

Abstract
The mechanistic underpinnings of the complex process of plant polysaccharide biosynthesis are poorly understood, largely due to the resistance of glycosyltransferase (GT) enzymes to structural characterization. In Arabidopsis thaliana, a glycosyl transferase family 37 (GT37) fucosyltransferase-1 (AtFUT1) catalyzes the regiospecific transfer of terminal 1,2-fucosyl residues to xyloglucan side chains – a key step in the biosynthesis of fucosylated sidechains of galactoxyloglucan. We unravel the mechanistic basis for fucosylation by AtFUT1 with a multipronged approach involving protein expression, X-ray crystallography, mutagenesis experiments and molecular simulations. Mammalian cell culture expressions enable sufficient production of the enzyme for X-ray crystallography, which reveals the structural architecture of AtFUT1 in complex with bound donor and acceptor substrate analogs. The lack of an appropriately positioned active site residue as a catalytic base leads us to propose an atypical water-mediated fucosylation mechanism facilitated by an H-bonded network, which is corroborated by mutagenesis experiments as well as detailed atomistic simulations.
Keywords: Arabidopsis thaliana, Fucosylation, Hemicellulose Synthesis, Fucosyltransferase, Reaction Mechanism
1. INTRODUCTION
Polysaccharides are the most abundant biopolymers on earth, largely due to the fact that they constitute the major components of the plant cell wall, which surrounds every plant cell. Apart from being an abundant source of dietary fiber and playing a major role in human nutrition, cell-wall-rich biomass derived from plants also represents an abundant and natural carbon resource that has immense potential as a feedstock for the large-scale production of renewable chemicals, fuels, and materials. Our efforts towards developing capabilities to fully realize this potential has led to the appreciation of the value of plant biomass, which has fostered significant interest in the enzymatic catalysts and their fundamental molecular mechanisms.
Although quite a few plant glycosyl transferases (GT) and glycan modifying enzymes have been identified, their structural characterization has been particularly challenging and elusive. The structure and mechanism of cellulose synthase, perhaps the most widely studied GT involved in cell wall biogenesis, is becoming clearer as a result of recent crystal structures and in crystallo enzymology of the bacterial cellulose synthase BcsA–BcsB complex.(Morgan et al., 2013, Morgan et al., 2014, Morgan et al., 2016) However, the plant cellulose synthase complex and other GTs involved in the biosynthesis of plant cell walls have until recently resisted crystallographic analysis.
Xyloglucans, major components of the plant cell wall, are structurally complex hemicellulosic polysaccharides, involved in cell growth and expansion, energy metabolism, and signaling.(Pauly and Keegstra, 2016) Xyloglucans consist of a β-(1,4)-D-glucan backbone decorated by regularly distributed side-chains characteristically initiated by α-D-Xylp residues at O-6 and often extended by addition of galactosyl and fucosyl substituents.(Tuomivaara et al., 2015) The xyloglucan common in gymnosperms and many angiosperms comprises cellotetraose repeating units bearing three α-D-Xylp substituents.(Vincken et al., 1997) This core structure is represented by the sequence XXXG using the widely accepted xyloglucan nomenclature,(Fry et al., 1993, Tuomivaara et al., 2015) where an uppercase G represents an unsubstituted β-D-Glcp backbone residue and X denotes a β-D-Glcp backbone residue appended at O-6 with a single α-D-Xylp. Xyloglucans in primary cell walls often bear the common diglycosyl side-chain β-D-Galp-(1,2)-α-D-Xylp, designated L, and the triglycosyl side-chain α-L-Fucp-(1,2)-β-D-Galp-(1,2)-α-D-Xylp, denoted by F, which combine to form repeating units such as XXFG and XLFG. To date, 24 unique, naturally occurring xyloglucan side-chain structures have been described.(Tuomivaara et al., 2015, Pauly and Keegstra, 2016)
Xyloglucan, like many eukaryotic glycan polymers, is synthesized by Golgi localized glycosyltransferases (GTs), which employ nucleotide-sugar donors to catalyze single monosaccharide extensions of glycan acceptor molecules. Sequence-based families of GTs have been identified and compiled in the Carbohydrate-Active Enzymes (CAZy) database.(Coutinho et al., 2003) The major GTs involved in synthesis of fucogalactoxyloglucan in the model plant Arabidopsis thaliana have been identified,(Zabotina et al., 2012, Pauly and Keegstra, 2016) and synthesis of this polysaccharide requires at least one β-(1,4)-D-glucan synthase (GT2), one or more α-1,6-D-xylosyltransferases (GT34), two distinct β-1,2-D-galactosyltransferases (GT47), and one α-1,2-fucosyltransferase (GT37). Fucosylation of xyloglucan in A. thaliana is catalyzed by AtFUT1, a type II membrane protein localized in the Golgi apparatus with a luminal catalytic domain. AtFUT1 uses an inverting mechanism to transfer a fucose (Fuc) residue from a β-linked guanosine 5′-diphospho-β-L-fucose (GDP-Fuc) donor exclusively to the galactose (Gal) residue of the L-side chain closest to the reducing end of the xyloglucan subunit, as in XXLG or XLLG, to form a side chain terminated by an α-linked Fuc, as in XXFG or XLFG. (Figure 1a).
Figure 1.
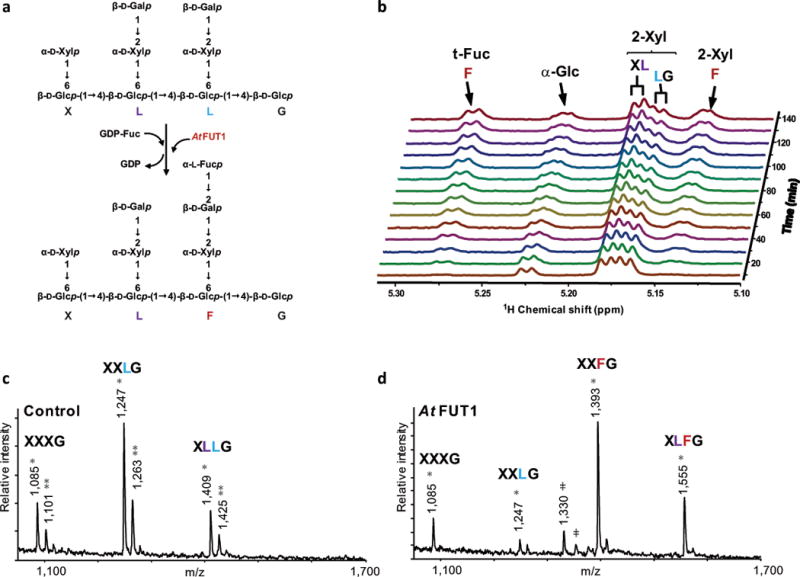
AtFUT1-catalyzed regiospecific transfer of a fucosyl residue from GDP-Fuc to the xyloglucan oligosaccharide XLLG. (a) In vitro fucosyltransferase scheme. (b) Real-time 1D 1H NMR analysis of products generated during the reaction catalyzed by AtFUT1 using GDP-Fuc as donor and XLLG as acceptor for 2.5 h. During the course of the reaction, the intensity of the signal for the galactosyl residue closer to the reducing end (L light blue) decreases. At the same time, the two signals of the fucosylated side chain (F, red) steadily increase. The signal of the galactosyl residue closer to the non-reducing end in the oligosaccharide (L, blue) remains unchanged. (c, d) In vitro fucosylation of fut1–3 xyloglucan. MALDI-TOF MS spectra of xyloglucan oligosaccharides generated by XEG treatment of fut1–3 xyloglucan before (c) and after (d) in vitro fucosylation by AtFUT1. Single asterisks (*) and double asterisks (**) indicate the [M + Na+]+ and [M + K+]+ adducts, respectively. Unknown peaks are also indicated (ǂ). Please see Table S1 for a complete list of calculated and observed m/z values for xyloglucan oligosaccharides.
Understanding glycosyltransferases at the molecular level, through a combination of structural and kinetic studies, is paramount for gaining fundamental insight into regiospecific glycopolymer biosynthesis. Crystal structures of eukaryotic glycosyltransferases, especially from plants, are highly underrepresented in structural databases despite their biological importance. This largely results from difficulties in expressing and purifying high-quality preparations of these eukaryotic enzymes in quantities sufficient for crystallization and diffraction analysis. This has created a significant gap in our understanding of the fundamental mechanistic underpinnings of plant glycosyltransferases, especially fucosyltransferases. In this study, we demonstrate a strategy for the functional expression, X-ray crystallography, kinetic and mutagenic analyses of the plant glycosyltransferase AtFUT1. The experimental crystal structure data lays the foundation for detailed molecular simulations of the ternary (i.e. enzyme-donor-acceptor) complex in an effort to unravel the molecular basis for the fucosylation reaction mechanism. Molecular dynamics (MD) simulations reveal the sustained presence of a crucial water molecule at the active site and present putative active site configurations that lead to the proposal of an atypical water mediated SN1-like reaction mechanism. The quantum mechanical calculations of this proposed reaction mechanism reveal a feasible transition state structure involving the active site water molecule that enables the phosphate group to act as a base. Further, our data lend insight into the biochemical defect present in the classic cell wall mutant murus 2 (mur2), demonstrating that the point mutation results in a disruption of the H-bonding network that AtFUT1 utilizes to stabilize the transition state to conduct the fucosylation reaction.
In a very recent study, Rocha et al. performed an independent structural characterization of the AtFUT1 enzyme in complex with GDP, acceptor XLLG and propose a plausible reaction mechanism.(Rocha et al., 2016) We compare and contrast our multi-pronged research findings on AtFUT1 to challenge the proposed mechanism wherein the amino acid residue Asp300 is suggested to act as a base for an SN2 catalyzed mechanism.
2. RESULTS AND DISCUSSION
2.1 Recombinant production in HEK cells yields active AtFUT1 that fucosylates XXLG with the same regiospecificity found in planta
The successful expression of plant GTs has been a major stumbling block for their structural elucidation.(Held et al., 2015, Attia and Brumer, 2016) Functional expression of eukaryotic GTs requires a proper folding environment that provides for disulfide bond formation and other post-translational modifications, such as glycosylation. To address this challenge, mammalian cell expression systems such as Human Embryonic Kidney (HEK) Cells, which provide an appropriate environment in the secretory pathway for folding and post-translational modification, are becoming increasingly popular for the heterologous overexpression of secreted proteins.(Subedi et al., 2015) For example, we have recently characterized enzymes involved in xylan biosynthesis after expressing them in HEK293 cells.(Urbanowicz et al., 2014) We have also used a derived cell line (HEK293S (GnTI−)) lacking N-acetylglucosaminyltransferase I (GnTI)(Reeves et al., 2002) to express GTs whose N-glycan substituents retain Man5-GlcNac2 structures that can be efficiently trimmed using endo- and exoglycosidases to generate chemically homogeneous proteins that are amenable to diffraction analysis.
Expression of the soluble catalytic domain of AtFUT1 was achieved by transient transfection of suspension culture HEK293 cells using a fusion protein strategy similar to prior studies on mammalian GTs.(Meng et al., 2013) A truncated form of AtFUT1, devoid of the NH2-terminal cytoplasmic tail and transmembrane domain, was generated as a fusion protein containing an NH2-terminal signal sequence followed by an 8xHis tag, an AviTag, “superfolder” GFP, the TEV protease recognition site, and amino acid residues 81–558 of AtFUT1 (termed GFP-AtFUT1). Expression and secretion of GFP-AtFUT1 in wild type HEK293 and HEK293S (GnTI−) cells yielded high levels of secreted recombinant fusion protein based on GFP fluorescence (~120 and ~100 mg/L, respectively).(Meng et al., 2013) The yields that we were able to achieve using HEK-based expression are exceedingly higher than the recombinant expression of HisΔ68-AtFUT1 carried out by Cicéron et al. using insect-cell cultures (2016) and used by Rocha et al. for structural analysis (2016), where they were only able to achieve 4 mg L−1 after extensive optimization.(Cicéron et al., 2016, Rocha et al., 2016) Expression of GFP-AtFUT1 in the glycosylation mutant HEK293S (GnTI−) cells limited N-glycosylation to Man5GlcNAc2 structures and allowed the cleavage of these glycans with endoglycosidase F1 (EndoF1), resulting in a single GlcNAc residue at the glycosylation site.
This workflow was employed for AtFUT1, analogous to prior mammalian GT structural studies,(Meng et al., 2013) wherein the initial recombinant GFP-AtFUT1 product was purified by Ni2+-NTA chromatography, yielding 93 mg of GFP-AtFUT1. This was followed by enzymatic removal of tag sequences and glycan structures from 90 mg of GFP-AtFUT1 by concurrent digestion with recombinant TEV protease and EndoF1. Purification of deglycosylated AtFUT1 was achieved first by an additional Ni2+-NTA step, yielding 33.2 mg of protein. This was followed by size exclusion chromatography, which resulted in quantities of purified protein (17.4 mg) sufficient for employing protein crystallography.
AtFUT1 is a xyloglucan specific glycosyltransferase that catalyzes the transfer of fucosyl residues exclusively to the O-2 of the galactosyl residue in side chains adjacent to the unsubstituted glucose (XXLG) in the xyloglucan backbone in vivo. To determine if the recombinant AtFUT1 has the same regiospecificity in vitro, we used real-time 1D 1H NMR to characterize the fucosylated products generated during the course of the reaction. The xyloglucan oligosaccharide XLLG (Figure 1a), composed of nine glycosyl residues and containing two identical galactose-containing L-side chains attached to the oligosaccharide backbone at different positions, was used as an acceptor. NMR analysis showed that the signal associated to the galactose closer to the reducing end of the oligosaccharides (XLLG) steadily decreased while signals corresponding to the fucosyl and xylosyl residues in side chain F appeared (Figure 1b). The signal for the other galactose (XLLG) remained unchanged during the course of the reaction. These results confirm that recombinant AtFUT1 catalyzes the fucosylation of xyloglucan oligosaccharides in vitro with the same regiospecificity that is found in planta.(Perrin et al., 1999) The Arabidopsis thaliana mutant fut1–3 (Salk_139678)(Alonso et al., 2003) contains a T-DNA insertion in the second exon of the AtFUT1 (At2g03220), resulting in plants that do not produce the AtFUT1 protein and are thus unable to add fucosyl residues to nascent xyloglucan. Non-fucosylated xyloglucan polymer was isolated from fut1–3 (Salk_139678) mutant plants,(Peña et al., 2012) and used as an acceptor substrate for recombinant AtFUT1, which complemented the xyloglucan chemotype of this mutant in vitro (Figure 1 c, d).
2.2 Crystal structures reveal atomistic architecture of AtFUT1
Here we present two structures of AtFUT1 – one in complex with GDP and the other with the model xyloglucan oligosaccharide acceptor XXLG. Data for our structures enabled refinements to resolutions of 1.79 Å for AtFUT1-XXLG and 1.90 Å for AtFUT1-GDP, with R- and Rfree-factors of 0.166/0.217 and 0.223/0.289 for AtFUT1-XXLG and AtFUT1-GDP, respectively (Table S2a). There are two molecules in the asymmetric unit in the case of AtFUT1-GDP and four molecules present for AtFUT1-XXLG. In addition to the GDP- and XXLG-molecules bound at the active site, multiple MES, glycerol and ethylene glycol molecules were also found on the surface of both structures (Figure 2). AtFUT1 exhibits a GT-B fold(Bourne and Henrissat, 2001) with two β/α/β Rossmann-like domains and hence does not contain a DXD motif or a bound metal ion to facilitate nucleotide sugar interaction, as found in enzymes that contain a GT-A fold.(Lairson et al., 2008) The crystal structures have significantly different cell dimensions, despite being grown under the same conditions. Sometimes, three different crystal forms could be observed in the same drop. However, due to crystal contacts, only the crystals described here were able to incorporate GDP or XXLG, respectively. Conformational validation of XXLG indicated no serious problems (Table S2b). Xylose 619, the furthest away sugar residue from active site, was found to be in a higher energy conformation. However, manual inspection of the electron density showed it to be correctly built. The cause of the strain most likely is the weak and noisy density in the bulk solvent region. These structures have been deposited to the Protein Data Bank (http://www.rcsb.org/) with the following PDB-codes: AtFUT1-XXLG: 5KOE, AtFUT1-GDP: 5KWK (release date: 2016-09-28).
Figure 2.
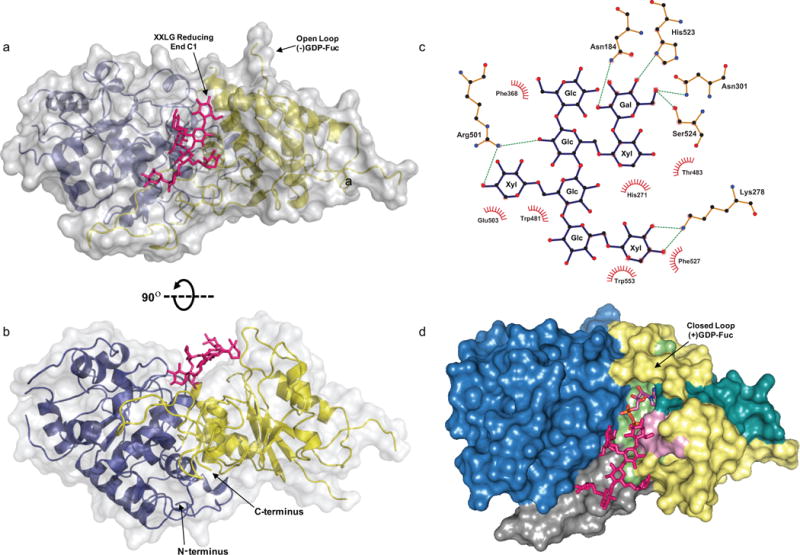
The FUT1-XXLG structure. (a–b) The secondary structures are displayed as cartoon models with transparent surfaces. The N- and C-terminal Rossmann-like folds are shown in blue and yellow, respectively. XXLG is labeled and shown as pink sticks. (c) LigPlot+ 2-D interaction map of the residues that interact with the oligosaccharide acceptor, XXLG. Glycosyl residues in XXLG are labeled. C1 of the reducing end Glc (XXLG) and C2 of Gal (XXLG), where fucosyl addition occurs, are indicated by arrows. (d) A surface-colored model of the structurally aligned donor and acceptor complexes of AtFUT1 is shown. The N-terminal and C-terminal Rossmann like domains are shown in blue and yellow, respectively and α-2/6-motif I (pink), α-2-motif-III (cyan), and α-6-motif III (green) are indicated. The C-terminal proline-rich region, specific to GT37 enzymes and predicted to be involved in acceptor substrate binding, is highlighted in grey.
The crystallographic studies presented here along with that of Rocha et al.’s (PDB IDs: 5KOR; release date: 2016-10-12) constitute the first ever structures of plant fucosyl transferases. While Rocha et al. describe the XLLG, GDP bound AtFUT1 structure, we describe crystal structures for AtFUT1 in complex with the glycosyl donor analog GDP (AtFUT1-GDP) and with a closely related oligosaccharide acceptor XXLG (AtFut1-XXLG). The ligands are observed to be located in a cleft formed between the two Rossmann-like domains (Figure 2,3). Similarly to what was reported by Rocha et al., the position of the fucosyl residue in AtFUT1-GDP (Figure 3) could not be determined reliably in the electron density maps, most likely due to partial hydrolysis of the fucosyl residue and/or the existence of multiple conformations (Appendix S1, Figure S1), including putative states in which the glycosidic bond is highly polarized or even partially dissociated. The existence of both XXLG and GDP bound structures enabled us to superimpose these structures (Figure 2d) and model the possible ternary complex (Materials & Methods: Preparation of the AtFUT1-GDP-Fuc-XXLG ternary complex).
Figure 3.
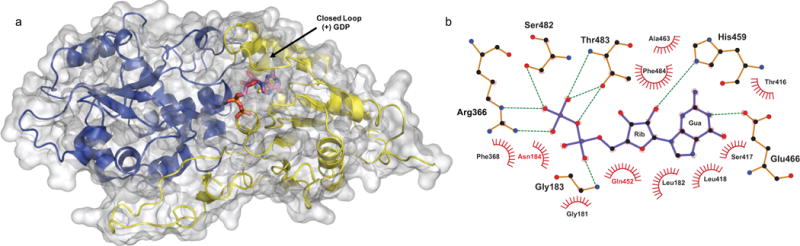
The FUT1-GDP structure. (a) The secondary structure is displayed as a cartoon model with transparent surfaces, and the N- and C-terminal Rossmann-like folds are shown in blue and yellow, respectively. GDP is labeled and shown as sticks with cyan carbon atoms, blue nitrogens, orange phosphates, and red oxygens. (b) LigPlot+ 2D ligand-protein interaction diagram of the AtFUT1-GDP complex. Hydrogen bonds are shown by green dotted lines. Red spoked arcs represent residues making nonbonded contacts with GDP, depicted in purple.
Figure 4 depicts the structural alignment of the AtFUT1 complex with GDP and XLLG by Rocha et al. (PDB ID 5KOR), AtFUT1 complexed with GDP (PDB ID 5KWK), and AtFUT1 in complex with XXLG (PDB ID 5KOE). The structural alignment of the 2 substrate bound structures in this study with the ternary complex from Rocha et al. resulted in similarities between the 3 structures as indicated by an overall Cα RMSDs of 0.44 Å and 0.36 Å, respectively. Upon closer inspection, we find that even the substrate binding modes for both the acceptor and donor are quite similar in these structures. The conformations of the GDP substrate in PDBs 5KOR and 5KWK are nearly identical, which is evident from the values of the α [56.1° vs. 51.1°], β [106.7° vs. 103.0°], γ [−90.44° vs. −77.61°] torsion angles.
Figure 4.
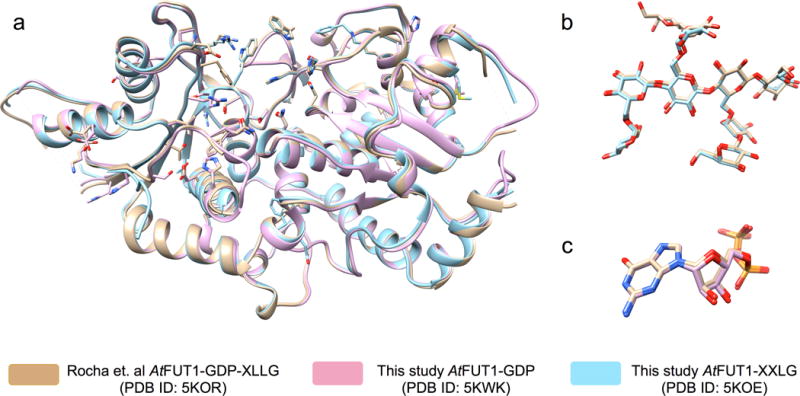
3-D structural alignment of substrate bound AtFUT1 complexes. a. Overall alignment of the protein backbone with side chains of important active site residues. The substrates have been hidden for clarity. The ternary complex from Rocha et al. (PDB ID: 5KOR) is shown in Brown, our GDP bound (PDB ID: 5KWK) and XXLG bound (PDB ID: 5KOE) AtFUT1 structures are shown in pink and blue respectively. b. Alignment of the acceptor substrate XLLG from PDB ID: 5KOR (carbon atoms shown in brown) and XXLG from PDB ID: 5KOE (carbon atoms shown in blue) c. Alignment of the Donor substrate GDP from PDB ID: 5KOR (carbon atoms shown in brown) and from PDB ID: 5KWK (carbon atoms shown in pink). The alignments performed using the MatchMaker tool in the UCSF Chimera visualization package.
2.3 Donor and acceptor binding interactions at the active site
AtFUT1 is classified in the CAZy database as a member of GT37; however, information regarding the structure or folding properties of this protein or family were unavailable until recently. All members of the fucosyltransferase superfamily utilize the same donor substrate, GDP-Fuc. While overall sequence similarity among different fucosyltransferase families is very low, phylogenetic studies have revealed relationships between α-(1,2)-, α-(1,6)- and protein-O-fucosyltransferases from eukaryotes and prokaryotes.(Oriol et al., 1999) According to the classification system developed by the CAZy database,(Coutinho et al., 2003) these enzymes are found in families GT11, GT23, GT37, GT65, GT68, and GT74. Amino acid sequence alignment of the GT37 family from A. thaliana has revealed several conserved motifs (Figure S2). These conserved motifs contain amino acids involved in binding the donor in α-(1,2)-, α-(1,6)- and protein O-fucosyltransferases.(Oriol et al., 1999, Lehner et al., 2015)
Members of the plant specific GT37 family contain three of these conserved regions (α-2/6-motif I, α-2-motif-III and α-6-motif III) (Figure 2d, Figure S2). Several of the residues in AtFUT1 that are now known to interact with GDP are conserved in the GT37 family, with the exception of Asp300 and Ser552, including Arg366, Glu466, Ser482, Thr483, and Tyr486, are located within these motifs (Figure S2).
The orientation of the guanine moiety is in the anti conformation with a torsion angle (χ) of 176.8°, with 3T2 (C2′-endo) puckering for the ribose. The pyrophosphate group of GDP shows atypical torsion angles (α, β, γ are 51.1°, 103°, and −77.7° respectively). This twisted conformation of the pyrophosphate has been observed for other fucosyltransferases including Helicobacter pylori α-(1,3)-fucosyltransferase (HpFucT), C. Elegans CePOFUT1 and the substrate bound structures of AtFUT1 by Rocha et al.(Sun et al., 2007, Lira-Navarrete et al., 2011) Our data further supports the observation that fucosyltransferases twist the pyrophosphate in their GDP complex substrates to conform to the active site. Notably, this ‘back-bent’ nucleotide sugar conformation has also been observed for O-GlcNAc transferase (OGT), which positions the sugar in close proximity to the α-phosphate of the donor that functions as the catalytic base.(Schimpl et al., 2012) In the crystal structure it is observed that the guanine moiety of GDP in AtFUT1 is stabilized by hydrogen bonds to Glu466, His459, and by Van der Waals interactions with multiple residues nearby (Figure 3b). The phosphodiester component of GDP is held in place by hydrogen bonds to Gly183, Arg366, Ser482 and Thr483. This observation is further corroborated in molecular dynamics trajectories of the ternary complex (AtFUT1-GDP-Fuc-XXLGXXLG), which establishes the involvement of these specific residues in facilitating GDP-Fuc and XXLGXXLG binding. The energetic analyses from MD trajectories also indicate that GDP-Fuc binding is predominantly driven by electrostatic interactions (Appendix S2, Table S3a). These binding interactions are nearly identical to those recently reported by Rocha et al in their crystal structure.(Rocha et al., 2016) Another interesting observation is that GDP is coordinated by more water-mediated contacts than direct contacts with AtFUT1 to facilitate the release of the spent donor. The binding of GDP and GDP-Fuc to fucosyltransferases has been described for other members of the fucosyltransferase superfamily(Brzezinski et al., 2012, Chen et al., 2012) and the conformation of the GDP in AtFUT1 is almost identical (Figure S3) to the one observed in NodZ by Brzezinski et al.(Brzezinski et al., 2012) Closer inspection of GDP-fucose binding in the catalytically competent FUT1 complex (Figure S3) shows that the oxygen atoms of the second phosphate moiety of GDP are at ideal hydrogen bonding distances from Arg366.
The binding of XXLG to AtFUT1 in the crystal structure is characterized by a small number of contacts (Figure 2c) relative to the size of the molecule, indicating the plasticity of the acceptor substrate. This plasticity is further illustrated by the near identical binding modes of XLLG and XXLG substrates between the structures elucidated here and by Rocha et al.(Rocha et al., 2016) Indeed, AtFUT1 is not affected by the presence of an L side-chain in the middle of the oligosaccharide (XLLG) for the regiospecific addition of the fucosyl moiety (Figure 1b), readily converting both XLLG or XXLG to XLFG or XXFG, respectively. In the crystal structure, the Gal residue is well stabilized at the active site with hydrogen bonds to Asn184, Asn301, His523 and Ser524 (Figure 2c), which is consistent with the binding interactions observed by Rocha et al.(Rocha et al., 2016) Additional residues observed to contribute to acceptor binding in MD simulations of the XXLGXXLG substrate include electrostatic contributors such as Asp300, Glu370, Arg501; as well as the aromatic residues, Trp481, Phe368, and Lys278 (Appendix S2, Table S3b). A notable observation from the MD simulation analysis is that, unlike the donor, the acceptor XXLGXXLG is stabilized by a combination of both electrostatic and Van der Waals interactions.
Interestingly, AtFUT1 is able to fucosylate the galacturonic acid (GalA) residue of the uncommon side-chain Y of XXYG to produce XXZG in planta, where Y is the diglycosyl side-chain (β-D-GalpA-(1,2)-α-D-Xylp) and Z is the triglycosyl side-chain (α-L-Fuc-(1,2)-β-D- GalpA -(1,2)-α-D-Xylp).(Peña et al., 2012) This indicates that the enzyme is able to recognize the two acceptor residues regardless of the oxidation state at C6. A closer look at Gal in the acceptor bound AtFUT1 structure points to several factors that support the feasibility of accommodating GalA for fucosylation. The environment around C6 is observed to be partially solvent exposed with the presence of putative stabilizing enzyme interactions at the active site with Asn301, Ser524 and His523 (Figure 5). However, the non-reducing end Y side-chain of the oligosaccharides YXXG and XYXG were never found to be fucosylated in vivo, showing again the importance of the side-chain location in the oligosaccharide for enzyme activity.
Figure 5.
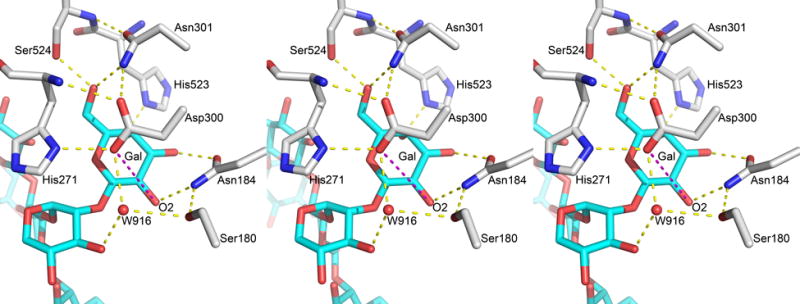
Environment of Asp300 in crystal structures. Figure shows the orientation of Asp300 and H-Bond network around Galactose2 sugar in stereo view (left and center depict a diverged-eye view while the center and right depict the cross-eyed view) from PDB ID: 5KOE. The important distance between Asp300OD2 and GalO2 is shown in magenta and is at a value of 5.4 Å. The other H-bonds are depicted in yellow dashed lines and are between 2.7 and 3.0 Å.
In contrast to Gal, the rest of the xyloglucan oligosaccharide has only two hydrogen bond partners, Lys278 and Arg501, coordinated to the first (XXLG) and second xylosyl (XXLG) containing side-chain structures from the non-reducing end, respectively. The galactose ring that accepts fucose during the reaction is positioned in proximity to the two alternative fucose positions near the GDP binding site (Figure S1). It is important to emphasize the significance of this crystallographic information about donor and acceptor bound AtFUT1, since it enables the next level of experimental and computational investigations, (described in subsequent sections), that are crucial to the unraveling of AtFUT1 mechanism.
2.4. Reaction Mechanism
The absence of fucosyltransferase ternary crystal structures (enzyme, acceptor and donor) with bound fucose has challenged the elucidation of an unequivocal mechanism for fucosylation. While it is known that the reaction is mediated via a direct nucleophilic substitution of the donor fucose residue (С1 of the fucose) by the acceptor (O2 of the galactose) with inversion at the anomeric center, two mechanisms for fucosylation have been proposed.(Lira-Navarrete et al., 2011, Kötzler et al., 2013) Human core α(1–6)-fucosyltransferase (FUT-8), belonging to the GT-B family, is proposed to employ a single-step SN2 mechanism(Kötzler et al., 2013) with base catalyzed deprotonation of the acceptor nucleophile, consistent with classical models for inverting glycosyltransferases.(Lairson et al., 2008) Similarly, CePOFUT2 and Helicobacter pylori fucosyltransferase (FucT), both belonging to the GT-B family, have a proximally located glutamic acid residue that has been implicated as the catalytic base that activates the nucleophile on the acceptor.(Sun et al., 2007, Valero-González et al., 2016) In contrast, Caenorhabditis elegans protein O-fucosyltransferase (CePOFUT1), also belonging to the GT-B family, is proposed to employ an SN1 like(Lira-Navarrete et al., 2011) mechanism, where the β–phosphate of the GDP-Fuc donor acts as the catalytic base to deprotonate the hydroxyl oxygen of the acceptor and, along with an associated Arg residue, facilitates glycoside bond cleavage and formation of a transient oxocarbenium ion that is subsequently attacked by the acceptor. The mechanisms in these enzymes are relevant to AtFUT1 since it belongs to the GT-B family and involves transfer of a fucose residue from a GDP-Fuc donor.
2.4.1 Sequence alignment, structure analysis, and in silico studies point to the lack of an active site catalytic base
The search for the residue that could serve as a probable catalytic base in the active site of AtFUT1 concluded with only one likely candidate – Asp300, which was thoroughly investigated by detailed analysis of crystal structures, molecular modeling and mutagenesis experiments (described further in section 2.5). However, this residue was found to be unsuitable for this role based on several reasons described in more detail below. First, Asp300 is not universally conserved throughout homologous proteins. Our sequence alignment shows that aspartic acid (Asp, D) is present in this position in only two of the ten members of the GT37 family in Arabidopsis. Further, the remaining eight proteins have asparagine (Asn, N) in the position equivalent to Asp300 in AtFUT1, which cannot serve as a catalytic base (Figure S2).
Secondly, one of the important prerequisites for Asp300 to act as a base is its proximity to the nucleophile (Gal O2). In analogous systems that employ an enzymatic residue as a base (O-GlcNac transferases), the base atom to nucleophile atom distance in catalytically competent enzyme-substrate complexes has been observed to be 3 Å.31 However, this assumes that the atoms in question can move closer to form the transition state complex. For proton transfer between two oxygen atoms the ideal distance is below 2.5 Å.(Scheiner and Harding, 1981, Hillenbrand and Scheiner, 1984, Lapid et al., 2005) In both available crystal structures of the AtFUT1-acceptor complex (PDB ID 5KOE and 5KOR), the Asp300 sidechain is observed in the same conformation and is well stabilized via three H-bonds to Asn301NH2, His271N and His271ND1 (Figure 5). Further, the Asp300OD2-GalO2 distance is 5.0 Å and 5.1 Å for two copies of the complex in 5KOR, and 5.4 Å in 5KOE. Such a long distance would preclude participation of Asp300 in catalysis as a base.
This is also supported by insights from the in silico AtFUT1 ternary complex constructed from the XXLG and GDP bound crystal structures (Materials & Methods: Preparation of the AtFUT1-GDP-Fuc-XXLG ternary complex) that enable the investigation of catalytically competent active site topologies via MD simulations. In our 70ns MD simulations, the Asp300OD2 to GalO2 distance averages at ~6.5 ± 1.2 Å and spans between a minimum of 3.1 Å and a maximum of 13 Å. Even at the minimum observed distance of 3.1 Å, the formation of the transition state is improbable and overall geometry is not favorable for the reaction to occur, since the GalO2 proton points in the direction opposite to Asp300OD2. The orientation of the Asp300 side chain is observed to be invariant throughout 70ns simulation, and the H-bond between Asp300OD1 and His271N remains intact. Given the unfavorable geometry that includes large distances for proton transfer, it is highly likely that AtFUT1-catalyzed fucosyltransfer is mediated by an alternative mechanism.
2.4.2 In silico studies suggest a water-mediated reaction mechanism
Figure 6a illustrates one of many frequently observed active site topologies in MD simulations that could plausibly be conducive for the fucosyl transfer reaction via an alternative mechanism. MD trajectories reveal the prolonged presence of critically positioned water molecules at the active site. In a recent study on CePOFUT1, dynamic water-mediated interactions were implicated to play an important role in substrate recognition.(Valero-González et al., 2016) In AtFUT1, a water molecule coordinated by Trp481O, β-galactose, and the β-phosphate oxygen is constantly observed at the active site during MD simulations, and is also present in the crystal structures (PDB IDs 5KWK, 5KOE and 5KOR(Rocha et al., 2016)). The active site topology shown in Figure 6a illustrates an instance wherein this water molecule is <3 Å from the acceptor nucleophilic site (Gal O2) and the α-phosphate oxygen, while the anomeric carbon of the Fuc residue of the donor is close enough to the acceptor nucleophile (~3.3 Å) to promote transfer via an SN1 mechanism. In light of the absence of a proximal catalytic base, and the extended distance between the C1 of the donor and the β-phosphate oxygen (~4.2 Å), we propose that this water molecule is involved in the catalytic mechanism of AtFUT1. As shown in Figure 6b, the tightly bound water molecule, observed in all crystal structures, is in an ideal position to act as a catalytic acid/base that protonates the β-phosphate of GDP-Fuc and deprotonates the O2 hydroxyl of the Gal residue, initiating a semi-concerted SN1-like mechanism. Protonation of the β-phosphate also stabilizes the negative charge of this leaving group.
Figure 6.
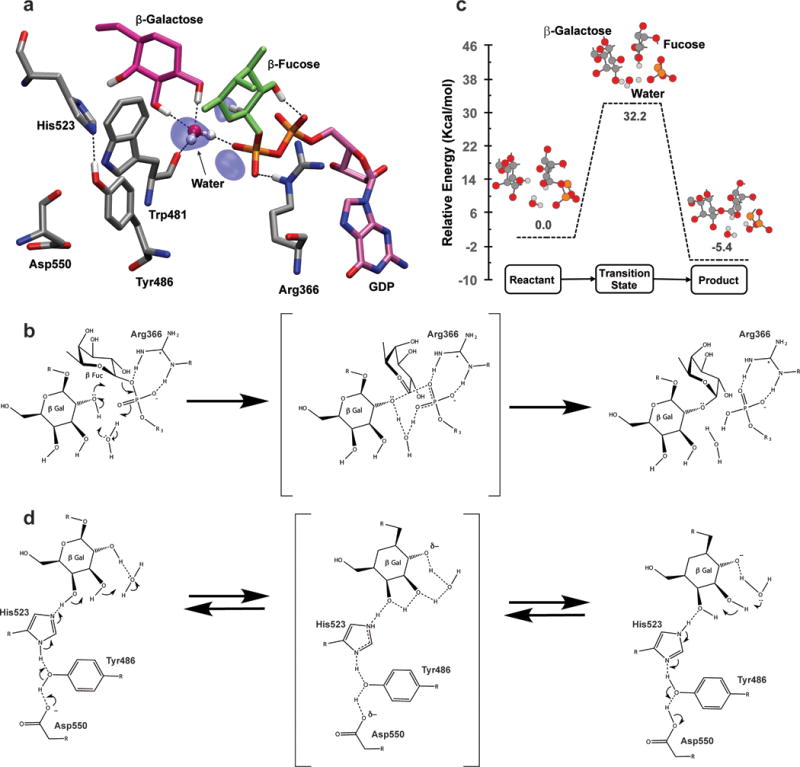
(a) MD snapshot of the FUT1 active site conducive for fucosylation. The acceptor residue (Gal) and donor residue (Fuc) are shown in licorice representation. Iso-surfaces indicate regions where water occupancy is 75% or greater over the 70 ns trajectory. Important active site residues – Trp481 (binds catalytic water), Arg366 (binds to phosphate oxygens and stabilizes the transition state), Asp550, Tyr486, His523, (in the extended H-bond network) are shown in licorice representation. Only hydrogens involved in H-bonding are shown. (b) Schematic describing the proposed water mediated reaction mechanism. (c) Potential energy surface for the reaction mechanism calculated at the M06-2x level of theory and 6-31G(d,p) basis set. The optimized states of the reaction for the reactive center of the QM system are indicated in ball and stick representation. (d) Putative role of the extended H-bond network in potentiating the activity of the catalytic water molecule.
The plausibility of this mechanism is corroborated by the discovery of a transition state (Figure 6c) that describes the AtFUT1 reaction path for the mechanism using quantum mechanical calculations (Materials & Methods: Quantum Mechanical Calculations, Figure S4). There are many features of this transition state that are of significant interest. Firstly, fucose is already partially released from the donor. We hypothesize that Arg366 facilitates this release by stabilizing the developing negative charge on the phosphate oxygen; consistent with its indispensable role in catalysis.(Lira-Navarrete et al., 2011, Kötzler et al., 2012) The fucose residue is stabilized with hydrogen bonds to the α and β phosphate oxygens. Secondly, the water acts as a proton shuttle, accepting a proton from the acceptor (O-2 hydroxyl) to generate the nucleophile while donating a proton to phosphate, which acts as a base. This explains the ability of FUT1 to facilitate fucosylation in spite of the lack of a proximally located basic amino acid at the active site. Finally, in the transition state, the partial release of the fucose residue is also observed to result in a ring-flattened (sp2 hybridized) sugar conformation of an oxocarbenium ion that enables nucleophilic attack and inversion of configuration. All these features of the transition state are remarkably akin to the mode of fucosylation proposed in CePOFUT1(Lira-Navarrete et al., 2011) and observed in the human α-(1–3)-fucosyltransferase (FUT5) via isotopic effects, which indicated that the fucose-phosphate glycosidic bond is cleaved prior to nucleophilic attack.(Murray et al., 1997) The QM-calculated activation barrier (Figure 6b), represents the upper limit for the reaction since all environmental factors that facilitate the reaction are not included in the interest of computational tractability.(Fleming and Pfaendtner, 2013)
2.5 Mutagenic and kinetic data point to the involvement of multiple residues in facilitating AtFUT1 fucosylation
To evaluate the catalytic activity of AtFUT1 and its mutant variants, an in vitro fucosyltransferase activity assay consisting of (GDP-Fuc) as an activated sugar donor and the model xyloglucan acceptor, XXLG (Figure 7) was used. Further, since GTs can also transfer sugar (from the nucleotide donor) to water in the absence of an acceptor, albeit at a low but significant rate, the enzymatic properties of AtFUT1 were evaluated by measuring both the nucleotide sugar hydrolase and fucosyltransferase activities for the WT enzyme and several mutant variants by measuring the production of GDP from GDP-Fuc (GDP-Glo™) in the presence and absence of XXLG (Figure 7). The kinetic parameters of the WT enzyme with GDP-Fuc (Table 1, Figure S5) are consistent with those previously reported for the FUT1 ortholog from Pisum sativum (PsFT1) and HisΔ68-AtFUT1 carried out by Cicéron et al. (2016).(Faik et al., 2000, Cicéron et al., 2016) Interestingly, although WT AtFUT1 is able to hydrolyze GDP-fucose, our data indicated that the rate of hydrolysis is at least 80-fold lower than fucosyltransfer (Figure 7).
Figure 7.
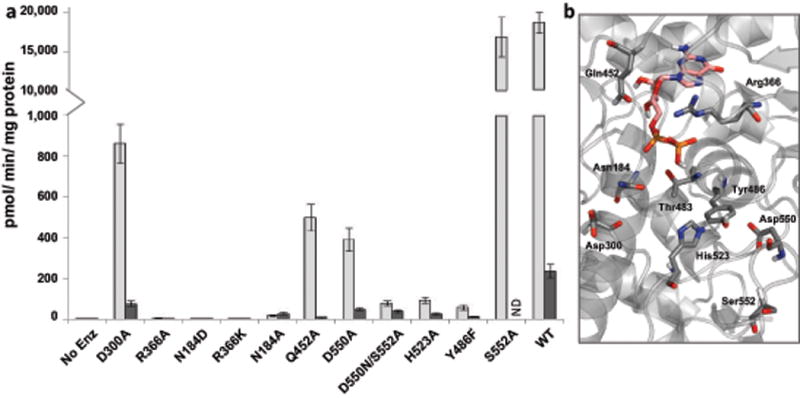
Relative enzymatic activity of AtFUT1 mutant variants compared with the WT AtFUT1. (a) Enzymatic properties of AtFUT1 were evaluated by measuring both the nucleotide sugar hydrolase and fucosyltransferase activities for the WT enzyme and several mutant variants. Activity was determined by measuring the production of guanidine diphosphate (GDP) from GDP-Fuc (100 μM) formed upon transfer of a fucosyl residue to water or a glycan acceptor in the absence (dark grey) or in the presence (light grey) of the acceptor substrate XXLG (250 μM), respectively. The data are the average from three independent assays and error bars indicate +/− S.D. (b) Close up view of the WT AtFUT1-GDP complex highlighting the residues selected for mutagenesis.
Table 1.
Kinetic Parameters for AtFUT1 WT and mutants.
| Donor Substrate | Donor Substrate | Acceptor Substrate | |
|---|---|---|---|
| Enzyme | kcat, min−1 | * KM, μM | † KM, μM |
| WT | 3.8 (±0.05) | 25.32 (±4.2) | 201.3 (±12.0) |
| N184A | NA | NA | NA |
| N184D | NA | NA | NA |
| D300A | 0.319 (±0.016) | 40.1 (±7.5) | 1703 (±161.8) |
| R366A | NA | NA | NA |
| R366K | NA | NA | NA |
| Q452A | 0.32 (±0.017) | 61.77 (±10.5) | 2304 (±121.6) |
| D550N | NA | NA | NA |
| D550A | 0.206 (±0.01) | 55.33 (±6.7) | 1441 (±65.26) |
| D550N/S552A | 0.04 (±0.002) | 90.79 (±14.8) | ND |
| T483A | NA | NA | NA |
| Y486F | 0.096 (±0.006) | 60.44 (±12.3) | 3334 (±145.8) |
| H523A | 0.074 (±0.005) | 84.28 (±18.3) | 1944 (±123.4) |
Experiments were performed in triplicate and values are expressed as the mean (± s.e.m).
NA, no activity detected (<10−4 of WT); ND, KM was too high to determine.
The donor substrate was GDP-Fuc and was varied (0–400 μM) in the presence of (750 μM) XXLG (acceptor substrate).
The donor substrate was GDP-Fuc (200 μM) and activity was measured with increasing amounts of acceptor XXLG (0–800 μM, or 0–4 mM for mutant variants). Figure S5 shows plots for the Michaelis Menten Kinetics.
The mutants that retained measurable fucosyltransferase activity were used for comparative analysis. Four enzyme variants, all close to the point mutation present in mur2 plants, resulted in decreased protein expression (D550N, D550A, S552A and D550N/S552A), with little to no protein being secreted (Materials & Methods Table 2). Mutant variants that correspond to amino acids that form H-bonds with the phospho-diester of the GDP-Fuc donor including R366K, R366A, N184D, and T483A expressed well but had complete or nearly complete loss (N184A is <0.1% of WT) of transferase activity (Figure 7, Materials & Methods: Table 2), indicating that they are very important for substrate-binding and/or catalysis. S552A displayed very similar activity to the wild-type enzyme while D300A, Q452A and D550A were diminished 20 to 50-fold (Figure 7). All of the mutant variants showed an increase in the KM values for the acceptor substrate (Table 1), indicating that modulation of residues that interact with the donor diminishes binding affinity for XXLG. Of particular interest is the fact that amongst all the mutants in close proximity to the donor, the D300A mutant retained the highest transferase activity. The observation that D300A does not abolish enzyme activity presents the crucial evidence for an alternative mechanism for fucosylation in AtFUT1 that does not involve Asp300 as the catalytic base.
Table 2.
Primer sequences for cloning and site directed mutagenesis for generation of constructs for protein expression.
| Enzyme Form | * GFP Fluor | Primer Name | Primer Sequence 5′-3′ |
|---|---|---|---|
| ǂWT | 1358 | FUT1_pDONR_GS-F | AACTTGTACTTTCAAGGCGGAGTTTTCCCAAATGTTA |
| FUT1_pDONR_GS-R | ACAAGAAAGCTGGGTCCTATACTAGCTTAAGTCCCCA | ||
| †R366A | 1719 | FUT1_R366A_F | GATTCAAGTAGCAGTTTTCGATGAAGACC |
| FUT1_R366_R | CCAATCTTCTCATCCGCATG | ||
| †R366K | 1495 | FUT1_R366K_F | GATTCAAGTAAAAGTTTTCGATGAAG |
| FUT1_R366_R | CCAATCTTCTCATCCGCATG | ||
| †N184D | 1112 | FUT1_N184D_F | CGGCTTAGGGGACAGGATACT |
| FUT1_N184_R | CTAAAAGAAATCCACACAACATATTTG | ||
| †N184A | 1049 | FUT1_N184A_F | CGGCTTAGGGGCCAGGATACTTTC |
| FUT1_N184_R | CTAAAAGAAATCCACACAACATATTTG | ||
| †Q452A | 1427 | FUT1_Q452A_F | AGAAGGTTATGCGCAGACCGAAAAAAAGATG |
| FUT1_Q452_R | TGGCTCGGCTGATGAACA | ||
| †D300A | 314 | FUT1_D300A_F | GTTAAAACAGCCAATTACTTTGTTCC |
| FUT1_D300_R | AATCAACCAAGGGACTTTC | ||
| †H523A | 1117 | FUT1_H523A_F | GCCTTGTTTCGCCTCGCCTCCATTC |
| FUT1_H523A_R | TCCATCGACATAGCCCGA | ||
| †Y486F | 799 | FUT1_Y486F_F | ACATTTGGATTTGTAGCTCAAGG |
| FUT1_Y486F_R | AGACCAAGCACTTGTCAC | ||
| †D550N | 55 | FUT1_D550N_F | ACATTGTGAGAATATCAGCTGG |
| FUT1_D550N_R | CTCACATGAGGAACTAGTG | ||
| †D550A | 99 | FUT1_D550A_F | CATTGTGAGGCTATCAGCTGG |
| FUT1_D550A_R | TCTCACATGAGGAACTAG | ||
| †S552A | 121 | FUT1_S552A_F | TGAGGATATCGCCTGGGGACTTAAG |
| FUT1_S552A_R | CAATGTCTCACATGAGGAAC | ||
| †D550N/S552A | 85 | FUT1_D550N/S470A_F | ACATTGTGAGAATATCGCCTGGGGACTTAAG |
| FUT1_S552A_R | CAATGTCTCACATGAGGAAC | ||
| †T483A | 1686 | FUT1_T483A _F | TGCTTGGTCTGCATTTGGATATG |
| FUT1_T483A _R | CTTGTCACAAGATTATCTGTC |
Underlined, italicized sequences denote the partial attB adapter sequences appended to primers used in the first round of PCR amplification.
The underlined nucleotide in the sequences of primers used to perform site-directed mutagenesis are the “mutated” bases.
Expression and secretion of the GFP-AtFUT1 fusion proteins in transiently transfected 293-F cells and was determined by measuring the relative fluorescence of the recombinant proteins secreted into the media.
The reaction catalyzed by AtFUT1 is a bi-substrate reaction that results in the production of two products, GDP and XXFG. To further substantiate the data obtained by measuring the rates of production of GDP from GDP-Fuc, the WT AtFUT1 enzyme and all of the mutant variants were also evaluated for their ability to transfer Fuc to XXLG, forming XXFG. These activity assays also utilized GDP-Fuc as a sugar donor and XXLG as an acceptor. After an extended incubation (16 h), the oligosaccharide products were analyzed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) and quantified by high performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD). MALDI-TOF MS analysis of the reaction products indicated that recombinant WT AtFUT1, and all of the mutant variants that retain activity, catalyze the transfer of a single fucosyl residue from GDP-Fuc onto XXLG, forming XXFG (Figure S6). This data is consistent with the results of the comparative mutant analysis carried out utilizing the GDP-Glo™ assay, confirming the robustness of these parallel approaches. HPAEC-PAD analysis of the reaction products after an extensive incubation indicated that the D300A mutant variant is indeed able to produce 27% of the amount of XXFG product, relative to the WT control (Figure S7). Our combined data, using various techniques, confirms that D300A does not abolish transferase activity and indicates that AtFUT1 utilizes an alternative mechanism for fucosylation that does not involve Asp300 as the catalytic base.
Our data afford conclusions that differ from those published by Rocha, et al.(Rocha et al., 2016) However, we base these conclusions on the considerable depth of our analyses. For example, we analyzed all enzyme variants using three independent, complimentary techniques under similar conditions. A highly purified xyloglucan oligosaccharide (XXLG) was used as a model acceptor substrate that only has a single galactose substituent for fucosylation. In contrast, Rocha et al. used polymeric xyloglucan from tamarind seeds in a radioactivity-based assay, and characterized neither the starting substrate nor the reaction products, making it difficult to evaluate the robustness of their assays. For comparative analysis, we first used a highly sensitive, commercial assay (GDP-Glo Glycosyltransferase Assay (Promega)) to measure the production of GDP from GDP-Fuc. In our hands, the “detection limit of the luminescent assay was 40 nM GDP with a linear response up to 20 μM” as indicated in the Materials and Methods. Next, we verified and quantified the conversion of XXLG to XXFG by direct structural analysis of the products of long reactions (~16 hr) using both MALDI-TOF MS (Figure S6) and HPAEC-PAD (Figure S7) in parallel. The measured activities of the wild type enzyme and the mutant variants were consistent using these three techniques. The mutant analysis performed by Rocha et al., was carried out using a single, radioactivity-based assay and presented as “Relative FUT1 activity (%)”. Rigorous comparison of the results obtained in the two studies would require metrics (such as the detection limits of their assay) that they have not listed. However, the one key difference between our assays and the assay performed by Rocha et al., is that their reactions contained much less enzyme (20 nM) relative to what we used (> 20 μM), which may have resulted in levels of activity below their detection limit for catalytically hindered FUT variants.
2.5.1 Conserved arginine plays an indispensable role in binding and catalysis
Putative roles for the conserved active site Arginine residue that interacts with the β-phosphate have been suggested in the context of substrate binding as well as catalysis in CePOFUT1, NodZ FUT and FUT8.(Lira-Navarrete et al., 2011, Brzezinski et al., 2012, Kötzler et al., 2012) In order to investigate the role of Arg366 in AtFUT1, the R366K mutant variant was also crystallized. The corresponding crystal structure was refined to R- and Rfree- factors of 0.253/0.327 at a 2.2 Å resolution. The AtFUT1-R366K crystal structure has been deposited to the PDB with the ID: 5KX6 (Table S2a). Structurally, this mutation is observed to cause the GDP molecule to partially swing out of the pocket and lose many of its previous contacts observed for the wild type enzyme (Figure S8). The orientation of the guanine moiety is in the anticlinal conformation with a torsion angle (χ) of −112.8°, and the ribose has a 2T3 (C2′-exo) pucker (when compared to its endo pucker in the WT enzyme; PDB ID-5KWK). This conformational shift in GDP leads to a mode of binding that is clearly not permissive for transfer of a fucosyl residue to XXLG, and demonstrates that Arg366 is needed to orient GDP-fuc to a catalytically competent conformation.
Apart from the fact that the absence of Arg366 at the active site significantly impacts GDP binding, the reduced number of hydrogen bonding contacts afforded by the Lys residue results in decreased stabilization of the transition state for the water mediated reaction. Unsurprisingly, QM calculations for the R366K mutant yielded no transition states. This partially explains the results of mutagenesis studies of the R366K and R366A variants in AtFUT1, both of which result in complete ablation of transferase activity.
2.5.2 An extended H-bonding network could facilitate acceptor deprotonation
The high transition state barrier observed in QM calculations suggests that the protein utilizes a transition state stabilization strategy to conduct the fucosylation reaction. An intriguing possibility for enabling a reduction in the energetic barrier could be an extended H-bond network consisting of three amino acids (His523, Tyr486 and Asp550) and a portion of the galactosyl residue (stabilized by Asn184) to promote the exchange of protons of the catalytic water molecule. As shown in Figure 6d, the negative charge on the Asp550 residue (bottom left) can hypothetically be delocalized and migrate through side chain ionization of Tyr486, His523, the O3 and O2 hydroxyls of the Gal residue to the catalytic water. This charge distribution is analogous to that in the transition state shown in Figure 6b, suggesting that the extended H-bond network could act as a reversible proton sink to make catalysis more energetically favorable. This hypothesis is supported by two important experimental observations. Firstly, mutations of the amino acids composing the H-bond network (e.g., D550A, Y486F, and H523A) significantly reduce AtFUT1 activity (>200-fold reduction in activity relative to WT) (Figure 7), even though two of these residues (Asp550 and Tyr486) do not make direct contact with either the acceptor or the donor substrates (Appendix S2, Table S3a,b). However, it should be noted that the mutations in the vicinity of Asp550, do result in decreased protein expression, which may indicate that observed loss in activity may be due protein destabilization in addition to its role in the H-bond network (Materials & Methods Table 2). In this context, the S552A variant, located in the same small region as Asp550, also has significantly decreased protein expression, but retains WT levels of fucosyltransferase activity. Notably, KM values for the nucleotide donor were only slightly increased by the mutations (1.5–3.3 fold, Table 1), indicating that they do not significantly impact substrate binding. However, kcat/KM values were significantly decreased (40–170-fold) pointing to their putative role in catalyzing fucosylation. Interestingly, the magnitude of decrease in kcat (Table 1) for each of these mutants is strongly correlated to the proximity of the residue to the reaction center. Secondly, the rate of hydrolysis of GDP-Fuc by AtFUT1 in the absence of the acceptor substrate is very low (Figure 7) as would be predicted by the contribution of the acceptor Gal residue in the H-bonding network. Consistent with this hypothesis is the observation that donor hydrolysis was not as severely affected by mutations affecting amino acids in the H-bond network as was the transferase activity. (Less than 25-fold reduction in GDP-Fuc hydrolysis was observed, compared to >200-fold reduction in fucosylation-Figure 7)
2.5.3 Loss of fucosyltransferase activity in the Arabidopsis mutant mur2
The cell wall defects in mur2 are due to a missense mutation in AtFUT1 that causes an almost complete loss of enzyme function and a reduction in xyloglucan fucosylation.(Vanzin et al., 2002) The mur2 mutant was originally identified in a screen of chemically mutagenized Arabidopsis plants based on neutral monosaccharide compositional analysis of plant cell walls.(Reiter et al., 1997) Two non-allelic mutant lines, mur2 and mur3, were identified that were approximately 50% deficient in cell wall fucose.(Reiter et al., 1997) Careful analysis of each L-fucose containing cell wall polymer in mur2 plants showed that they produce xyloglucan that contains less than 2% of the wild-type amount of fucose. The mur2 mutants did not show any obvious physical or developmental aberrations and responded similarly to cold, heat, and salt stress, relative to wild-type plants.(Reiter et al., 1997, Vanzin et al., 2002) Ultrastructural analysis of mur2 plants by scanning electron microscopy (SEM) indicated that they produce underdeveloped trichome papillae. Interestingly, mur3 also has a weak papillae phenotype, and AtMUR3 was later shown to act as a galactosyltransferase that transfers galactose to the third xylose residue in the XXXG xyloglucan core structure, which is the biosynthetic step directly prior to the addition of L-fucose by AtFUT1.(Madson et al., 2003)
The fucosylation chemotype of mur2 is due to a point mutation that results in the replacement of Asp550 with an asparagine (D550N) near the catalytic domain.(Vanzin et al., 2002) We designed several mutants to determine the biochemical basis of this mutation. An equivalent (D550N) mutation in the recombinant product did not secrete any soluble protein for analysis (Materials & Methods: Table 2). Interestingly, the D550N mutation creates an Asn-linked glycosylation sequon (Apweiler et al., 1999) at the mutation site (NxS sequon site at Asn550-Ile551-Ser552).(Vanzin et al., 2002) To test the hypothesis that glycosylation at the D550N mutation site leads to enzyme inactivation in vivo, a series of mutant variants were produced at the Asp550 and Ser552 positions (D550A, S552A and D550N/S552A). Each mutant, except D550N, resulted in soluble, secreted fusion proteins at ~20-fold reduced levels compared to wild type AtFUT1 (Materials & Methods: Table 2). While the D550A mutant exhibited reduced activity, a double mutant (D550N/S552A) that eliminates the N-glycosylation motif exhibited only 1.5% activity relative to WT, while the S552A single mutant had activity similar to the wild type enzyme (Figure 7). The drastic reduction of catalytic efficiency (kcat/Km) for the D550N/S552A double mutant (340-fold, Table1) together with its modest increase in Km, suggests that disruption of the H-bonding network has a more pronounced effect on catalysis than on protein folding and stability. Although decreased expression or stability of the mur2 protein may contribute to the mur2 phenotype in planta, the severely decreased catalytic efficiency of the mutant protein is sufficient to cause the phenotype.
3. CONCLUSIONS
Overall, these findings present the initial foray to unravel the enzymatic mechanisms of plant cell wall hemicellulose biosynthesis. We demonstrate protocols developed for the expression and purification of plant GTs in mammalian cell cultures followed by their successful structural characterization by crystallography. The structure of AtFUT1 in complex with substrate analogs forms the basis for further enzymatic characterization and detailed computational investigations at the molecular level. Our modeled ternary structures, combined with mutagenesis data, also reveal a surprising result: there is an apparent lack of proximal residues that could serve as a catalytic base for fucosyltransfer by AtFUT1. Further investigations using MD and quantum computations indicate that AtFUT1 may use an atypical water-mediated mechanism with the potential contribution of an H-bonding network for acceptor nucleophile activation. The proposed mechanism coupled with the H-bonding network is consistent with mutagenic experimental data and opens the door for the rationalization of severely decreased fucosylation activity in the Arabidopsis mur2 mutant. Additional computational (e.g. constant pH MD simulations and QM/MM studied to incorporate protein and solvent structure effects) and experimental (structural and kinetic isotope analyses) studies have the potential to evaluate the proposed mechanism and provide hypotheses for further empirical testing. Finally, the structure of the acceptor complex also provides key information required to understand the regiospecificity of AtFUT1. Thus, these results establish a starting point for dissecting the complex enzymology of plant polysaccharide biosynthesis and lay a foundation for future studies. The fundamental insights gained from this study will play a crucial role in developing capabilities to develop design schema to engineer glycosyltransferases to make specific modifications of plant cell wall structures. The availability of protein–ligand structural information can also be used to guide the design of glycosyltransferase inhibitors to develop new, highly specific herbicides. Thus, this study lays out an exemplar to expand our knowledgebase on these and other plant glycosyltransferases.
4. MATERIALS & METHODS
Generation of Constructs for Protein Expression
A truncated coding region sequence of AtFUT1 (amino acids 81–558) was amplified from cDNA prepared from leaves of wild-type A. thaliana (Col-0) and used as a template.(Urbanowicz et al., 2012) All reactions were carried out using Phusion High-Fidelity DNA Polymerase (Thermo Scientific). To create Gateway entry clones, attB-PCR products were generated using two-step adapter PCR as described in the Gateway Technology Manual (Life Technologies). The following primer pairs were used for gene-specific amplification of the FUT sequence: FUT1_pDONR_GS-F,
5′-AACTTGTACTTTCAAGGCGGAGTTTTCCCAAATGTTA-3′/FUT1_pDONR_GS-R,
5′-ACAAGAAAGCTGGGTCCTATACTAGCTTAAGTCCCCA-3′; Underlined sequences denote the partial attB adapter sequences appended to the primers used in the first round of PCR amplification and the bold sequence indicates the inserted STOP codon. A second set of universal primers, attB_Adapter-F, 5′- GGGGACAAGTTTGTACAAAAAAGCAGGCTCTGAAAACTTGTACTTTCAAGGC-3′ / attB_Adapter-R, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTC-3′ was used to complete the attB recombination region and insert a tobacco etch virus (TEV) protease cleavage site. The completed attB-PCR products were cloned into the pDONR221 plasmid vector (Life Technologies) using Gateway BP Clonase II Enzyme Mix (Life Technologies) according to the manufacturer’s instructions. The correct amplification and integration of the DNA sequence was confirmed by sequence analysis using M13 universal primers, M13 (−20) Forward and M13 Reverse. To create expression clones, the entry clones were recombined into a Gateway adapted version of the pGEn2 mammalian expression vector (pGEn2-DEST), using Gateway LR Clonase II Enzyme Mix (Life Technologies). The resulting expression construct encodes a fusion protein comprised of an NH2-terminal signal sequence, an 8xHis tag, an AviTag recognition site, the “superfolder” GFP (sfGFP) coding region, the seven amino acid recognition sequence of the tobacco etch virus (TEV) protease, followed by residues 81–558 of A. thaliana FUT1. Mutated variants of AtFUT1 were generated by site-directed mutagenesis using the Q5® Site-Directed Mutagenesis Kit (New England Biolabs) according to the manufacturer’s instructions using the pGEN2-DEST construct as a template. Oligonucleotide primers used to generate the base changes are listed in Table 2. The introduction of mutations was confirmed by sequencing. For transfection, plasmids were purified using the PureLink HiPure Plasmid Filter Maxiprep Kit (Life Technologies).
Protein expression and purification
Recombinant enzyme expression was accomplished by transient transfection of HEK293 cells (FreeStyle 293-F cells, Life Technologies, Grand Island, NY) or HEK293S GnTI− cells (catalog number CRL-3022, ATCC, Manassas, VA) as previously described.(Barb et al., 2012) Briefly, HEK293 cells were maintained in serum free Freestyle 293 expression medium (Life Technologies) and HEK293S GnTI− cells were cultured in an Ex-cell 293 serum-free medium (Sigma, St. Louis, MO). Transfections were initiated at cell densities of 2.5 × 106 by addition of 4 μg/mL of the respective expression plasmid and 9 μg/mL polyethylenimine (linear 25 kDa PEI, Polysciences, Inc., Warrington, PA) to the suspension culture. For transfections in HEK293 cells, the cultures were diluted 1:1 with Freestyle 293 expression medium containing valproic acid (2.2 mM final) 24 h after transfection. For HEK293S GnTI- cells, the cultures were diluted 1:1 with ESF serum-free medium (Expression Systems, Davis, CA) containing valproic acid (2.2 mM final) 24 h after transfection. Protein production was continued for a further 5 d at 37°C.
Culture medium was harvested and clarified by sequential centrifugation at 1,500 rpm for 15 min, 2,500 rpm for 15 min, 4,000 rpm for 15 min and 12,000 rpm for 30 min then passed through a 5 μM filter (Pall Corporation). All chromatography experiments were carried out on an ÄKTA FPLC System (GE Healthcare). The media was adjusted to contain HEPES (50 mM, pH 7.2), sodium chloride (400 mM), and imidazole (20 mM) prior to column loading. Small scale purification of His8-GFP tagged enzymes secreted into the culture medium by HEK293 cells was performed using HisTrap HP columns (GE Healthcare) according to the manufacturer’s instruction. To eliminate the possibility of protein contamination, purification of the wild type enzyme and each variant was carried out on individual 1 ml HisTrap HP columns. Prior to use, a blank run was performed on each new column to remove any weakly bound Ni2+ ions. Briefly, columns were washed with five column volumes (CV) of 100% Buffer A at 1 ml/min, followed by a linear gradient of Buffer A to 100% Buffer B for 5 CV. Then, the columns were washed with 100% Buffer B for 5 CV, followed by a liner gradient of 100% Buffer B to 100% Buffer A in 5 CV, prior to re-equilibration with Buffer A. All columns were washed with water containing 0.04% sodium azide after use and stored at 4°C in 20% ethanol between uses. For scaled up purification, adjusted media was loaded onto a HisPrep FF 16/10 column (GE Healthcare) equilibrated with Buffer A (50 mM HEPES, pH 7.2, 0.4 M sodium chloride and 20 mM imidazole). The column was washed and eluted with a step gradient, consisting of five CV per condition of Buffer A to Buffer B (50 mM HEPES, pH 7.2, 0.4 M sodium chloride and 500 mM imidazole). These were three sequential wash steps of 0%, 10% and 20% Buffer B, followed by two elution steps of 60% and 100% Buffer B. Fractions containing GFP fluorescence (60% Buffer B elution) were collected and pooled. The HisPrep FF 16/10 column used for the first round of immobilized metal affinity chromotography was stripped and recharged between each purification cycle according to the manufacturer’s instruction. Recombinant His-tagged EndoF1(Kwan et al., 2005) and His-tagged GFP-TEV protease(Wu et al., 2010) were produced in Escherichia coli. For removal of the N-terminal 8xHis-AviTag-sfGFP fusion tag, the purified His8-sfGFP-AtFUT1 preparation was treated with recombinant His-tagged GFP-TEV protease and His-tagged EndoF1 at ratios of 1:40 and 1:20, respectively, relative to AtFUT1. The digestion was incubated at 4°C overnight, followed by 15-fold dilution with 25 mM HEPES, 300 mM NaCl, pH 7.0. The sample was then passed through a second, separate HisPrep FF 16/10 column (GE Healthcare) to remove the cleaved N-terminal fusion tag, His-tagged GFP-TEV and His-tagged EndoF1. The second HisPrep FF 16/10 column, used solely for the tag removal step, was stripped and recharged between each purification cycle according to the manufacturer’s instruction. The unbound protein containing tag-free AtFUT1 was concentrated and further purified by size exclusion chromatography using Superdex 75 resin and 20 mM Tris pH 7, 100 mM NaCl as the elution buffer.
The purity of the proteins was confirmed by SDS-PAGE, Coomassie Brilliant Blue R-250 (Bio-Rad) staining, and in-gel GFP fluorescence. For activity assays, proteins were buffer exchanged into HEPES sodium salt-HCl (75 mM), pH 6.8, using a PD-10 gel filtration column (GE Healthcare) and then concentrated using a 30 kDa molecular weight cut-off Amicon Ultra centrifugal filter device (Millipore). To examine the divalent metal-dependence of FUT1, metal-depleted enzyme was prepared by dialysis against HEPES sodium salt-HCl (75 mM), pH 6.8 buffer containing 5 g/L Chelex-100 (Bio-Rad) at 4°C for a minimum of 6 h. For real-time NMR experiments, purified proteins were exchanged into potassium bicarbonate buffer (50 mM), pH 6.8, prepared with D2O (99.98%, Cambrige Isotope Laboratories) by diafiltration using a 30 kDa molecular weight cut-off Amicon Ultra centrifugal filter device. Protein concentration was determined by the Bio-Rad protein assay.
Crystallization
AtFUT1 co-crystals with GDP-fucose (AtFUT1-GDP) and XXLG (AtFUT1-XXLG) were initially obtained with sitting drop vapor diffusion using a 96-well plate with PEG ion HT screen from Hampton Research (Aliso Viejo, CA). 50 μL of well solution was added to the reservoir and drops were made with 0.2 μL of well solution and 0.2 μL of protein solution using a Phoenix crystallization robot (Art Robbins Instruments, Sunnyvale, CA). The crystals for all mutants were grown at 20°C using screens containing 0.1 M MES pH 6.0 to 7.0 and 16% to 23% w/v PEG 3350. The protein solutions contained 7 mg/mL of protein, 20 mM Tris pH 7, 100 mM NaCl and either 5 mM XXLG or 5 mM GDP-fucose. AtFUT1-XXLG crystals were soaked in well solution with 0.7 M KCL before flash freezing and with AtFUT1-GDP crystals 10% glycerol and 10% ethylene glycol were used for additional cryo protection. One crystal was briefly soaked in a 2 μL drop of well solution with 0.5 M KI before flash freezing and data collection to get an iodine derived crystal.
Data collection and processing
The AtFUT1 crystals were flash frozen in a nitrogen gas stream at 100 K before home source data collection using an in-house Bruker X8 MicroStar X-Ray generator with Helios mirrors and Bruker Platinum 135 CCD detector. Data were indexed and processed with the Bruker Suite of programs version 2014.9 (Bruker AXS, Madison, WI).
Structure Solution and Refinement
Intensities were converted into structure factors and 5% of the reflections were flagged for Rfree calculations using programs F2MTZ, Truncate, CAD and Unique from the CCP4 package of programs.(Winn et al., 2011) The program Crank(Pannu et al., 2011) with SAD phasing was used to solve a partial model of AtFUT1 using the KI derivative with manually picking sites and assigning them to be sulphur or iodine. MOLREP(Vagin and Teplyakov, 2010) version 11.2.08 was used for molecular replacement using the partial AtFUT1 structure as the search model. Refinement and manual correction was performed using REFMAC5(Murshudov et al., 2011) version 5.8.0073 and Coot(Emsley et al., 2010) version 0.7.2. Program Privateer MkIII was used to validate the geometry of carbohydrate structures(Agirre et al., 2015a, Agirre et al., 2015b). The MOLPROBITY method(Chen et al., 2010) was used to analyze the Ramachandran plot and root mean square deviations (rmsd) of bond lengths and angles were calculated from ideal values of Engh and Huber stereo chemical parameters.(Engh and Huber, 1991) Wilson B-factor was calculated using CTRUNCATE version 1.15.10.(Winn et al., 2011) The data collection and refinement statistics are shown in Table S1.
Structure Analysis
Programs Coot(Emsley et al., 2010) and PyMOL (http://www.pymol.org) were used for comparing and analyzing structures. Figures 2, 3, 5, 7, S1, S3 and S5 were created using PyMOL.
Preparation of the AtFUT1-GDP-Fuc-XXLG ternary complex
In order to explore the AtFUT1 reaction mechanism, the ternary complex (AtFUT1:XXLG:GDP-Fuc) was modeled by starting with crystallographic model for the enzyme in complex with GDP (Chain B of PDB ID 5KWK), in which the flexible loop is folded over GDP. Since we could not identify a single, energetically accessible conformation for the fucosyl residue in this complex that was consistent with electron density data, the conformation of the fucosyl residue was modeled on the basis of energetic considerations. The complex of AtFUT1 with XXLG (Chain A of PDB ID: 5KOE) was then aligned with the enzyme in complex with GDP (Chain B of PDB ID: 5KWK) solely in order to place the acceptor substrate (XXLG) in the active site of the complex with GDP. As fucosylation by AtFUT1 occurs after backbone elongation and addition of the Xyl and Gal side chain residues, it likely acts in vivo on nascent xyloglucan molecules that are significantly larger than XXLG. Thus, a model of the XXLG “dimer” (i.e., XXLGXXLG) was generated by replicating the structure of XXLG in the AtFUT1 active site and manually translating and rotating this copy to a position where a glycosidic bond from C1 of the reducing Glc residue of one copy to O4 of the Glc at the non-reducing end of the other copy could be formed. This structure was then placed in the active site of the ternary complex by placing one of the copies with XXLG in the location that was determined as described above. Since the dimer has two copies of XXLG, either of which can fit into the active site, this resulted in two different models, in which the oligosaccharide in the active site is extended at either the reducing end or at the non-reducing end. The resulting ternary complexes were then energy minimized for use as starting points for molecular dynamics (MD) calculations. PDB coordinates of the MD equilibrated ternary complex structure are provided as part of Supporting Information Data (Data S1).
Molecular Dynamics Simulations
Initially, MD simulations of the following systems were conducted, AtFUT1 with GDP-Fuc and a. XXLG, b. XXLGXXLG reducing end, c. XXLGXXLG non-reducing end, to evaluate which acceptor configuration maintains substrate stability at the active site. The protonation states for titratable residues on the protein were assigned manually upon careful inspection of the protein environment. The residues in the loop 398–406 were not modeled in the crystal structure due to weak and scattered electron density indicating significant flexibility of that region. This loop was modeled computationally via multiple minimization routines. Water molecules observed in the crystal structure were retained when constructing solvated systems.
The CHARMM 36 force field was used to build the protein,(Best et al., 2012) the xyloglucan acceptor and the GDP-Fuc donor,(Guvench et al., 2008) while the TIP3P forcefield was used to model the water.(Jorgensen et al., 1983) The simulation protocol involved minimization and short MD runs in-vacuo of the enzyme-substrate(s) complex with crystal waters followed by creation of a solvated water box. The protein was solvated with a buffer of 12 Å. Ions were added to ensure a charge neutral system. The solvated charge neutral system was equilibrated in the Iso-baric isothermal (NPT) ensemble at a temperature of 300 K. The equilibration protocol involved initial restrains of the protein backbone that were gradually released over sequential 100 ps runs. This was followed by a 100 ns unrestrained production run in the isochoric isothermal ensemble (NVT) at 300 K for each system. All MD simulations were performed within the CHARMM MD package (version c41)(Brooks et al., 2009) using the DOMDEC engine.(Hynninen and Crowley, 2014) The simulations were performed using a 1 fs time step, constrained bonds to hydrogen atoms, periodic boundary conditions, a non-bonded cutoff of 11 Å, and the Particle-Mesh Ewald method for long-range electrostatics.(Darden et al., 1993) The production runs were conducted on the AtFUT1 with GDP-Fuc and XXLGXXLG reducing end bound at the active site (Appendix S3).
Quantum Mechanical Calculations
All calculations were performed using the Gaussian09 package (Revision D.01).(Frisch et al.) A reduced system consisting of reactive moieties (Figure S4) at the active site was constructed based on a putative reactant configuration observed in MD simulations. A water molecule coordinated by the phosphate oxygen and the galactose O2 hydroxyl group, which is observed in both MD simulations and the crystal structure, was included for QM calculations. Geometry optimizations and frequency calculations were performed without any constraints at the M06-2× level of theory, and 6–31g(d,p) basis set.(Rassolov et al., 2001, Zhao and Truhlar, 2008) The M06-2x is a hybrid DFT functional well known for accurate calculations for main group chemistry, accounting non-covalent interactions, and has been successfully employed to evaluate glycosyl transferase and other enzymatic mechanisms.(Tvaroška et al., 2012, Bharadwaj et al., 2015) The transition state (TS) search was performed over the galactose O2 and fucose C1 distance as the reaction coordinate. Frequency calculations were used to characterize the nature of TS, which is characterized by a single imaginary frequency, followed by Intrinsic Reaction Coordinate (IRC) calculations to ensure that the TS connected the reactant and product along the reaction path.(Fukui, 1981) The terminal points on the IRC path were utilized to obtain the optimized reactant and product conformations that were characterized by no imaginary frequency. The frequency analyses formed the basis for the estimation of reaction barriers and thermodynamics. The QM optimized coordinates for the reactant (Data S2), product (Data S3) and transition state (Data S4) configurations are available as part of the Supporting Information Data Files.
Mutagenesis and Kinetic Analysis
The amount of GDP formed as a by-product of the fucosyltransferase reaction was determined using the GDP-Glo™ Glycosyltransferase Assay (Promega) according to the manufacturer’s instructions. Standard fucosyltransferase reactions (5–10 μL) analyzed by the GDP-Glo™ Glycosyltransferase Assay consisted of HEPES sodium salt-HCl (75 mM), pH 6.8, GDP-Fuc (100 μM) as donor, XXLG (250 μM) as an acceptor and purified AtFUT1 and variants (200 ng). Assays were initiated with the addition of enzyme and carried out for indicated times at 28°C. For kinetic analysis, higher concentrations of enzymes were used for mutant variants (200 ng, WT; 483 ng, D300A and Q452A; 967 ng, D550A; 1697 ng, Y486F, H523A and D550N/S552A). For acceptor kinetics, reactions were carried out with XXLG (0–800 μM, or 0–4 mM for mutant variants) and GDP-Fuc (200 μM) as a donor. For kinetics analysis of the donor substrate, reactions were carried out with GDP-Fuc (0–100 μM, or 0–400 μM for mutant variants) and a constant concentration of XXLG (750 μM) as an acceptor. Reactions were performed at 28°C for up to 20 min and were stopped at the indicated times by flash freezing in liquid nitrogen. For detection of GDP, reactions were mixed with an equal volume of GDP-Glo™ Detection Reagent (5–10 μL) in white polystyrene, low-volume, 384-well assay plates (Corning Incorporated) and incubated for 60 min at 28°C. After incubation, luminescence measurements were performed using a multifunctional microplate reader (POLARstar OPTIMA, BMG Labtech). A standard curve was used for quantification of GDP produced, and the detection limit of the luminescent assay was 40 nM GDP with a linear response up to 20 μM. The steady state parameters kcat, Km and Vmax were calculated by fitting the initial velocities to the Michaelis-Menten equation using nonlinear curve fitting in GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Determination of Fucosyltransferase Activity by HPAEC-PAD and MALDI-TOF MS
The oligosaccharide acceptor, XXLG, was generated and purified according to Tuomivaara.(Tuomivaara et al., 2015) GDP-Fuc was purchased from Promega Corporation (USA). Fucosyltransferase activity was routinely assayed by using GDP-Fuc and the xyloglucan oligosaccharide XXLG. The standard fucosyltransferase assay (15 μL) was performed in HEPES sodium salt-HCl (75 mM), pH 6.8 containing AtFUT1 (200 ng/5 μl of reaction volume), GDP-Fuc (0.25–0.5 mM) as donor and XXLG (0.25–0.5 mM) as acceptor, without the addition of exogeneous metals. Reactions were incubated at 28°C for the indicated times. The reaction products were analyzed by Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-TOF MS) using a Microflex LT spectrometer (Bruker). Aliquots (5 μL) of the reaction mixture were incubated with one μL of a suspension of Dowex-50 cation exchanger resin in water for 1 h. After centrifugation, one μL aliquots of the supernatants were mixed with an equal volume of matrix solution (20 mg/mL 2,5-dihydroxbenzoic acid in aqueous 50% methanol) on the target plate. The positive-ion spectra were recorded and at least 200 laser shots were summed to generate each spectrum.
HPAEC-PAD analysis was performed with a Dionex ICS-3000 chromatography system (Thermo Scientific) equipped with an autosampler and a pulsed amperometric detector (PAD). A portion of the reaction (10 μL) was analyzed using a CarboPac PA1 column (Thermo Scientific) according to the methods by.(Tuomivaara et al., 2015) A standard curved of XXLG was used for quantification.
NMR Experiments
NMR experiments were recorded at 25°C on a Varian Inova NMR spectrometer (Agilent, Santa Clara, CA) operating at 600 MHz, equipped with a 5-mm NMR cold probe. To determine the regiospecificity of AtFUT1, the reaction catalyzed by the enzyme in vitro was monitored using real-time 1H NMR. The assay was performed in potassium bicarbonate (50 mM), pH 6.8, in D2O containing purified xyloglucan oligosaccharide XLLG (0.4 mM), GDP-Fuc (0.4 mM) and purified AtFUT1 (4.7 μM). Data acquisition was initiated 5 min after the reaction components were mixed. The 1D 1H spectra consisted of 16 transients that were acquired with water pre-saturation every 5 min over 2.5 h.
Supplementary Material
Details of the GDP/GDP-Fuc binding and positive electron density in the FUT1-GDP structure.
Molecular dynamics reveals active site residues stabilizing GDP-Fuc and XXLGXXLG binding.
Molecular Dynamics of different ternary complexes.
Snapshot of MD Equilibrated ternary complex-AtFUT1-GDPf-XXLG.
Optimized Geometries for the Product QM System.
Optimized Geometries for the Reactant QM System.
Transition State Geometries for the QM System.
Stereo figure of the GDP binding site of FUT1-GDP.
Amino acid sequence alignment of the GT37 family.
The AtFUT1-GDP and NodZ structures superimposed.
Reduced system considered for QM calculations.
Michaelis-Menten kinetics of AtFUT1 and catalytic variants.
MALDI-TOF MS Analysis of AtFUT1 variants.
AtFUT1 catalyzed transfer of fucosyl residues from GDP-Fuc onto the model xyloglucan oligosaccharide acceptor substrate XXLG.
Conformational shift of GDP in the R366K mutant.
Theoretical and observed mass to charge ratios (m/z) of xyloglucan oligosaccharides.
X-ray data collection and refinement statistics.
Carbohydrate conformational validation by Privateer.
Average interaction energies of GDP-Fuc with AtFUT1 amino acid residues.
Average interaction energies of XXLGXXLG with AtFUT1 amino acid residues.
Significance Statement.
The fucosyl transferase (AtFUT1) enzyme in Arabidopsis thaliana is an important cog in the machinery of plant cell wall biosynthesis where it catalyzes the final step of the synthesis of xyloglucan. Multiple approaches involving protein expression, structural and biochemical characterization, molecular simulations and mutagenesis experiments challenge the current proposed reaction mechanism and reveal an unconventional water-mediated mechanism by which AtFUT1 catalyzes xyloglucan fucosylation.
Acknowledgments
This work was funded by The BioEnergy Science Center (BESC), a US Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the U.S. Department of Energy Office of Science. We thank the Center for Plant and Microbial Complex Carbohydrates (DEFG0296ER20097) for equipment support. This research was supported by NIH grants P41GM103390 to K.W.M. and P01 GM107012 to Dr. G.J. Boons (University of Georgia). We are also grateful for the access and use of NREL Computational Sciences resources (Peregrine) supported by the DOE Office of EERE under contract number DE-AC36-08GO28308 and the Extreme Science and Engineering Discovery Environment (XSEDE)(Towns et al., 2014), which is supported by National Science Foundation grant number ACI-1053575.
Footnotes
Code Availability
For Molecular dynamics CHARMM MD package (Ver. C41) is available for free at https://www.charmm.org/charmm/program/obtaining-charmm/
For quantum mechanical calculations the Gaussian09 software package (Revision D.01) may be obtained for a license fee at http://www.gaussian.com/g_prod/g09.html.
Programs Coot(Emsley et al., 2010) is available free at http://www2.mrc-lmb.cam.ac.uk/Personal/pemsley/coot/. PyMOL is available as an open-source software package or for a fee as a supported binary at http://www.pymol.org.
Accession codes. Protein Data Bank: The coordinates and structure factors for the AtFUT1-GDP, AtFUT1-XXLG, R366K AtFUT1-GDP structures are deposited under the accession codes 5KOE, 5KWK and 5KX6 respectively.
AUTHOR CONTRIBUTIONS
BRU devised experiments, carried out molecular biology, protein purification and enzyme activity assays. MP carried out NMR experiments. STT prepared oligosaccharide substrates. SW and JY did protein expression. X-ray crystallography was performed and analyzed by VVL and MA. VSB setup, performed and analyzed all the molecular simulations (MD and QM). VSB and BRU interpreted the data and wrote the manuscript with input from MC, MH, KM and WY.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
References
- Agirre J, Davies G, Wilson K, Cowtan K. Carbohydrate anomalies in the PDB. Nat Chem Biol. 2015a;11:303. doi: 10.1038/nchembio.1798. [DOI] [PubMed] [Google Scholar]
- Agirre J, Iglesias-Fernandez J, Rovira C, Davies GJ, Wilson KS, Cowtan KD. Privateer: software for the conformational validation of carbohydrate structures. Nat Struct Mol Biol. 2015b;22:833–834. doi: 10.1038/nsmb.3115. [DOI] [PubMed] [Google Scholar]
- Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, Shinn P, Stevenson DK, Zimmerman J, Barajas P, Cheuk R. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science. 2003;301:653–657. doi: 10.1126/science.1086391. [DOI] [PubMed] [Google Scholar]
- Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochimica et Biophysica Acta (BBA)-General Subjects. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- Attia MA, Brumer H. Recent structural insights into the enzymology of the ubiquitous plant cell wall glycan xyloglucan. Current Opinion in Structural Biology. 2016;40:43–53. doi: 10.1016/j.sbi.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Barb AW, Meng L, Gao Z, Johnson RW, Moremen KW, Prestegard JH. NMR characterization of immunoglobulin G Fc glycan motion on enzymatic sialylation. Biochemistry. 2012;51:4618–4626. doi: 10.1021/bi300319q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD., Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. Journal of chemical theory and computation. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj VS, Vyas S, Villano SM, Maupin CM, Dean AM. Unravelling the impact of hydrocarbon structure on the fumarate addition mechanism–a gas-phase ab initio study. Physical Chemistry Chemical Physics. 2015;17:4054–4066. doi: 10.1039/c4cp04317k. [DOI] [PubMed] [Google Scholar]
- Bourne Y, Henrissat B. Glycoside hydrolases and glycosyltransferases: families and functional modules. Current opinion in structural biology. 2001;11:593–600. doi: 10.1016/s0959-440x(00)00253-0. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL, MacKerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S. CHARMM: the biomolecular simulation program. Journal of computational chemistry. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzezinski K, Dauter Z, Jaskolski M. Structures of NodZ α1, 6-fucosyltransferase in complex with GDP and GDP-fucose. Acta Crystallographica Section D: Biological Crystallography. 2012;68:160–168. doi: 10.1107/S0907444911053157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CI, Keusch JJ, Klein D, Hess D, Hofsteenge J, Gut H. Structure of human POFUT2: insights into thrombospondin type 1 repeat fold and O‐fucosylation. The EMBO journal. 2012;31:3183–3197. doi: 10.1038/emboj.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicéron F, Rocha J, Kousar S, Hansen SF, Chazalet V, Gillon E, Breton C, Lerouxel O. Expression, purification and biochemical characterization of AtFUT1, a xyloglucan-specific fucosyltransferase from Arabidopsis thaliana. Biochimie. 2016;128:183–192. doi: 10.1016/j.biochi.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Coutinho PM, Deleury E, Davies GJ, Henrissat B. An evolving hierarchical family classification for glycosyltransferases. Journal of molecular biology. 2003;328:307–317. doi: 10.1016/s0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. The Journal of chemical physics. 1993;98:10089–10092. [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engh RA, Huber R. Accurate Bond and Angle Parameters for X-Ray Protein-Structure Refinement. Acta Crystallographica Section A. 1991;47:392–400. [Google Scholar]
- Faik A, Bar-Peled M, DeRocher AE, Zeng W, Perrin RM, Wilkerson C, Raikhel NV, Keegstra K. Biochemical characterization and molecular cloning of an α-1, 2-fucosyltransferase that catalyzes the last step of cell wall xyloglucan biosynthesis in pea. Journal of Biological Chemistry. 2000;275:15082–15089. doi: 10.1074/jbc.M000677200. [DOI] [PubMed] [Google Scholar]
- Fleming KL, Pfaendtner J. Characterizing the catalyzed hydrolysis of β-1, 4 glycosidic bonds using density functional theory. The Journal of Physical Chemistry A. 2013;117:14200–14208. doi: 10.1021/jp4081178. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JJA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09 Rev C.01: Gaussian, Inc. [Google Scholar]
- Fry SC, York WS, Albersheim P, Darvill A, Hayashi T, Joseleau JP, Kato Y, Lorences EP, Maclachlan GA, McNeil M. An unambiguous nomenclature for xyloglucan‐derived oligosaccharides. Physiologia Plantarum. 1993;89:1–3. [Google Scholar]
- Fukui K. The path of chemical reactions - the IRC approach. Accounts of Chemical Research. 1981;14:363–368. [Google Scholar]
- Guvench O, Greene SN, Kamath G, Brady JW, Venable RM, Pastor RW, Mackerell AD. Additive empirical force field for hexopyranose monosaccharides. Journal of computational chemistry. 2008;29:2543–2564. doi: 10.1002/jcc.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held MA, Jiang N, Basu D, Showalter AM, Faik A. Plant cell wall polysaccharides: structure and biosynthesis. Polysaccharides: Bioactivity and Biotechnology. 2015:3–54. [Google Scholar]
- Hillenbrand EA, Scheiner S. Effects of molecular charge and methyl substitution on proton transfer between oxygen atoms. Journal of the American Chemical Society. 1984;106:6266–6273. [Google Scholar]
- Hynninen AP, Crowley MF. New faster CHARMM molecular dynamics engine. Journal of computational chemistry. 2014;35:406–413. doi: 10.1002/jcc.23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. The Journal of chemical physics. 1983;79:926–935. [Google Scholar]
- Kötzler MP, Blank S, Bantleon FI, Spillner E, Meyer B. Donor substrate binding and enzymatic mechanism of human core α1, 6-fucosyltransferase (FUT8) Biochimica et Biophysica Acta (BBA)-General Subjects. 2012;1820:1915–1925. doi: 10.1016/j.bbagen.2012.08.018. [DOI] [PubMed] [Google Scholar]
- Kötzler MP, Blank S, Bantleon FI, Wienke M, Spillner E, Meyer B. Donor assists acceptor binding and catalysis of human α1, 6-fucosyltransferase. ACS chemical biology. 2013;8:1830–1840. doi: 10.1021/cb400140u. [DOI] [PubMed] [Google Scholar]
- Kwan EM, Boraston AB, McLean BW, Kilburn DG, Warren RAJ. N-Glycosidase–carbohydrate-binding module fusion proteins as immobilized enzymes for protein deglycosylation. Protein Engineering Design and Selection. 2005;18:497–501. doi: 10.1093/protein/gzi055. [DOI] [PubMed] [Google Scholar]
- Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Biochemistry. 2008;77:521. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- Lapid H, Agmon N, Petersen MK, Voth GA. A bond-order analysis of the mechanism for hydrated proton mobility in liquid water. The Journal of chemical physics. 2005;122:014506. doi: 10.1063/1.1814973. [DOI] [PubMed] [Google Scholar]
- Lehner A, Menu-Bouaouiche L, Dardelle F, Le Mauff F, Driouich A, Lerouge P, Mollet JC. In silico prediction of proteins related to xyloglucan fucosyltransferases in Solanaceae genomes. Plant signaling & behavior. 2015;10:e1026023. doi: 10.1080/15592324.2015.1026023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira-Navarrete E, Valero-González J, Villanueva R, Martínez-Júlvez M, Tejero T, Merino P, Panjikar S, Hurtado-Guerrero R. Structural insights into the mechanism of protein O-fucosylation. PLoS One. 2011;6:e25365. doi: 10.1371/journal.pone.0025365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madson M, Dunand C, Li X, Verma R, Vanzin GF, Caplan J, Shoue DA, Carpita NC, Reiter WD. The MUR3 gene of Arabidopsis encodes a xyloglucan galactosyltransferase that is evolutionarily related to animal exostosins. The Plant Cell. 2003;15:1662–1670. doi: 10.1105/tpc.009837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Forouhar F, Thieker D, Gao Z, Ramiah A, Moniz H, Xiang Y, Seetharaman J, Milaninia S, Su M. Enzymatic Basis for N-Glycan Sialylation STRUCTURE OF RAT α2, 6-SIALYLTRANSFERASE (ST6GAL1) REVEALS CONSERVED AND UNIQUE FEATURES FOR GLYCAN SIALYLATION. Journal of Biological Chemistry. 2013;288:34680–34698. doi: 10.1074/jbc.M113.519041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JLW, McNamara JT, Fischer M, Rich J, Chen HM, Withers SG, Zimmer J. Observing cellulose biosynthesis and membrane translocation in crystallo. Nature. 2016 doi: 10.1038/nature16966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JLW, McNamara JT, Zimmer J. Mechanism of activation of bacterial cellulose synthase by cyclic di-GMP. Nature structural & molecular biology. 2014;21:489–496. doi: 10.1038/nsmb.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JLW, Strumillo J, Zimmer J. Crystallographic snapshot of cellulose synthesis and membrane translocation. Nature. 2013;493:181–186. doi: 10.1038/nature11744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray BW, Wittmann V, Burkart MD, Hung SC, Wong CH. Mechanism of human α-1, 3-fucosyltransferase V: glycosidic cleavage occurs prior to nucleophilic attack. Biochemistry. 1997;36:823–831. doi: 10.1021/bi962284z. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oriol R, Mollicone R, Cailleau A, Balanzino L, Breton C. Divergent evolution of fucosyltransferase genes from vertebrates, invertebrates, and bacteria. Glycobiology. 1999;9:323–334. doi: 10.1093/glycob/9.4.323. [DOI] [PubMed] [Google Scholar]
- Pannu NS, Waterreus WJ, Skubak P, Sikharulidze I, Abrahams JP, de Graaff RA. Recent advances in the CRANK software suite for experimental phasing. Acta Crystallogr D Biol Crystallogr. 2011;67:331–337. doi: 10.1107/S0907444910052224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly M, Keegstra K. Biosynthesis of the Plant Cell Wall Matrix Polysaccharide Xyloglucan*. Annual review of plant biology. 2016;67:235–259. doi: 10.1146/annurev-arplant-043015-112222. [DOI] [PubMed] [Google Scholar]
- Peña MJ, Kong Y, York WS, O’Neill MA. A galacturonic acid–containing xyloglucan is involved in Arabidopsis root hair tip growth. The Plant Cell. 2012;24:4511–4524. doi: 10.1105/tpc.112.103390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RM, DeRocher AE, Bar-Peled M, Zeng W, Norambuena L, Orellana A, Raikhel NV, Keegstra K. Xyloglucan Fucosyltransferase, an Enzyme Involved in Plant Cell Wall Biosynthesis. Science. 1999;284:1976–1979. doi: 10.1126/science.284.5422.1976. [DOI] [PubMed] [Google Scholar]
- Rassolov VA, Ratner MA, Pople JA, Redfern PC, Curtiss LA. 6‐31G* basis set for third‐row atoms. Journal of Computational Chemistry. 2001;22:976–984. [Google Scholar]
- Reeves PJ, Callewaert N, Contreras R, Khorana HG. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proceedings of the National Academy of Sciences. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter WD, Chapple C, Somerville CR. Mutants of Arabidopsis thaliana with altered cell wall polysaccharide composition. The Plant Journal. 1997;12:335–345. doi: 10.1046/j.1365-313x.1997.12020335.x. [DOI] [PubMed] [Google Scholar]
- Rocha J, Cicéron F, de Sanctis D, Lelimousin M, Chazalet V, Lerouxel O, Breton C. Structure of Arabidopsis thaliana FUT1 Reveals a Variant of the GT-B Class Fold and Provides Insight into Xyloglucan Fucosylation. The Plant Cell. 2016 doi: 10.1105/tpc.16.00519. tpc-00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiner S, Harding LB. Proton transfers in hydrogen-bonded systems. 2. Electron correlation effects in (N/sub 2/H/sub 7/)/sup+ J Am Chem Soc. 1981;103 (United States) [Google Scholar]
- Schimpl M, Zheng X, Borodkin VS, Blair DE, Ferenbach AT, Schüttelkopf AW, Navratilova I, Aristotelous T, Albarbarawi O, Robinson DA. O-GlcNAc transferase invokes nucleotide sugar pyrophosphate participation in catalysis. Nature chemical biology. 2012;8:969–974. doi: 10.1038/nchembio.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subedi GP, Johnson RW, Moniz HA, Moremen KW, Barb A. High Yield Expression of Recombinant Human Proteins with the Transient Transfection of HEK293 Cells in Suspension. JoVE (Journal of Visualized Experiments) 2015:e53568–e53568. doi: 10.3791/53568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HY, Lin SW, Ko TP, Pan JF, Liu CL, Lin CN, Wang AHJ, Lin CH. Structure and Mechanism of Helicobacter pylori Fucosyltransferase A BASIS FOR LIPOPOLYSACCHARIDE VARIATION AND INHIBITOR DESIGN. Journal of Biological Chemistry. 2007;282:9973–9982. doi: 10.1074/jbc.M610285200. [DOI] [PubMed] [Google Scholar]
- Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, Hazlewood V, Lathrop S, Lifka D, Peterson GD. XSEDE: accelerating scientific discovery. Computing in Science & Engineering. 2014;16:62–74. [Google Scholar]
- Tuomivaara ST, Yaoi K, O’Neill MA, York WS. Generation and structural validation of a library of diverse xyloglucan-derived oligosaccharides, including an update on xyloglucan nomenclature. Carbohydrate research. 2015;402:56–66. doi: 10.1016/j.carres.2014.06.031. [DOI] [PubMed] [Google Scholar]
- Tvaroška I, Kozmon S, Wimmerová M, Koča J. Substrate-assisted catalytic mechanism of O-GlcNAc transferase discovered by quantum mechanics/molecular mechanics investigation. Journal of the American Chemical Society. 2012;134:15563–15571. doi: 10.1021/ja307040m. [DOI] [PubMed] [Google Scholar]
- Urbanowicz BR, Peña MJ, Moniz HA, Moremen KW, York WS. Two Arabidopsis proteins synthesize acetylated xylan in vitro. The Plant Journal. 2014;80:197–206. doi: 10.1111/tpj.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanowicz BR, Pena MJ, Ratnaparkhe S, Avci U, Backe J, Steet HF, Foston M, Li H, O’Neill MA, Ragauskas AJ, Darvill AG, Wyman C, Gilbert HJ, York WS. 4-O-methylation of glucuronic acid in Arabidopsis glucuronoxylan is catalyzed by a domain of unknown function family 579 protein. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14253–14258. doi: 10.1073/pnas.1208097109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Valero-González J, Leonhard-Melief C, Lira-Navarrete E, Jiménez-Osés G, Hernández-Ruiz C, Pallarés MC, Yruela I, Vasudevan D, Lostao A, Corzana F. A proactive role of water molecules in acceptor recognition by protein O-fucosyltransferase 2. Nature chemical biology. 2016;12:240–246. doi: 10.1038/nchembio.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanzin GF, Madson M, Carpita NC, Raikhel NV, Keegstra K, Reiter WD. The mur2 mutant of Arabidopsis thaliana lacks fucosylated xyloglucan because of a lesion in fucosyltransferase AtFUT1. Proceedings of the National Academy of Sciences. 2002;99:3340–3345. doi: 10.1073/pnas.052450699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincken JP, York WS, Beldman G, Voragen AG. Two general branching patterns of xyloglucan, XXXG and XXGG. Plant Physiology. 1997;114:9. doi: 10.1104/pp.114.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wu D, Lu Z, Chen W, Hu X, Ding Y. A novel method for high-level production of TEV protease by superfolder GFP tag. BioMed Research International. 2010;2009 doi: 10.1155/2009/591923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabotina OA, Avci U, Cavalier D, Pattathil S, Chou YH, Eberhard S, Danhof L, Keegstra K, Hahn MG. Mutations in multiple XXT genes of Arabidopsis reveal the complexity of xyloglucan biosynthesis. Plant physiology. 2012;159:1367–1384. doi: 10.1104/pp.112.198119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Truhlar D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Account. 2008;120:215–241. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of the GDP/GDP-Fuc binding and positive electron density in the FUT1-GDP structure.
Molecular dynamics reveals active site residues stabilizing GDP-Fuc and XXLGXXLG binding.
Molecular Dynamics of different ternary complexes.
Snapshot of MD Equilibrated ternary complex-AtFUT1-GDPf-XXLG.
Optimized Geometries for the Product QM System.
Optimized Geometries for the Reactant QM System.
Transition State Geometries for the QM System.
Stereo figure of the GDP binding site of FUT1-GDP.
Amino acid sequence alignment of the GT37 family.
The AtFUT1-GDP and NodZ structures superimposed.
Reduced system considered for QM calculations.
Michaelis-Menten kinetics of AtFUT1 and catalytic variants.
MALDI-TOF MS Analysis of AtFUT1 variants.
AtFUT1 catalyzed transfer of fucosyl residues from GDP-Fuc onto the model xyloglucan oligosaccharide acceptor substrate XXLG.
Conformational shift of GDP in the R366K mutant.
Theoretical and observed mass to charge ratios (m/z) of xyloglucan oligosaccharides.
X-ray data collection and refinement statistics.
Carbohydrate conformational validation by Privateer.
Average interaction energies of GDP-Fuc with AtFUT1 amino acid residues.
Average interaction energies of XXLGXXLG with AtFUT1 amino acid residues.