antigen-presenting cells (APC), cells were pretreated with Fc block (anti-CD16/32), washed, and stained with Live/Dead Aqua. Cells were then stained for F4/80, CD45, CD11b, CD11c, PD-1, and TIM-3. Appropriate isotype controls were used and cells were acquired on the LSR II flow cytometer (BD). All FACS data were analyzed using BD FACSDiva software. Nonviable cells were excluded by forward versus side scatter analysis and Live/Dead Aqua (Invitrogen) staining.
Depletion studies
Tumor-bearing mice received intraperitoneal injections of either anti-CD4 (clone GK1.5, Bio X Cell) or anti-CD8 (clone 2.43, Bio X Cell) at 200 mg per animal on postimplantation days 4, 5, 6, 16, and 21.
Flank rechallenge
All animals that demonstrated long-term survival (100 days postimplantation) were challenged with subcutaneous right flank injection of 2 × 106 GL261-luc2 cells suspended in 100 mL of PBS and Geltrex basement membrane matrix (Life Technologies) in a 1:1 ratio. Four naïve mice were also injected as controls. Tumor presence was assessed by bioluminescent imaging on postrechallenge day 10. Tumor volumes were measured every 7 days, and mice were euthanized once tumors reached 1,000 mm3.
IHC for human TIM-3
Formalin-fixed paraffin-embedded (FFPE) primary glioblastoma multiforme samples were obtained from an institutional brain tumor tissue bank authorized by the Institutional Review Board of Johns Hopkins University (Baltimore, MD). IHC for TIM-3 was performed on 8 randomly selected samples using a primary mouse anti-human mAb (clone F38-2E2, eBioscience) at a concentration 1.5 μg/mL, following an antigen retrieval of 10 minutes in citrate buffer, pH 6.0 at 120°C. A secondary anti-mouse IgG1 antibody was used at a concentration of 1.0 μg/mL. Amplification was performed using PerkinElmer biotin tyramide signal amplification and Dako streptavidin horseradish peroxidase. Signal was visualized by 3,3′-diaminobenzidine staining. All slides were reviewed by a board-certified pathologist (P.C. Burger).
Statistical analysis
Survival was analyzed by Kaplan–Meier method and compared by log-rank test. Unpaired t test was used to make comparisons between two independent groups. Comparisons between groups were presented as mean ± SEM. All data were analyzed using GraphPad Prism 6 and values of P < 0.05 were considered statistically significant.
Results
Tumor-bearing mice display a higher frequency of TIM-3 expression on lymphocytes compared with naïve mice
To our knowledge, no data have been published on TIM-3– bearing immune cells in a murine glioma model. We first investigated the presence of TIM-3 in peripheral and cerebral tissues at physiologic baseline. Lungs, livers, lymph nodes, spleens, and brains were harvested from 6 naïve wild-type (non–tumor bearing) mice, and lymphocyte populations were isolated from each organ. Flow cytometric analysis demonstrated that less than 0.5% of all CD4 and CD8 T cells in each of these organs expressed TIM-3 on their surfaces (Fig. 1A and B and Supplementary Fig. S2). Notably, in the brain, only 0.23 ± 0.06% of all CD4 and 0.08 ± 0.05% of all CD8 T cells expressed TIM-3 on their cell surface.
Figure 1.
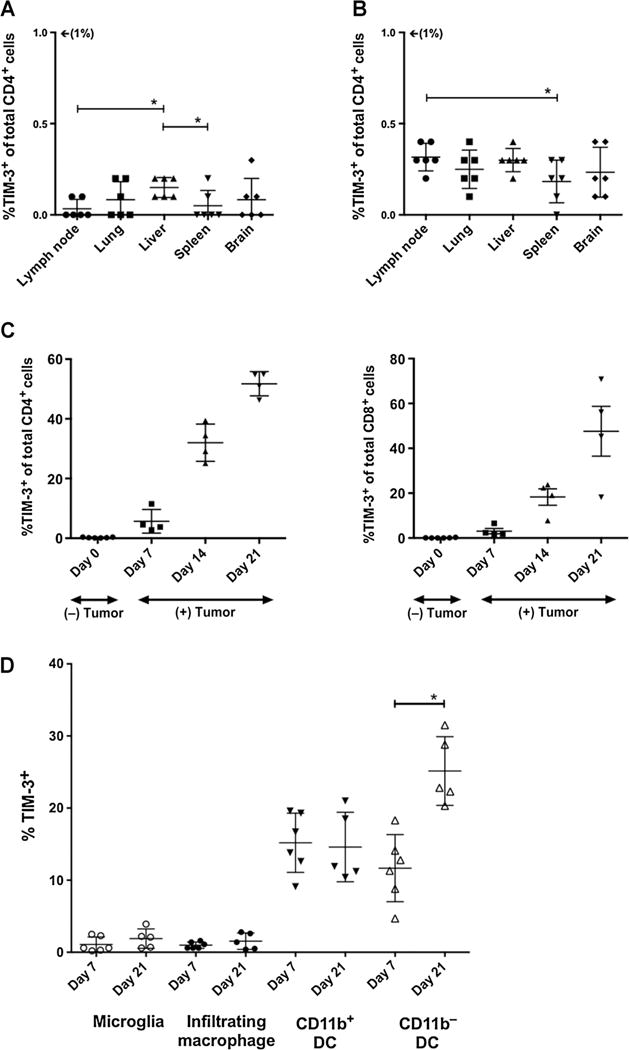
TIM-3 expression on peripheral and CNS lymphocytes. In naïve, non-tumor–bearing mice, less than 0.5% of all CD4+ (A) and CD8+ T cells (B) isolated from peripheral lymph nodes, lungs, livers, spleens, and brains had surface expression of TIM-3. C, Compared with naïve mice, tumor-bearing mice demonstrate a progressive increase in TIM-3 expression. From days 7, 14, and 21 postimplantation, the percentage of CD4+ T cells expressing TIM-3 rose from 5.65 ± 1.98% to 32 ± 3.16% to 51.75 ± 2.03%, respectively (P < 0.05). The percentage of CD8+ T cells rose from 3.05 ± 0.05% to 18.28 ± 3.62% to 47.60 ± 11.11%, respectively (P < 0.05). D, F4/80+CD45dimCD11bhi microglia and F4/80+CD45hiCD11c−CD11bhi brain-infiltrating macrophages demonstrate negligible TIM-3 expression on days 7 or 21. F4/80+CD45hiCD11c+CD11b+ DCs demonstrated stable, low expression of TIM-3, whereas F4/80−CD45dimCD11c+CD11b− DCs showed a statistically significant increase by day 21. All experiments repeated in duplicate with ≥4 mice per arm. P values were determined by unpaired t tests; *, P < 0.05. Comparisons within groups were presented as mean ± SEM.
We then harvested BILs at postimplantation days 7, 14, and 21 to investigate changes in TIM-3 expression over time in glioma-bearing mice. Compared with naïve animals, we observed a statistically significant increase in TIM-3 expression on BILs at each time point (Fig. 1C). By day 7, 5.65 ± 1.98% of all infiltrating CD4 T cells expressed TIM-3, compared with 0.23 ± 0.06% at baseline (P = 0.008). By day 14, mean frequency rose to 32 ± 3.16% (vs. day 7, P = 0.0004) and by day 21, mean frequency was 51.75 ± 2.03% (vs. day 14, P = 0.002). Infiltrating CD8 T cells demonstrated a similar trend of TIM-3 upregulation, with mean frequencies of 3.05 ± 0.05%, 18.28 ± 3.62%, and 47.60 ± 11.11% at days 7, 14, and 21, respectively (P < 0.05). Together, these data demonstrate that TIM-3+ CD4 and CD8 T cells comprise a large population of BILs in advanced murine gliomas and could potentially be targeted by anti-TIM-3–blocking antibodies.
We also examined TIM-3 expression in various myeloid-derived APCs in the brain. By gating on CD45+ cells, we were able to distinguish 4 major APC populations in the brain after tumor implantation: microglia (F4/80+CD45dimCD11bhi), infiltrating macrophages (F4/80+CD45hiCD11c−CD11bhi), myeloid DCs (F4/80+CD45hiCD11c+CD11b+), and lymphoid DCs (F4/80−CD45dimCD11c+CD11b−; Supplementary Fig. S3). Microglia and infiltrating macrophages demonstrated the lowest frequencies of TIM-3 expression on day 7 after tumor implantation at 1.08 ± 0.42% and 0.98 ± 0.18%, respectively (Fig. 1D). Myeloid-derived DCs were observed to have a higher rate of TIM-3 expression at 15.18 ± 1.68% on day 7. None of these groups demonstrated a statistically significant increase in frequency by day 21. In contrast, lymphoid-derived DCs expressed TIM-3 at a rate of 11.67 ± 1.90% on day 7, with an increase to 25.14 ± 2.13% by day 21 (P = 0.001). These data suggest that only specific subsets of APCs express or upregulate TIM-3 in the setting of glioma.
Frequency of PD-1 and TIM-3 coexpression on BILs increases with time
PD-1+TIM-3+ T cells have been shown to comprise a predominant fraction of BILs in various solid tumors and represent the most severely exhausted T-cell phenotype as defined by a failure to proliferate or produce cytokines such as IL2, TNF, and IFNγ (11). We hypothesized that PD-1 and TIM-3–coexpressing BILs are present in gliomas and represent a target for dual checkpoint blockade therapy. To test this hypothesis, we harvested BILs from glioma-bearing mice on day 7, 14, and 21 and assessed PD-1 and TIM-3 surface expression (Fig. 2 and Supplementary Fig. S4). We observed that on day 7, the majority of CD4 T cells, CD8 T cells, and FoxP3+ Tregs were PD-1−TIM-3− with only 2.93 ± 1.23%, 1.83 ± 0.97%, and 2.70 ± 0.44%, respectively, coexpressing both checkpoints. However, we observed a reversal of this pattern on day 21, with rates of coexpression rising to 51.15 ± 2.01%, 46.20 ± 11.03%, and 64.30 ± 1.48% on CD4 T cell, CD8 T cell, and Tregs, respectively. In the intermittent period, a large peak in PD-1+TIM-3− BILs were seen. These cells could represent a precursor to PD-1+TIM-3+ T cells, or a separate, transient population of exhausted T cells.
Figure 2.
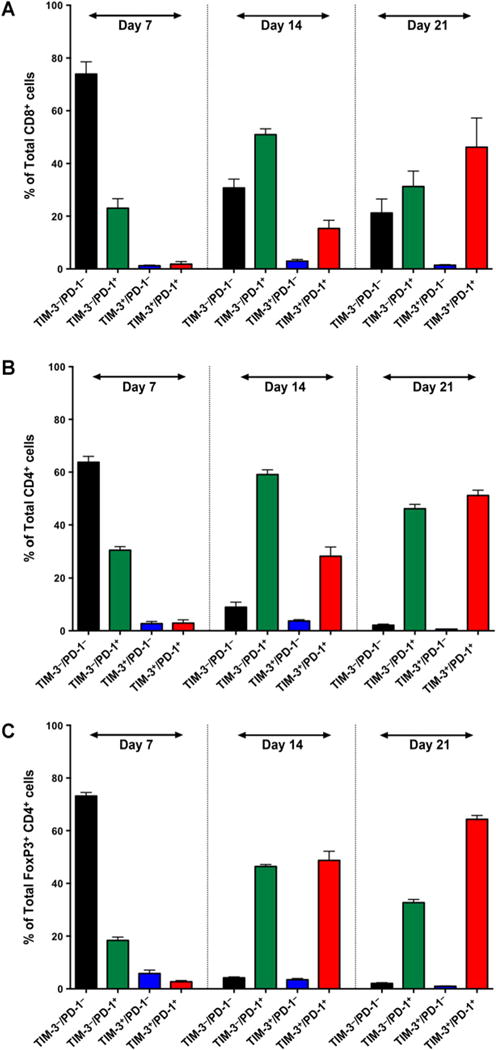
Frequency of TIM-3 and PD-1 coexpression on BILs over time. On day 7, the majority of CD8+ T cells (A) CD4+ T cells (B), and FoxP3+CD4+ Tregs (C) were TIM-3−PD-1−. By day 14, the majority of effector T cells were single positive for PD-1, whereas regulatory T cells had an equal rate of PD-1 single expression or PD-1 and TIM-3 coexpression. By day 21, the majority of all three lymphocyte subsets were TIM-3+PD-1+. Experiments were run in duplicate with ≥4 mice per arm. Comparisons within groups were presented as mean SEM.
Targeting PD-1 and TIM-3 results in long-term survival
Having confirmed TIM-3 and PD-1 expression on glioma-infiltrating T cells, we hypothesized that dual blockade with anti-PD-1 and anti-TIM-3 antibody would result in longer survival compared with either monotherapy and that the addition of SRS would further improve survival as previously demonstrated with anti-PD-1 and SRS combination therapy (3). Using our in vivo mouse model, we compared monotherapy with SRS alone, anti-PD-1 alone, or anti-TIM-3 alone versus dual therapy with anti-PD1 + SRS, anti-TIM-3 + SRS, or anti-PD-1 + anti-TIM-3 versus triple therapy with anti-PD-1 + anti-TIM-3 + SRS (Fig. 3). A single dose of SRS was administered prior to anti-PD-1 to induce an inflammatory response and prime the tumor microenvironment for immune-based therapy. The timing for each treatment modality was based on a previously validated radiation administration schedule that took into consideration the rate of tumor growth and survival expectancy in our tumor model (19). TIM-3 dosing and timing was based on a treatment schedule suggested by Ngiow and colleagues, which emphasized the importance of early delivery of TIM-3 antibody (20). Tumor growth was assessed on day 7 and 21 using bioluminescent IVIS imaging. We determined that neither anti-TIM-3 nor SRS alone had significant treatment effect, whereas anti-PD-1 improved median survival (33 days) compared with control (22 days, P < 0.0001). The addition of anti-TIM-3 to SRS therapy increased median survival to 100 days (P = 0.017) compared with 27 days with SRS alone (P < 0.0001) and resulted in 63.2% long-term survival (defined as survival at 100 days postimplantation). Adding anti-TIM-3 to anti-PD-1 therapy improved median survival from 33 days (anti-PD-1 alone) to 100 days (anti-TIM-3 + anti-PD-1) and improved overall survival (OS) from 27.8% to 57.9%, respectively, although these numbers did not achieve statistical significance (P = 0.103). However, the addition of anti-TIM-3 to anti-PD-1 and SRS (triple therapy) resulted in an OS of 100% (P = 0.004).
Figure 3.
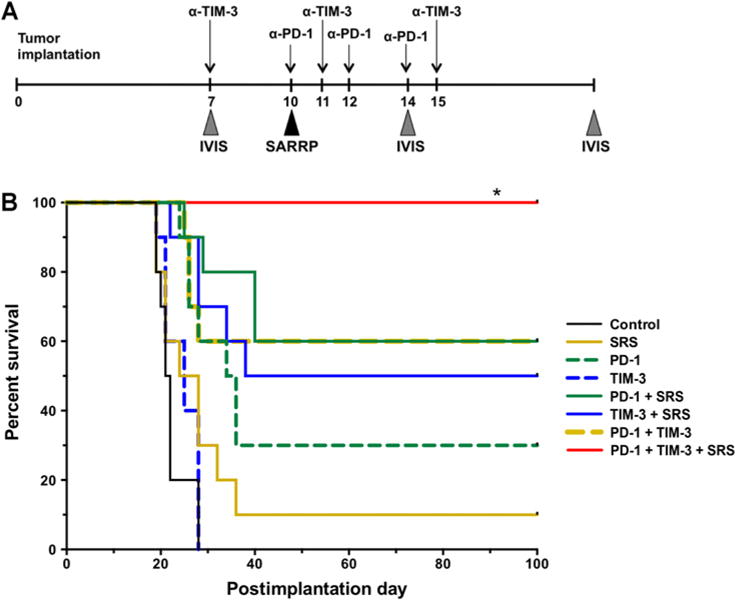
Survival experiments. A, Experimental setup and treatment schedule is depicted. Anti-PD-1 antibody was administered on days 10, 12, and 14. Anti-TIM-3 antibody was given on days 7, 11, and 15. 10 Gy of radiation was delivered by a SARRP instrument. SARRP, Small Animal Radiation Research Platform. B, Kaplan–Meier curve. P < 0.05 between triple therapy group (anti-PD-1 + anti-TIM-3 + SRS) and all other arms. Survival experiment was repeated in triplicate and a representative repeat was presented, n =10. Survival was analyzed by Kaplan–Meier method and compared by log-rank Mantel–Cox test. *, P < 0.05.
Combination therapy improves immune profile of tumor microenvironment
To investigate the mechanisms underlying the observed treatment effects, brains were harvested on day 21 and assessed for markers of immune activation. The ratio of CD8 effector T cells versus Tregs has been correlated with treatment efficacy in several tumor models (21, 22). In our in vivo experiments, all treatment arms except anti-TIM-3 + SRS showed a statistically significant increase in CD8 T cell to FoxP3+ Treg ratio compared with control animals (P < 0.05; Fig. 4A). The highest ratio was observed in the anti-PD-1 only, anti-PD-1 and anti-TIM-3, and triple therapy treatment arms, with no significant difference among these groups. Notably, the addition of SRS to anti-TIM-3 therapy did not significantly increase this ratio. The frequency of CD4+FoxP3+ Treg was also assessed (Fig. 4B). A significant decrease in Treg frequency was observed in all arms except anti-TIM-3 alone and anti-TIM-3 + SRS. The lowest frequencies were seen in the anti-PD-1 alone, anti-PD-1 + SRS, anti-PD-1 + anti-TIM-3, and triple therapy groups, with no significant difference among these groups. These data suggest that PD-1 blockade is the major mechanism responsible for these observed effects on BIL populations.
Figure 4.
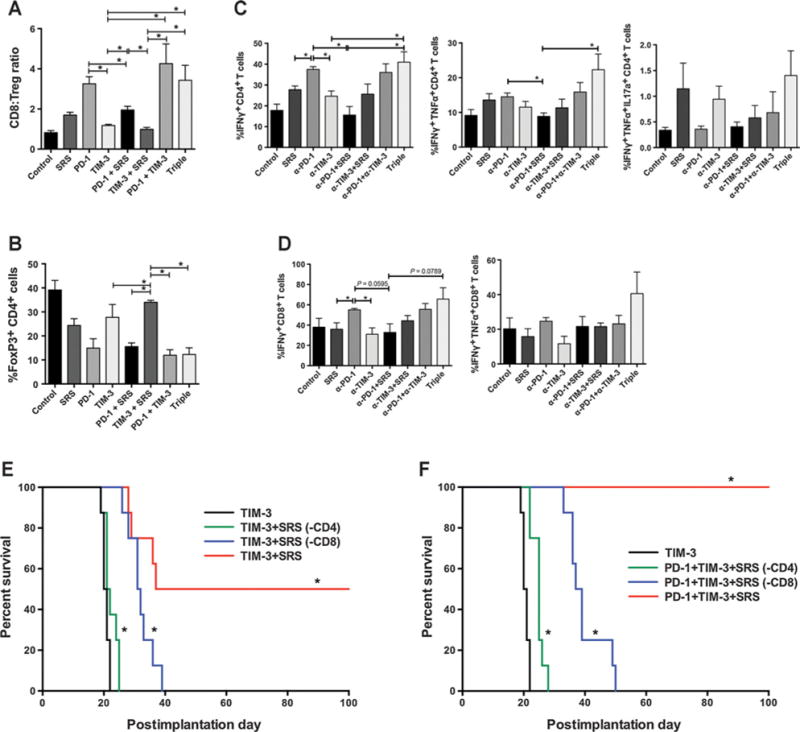
Immune analysis. A, CD8 effector to FoxP3+ T regulatory cell ratios are highest in treatment arms that include anti-PD-1. B, Dual checkpoint blockade and triple therapy show a trend toward the lowest percentage of FoxP3+ CD4 T cells. Triple therapy shows a trend toward highest mono- and polyclonal cytokine production by CD4 (C) and CD8 (D) T cells compared with all the other arms. Immune profiling experiments were repeated in duplicates with ≥3 mice per arm (A–D). E, CD4 T-cell depletion abrogated the treatment effect of anti-TIM-3 + SRS with no significant difference in survival between the TIM-3 only and TIM-3 + SRS(-CD4) arms. CD8 depletion [TIM-3 +SRS(-CD4)] also resulted in diminished treatment effect (0% OS) but had significantly improved survival compared with animals treated with anti-TIM-3 alone with median survival of 31.5 and 20.5 days, respectively (P < 0.001). F, CD4 and CD8 depletion also abrogated the survival benefits of triple therapy (0% OS), with median survival of 25.0 and 38.0 days, respectively. Depletion experiment was repeated in duplicate, and a representative repeat was presented, n = 8. Survival was analyzed by Kaplan–Meier method and compared by log-rank Mantel–Cox test. *, P < 0.05.
BIL activity and cytokine production was also assessed in each treatment arm (Fig. 4C and D). The frequency of IFNγ-producing CD4 cells was significantly increased in the triple therapy group compared with anti-TIM-3 alone (P = 0.04) and anti-PD-1 + SRS (P = 0.016). We observed a similar trend of increased rate of IFNγ-producing CD8 cells when adding TIM-3 blockade to anti-PD-1 + SRS therapy (P = 0.079); the addition of SRS to anti-TIM-3 also increased the frequency of IFNγ+TNFα+ effector T cells. We also noted a trend toward more polyfunctional CD4 and CD8 T cells in the triple therapy arms (IFNγ+TNFα+ or IFNγ+TNFα+IL17a+). Our results demonstrate a pattern of improved cytokine profile for both CD8 and CD4 T cells with the addition of anti-TIM-3 antibody (triple therapy) to anti-PD-1 + SRS.
Previous data have shown that the influx of CD8 T cells is responsible for the treatment effect of anti-PD-1 + SRS (3). To better elucidate the mechanism mediating the therapeutic efficacy of adding SRS to anti-TIM-3 antibody, we depleted mice of CD4 of CD8 T cells before treatment (Fig. 4E). In mice depleted of CD4 cells, the survival benefit of combination therapy was abrogated, with no significant difference between anti-TIM-3 alone and anti-TIM-3 + SRS. In contrast, mice depleted of CD8 cells demonstrated a marginal improvement in median survival compared with the TIM-3 monotherapy arm (P < 0.0001). However, the treatment response was significantly less robust than in nondepleted animals, with a long-term survival (≥100 days) of 0%. These data indicate that both CD4 and CD8 lymphocytes play a critical role in the observed difference in OS between anti-TIM-3 monotherapy and anti-TIM-3 + SRS dual therapy arms.
To demonstrate that both CD4 and CD8 T cells are required to mediate the effects of triple therapy, we treated CD4 or CD8-depleted mice with anti-PD-1, anti-TIM-3, and SRS (Fig. 4F). Compared with nondepleted mice treated with anti-TIM-3 alone (median survival 20.5 days), both CD4- and CD8-depleted mice demonstrated a marginal but statistically significant improvement with triple therapy with 0% long-term survival (≥100 days) but median survivals of 25 and 38 days, respectively.
Long-term survivors demonstrate immune memory
Long-term survival was defined as 100 days postimplantation. “Cured” mice were tested for durable immune memory by tumor rechallenge on day 100. Using naïve mice as controls, all surviving mice were rechallenged with injections of GL261-Luc in the right flank. Hundred percent of the naïve mice developed flank tumors (Fig. 5A). However, none of the long-term survivors were found to have established tumors by day 30 after flank rechallenge, as confirmed by bioluminescent imaging on day 10 (Fig. 5B). These survival patterns indicate that the cured mice have long-term immunologic memory against the GL261-Luc cells.
Figure 5.
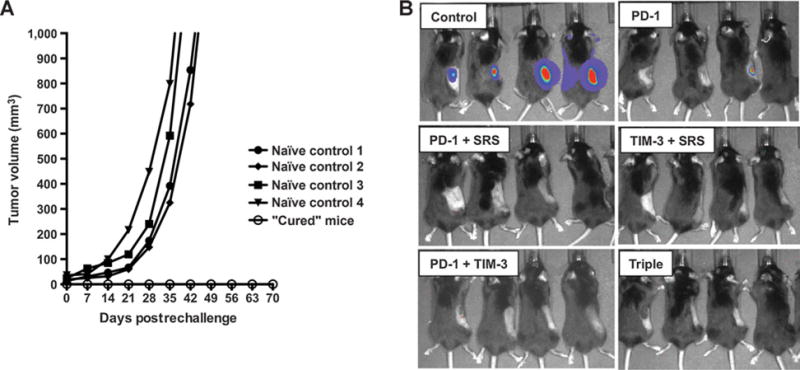
Long-term survivors have durable immune memory. A, Mice with no tumor by postimplantation day 100 were rechallenged with 2 × 106 GL261-luc2 cells in the right flank and compared with 4 naïve control animals. Control mice developed flank tumors of 1,000 mm3 volume by postrechallenge week 7. None of the long-term survivors developed flank tumors by week 10. B, IVIS imaging on day 10 demonstrates strong bioluminescence in naïve controls, and no signal in the long-term survivors, with the exception of one mouse in the PD-1–treated group that showed weak signal. By day 14, the signal was no longer detectable in this animal (not shown). Rechallenge experiments were repeated in duplicate using all remaining survivors.
Tumor-infiltrating immune cells in primary glioblastoma multiforme express TIM-3
TIM-3 expression in human tumors was assessed using IHC on FFPE-embedded primary glioblastoma multiforme tissue samples from 8 individual patients (Fig. 6) Five of the 8 samples were positive for TIM-3 expression on perivascular or TILs. Four of the samples were also found to have a staining pattern consistent with positive tumor cells. Two samples demonstrated lysosomal staining in cells of unidentified type, and one showed linear staining that was read as possible activated microglia. These findings indicate that immune infiltrates in human glioblastoma multiforme express TIM-3 and represent a potential clinical target for anti-TIM-3 therapy.
Figure 6.
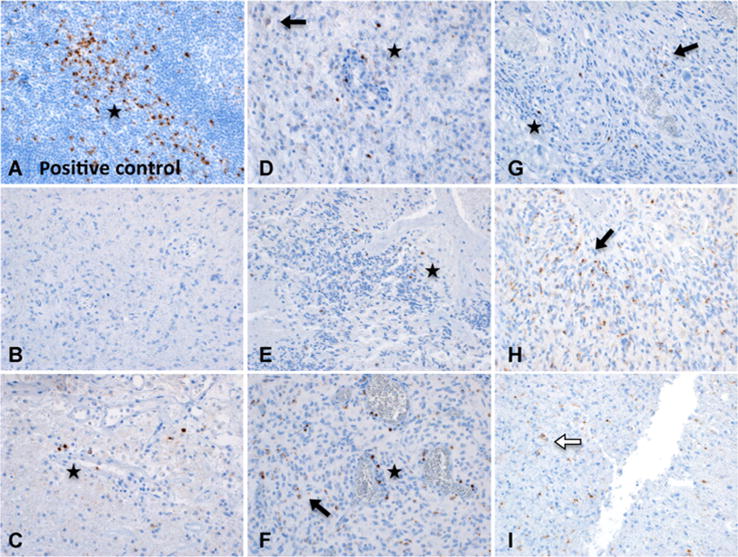
TIM-3 expression in primary human brain tumor samples (magnification, 100×). B–I, Representative patterns of TIM-3 staining in FFPE-embedded primary glioblastoma multiforme specimens from 8 patients. Star, lymphocyte staining pattern; black arrow, tumor cell pattern; white arrow, lysosomal staining pattern. A, Tonsil (positive control) showing strong staining for TIM-3. B–I, Representative patterns of TIM-3 staining in human primary glioblastoma multiforme. B, Negative for TIM-3 expression. C, Perivascular positive lymphocytes, very diffuse staining is most likely nonspecific. D, Staining of cells in a lymphocytic pattern, one diffuse cytoplasmic tumor cell. E, Potential TIL, most likely lysosomal staining in cells of unidentified type. F, Tumor cells as well as scattered, intensely stained perivascular cells consistent with lymphocytes. Linear staining may be a process of activated microglia or tumor cell. G, Tumor cell staining pattern with potential lymphocyte (left, most intense stain). Cells with diffuse dots could be either neoplastic or inflammatory, but not possible to differentiate positively. H, Multiple intense staining in tumor cells. I, Nonlymphocytic, globular, lysosomal staining pattern in cells of unidentified type.
Discussion
TIM-3 has been described as an inhibitory checkpoint molecule and a promising target for immunotherapy, but its role in intracranial tumorigenesis has not yet been extensively characterized. Here, we have shown that TIM-3 is upregulated in the setting of glioma and that treatment with dual anti-PD-1 and anti-TIM-3 checkpoint blockade plus SRS results in a dramatic treatment effect. Our results suggest that targeting multiple checkpoints may be a superior strategy for anti–glioblastoma multiforme immunotherapy.
A recent study of clinical blood samples found that newly diagnosed glioma patients had significantly higher levels of TIM-3 expression on their blood leukocytes as compared with healthy controls. In addition, a higher frequency of TIM3+ CD8 T cells was positively correlated with a higher tumor grade and negatively correlated with Karnofsky scores (13). In our in vivo experiments, we isolated BILs from the brains of orthotopic glioma-bearing mice and observed a significant increase in TIM-3 expression on brain-infiltrating CD4 and CD8 cells compared to naïve, non-tumor–bearing control animals.
Anti-TIM-3 monotherapy
Having demonstrated that TIM-3 is upregulated in our glioma model, we hypothesized that targeted blockade could restore antitumor activity and result in tumor regression in glioma-bearing mice. The results of our survival experiments showed that there was no significant difference in median and OS between animals receiving no treatment versus anti-TIM-3 monotherapy. By postimplantation day 21, the monotherapy and control groups were indistinguishable by clinical features, such as hunched posture, lethargy, and weight loss. This is not a novel finding, as several previous studies have shown that anti-TIM-3 alone has little to no therapeutic activity against in several tumors models such as CT26 colon tumors (11), MCA-induced sarcomas (23), and ID8 ovarian cancer models (24).
Anti-TIM-3 + PD-1
Whereas anti-TIM-3 alone was insufficient for treatment response, the combination of anti-TIM-3 and anti-PD-1 antibody resulted in a significant survival benefit (0% vs. 57.9% OS). PD-1 and TIM-3–coexpressing T cells have been described as a severely incapacitated subset of BILs (11, 12, 25). The frequency of PD-1+TIM-3+ vaccine-induced CD8 T cells was found to negatively correlate with in vivo expansion of effector lymphocytes in metastatic melanoma patients. Dual blockade with anti-PD-1 and anti-TIM-3 antibodies improved proliferation and cytokine production in vitro (12). Using multiple solid tumor models, Sakuishi and colleagues demonstrated that PD-1 and TIM-3 coexpression denotes a more severely exhausted phenotype of CD8 T cells compared with PD-1 expression alone. Dual blockade in vivo effectively restored IFNγ production and controlled tumor growth (11). TIM-3 and PD-1 coexpression has also been reported on tumor-infiltrating Tregs (26), but in contrast to effector T cells, TIM-3 may function as a positive regulator on FoxP3+ Tregs and be a marker for a Treg population that is highly effective in inhibiting the TH1 and TH17 immune responses (27). Our data demonstrated a clear trend toward increasing frequency of PD-1+TIM-3+ CD8, CD4, as well as FoxP3+ T cells during the first 3 weeks after GL261-luc tumor implantation. These findings suggest that upregulation of checkpoints contributed to an increasingly immunosuppressive tumor microenvironment. Rescue of this severely tolerized population of antitumor T cells with dual blockade may be an explanation for the increased survival with dual checkpoint blockade.
Anti-TIM-3 + SRS
Dual therapy with anti-TIM-3 and SRS also resulted in an improved survival compared with anti-TIM-3 alone (63.16% vs. 0% OS). Although the mechanism underlying the synergistic response of immunotherapy and radiation has not been completely elucidated, our in vivo experiments demonstrated a clear survival benefit conferred by the use of SRS with checkpoint inhibition. These findings are supported by mounting evidence in the literature that radiation can improve the efficacy of conventional chemotherapy (3) as well as targeted immunotherapies (3, 28). It has been hypothesized that the cytocidal effects of local radiation allow for tumor antigen release and subsequent activation of the innate and adaptive immune response (3, 29, 30). Radiotherapy has also been associated with the abscopal effect, that is, the immune-mediated eradication of tumors distant from the site of radiation (31, 32). Lymphodepleting whole-body radiation (WBR) has also been posited as a way to “prime” the body for subsequent checkpoint therapy, as demonstrated by Jing and colleagues in a recent study of a 5T33 murine multiple myeloma model (33). Having previously shown that anti-PDL-1–mediated antitumor effect was significantly improved if administered after nonmyeloablative radiation (34), the authors reported that blocking both PDL-1 and TIM-3 resulted in a significant survival benefit compared with anti-PDL-1 or anti-TIM-3 monotherapy when administered after sublethal WBR (500cGy; ref. 33).
A single high dose of radiation (30 Gy) has also been shown to significantly improve the immune profile of CT26 colon tumors (35). Irradiated tumors were found to contain a 10-fold higher ratio of effector T cells to immunosuppressive myeloid-derived suppressor cells compared with nonirradiated tumors. However, the frequency of TIM-3 and/or PD-1 expression on T cells remained the same, suggesting that although radiation facilitates immune cell enrichment in tumors, BILs remain inactivated within the tumor microenvironment. These data are consistent with our own, supporting the hypothesis that radiation increases T-cell trafficking to the tumor, whereas anti-PD-1 and anti-TIM-3 reverse tumor-mediated BIL exhaustion, resulting in a more robust antitumor effect.
Triple therapy
Finally, triple therapy with anti-TIM-3, anti-PD-1, and SRS resulted in an OS of 100%. These findings suggest that the individual treatment modalities are complementary and that the multimodal activation of the immune system results in a robust antitumor response and high rates of long-term survivors. Flank rechallenge of all long-term survivors (>100 days) demonstrated that all “cured” animals could reject GL261-luc cells, implying that these mice possessed durable immune memory. This phenomenon has been previously described by Zeng and colleagues (3) in long-term survivor mice treated with anti-PD-1 + SRS. Our experiments further suggest that treatment with anti-TIM-3 does not abrogate this memory formation.
Immune profiling
To characterize the immune effects of each treatment, we conducted a series of flow cytometry–based analyses to compare the different treatment arms. Effector:regulatory BIL ratios have been associated with treatment efficacy and may be used as an indirect measure of antitumor activity (22, 36). Increased effector T-cell infiltration is also positively correlated with survival (37), while higher levels of Tregs negatively impact prognosis in glioblastoma multiforme patients (38). We demonstrated an overall trend toward a superior immune profile with triple therapy, as demonstrated by higher CD8:Treg ratios, lower frequency of FoxP3+ Tregs, and higher production of IFNγ, TNFα, and IL17a inflammatory cytokines. More specifically, we observed the highest CD8 T cell-to-Treg ratios as well as the lowest frequency of FoxP3+ CD4 T cells (Tregs) in the anti-PD-1 alone, anti-PD-1 + SRS, anti-PD-1 + anti-TIM-3, and triple therapy groups. There was no statistical difference between the anti-TIM-3 treatment arm and the control group, which was expected given that we did not see a difference in os between these two arms. Adding anti-TIM-3 to anti-PD-1 similarly did not improve the effector to regulatory cell ratios.
Another measure of effector T-cell activity is proinflammatory cytokine expression, and previous studies have shown that inhibitory checkpoints, such as PD-1 and TIM-3, negatively regulate IFNγ, TNFα, and IL17a secretion (12, 39). Polyfunctional T cells (secreting two or more different cytokines) also represent a more potent and durable subset of effector lymphocytes (40, 41); presence or increase in these multicytokine-secreting T cells may therefore reflect an improvement in the quality of immune response. Combining anti-TIM-3 with anti-PD-1 antibody did not significantly increase CD4 or CD8 T cell IFNγ production as compared with PD-1 blockade alone. However, when compared with anti-PD-1 + SRS dual therapy, triple therapy with anti-PD-1 + SRS + anti-TIM-3 demonstrated a trend toward increased IFNγ, TNFα, and IL17a production by CD4 or CD8 cells. This improvement in proinflammatory cytokine profile correlates with improved treatment effects and OS. One limitation of this experiment is the lack of a comprehensive understanding of the pro- and anti-inflammatory cytokine interactions in the tumor micro-environment, especially in the setting of radiation. Further study of the presence and levels of additional cytokines, such as TGFβ that has been shown to have both pro- and anti-inflammatory properties (42), may provide additional insights into the mechanism and limitations of our treatment strategy.
To elucidate the mechanism for tumor regression in mice that did not receive anti-PD-1 antibody, we depleted mice of CD4 or CD8 cells before treating with anti-TIM-3 and SRS. In our experiments, the survival benefit of adding SRS to TIM-3 blockade was completely abrogated by CD4 depletion, with no significant difference between median and OS between anti-TIM-3 + SRS + anti-CD4 versus anti-TIM-3 alone. CD8 depletion resulted in a partially abrogated effect, with a minimal improvement in median survival and 0% OS. Interestingly, these findings differ from those published by Zeng and colleagues for anti-PD-1 + SRS (3). Whereas CD8 depletion resulted in complete abrogation of the anti-PD-1 plus SRS antitumor activity, it appears that CD4 T cells may play a more prominent role in the anti-TIM-3 plus SRS response. Therefore, as might be expected, both populations were shown to be critical for the response to triple therapy with anti-TIM-3 + anti-PD-1 + SRS as demonstrated in our second set of depletion experiments.
Myeloid contributions
A previous study by Ngiow and colleagues defined the mechanism of TIM-3 blockade as both CD4 and IFNγ+CD8 T cell mediated, with a possible role for CD11b+ host DCs (20). Our results show that while TIM-3 is expressed at very low levels on microglia and brain infiltrating macrophages, CD11c+ DCs expressed TIM-3 at a significantly higher rate. We also found that these cells could be further categorized as CD11bhi or CD11blow DCs. Notably, the CD11blow population was seen to upregulate surface TIM-3 during the first 3 weeks after tumor implantation, while the CD11bhi DCs maintained a relatively constant level of expression during this time. TIM-3’s function on APCs is not understood and the literature yields inconsistent results confounded by differences in pathology, timing, and anatomic location (43–45). For instance, Anderson and colleagues found that TIM-3 had opposing functions on the innate and adaptive immune system, such that CD11b+CD11c+ DC-derived TIM-3 promoted TNFα secretion and TH1 response in the setting of experimental autoimmune encephalomyelitis (43). Their observation that TIM-3 expression was restricted to CD11b+ DCs and not CD11b+ macrophages was consistent with our own findings. However, Chiba and colleagues reported that TIM-3 activation on tumor-infiltrating DCs suppressed the innate immune response and attenuated the efficacy of targeted immunotherapies (44). The observed CD45+CD11c+CD11blow phenotype correlates with previous descriptions of “lymphoid DCs” (46), but the function of these particular APCs in the brain is not fully known. From a clinical standpoint, the efficacy of TIM-3 blockade has been shown to have only a minor dependence on CD11c+ DC activity (23). Therefore, the relevance and implications of TIM-3 upregulation on APCs will require further investigation.
TIM-3 expression in human glioblastoma multiforme
Human studies have previously confirmed the presence of TIM-3 in the setting of hepatocellular, cervical, colorectal, and ovarian cancers, as well as melanoma and leukemia (47–49). However, the presence of TIM-3 in human glioblastoma multiforme has not yet been clearly confirmed by IHC. A recent study by Liu and colleagues found that Gal-9 (the activating ligand for TIM-3) was expressed at increased level in glioma patients’ brain tissues as compared with noncancerous tissue from control patients (14). Furthermore, Gal-9 expression was significantly higher in grade 4 gliomas than in lower grade gliomas (grades 2–3), and levels of expression were associated with TIM-3 expression on CD4 and CD8 TILs.
In our final experiments, we found that 7 of 8 primary glioblastoma multiforme tissue samples stained positive for an anti-TIM-3 mAb stain. Five of the samples showed TIM-3–positive lymphocytes, four revealed a tumor cell staining pattern, and two were suspicious for TIM-3 positivity on microglia or other unidentified lysosome-containing immune cells. The IHC results are evidence that TIM-3 is expressed in human glioblastoma multiforme, and taken together with studies confirming Gal-9 expression on glioma tumor cells (14), these findings suggest that TIM-3 blockade may be an effective immunotherapeutic strategy in glioblastoma multiforme and provide a rationale for clinical translation of anti-TIM-3 therapies.
To our knowledge, this is the first preclinical investigation on the antitumor effects of TIM-3 blockade with SRS and/or anti-PD-1 in the setting of established glioma. Using a syngeneic orthotopic murine glioma model, we have shown that severely exhausted PD-1+TIM-3+ BILs accumulate in intracranial tumors in a time-dependent manner and that combination radiation and dual immune checkpoint blockade results in a significant increase in survival. Our study demonstrates the presence of TIM-3–expressing targets in human glioblastoma multiforme and provides preclinical evidence for a novel treatment combination that has potential to improve the antitumor immune response and result in durable immunity. Further study in the preclinical and clinical settings will be needed to assess the utility and efficacy of this combination therapy.
Supplementary Material
Translational Relevance.
To our knowledge, this is the first preclinical investigation on the antitumor effects of TIM-3 blockade with stereotactic radiosurgery and/or anti-PD-1 in the setting of established glioma. Using a syngeneic orthotopic murine glioma model, we demonstrate that severely exhausted PD-1+TIM-3+ lymphocytes accumulate in intracranial tumors in a time-dependent manner and that combination radiation and dual immune checkpoint blockade results in a significant increase in survival. Our study demonstrates the presence of TIM-3–expressing targets in human glioblastoma multiforme, provides preclinical evidence for a novel treatment combination that has potential to improve the antitumor immune response and result in durable immunity, and has direct implications for a clinical trial.
Acknowledgments
We thank T. Kochel for her support in conducting immunostaining experiments and sharing materials and space; B. Francica for providing anti-PD-1 antibodies from hybridoma; L. Blosser for flow cytometry support; and A. Bradford for scientific input.
Grant Support
This work was supported by Medical Student Research Fellowships from Howard Hughes Medical Institute (to J.E. Kim and M.A. Patel) and Bristol-Myers Squibb.
P.T. Tran is a consultant/advisory board member for Regeneron. J.M. Taube reports receiving commercial research grants from and is a consultant/advisory board member for Bristol-Myers Squibb. C.G. Drake reports receiving commercial research grants from Bristol-Myers Squibb; holds ownership interest (including patents) in Compugen, NexImmune, Potenza Therapeutics, and Tizona Biotech; and is a consultant/advisory board member for AZ Mediummune, Bristol-Myers Squibb, Compugen, Genentech, Merck, Potenza Therapeutics, and Tizona. H. Brem is a consultant/advisory board member for Accelerating Combination Therapies, AsciepiX, Camden Partners, Perosphere, and stemGen. M. Lim is a consultant/advisory board member for Aegenus, Bristol-Myers Squibb, Merck, and Oncorus; reports receiving commercial research grants from Aegenus and Bristol-Myers Squibb; and other commercial research support from Accuray, Aegenus, Altor, Arbor, Bristol-Myers Squibb, Celldex, and Immunocellular.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: J.E. Kim, M.A. Patel, C.M. Jackson, A.M. Martin, C.G. Drake, H. Brem, M. Lim
Development of methodology: J.E. Kim, M.A. Patel, H. Xu, D. Mathios, H. Brem, M. Lim
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): J.E. Kim, M.A. Patel, A. Mangraviti, E.S. Kim, D. Theodros, E. Velarde, A. Liu, E.W. Sankey, A. Tam, T. Garzon-Muvdi, B.M. Tyler, P.C. Burger, M. Lim
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J.E. Kim, D. Mathios, J.M. Taube, C.G. Drake, H. Brem, M. Lim
Writing, review, and/or revision of the manuscript: J.E. Kim, M.A. Patel, A. Mangraviti, E.S. Kim, D. Theodros, E. Velarde, A. Liu, D. Mathios, C.M. Jackson, M. Sheu, B.M. Tyler, P.T. Tran, X. Ye, A. Olivi, J.M. Taube, P.C. Burger, C.G. Drake, H. Brem, D.M. Pardoll, M. Lim
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): J.E. Kim, S. Harris-Bookman, A. Olivi, H. Brem, M. Lim
Study supervision: D. Mathios, X. Ye, M. Lim
Reprints and Subscriptions To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at pubs@aacr.org.
References
- 1.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 2.Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, et al. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the united states. Clin Cancer Res. 2010;16:2443–9. doi: 10.1158/1078-0432.CCR-09-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–9. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. 2013;19:4917–24. doi: 10.1158/1078-0432.CCR-12-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.See AP, Han JE, Phallen J, Binder Z, Gallia G, Pan F, et al. The role of STAT3 activation in modulating the immune microenvironment of GBM. J Neurooncol. 2012;110:359–68. doi: 10.1007/s11060-012-0981-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson C, Ruzevick J, Amin AG, Lim M. Potential role for STAT3 inhibitors in glioblastoma. Neurosurg Clin N Am. 2012;23:379–89. doi: 10.1016/j.nec.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Heimberger AB, Sun W, Hussain SF, Dey M, Crutcher L, Aldape K, et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro Oncol. 2008;10:98–103. doi: 10.1215/15228517-2007-046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu C, Anderson AC, Kuchroo VK. TIM-3 and its regulatory role in immune responses. Curr Top Microbiol Immunol. 2011;350:1–15. doi: 10.1007/82_2010_84. [DOI] [PubMed] [Google Scholar]
- 11.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175–86. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han S, Feng S, Xu L, Shi W, Wang X, Wang H, et al. Tim-3 on peripheral CD4 (+) and CD8(+) T cells is involved in the development of glioma. DNA Cell Biol. 2014;33:245–50. doi: 10.1089/dna.2013.2306. [DOI] [PubMed] [Google Scholar]
- 14.Liu Z, Han H, He X, Li S, Wu C, Yu C, et al. Expression of the galectin-9-tim-3 pathway in glioma tissues is associated with the clinical manifestations of glioma. Oncol Lett. 2016;11:1829–34. doi: 10.3892/ol.2016.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–96. [PubMed] [Google Scholar]
- 16.Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113:3821–30. doi: 10.1182/blood-2008-10-185884. [DOI] [PubMed] [Google Scholar]
- 17.Wong J, Armour E, Kazanzides P, Iordachita I, Tryggestad E, Deng H, et al. High-resolution, small animal radiation research platform with x-ray tomographic guidance capabilities. Int J Radiat Oncol Biol Phys. 2008;71:1591–9. doi: 10.1016/j.ijrobp.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng H, Kennedy CW, Armour E, Tryggestad E, Ford E, McNutt T, et al. The small-animal radiation research platform (SARRP): Dosimetry of a focused lens system. Phys Med Biol. 2007;52:2729–40. doi: 10.1088/0031-9155/52/10/007. [DOI] [PubMed] [Google Scholar]
- 19.Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS One. 2014;9:e101764. doi: 10.1371/journal.pone.0101764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 21.Waitz R, Solomon SB, Petre EN, Trumble AE, Fasso M, Norton L, et al. Potent induction of tumor immunity by combining tumor cryoablation with anti-CTLA-4 therapy. Cancer Res. 2012;72:430–9. doi: 10.1158/0008-5472.CAN-11-1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rizzuto GA, Merghoub T, Hirschhorn-Cymerman D, Liu C, Lesokhin AM, Sahawneh D, et al. Self-antigen-specific CD8+ T cell precursor frequency determines the quality of the antitumor immune response. J Exp Med. 2009;206:849–66. doi: 10.1084/jem.20081382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71:3540–51. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 24.Guo Z, Cheng D, Xia Z, Luan M, Wu L, Wang G, et al. Combined TIM-3 blockade and CD137 activation affords the long-term protection in a murine model of ovarian cancer. J Transl Med. 2013;11:215. doi: 10.1186/1479-5876-11-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117:4501–10. doi: 10.1182/blood-2010-10-310425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakuishi K, Ngiow SF, Sullivan JM, Teng MW, Kuchroo VK, Smyth MJ, et al. TIM3FOXP3 regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. 2013;2:e23849. doi: 10.4161/onci.23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur J Immunol. 2014;44:2703–11. doi: 10.1002/eji.201344392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barker CA, Postow MA. Combinations of radiation therapy and immunotherapy for melanoma: A review of clinical outcomes. Int J Radiat Oncol Biol Phys. 2014;88:986–97. doi: 10.1016/j.ijrobp.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demaria S, Bhardwaj N, McBride WH, Formenti SC. Combining radiotherapy and immunotherapy: a revived partnership. Int J Radiat Oncol Biol Phys. 2005;63:655–66. doi: 10.1016/j.ijrobp.2005.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–34. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379–88. doi: 10.1158/1078-0432.CCR-09-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silk AW, Bassetti MF, West BT, Tsien CI, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2:899–906. doi: 10.1002/cam4.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jing W, Gershan JA, Weber J, Tlomak D, McOlash L, Sabatos-Peyton C, et al. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. J Immunother Cancer. 2015;3:2. doi: 10.1186/s40425-014-0043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kearl TJ, Jing W, Gershan JA, Johnson BD. Programmed death receptor-1/programmed death receptor ligand-1 blockade after transient lymphodepletion to treat myeloma. J Immunol. 2013;190:5620–8. doi: 10.4049/jimmunol.1202005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filatenkov A, Baker J, Mueller A, Kenkel JA, Ahn GO, Dutt S, et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res. 2015;21:3727–39. doi: 10.1158/1078-0432.CCR-14-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grauer OM, Nierkens S, Bennink E, Toonen LW, Boon L, Wesseling P, et al. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J Cancer. 2007;121:95–105. doi: 10.1002/ijc.22607. [DOI] [PubMed] [Google Scholar]
- 37.Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res. 2011;17:4296–308. doi: 10.1158/1078-0432.CCR-10-2557. [DOI] [PubMed] [Google Scholar]
- 38.Jacobs JF, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJ, Wesseling P, et al. Prognostic significance and mechanism of treg infiltration in human brain tumors. J Neuroimmunol. 2010;225:195–9. doi: 10.1016/j.jneuroim.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 39.Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009;39:2492–501. doi: 10.1002/eji.200939274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilde S, Sommermeyer D, Leisegang M, Frankenberger B, Mosetter B, Uckert W, et al. Human antitumor CD8+ T cells producing Th1 poly-cytokines show superior antigen sensitivity and tumor recognition. J Immunol. 2012;189:598–605. doi: 10.4049/jimmunol.1102165. [DOI] [PubMed] [Google Scholar]
- 41.Ding ZC, Huang L, Blazar BR, Yagita H, Mellor AL, Munn DH, et al. Polyfunctional CD4(+) T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood. 2012;120:2229–39. doi: 10.1182/blood-2011-12-398321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M, Kleber S, Rohrich M, Timke C, Han N, Tuettenberg J, et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011;71:7155–67. doi: 10.1158/0008-5472.CAN-11-1212. [DOI] [PubMed] [Google Scholar]
- 43.Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of tissue inflammation by the immune receptor tim-3 expressed on innate immune cells. Science. 2007;318:1141–3. doi: 10.1126/science.1148536. [DOI] [PubMed] [Google Scholar]
- 44.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13:832–42. doi: 10.1038/ni.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suter T, Biollaz G, Gatto D, Bernasconi L, Herren T, Reith W, et al. The brain as an immune privileged site: Dendritic cells of the central nervous system inhibit T cell activation. Eur J Immunol. 2003;33:2998–3006. doi: 10.1002/eji.200323611. [DOI] [PubMed] [Google Scholar]
- 46.Hesske L, Vincenzetti C, Heikenwalder M, Prinz M, Reith W, Fontana A, et al. Induction of inhibitory central nervous system-derived and stimulatory blood-derived dendritic cells suggests a dual role for granulocyte-macrophage colony-stimulating factor in central nervous system inflammation. Brain. 2010;133:1637–54. doi: 10.1093/brain/awq081. [DOI] [PubMed] [Google Scholar]
- 47.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175–86. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng J, Chan PL, Liu Y, Qin G, Xiang Z, Lam KT, et al. ICOS regulates the generation and function of human CD4+ treg in a CTLA-4 dependent manner. PLoS One. 2013;8:e82203. doi: 10.1371/journal.pone.0082203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S, et al. A TIM-3/gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Stem Cell. 2015;17:341–52. doi: 10.1016/j.stem.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 50.Verhaegen F, Granton P, Tryggestad E. Small animal radiotherapy research platforms. Phys Med Biol. 2011;56:R55–83. doi: 10.1088/0031-9155/56/12/R01. [DOI] [PubMed] [Google Scholar]
- 51.Ford E, Purger D, Tryggestad E, McNutt T, Christodouleas J, Rigamonti D, et al. A virtual frame system for stereotactic radiosurgery planning. Int J Radiat Oncol Biol Phys. 2008;72:1244–9. doi: 10.1016/j.ijrobp.2008.06.1934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.