

Abstract
Background
During asexual reproduction the embryogenic callus can differentiate into a new plantlet, offering great potential for fostering in vitro culture efficiency in plants. The immature embryos (IMEs) of wheat (Triticum aestivum L.) are more easily able to generate embryogenic callus than mature embryos (MEs). To understand the molecular process of embryogenic callus formation in wheat, de novo transcriptome sequencing was used to generate transcriptome sequences from calli derived from IMEs and MEs after 3d, 6d, or 15d of culture (DC).
Results
In total, 155 million high quality paired-end reads were obtained from the 6 cDNA libraries. Our de novo assembly generated 142,221 unigenes, of which 59,976 (42.17%) were annotated with a significant Blastx against nr, Pfam, Swissprot, KOG, KEGG, GO and COG/KOG databases. Comparative transcriptome analysis indicated that a total of 5194 differentially expressed genes (DEGs) were identified in the comparisons of IME vs. ME at the three stages, including 3181, 2085 and 1468 DEGs at 3, 6 and 15 DC, respectively. Of them, 283 overlapped in all the three comparisons. Furthermore, 4731 DEGs were identified in the comparisons between stages in IMEs and MEs. Functional analysis revealed that 271transcription factor (TF) genes (10 overlapped in all 3 comparisons of IME vs. ME) and 346 somatic embryogenesis related genes (SSEGs; 35 overlapped in all 3 comparisons of IME vs. ME) were differentially expressed in at least one comparison of IME vs. ME. In addition, of the 283 overlapped DEGs in the 3 comparisons of IME vs. ME, excluding the SSEGs and TFs, 39 possessed a higher rate of involvement in biological processes relating to response to stimuli, in multi-organism processes, reproductive processes and reproduction. Furthermore, 7 were simultaneously differentially expressed in the 2 comparisons between the stages in IMEs, but not MEs, suggesting that they may be related to embryogenic callus formation. The expression levels of genes, which were validated by qRT-PCR, showed a high correlation with the RNA-seq value.
Conclusions
This study provides new insights into the role of the transcriptome in embryogenic callus formation in wheat, and will serve as a valuable resource for further studies addressing embryogenic callus formation in plants.
Electronic supplementary material
The online version of this article (10.1186/s12870-017-1204-2) contains supplementary material, which is available to authorized users.
Keywords: Transcriptome, Embryo culture, Embryogenic callus, Immature embryo, Mature embryo, Wheat (Triticum aestivum L.)
Background
Efficient plant regeneration from in vitro cultured cells and tissues is a prerequisite requirement for the successful application of plant genetic engineering, which has been incorporated into the improvement and breeding of many crops [1]. However, the regeneration efficiency of a great number of plant species is still low under in vitro conditions. Embryogenic callus formation offers great potential for fostering in vitro culture efficiency whereby somatic cells are induced to differentiate embryogenic cells and form somatic embryos that develop into new plants [2, 3]. The ability to initiate embryogenic cultures is controlled by the intrinsic and external factors of the explants, including the genotype, the developmental stage, and the different tissue of the explants, as well as the factors associated with the medium as perceived by the complementary sensors in cells, including stress, phytohormones, and the artificial nutritional environment in the culture process [4–6]. In plants, the tissue culture response (TCR), regenerative property, regeneration response, and somatic embryogenesis are all traits that are used to study tissue culture.
To understand the molecular mechanisms of gene regulation in in vitro culture, the expression of the somatic embryogenesis-related genes and signaling regulation has been researched in many species, including Cyclamen persicum [7], oil palm [8], cotton [9, 10], Arabidopsis [11], soybean [12] and chicory [13], using different approaches including cDNA microarray, in situ hybridization (ISH), and suppression subtractive hybridization (SSH). Transcriptional profiling has shown that many genes showing differential expression during somatic embryogenesis (SE) in cereals may be associated with embryogenic callus formation. Some transcription factor (TF) genes such as the LEC genes, including Arabidopsis LEC1 and L1 L, encode HAP3-related transcription factors [14–16], while Arabidopsis LEC2, FUS3, and ABI3, as well as maize Viviparous1 (Vp1), encode the B3 domain transcription [17, 18]. Some members of the AP2/ERF family [19] encoded by BBM [20, 21], AGL15 [22], and WUSCHEL (WUS) genes [23], are essential for maintaining embryogenic competency of plant somatic cells.
The bread wheat (Triticum aestivum L.) is one of the most important human food crops. Considerable effort and progress has been made in tissue culture system optimization over the last few decades, however tissue culture efficiency is still low and lags behind that of other plants such as rice and maize [24, 25]. A number of studies on wheat embryogenic callus formation-associated genes have been performed using Northern and semi qRT-PCR analyses [26, 27]. Considerable data have been accumulated regarding the influence upon TCR, but with a limited knowledge of the genes involved therein.
Next-generation sequencing (NGS) technology has high sensitivity and includes both low- and high-level gene expression [28–30], and has been increasingly applied in numerous plants to elucidate development [31], senescence [32, 33], effects of different factors on plant growth [32, 33], and determine transcriptome changes in response to various abiotic stresses such as cold [34], salt [35], drought [36], water [37], and has also been used in wheat transcriptome studies regarding response to stress or disease [38, 39]. In terms of in vitro culturing in plants, NGS has been successfully used in cotton [40], Picea balfouriana [41], camphor tree [42], Maize [43], and in Ramie [44] for transcriptome analysis. However, few reports have been found regarding the transcriptome in wheat in vitro culture at the genomic scale. Thus, the study of gene expression patterns and functioning during callus formation could provide a molecular basis for embryogenic callus formation in wheat. However, the large size and polyploid complexity of the wheat genome [45], and the difficulties of tissue culture, have been obstacles to the study of the genes involved in wheat embryogenic callus formation. Using Illumina deep sequencing, we compared the transcriptome expression changes between IMEs and MEs for in vitro culture at 3d, 6d, and 15d, and between developmental stages in IMEs and MEs, combined with the functional annotation of the DEGs, especially of those that are potentially involved in embryogenic callus formation. This will provide novel insights into wheat embryogenic callus formation and the transcriptome data presented here will serve as a valuable resource for future studies of the genes and gene regions involved.
Methods
Plant material and tissue culture
The bread wheat cultivar Zhoumai 18 was grown in the Zhengzhou experimental field (longitude E 113.65°, latitude N 34.76°) of the Henan Agricultural University of China during the 2011–2012 cropping season. Harvested seeds were planted in the same experimental field in October 5 2012, after which they were transplanted into a greenhouse in January 15 2013 under the following controlled environmental conditions: 75% relative humidity, 26/20 °C day/night temperature, 12−/12-h light/dark photoperiod, and a light intensity of 10,000 lx. Anthesis was recorded in February of 2013 when 50% of the plants had reached the flowering stage.
Immature seeds from the main spikes were harvested 14 days after pollination. The mature seeds were soaked in running tap water for 4 h. Immature and mature seeds were surface-sterilized for 30 s in 75% (v/v) ethanol, followed by 6 min immersion in 0.1% (m/v) mercuric chloride solution with agitation, and then rinsed 4 times with sterilized distilled water. The IMEs and MEs were extracted from the sterilized seeds on a clean bench and explants were placed with the scutellum upwards in sterile Petri dishes containing solid Murashige and Skoog (MS) medium (MS basal salts, Gamborg’s B5 vitamins, and 2 mg L−1 2, 4-Dichlorophenoxyacetic acid). They were then grown in a growth chamber at 22–24 °C in the dark. The MEs and IMEs were cultured in 3 biological replicates, each replicate consisting of 15 plates and each plate containing 10 embryos. The embryos were harvested at 3 DC, 6 DC, and 15 DC from 5 plates in each of the 3 replicates, after which they were snap-frozen in liquid nitrogen and stored at −80 °C until RNA extraction.
RNA extraction, RNA-seq library construction and Illumina sequencing
Total RNA was extracted from the IME and ME callus in the 3 biological replicates using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, and then characterized on a 1% agarose gel. The purity of the RNA was examined using a NanoDrop 2000 spectrophotometer (IMPLEN, CA, USA), and the integrity was examined using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). The RNA samples of the 3 biological replicates were mixed in equal amounts and used for the construction of libraries by the Biomarker Biotechnology Corporation (Beijing, China). A total of 3 μg RNA for each sample was used as input material for cDNA library construction. The mRNA was enriched and purified with oligo (dT)-rich magnetic beads and then broken into short fragments. Taking these cleaved mRNA fragments as templates, first strand cDNA was synthesized and primed by oligo-dT and the second strand cDNA was synthesized using random primers. The resulting cDNAs were then subjected to end-repair and phosphorylation using T4 DNA polymerase and Klenow DNA polymerase.
Following this, an “A” base was inserted as an overhang at the 3′ ends of the repaired cDNA fragments and Illumina paired-end solexa adaptors were subsequently ligated to these cDNA fragments to distinguish the different sequencing samples. To select a size range of templates for downstream enrichment, the products of the ligation reaction were purified and visualized on a 2% agarose gel. Next, PCR amplification was performed to enrich the purified cDNA template.
RNA sequencing was performed on an Illumina HiSeq (TM) 2000 (San Diego, CA, USA). The RNA-seq read data were deposited in the NCBI Sequence Read Archive (SRP093588).
De novo assembly and functional annotation
Clean, high quality reads were obtained after filtering the adaptor sequences and reads with ambiguous “N” bases and with a base quality less than Q30 (and more than 10% base quality less than Q20). The clean reads were assembled into contigs using the Trinity method (Grabherr MG, et al., 2011) using an optimized k-mer length (25-mer). Subsequently, the contigs were assembled into transcripts according to the paired-end information, after which they were clustered based on sequence similarity. Finally, the longest transcript in each cluster was regarded as a unigene, and the singletons were combined together as total unigenes. All unigenes were mapped to the reference sequences (wheat unigenes from NCBI) and allowed no more than one nucleotide mismatch. For annotation, all unigenes were used for the blast search and annotation against a series of public databases using BLASTx (E-value–5 ≤ 10), including the NCBI non-redundant (Nr) protein databases (http://www.ncbi.nlm.nih.gov), the Swiss-Prot protein database (http://www.expasy.ch/sprot), the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (http://www.genome.jp/kegg), the Cluster of Orthologous Groups (COG) database (http://www.ncbi.nlm.nih.gov/COG), and the pfam database. Functional classification by gene ontologies (GO) of all unigenes was performed using Blast2GO software.
Identification of differentially expressed genes
To quantify the gene expression level, the number of mapped clean reads was estimated using RSEM [46] for each sample, and the normalized expression values RPKM (reads per kilo base of exon model per million mapped reads) of each unigene in the 6 libraries were used as the value of gene expression levels [47]. To determine significant differences in gene expression, we used threshold criteria based on the FDR statistical method and compared the normalized expression values RPKM using a threshold value of P ≤ 0.001 and |log2 Ratio| ≥ 1 based on the FDR < 0.05.
Analysis of transcription factors
Putative transcription factors were identified by BLASTx against the Plant Transcription Factor Database (PlnTFDB) 3.0 [48] using the bread wheat Transcription Factor (http://planttfdb.cbi.pku.edu.cn/index.php?sp=Tae), Arabidopsis Transcription Factor (http://planttfdb.cbi.pku.edu.cn/index.php?sp=Ath) and the alignment results of orthologous in the NCBI. Subsequently, the differentially expressed TFs were picked out from the DEGs in the IME vs. ME comparison, and between the stages in IMEs and MEs.
Unigene quantification by real-time PCR.
A total of 32 unigenes were selected for expression profile validation by qRT-PCR. Reverse transcription (RT) reactions were performed in 3 independent biological replicates with RNA that was individually extracted from 3 independent biological samples of six types of callus, performed using a PrimeScript® RT reagent Kit with gDNA Eraser (Perfect Real Time; Takara, China). The first genomic DNA elimination reaction was conducted in a final volume of 10 μL including 2.0 μL of 5× gDNA Eraser Buffer, 1.0 μL of gDNA Eraser, 2.0 μL of total RNA, and 5 μL of RNase Free dH2O. Reactions were incubated at 42 °C for 2 min, and then maintained at 4 °C. The RT reactions (10.0 μL) were then used for the SYBR® Green qPCR assay in a 20-μL reaction mixture that included 4.0 μL of 5 × PrimeScript® Buffer 2 (for real time), 1.0 μL of PrimeScript® RT Enzyme Mix I, 1.0 μL of RT Primer Mix, and 4.0 μL of RNase Free dH2O. The reactions were incubated at 37 °C for 15 min, followed by 85 °C for 5 s, after which they were maintained at 4 °C.
Real-time PCR was performed on a Bio-Rad CFX96TM Real-Time System with SYBR® Premix Ex Taq II™ (Takara, China). Each reaction included 2 μL of product from the diluted RT reactions (cDNA solution), 1.0 μL of each primer (Additional file 1), 12.5 μL of SYBR® Premix Ex TaqTM II (2×), and 8.5 μL of RNase free water. All qRT-PCR reactions were incubated in a 96-well plate at 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s, 60 °C for 30 s, and 72 °C for 30 s. The actin gene (GenBank: AB181991) was used as the endogenous control. All reactions were run in triplicate. The specificity of each primer pair was verified by agarose gel electrophoresis and melting curve analysis. The relative expression levels of genes were calculated using the 2-ΔΔCt method [49]. The gene sample in the ME library along with the CT value was selected as the calibrator, in which the expression level was set as 1.0. The relative expression levels of the same genes in the corresponding IME library were then normalized by comparison.
Results
Illumina sequencing and de novo assembly
With regards to the transcriptional analysis, the calli from IMEs and MEs cultured in vitro at 3d, 6d and 15d were selected, resulting in a total of 6 cDNA libraries (IME3, IME6, IME15 and ME3, ME6, ME15) that were constructed and sequenced using the Illumina HiSeq™ 2000 platform. Following data cleaning and quality checking, 155,192,839 reads (31.34 G) with Q30 values ≥80.00% were obtained from the six libraries. Among the clean reads, 100% had quality scores at Q20 level (Table 1).
Table 1.
Output statistics of the total reads from 6 libraries
| Library | Total reads (n) | Total nucleotides (bp) | Q20 percentage | GC percentage | Q30 percentage |
|---|---|---|---|---|---|
| IME3 | 28,775,750 | 5,808,494,474 | 100.00% | 56.35% | 80.18% |
| IME6 | 24,767,646 | 5,002,204,197 | 100.00% | 56.47% | 80.13% |
| IME15 | 25,514,989 | 5,153,360,463 | 100.00% | 55.88% | 80.00% |
| ME3 | 25,841,219 | 5,219,396,740 | 100.00% | 56.10% | 80.05% |
| ME6 | 25,745,846 | 5,200,136,819 | 100.00% | 55.88% | 80.20% |
| ME15 | 24,547,389 | 4,957,729,251 | 100.00% | 55.73% | 80.22% |
ME3, ME6, and ME15 are mature embryos at 3 d, 6 d, and 15 d of culture, respectively
IME3, IME6, and IME15 are immature embryos at 3 d, 6 d, and 15d of culture, respectively
The high-quality reads were then merged and de novo assembled using the Trinity platform software, resulting in the generation of 331,201 transcripts with an average length of 1009.69 bp and a N50 length of 1639 bp. The total length of all transcripts was approximately 334 Mb. These transcripts were further subjected to cluster and assembly analyses, resulting in 142,221 unigenes with an average length of 657 bp and an N50 value of 1001 bp, of which 23,875 unigenes were more than 1kbin length (Table 2, Additional file 2). Open Reading Frames (ORFs) were analyzed by Getorf from the EMBOSS package and 141,393 unigenes (99.42%) had ORFs with a start codon (Table 3).
Table 2.
Overview of the de novo sequence assembly for wheat callus
| Length range | Contigs | Transcripts | Unigenes |
|---|---|---|---|
| <300 | 9,122,919 (98.65%) | 67,091 (20.26%) | 49,059 (34.49%) |
| 300–500 | 60,061 (0.65%) | 69,816 (21.08%) | 42,416 (29.82%) |
| 500–1000 | 38,359 (0.41%) | 75,986 (22.94%) | 26,871 (18.89%) |
| 1000–2000 | 18,524 (0.20%) | 74,109 (22.38%) | 15,378 (10.81%) |
| > 2000 | 7933 (0.09%) | 44,199 (13.35%) | 8497 (5.97%) |
| Total number | 9,247,796 | 331,201 | 142,221 |
| Total length | 554,005,887 | 334,411,704 | 93,394,731 |
| N50 length | 49 | 1639 | 1001 |
| Mean length | 59.91 | 1009.69 | 656.69 |
Table 3.
ORF length of the wheat unigene
| Length range | Unigene ORF |
|---|---|
| 0–300 | 104,094 (73.62%) |
| 300–500 | 16,493 (11.66%) |
| 500–1000 | 11,060 (7.82%) |
| 1000–2000 | 7326 (5.18%) |
| 2000+ | 2420 (1.71%) |
| Total number | 141,393 |
| Total length | 47,224,473 |
| N50 length | 558 |
| Mean length | 333.99 |
Functional annotation of unigenes
The unigenes were annotated by means of a Blastx search against the public databases. Among all 142,221 unigenes, 59,976 (42.17%) were successfully annotated by at least one database in 7 databases, i.e., Nr, Pfam, Swissprot, KOG, KEGG, GO and COG (Table 4). Furthermore, 26,529 (18.65%), 29,587 (20.08%) and 14,727 (10.36%) unigenes were annotated in the Swiss-Prot database, Pfam, and KEGG database, respectively (Additional file 3). In addition, 58,723 unigenes (41.29%) had significant matches in the NCBI non-redundant protein (Nr) database with an E-value cut-off of 10−5 (Table 2, Fig. 1), and 27,832 (47%) of them had an E-value lower than 1.0 e-50 (Fig. 1a). With respect to species, the top matched plant species included Aegilops tauschii (18,301, 31%), followed by Triticum urartu (11,324, 19%), Hordeum vulgare (8091, 14%) and Triticum aestivum (3869, 7%; Fig. 1b).
Table 4.
The number and distribution of unigenes annotated in the databases
| Database | COG | GO | KEGG | KOG | Pfam | Swissprot | nr | All |
|---|---|---|---|---|---|---|---|---|
| Unigene no. | 11,513 | 34,761 | 14,727 | 24,345 | 29,587 | 26,529 | 58,723 | 59,976 |
| Percentage (%) | 8.10 | 24.44 | 10.36 | 17.12 | 20.80 | 18.65 | 41.29 | 42.17 |
Fig. 1.
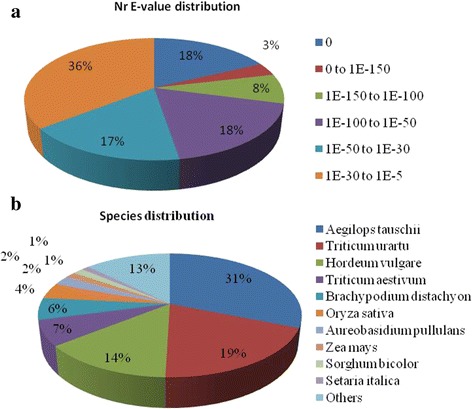
Characteristics of the homology search of unigenes against the Nr database. a. E-value distribution; b. Best hit species distribution. The cut-off values for the BLAST search were set at 1.0e−10
To further reveal their functions, gene ontology (GO) assignments were performed using Blast2GO, with the result that 34,761 (24.44%) unigenes received at least one GO term and were classified into 3 major functional categories: cellular component, molecular function, and biological process (E-value <1E-5). For the biological process category, the majority of unigenes were annotated with “metabolic process” (21,814 unigenes), “cellular process” (18,859 unigenes) and “single-organism process” (13,313 unigenes; Fig. 1). Within the molecular function category, the highly represented GO terms included “binding” (19,497 unigenes), “catalytic activity” (17,665 unigenes) and “transporter activity” (1811 unigenes), while “cell part” (22,229 unigenes), “cell” (22,171 unigenes), and “organelle” (19,617 unigenes) were highly represented in the cellular component category (Fig. 2, Additional file 4a).
Fig. 2.
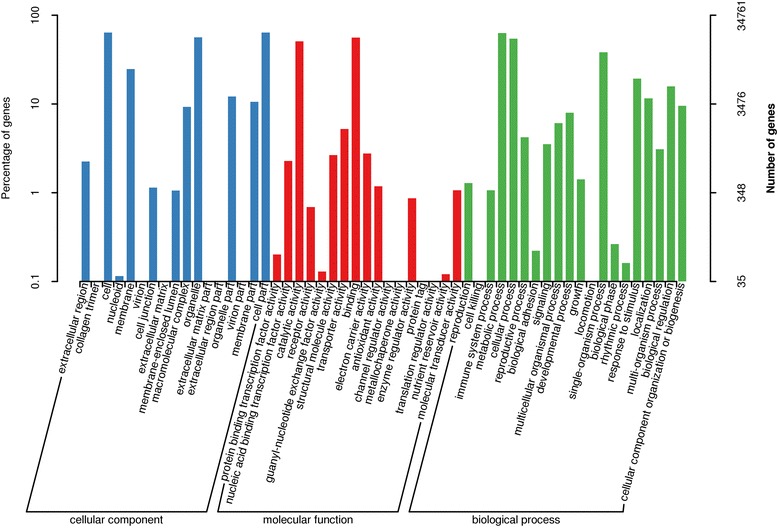
GO annotation of assembled unigenes by Blast2GO in wheat callus. A total of 34,761 unigenes were assigned to at least one GO term in 3 categories: biological process, molecular function, and cellular component. The x-axis indicates the sub-categories and the y-axis indicates the number of unigenes
The search against the COG database resulted in 11,513 (9.79%) unigenes assigned to COG classification and divided into 25 COG categories, with “general function prediction only” (3129), “replication, recombination and repair” (2004) and “transcription” (1548) being the 3 most highly represented (Fig. 3; Additional file 4b). Metabolic pathway analysis was conducted using the KEGG orthology (KO) categories system. Consequently, 14,727 (10.36%) unigenes were assigned to 128 pathways and divided into 5 classes. In the primary pathway hierarchy, “metabolism” and “genetic information processing” were dominant (Fig. 4; Additional file 4c).
Fig. 3.
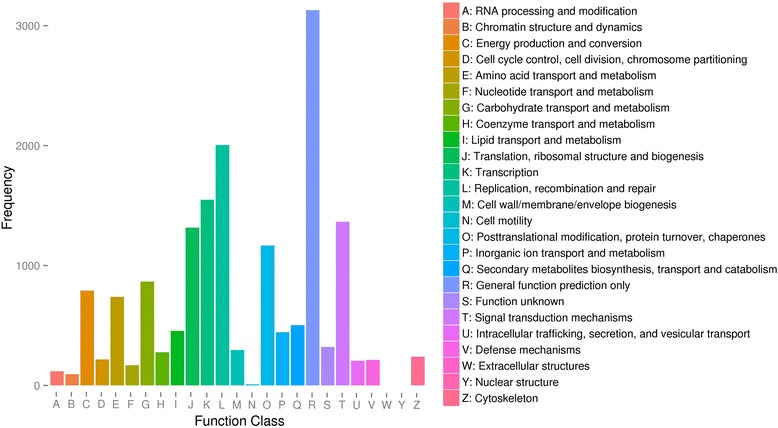
COG assignments of assembled unigenes in wheat callus. Out of 142,221 de novo assembled unigenes, 14,727 were assigned to 25 COG categories
Fig. 4.
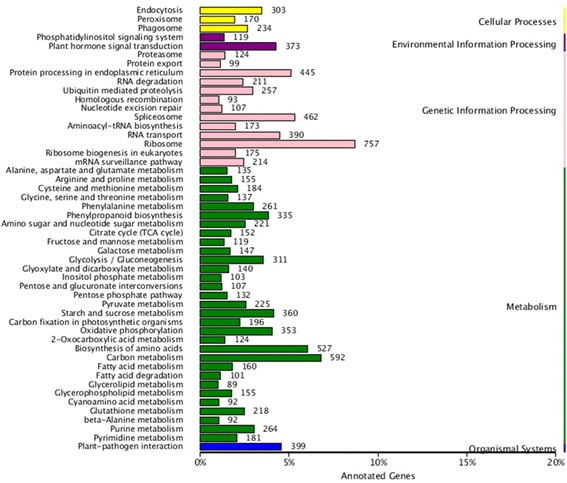
KEGG annotation of assembled unigenes in wheat callus. Distribution of the number of genes expressed in the various metabolic pathways. a cellular processes; (b) environmental information processing; (c) genetic information processing; (d) metabolism; (e) organismal systems
Investigation of differentially expressed genes and functional categorization
The expression levels of each unigene in the6 libraries were normalized and calculated using RPKM [47]; Additional file 5). Differentially expressed genes (DEGs) analysis in the comparisons of IME vs. ME at the 3 corresponding stages resulted in a total of 5194 DEGs in all three comparisons, with 3181, 2085 and 1468 DEGs at 3, 6 and 15 DC, respectively (Fig. 5a; Additional file 6a–c). With regards to the comparisons between developmental stages 4731 DEGs were identified (Fig. 5a; Additional file 7a-d), of which 2937 overlapped with the 5194 DEGs in the IME vs. ME comparisons (Additional file 7e).
Fig. 5.
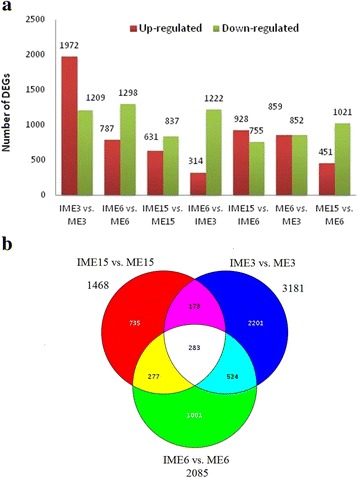
Histogram and Venn diagram of the differentially expressed genes (DEGs) in comparisons of IME vs. ME and between stages in IMEs and MEs during callus formation in wheat callus. a Histogram showing the number of DEGs up- or down-regulated in each comparison. b Venn diagrams showing similarly or distinctly regulated genes in the three comparisons of IME vs. ME
In the IME vs. ME comparisons, 283 DEGs overlapped in all 3 comparisons, of which 266 were annotated in the 7 databases mentioned above (Fig. 5b; Additional file 6d–e). The degree of DEG overlap between the comparisons during wheat embryogenic callus formation isindicated in Fig. 5b. Functional classification indicated that 2579, 1878, and 1271 DEGs could be annotated in at least one of the 7 databases at 3, 6, and 15 DC, respectively, while 1873, 1433, and 919 DEGs could be assigned to at least one GO term at 3, 6, and 15 DC, respectively (Table 5; Additional file 8a–c).
Table 5.
The number of differentially expressed unigenes annotated
| ALL | COG | GO | KEGG | KOG | Pfam | SWISS | Nr | |
|---|---|---|---|---|---|---|---|---|
| IME3 vs. ME3 (no.) | 2579 | 515 | 1873 | 713 | 1013 | 1875 | 1720 | 2545 |
| % | 81.08 | 16.19 | 58.88 | 22.41 | 31.85 | 58.94 | 54.07 | 80.01 |
| IME6 vs. ME6 (no.) | 1878 | 387 | 1433 | 542 | 722 | 1416 | 1265 | 1875 |
| % | 90.07 | 18.56 | 68.73 | 26.00 | 34.63 | 67.91 | 60.67 | 89.93 |
| IME15 vs. ME15 (no.) | 1271 | 241 | 919 | 333 | 431 | 917 | 831 | 1261 |
| % | 86.58 | 16.42 | 62.60 | 22.68 | 29.36 | 62.47 | 56.61 | 85.90 |
| IME6 vs.IME3 (no.) | 1302 | 233 | 916 | 344 | 489 | 950 | 830 | 1284 |
| % | 84.77 | 15.17 | 59.64 | 22.40 | 31.84 | 61.85 | 54.04 | 83.59 |
| IME15 vs. IME6 (no.) | 1450 | 282 | 1035 | 396 | 514 | 1037 | 956 | 1430 |
| % | 86.16 | 16.76 | 61.50 | 23.53 | 30.54 | 61.62 | 56.80 | 84.97 |
| ME6 vs. ME3 (no.) | 1525 | 422 | 1063 | 546 | 692 | 1095 | 956 | 1519 |
| % | 89.13 | 24.66 | 62.13 | 31.91 | 40.44 | 64.00 | 55.87 | 88.78 |
| ME15 vs. ME6 (no.) | 1274 | 331 | 897 | 416 | 526 | 925 | 789 | 1260 |
| % | 86.55 | 22.49 | 60.94 | 28.26 | 35.73 | 62.84 | 53.60 | 85.60 |
ME3, ME6, and ME15 are mature embryos at 3 d, 6 d, and 15 d of culture, respectively
IME3, IME6, and IME15 are immature embryos at 3 d, 6 d, and 15d of culture, respectively
In the comparisons between stages in IMEs and MEs, 1302, 1450, 1525 and 1274 DEGs could be annotated in at least one of the 7 databases in the IME6 vs. IME3, IME15 vs. IME6, ME6 vs. ME3, and ME15 vs. ME6 comparisons, respectively. Those assigned to GO terms included 1916, 1035, 1063, and 897 across the 4 comparisons, respectively (Table 5; Additional file 8d–g). In the biological process category of GO assignment, metabolic processes, cellular process and single-organism process were the 3 top groups in each comparison (Fig. 6a). With respect to the molecular function category, the assignments were mainly attributed to catalysis and binding (Fig. 6b).
Fig. 6.
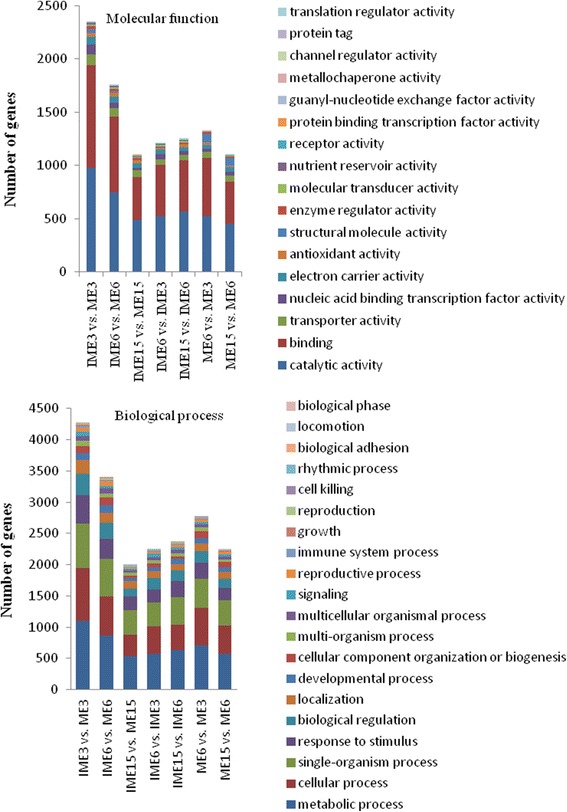
Molecular functions and biological processes of DEGs in comparisons of IME vs. ME and between stages in IMEs and MEs during callus formation based on gene ontology categories
Of the KEGG pathway annotation of DEGs in the IME vs. ME comparisons, 473 unigenes were assigned to 97 KEGG pathways in IME3 vs. ME3, while 335 unigenes were assigned to 89 KEGG pathways in IME6 vs. ME6, and 202 unigenes were assigned to 82 KEGG pathways in the IME15 vs. ME15 comparison (Additional file 9a–c).
Pathway enrichment analysis revealed that the most representative unigenes included phenylpropanoid biosynthesis, plant-pathogen interaction, starch and sucrose metabolism, and plant hormone signal transduction in the IME3 vs. ME3 comparison (Additional file 9 a). In the IME6 vs. ME6 comparison, the 4 most represented unigenes included phenylpropanoid biosynthesis, phenylalanine metabolism, starch and sucrose metabolism, and plant hormone signal transduction (Additional file 9b). In the IME15 vs. ME15 comparison, phenylpropanoid biosynthesis and phenylalanine metabolism were the prevailing categories (Additional file 9c).
In the comparisons between stages in the IMEs and MEs libraries, 228 unigenes were assigned to 79 KEGG pathways in the IME6 vs. IME3 comparison, while 253 were assigned to 86 KEGG pathways in the IME15 vs. IME6 comparison. Additionally, 393 were assigned to 93 KEGG pathways in the ME6 vs. ME3 comparison, and 326 to 84 KEGG pathways in the ME15 vs. ME6 comparison (Additional file 9d–g).
Differently expressed TFs
TFs possess important functions in embryogenic callus formation. All 1787 TFs were identified in this study (1748 from Triticum aestivum L., 23 from Arabidopsis thaliana L., and 16 from public databases; Additional file 10a). The DEGs were classified into TF families and the results indicated that there were 271 significantly differentially expressed TFs in IME vs. ME comparisons (Fig. 7a; Additional file 10b). In the IME3 vs. ME3 comparison, 133 out of the 191 TFs showed up-regulation, while 58 showed down-regulation (Fig. 7e). The prevailing family was the Apetala2 (AP2) /Ethylene Responsive Factor (ERF) family with 36 (30 up-regulated) unigenes, followed by the MYB family with 23 unigenes (16 up-regulated), the bHLH family with 18 unigenes (11 up-regulated), the WRKY family with 15 unigenes (14 up-regulated), and the NAC family with 10 unigenes (6 up-regulated; Fig. 7b).
Fig. 7.
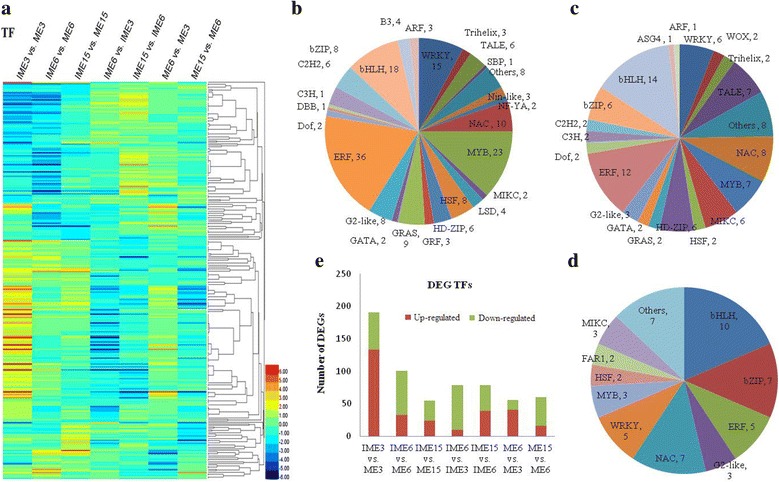
Analyses of differentially expressed TFs during wheat callus development. a A heat map of 271 differentially expressed TFs. Classification of TFs assigned in the three comparisons of IME3 vs. ME3 (b), IME6 vs. ME6 (c), and IME15 vs. ME15 (d). e Histogram showing the number of differentially expressed TFs up- or down-regulated in the different comparisons. IME3, IME6, and IME15 indicate calli from immature embryos at 3d, 6d, and 15d of culture, respectively. ME3, ME6, and ME15 indicate calli from mature embryos at 3d, 6d and 15d of culture, respectively
In the IME6 vs. ME6 comparison, 101 TFs were differentially expressed with 33 being up-regulated and 68 being down-regulated (Fig. 7e). These included the bHLH family with 14 unigenes (2 up-regulated), followed by the AP2/ERF family with 12 unigenes (7 up-regulated), the NAC family with 8 unigenes (8 down-regulated), and the MYB family with 7 unigenes (1 up-regulated; Fig. 7c). In the IME15 vs. ME15 comparison in which the somatic embryo became visible in IME, there were 55 differentially expressed TFs of which 24 were up-regulatedand 31 down-regulated (Fig. 7e). The predominant TF families included bHLH with 10 unigenes (2 up-regulated), followed with 7 unigenes by NAC (1 up-regulated) and bZIP (5 up-regulated), 5 unigenes of the WRKY (5 up-regulated) and ERFs (3 up-regulated; Fig. 7d).
Comparisons between stages in the IMEs and MEs libraries provided us with 194 significantly differentially expressed TFs in total, with 87, 79, 56, and 60 in IME6 vs. IME3, IME15 vs. IME6, ME6 vs. ME3, and ME15 vs. ME6, respectively, and the up-regulated genes were 10, 39, 41, and 16, respectively (Fig. 7e). Additionally, 150 of the 271 differentially expressed TFs in IMEs vs. MEs comparisons overlapped with the 194 differentially expressed TFs in the comparisons between the stages in IMEs and MEs. Furthermore, 10 of the 271 differentially expressed TFs of IMEs vs. MEs overlapped in all 3 comparisons (Table 6). Five of the 10 TFs were selected for expression level validation by qRT-PCR and the results indicated that they were all highly associated with the RNA-Seq value (Fig. 11).
Table 6.
Information of DEGs overlapped in IME vs. ME at three stages that are possibly involved in embryogenic callus formation and DEGs for qRT-PCR
| Category | ID | Annotation | qRT-PCR | Overlapped between stages in IMEs not in MEs | Bio- process |
|---|---|---|---|---|---|
| Bio-processes-involved genes (BPGs) | comp120922_c0 | Indole-3-glycerol phosphate lyase, chloroplastic; indole synthase | response to stimulus;in multi-organism process | ||
| comp129240_c0 | Cellulose synthase | ||||
| comp129268_c1 | Mobile element transfer protein; POT family | ||||
| comp128278_c0 | Cellulose synthase | reproductive process | |||
| comp127468_c0 | S-locus glycoprotein family | yes | in multi-organism process; reproductive process | ||
| comp115979_c0 | beta-expansin TaEXPB2 [Triticum aestivum] | in multi-organism process; reproductive process; reproduction | |||
| comp122548_c0 | Expansin-B6 [Triticum urartu] | ||||
| comp124996_c0 | Expansin EXPB8 [Triticum aestivum] | ||||
| comp126585_c2 | Expansin-B18 | ||||
| comp131969_c0 | Expansin EXPB9 [Triticum aestivum] | ||||
| comp123173_c0 | Aldose reductase [Triticum urartu] | response to stimulus; reproductive process | |||
| comp133735_c0 | Multicopper oxidase;; Laccase-19 | yes | |||
| comp139514_c1 | RNA recognition motif; Glycine-rich RNA-binding protein | ||||
| comp117215_c0 | Salt tolerant protein [Triticum aestivum] | response to stimulus | |||
| comp118386_c0 | hypothetical protein F775_08244 [Aegilops tauschii] | ||||
| comp122412_c0 | Pathogenesis-related protein / dehydrase and lipid transport | ||||
| comp123242_c0 | predicted protein [Hordeum vulgare subsp. vulgare] | ||||
| comp124683_c0 | NmrA-like family; NADH(P)-binding; Isoflavone reductase-like protein | ||||
| comp125897_c0 | GDSL/SGNH-like Acyl-Esterase family; Protein ESKIMO 1 | ||||
| comp127496_c0 | Glutaredoxin; Monothiol glutaredoxin-S5 (Rice) | ||||
| comp129379_c0 | AnnexinD4 [Aegilops tauschii] | ||||
| comp129891_c0 | Pirin-like protein | ||||
| comp130209_c0 | Cytochrome P450 | ||||
| comp131119_c0 | DREPP plasma membrane polypeptide; Salt stress root protein | Yes | |||
| comp131357_c0 | short chain dehydrogenase;11-beta-hydroxysteroid dehydrogenase 1B | ||||
| comp132259_c0 | Glycosyl transferases group 1; Sucrose synthase 1 | ||||
| comp132259_c1 | Sucrose synthase 2 [Triticum urartu] | ||||
| comp132409_c0 | Glycosyl hydrolases family 17 | ||||
| comp132805_c0 | Glycogenin-2 [Triticum urartu] | ||||
| comp134609_c0 | ATPase family associated with various cellular activities | ||||
| comp135852_c0 | Multicopper oxidase;; Putative laccase-5 (Rice) | ||||
| comp137996_c3 | Fructan 6-exohydrolase [Triticum aestivum] | ||||
| comp138064_c0 | Ethylene insensitive 3 | ||||
| comp138118_c0 | Senescence-associated protein | ||||
| comp140305_c0 | Universal stress protein family | ||||
| comp140523_c1 | Glycosyl hydrolases family 18; Xylanase inhibitor protein 1 | ||||
| comp141536_c0 | Aconitase family (aconitate hydratase) | Yes | Yes | ||
| comp141781_c0 | hypothetical protein TRIUR3_11299 [Triticum urartu] | ||||
| comp142326_c1 | RRM in Demeter;Transcriptional activator DEMETER | Yes | |||
| Unknown genes that overlapped between stages in IMEs not in MEs | comp120934_c0 | Xyloglucan endotransglucosylase/hydrolase (XTH) protein 22 | Yes | Yes | |
| comp126349_c0 | Xyloglucanendo-transglycosylase (XET) C-terminus | Yes | Yes | ||
| comp128901_c0 | Root cap | Yes | Yes | ||
| comp128978_c0 | Cortical cell-delineating protein | Yes | Yes | ||
| comp134128_c0 | U-box domain;E3 ubiquitin-protein ligase | Yes | Yes | ||
| comp136525_c0 | glycerol-3-phosphate acyltransferase 2 | Yes | Yes | ||
| comp142930_c0 | Reverse transcriptase Integrase core domain;gag-polypeptide of LTR copia-type | Yes | Yes | ||
| Overlapped TFs | comp127465_c1 | YABBY | Yes | ||
| comp131637_c0 | bHLH | Yes | |||
| comp134166_c0 | bZIP | Yes | |||
| comp118204_c0 | MIKC | ||||
| comp131990_c0 | LSD | ||||
| comp140577_c0 | G2-like | ||||
| comp125594_c0 | ERF | Yes | |||
| comp126788_c0 | ERF | Yes | Yes | ||
| comp128506_c0 | ERF | Yes | |||
| comp129826_c0 | NAC | ||||
| Overlapped SSEGs | comp111117_c0 | Disease resistance protein | |||
| comp123801_c0 | Disease resistance protein | ||||
| comp136600_c0 | Disease resistance protein | ||||
| comp78404_c0 | Disease resistance protein | ||||
| comp131631_c0 | Disease resistance protein | ||||
| comp132305_c0 | zinc finger | ||||
| comp132823_c2 | zinc finger | Yes | |||
| comp136499_c1 | zinc finger | ||||
| comp136627_c0 | zinc finger | ||||
| comp124435_c0 | LTP | ||||
| comp141004_c1 | LTP | ||||
| comp146809_c0 | LTP | Yes | |||
| comp126873_c0 | LEA | ||||
| comp113034_c0 | LEA | ||||
| comp113890_c0 | LEA | ||||
| comp129476_c0 | LEA | Yes | |||
| comp131074_c0 | LEA | ||||
| comp131947_c0 | LEA | ||||
| comp136871_c0 | LEA | ||||
| comp117547_c0 | Calcium-dependent | ||||
| comp120829_c0 | Calcium-dependent | ||||
| comp121468_c3 | Calcium-dependent | ||||
| comp122478_c2 | GST | ||||
| comp134985_c3 | GST | ||||
| comp162414_c0 | Auxin-induced protein | ||||
| comp125890_c1 | Peroxidase | ||||
| comp126327_c4 | Peroxidase | ||||
| comp128155_c1 | Peroxidase | ||||
| comp129605_c0 | Peroxidase | ||||
| comp129803_c0 | Peroxidase | Yes | |||
| comp130824_c1 | Peroxidase | Yes | |||
| comp130824_c2 | Peroxidase | ||||
| comp138541_c1 | Peroxidase | ||||
| comp140208_c0 | HSP | ||||
| comp124012_c0 | Germin-like protein | Yes | |||
| DEGs not overlapped that for qRT-PCR | comp116720_c1 | Heat shock 70 kDa protein | Yes | ||
| comp125045_c0 | Oleosin 16 kDa | Yes | |||
| comp125097_c0 | Zinc finger CCCH domain-containing protein | Yes | |||
| comp125797_c0 | Auxin-induced protein 1 | Yes | |||
| comp127206_c0 | WAT1-related protein | Yes | |||
| comp128173_c1 | GDSL esterase/lipase At2g40250 (Precursor) | Yes | |||
| comp128869_c0 | B3 domain-containing protein | Yes | |||
| comp131743_c0 | – | Yes | |||
| comp133896_c0 | Desiccation-related protein | Yes | |||
| comp134203_c0 | Cytochrome P450 71C3 | Yes | |||
| comp137615_c0 | Antimicrobial peptide 2d (Precursor) | Yes |
Fig. 11.
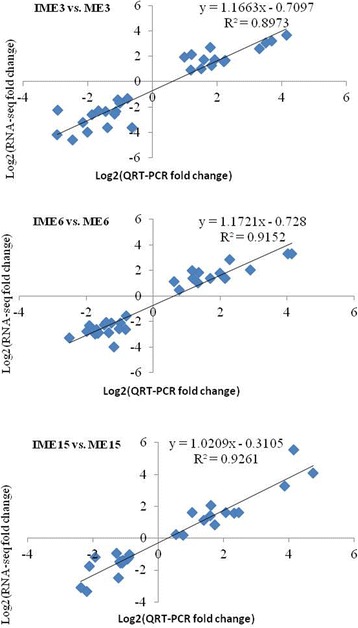
Correlation analysis of log2 fold change values obtained from RNA-Seq and qRT-PCR for 32 DEGs in the comparisons of IME vs. ME. RNA-Seq fold change refers to the ratios of RPKM values of IME to ME for selected genes, while Q-PCR fold change is the relative quantity of IME normalized to expression level of ME
Somatic embryogenesis-related genes in wheat callus
With respect to embryogenic callus formation, except for TFs, there were 346 DEGs identified in at least one of IME vs. ME comparisons including stress related genes or homologues to the previously annotated genes that were potentially somatic embryogenesis related genes, such as calcium-dependent protein kinase (CDPK)/calmodulin (CAM) [50, 51], germin-like protein (GLP) [52, 53], glutathione s-transferase (GST) [54], late embryogenesis abundant (LEA) proteins [55, 56], zinc finger [57, 58], non-specific lipid-transfer protein (LTP) [59], heat-shock protein (HSP) [60], indole acetic acid induced protein (Aux/IAA), and other aux related genes [4] (Fig. 8a). Of the 346 stress and somatic embryogenesis related genes (SSEGs), 35 overlapped in the 3 comparisons of IME vs. ME (Table 6). Five of the 35 SSEGs were selected for expression level validation by qRT-PCR and the results indicated that they were all highly correlated with the RNA-Seq value (Fig. 11). Auxin genes play an important role in plant cell dedifferentiation and redifferentiation [4, 61]. In this study, 44 DEGs (38 from the 346 SSEGs and 6 from the 271 TFs) were related to auxin, i.e., auxin-responsive protein (10), auxin efflux carrier component (2), auxin-induced protein (9), auxin-repressed protein (2), auxin transporter-like protein (2), auxin response factor (ARF, 4), small auxin-up RNAs (SAURs) family protein (7), GH3 auxin-responsive promoter (6), indole-3-acetaldehyde oxidase (1), and other auxin-related proteins were identified in IME vs. ME comparisons (Additional file 11a). The expression of 346 SSEGs in the 6 libraries is shown in Fig. 8b. Interestingly, 217 of them overlapped with the comparisons between stages in IME and/or ME (Additional file 11b).
Fig. 8.
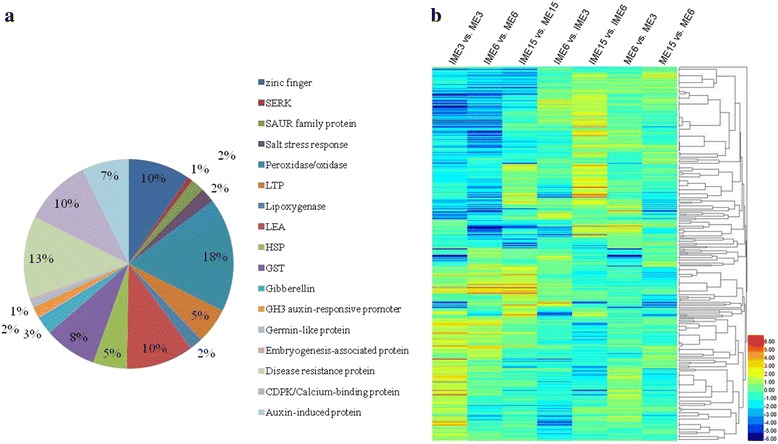
Analyses of differentially expressed somatic embryogenesis related genes during wheat callus development. a Expression of 346 differentially expressed somatic embryogenesis related genes. b The distribution of 346 somatic embryogenesis related genes in comparisons of IME vs. ME at the 3 stages. Numbers represent the percentage of genes out of the differentially expressed genes in each comparison
The KEGG pathways associated with plant hormone signal transduction in IME vs. ME comparisons, with regards to auxin related genes, included GH3 and SAURs which were down-regulated at 15d, as well as GH3 which was up-regulated at 3d and 6d. SAUR was mix regulated (up and down) at 3d and 6d, and AUX1 was down-regulated at 3d and up-regulated at 6d. In the comparisons between stages in IMEs, ARF was down-regulated in IME6 vs. IME3 and in IME15 vs. IME6, while GH3 was down-regulated in the IME15 vs. IME6 comparison, and most of the auxin related genes are down-regulated during embryogenic callus formation from 3d of callus initiation to stage 6d of visible embryogenic callus, and to the somatic embryo generation at 15d in IME (Additional file 12). This implies that the majority of the auxin related genes are regulated in an intricate program in embryogenic callus formation.
Expression and GO analysis of the common DEGs in IME vs. ME comparisons
Of the 5194 DEGs in IMEs vs. MEs, 283 were simultaneously differentially expressed in all the three comparisons. Of the 283 overlapping DEGs, 35 overlapped in the above mentioned 346 SSEGs, while 10 overlapped in the above-mentioned 271 TFs. The expressions of the 283 DEGs were clustered into 6 groups in the IME libraries (Fig. 9a).
Fig. 9.
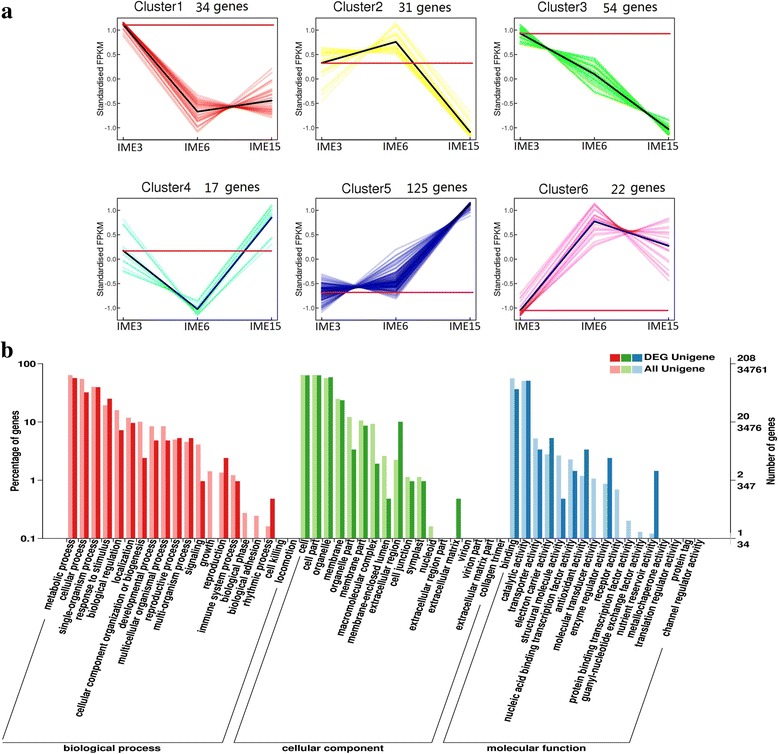
Analyses of 283 common differentially expressed genes in IME vs. ME at three stages during embryogenic callus formation in wheat. a Cluster analysis of 283 common DEGs in IME vs. ME at three stages based on K-means method. 283 common DEGs were divided into 6 distinct temporal expression profiles in IME. b GO Functional categorization of 283 common differentially expressed genes based on gene ontology
GO function analysis of the 283 overlapped DEGs indicated that 208 unigenes received at least one GO term and were classified into 3 major functional categories: cellular component, molecular function, and biological process. For the biological process category, the majority of unigenes were annotated with metabolic process (99 unigenes), single-organism process (78 unigenes) and cellular process (66 unigenes; Fig. 9b). The DEGs related to processes of response to stimulus (52 unigenes), multi-organism process (11 unigenes), reproductive process (11 unigenes), and reproduction (5 unigenes) possessed a significantly higher percentage in the 283 overlapped DEGs than in all unigenes (Fig. 9b). The 52 unigenes related to response to stimulus process included 18 of the 35 SSEGs and 2 of the 10 TFs, suggesting that somatic embryogenic formation was intimately related to the response to stimulus process. Therefore, the remaining 32 unigenes may also be involved in somatic embryogenic formation. With regards to multi-organism processes, reproductive processes and reproduction in biological processes, 11, 11 and 5 unigenes were further merged into 16 unigenes due to the involvement of some genes in multiple functions. Three of the merged 16 unigenes belonged to 35 SSEGs and the 10 TFs, and the remaining 13 genes may also be involved in somatic embryogenic formation. Of the 13 unigenes, 5 (comp115979_c0, comp122548_c0, comp124996_c0, comp126585_c2, comp131969_c0) were simultaneously involved in all 3 processes. Furthermore, the 32 and 13 unigenes possibly involved in somatic embryogenic formation were merged into 39 unigenes by the bio-process terms of the GO analysis (Table 6). Four of the 39 bio-processes-involved unigenes (BPGs) were selected to perform expression level analysis by qRT-PCR (Fig. 10). The results indicated that their expression levels were highly associated with the RNA-Seq value (Fig. 11).
Fig. 10.
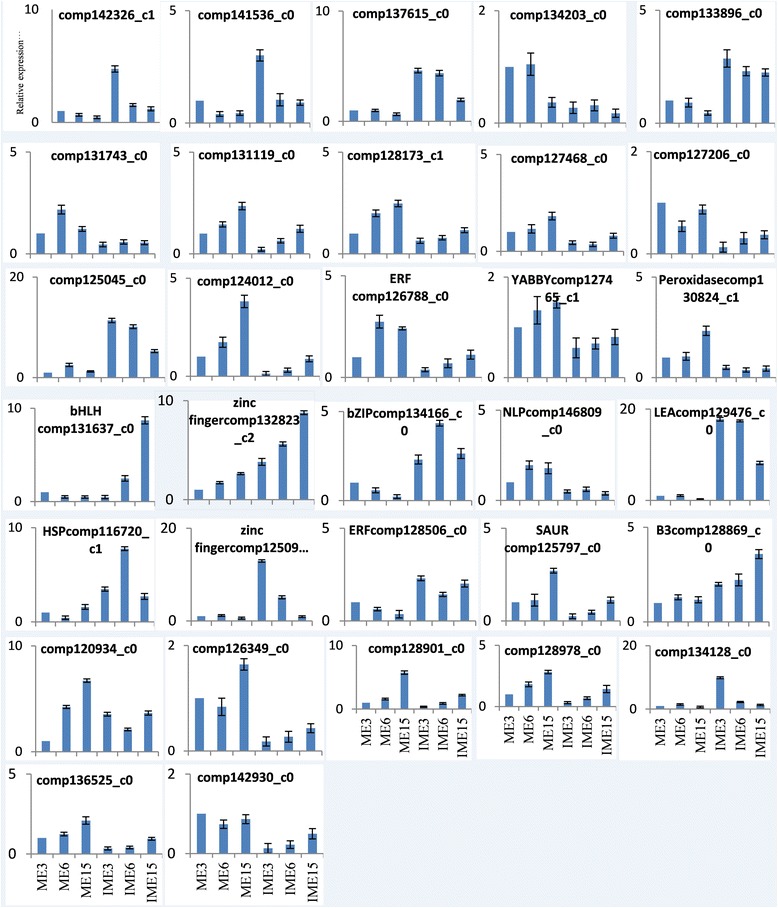
Relative mRNA levels of 32 DEGs in wheat callus were determined by quantitative RT-PCR analyses. Genes were normalized to the actin gene (GenBank: AB181991). Experiments were repeated in triplicate. Error bars represent one standard deviation (SD). Relative expression level of genes in IME were presented as fold-change (mean ± SD) compared according to ME of 1.0
Of all 283 overlapping DEGs in all 3 comparisons of IME vs. ME, 12 containing 5 annotated genes (1 from the 35 SSEGs, 2 from the 10 TFs, and 2 from the 39 BPGs mentioned above) and 7 unknown genes (Table 7) showed significant differences in the different stages of IMEs, but did not show any significant difference in the different stages of MEs, indicating that the 7 unknown genes might be related to embryogenic callus formation. The qRT-PCR results validated that the 7 unknown genes and one (comp126788_c0) of the 5 annotated genes showed highly consistent expression levels with RNA-Seq (Fig. 10). Therefore, expression comparative analysis and function analysis showed that the above-mentioned 10 IFs, 35 SSEGs, 39 BPGs and 7 unknown DEGs might contribute to embryogenic callus formation.
Discussion
Bread wheat is one of the major human food crops and has been subject to the application of biotechnology for crop improvement. High plant regeneration efficiency forms the basis for biotechnology by genetic transformation. Numerous studies have aimed to improve regeneration efficiency in wheat by optimizing the culture conditions, the explant physiological traits, and the genotypes. Unfortunately, the regeneration efficiency remains poor. Since embryogenic calli can be harnessed to generate plants with improved tissue culture efficiency, illustration of the molecular mechanism of embryogenic callus formation is helpful for manipulation of the regeneration process.
The expression of the somatic embryogenesis-related genes and signaling regulation has been researched in plants by comparative analysis between the somatic embryos and zygotic embryos [4, 42, 62] or during somatic embryogenesis [43] using NGS. In the present study, we obtained the transcriptome of the embryogenic calli derived from IMEs and the non-embryogenic calli derived from MEs following culturing for 3d, 6d, and 15d. The de novo assembly generated 142,221 unigenes and the comparisons of IME vs. ME at 3 stages provided us with 5194 DEGs; more DEGs were identified in the comparisons at 3 DC and 6 DC than at 15 DC. Additionally, 4731 DEGs were identified in the comparisons between the stages in IMEs and MEs, suggesting that the regulatory pathway exists in embryogenic callus formation in bread wheat.
Previous studies have identified genes that are potentially involved in somatic embryogenesis, including some genes that are related to stress response [4–6, 43] and auxin processes [40]. Several studies have revealed that stress plays a key role in somatic embryogenesis [63]. In the 3 comparisons of IME vs. ME, 346 SSEGs of DEGs were identified including HSP, GLPs, LEA, CDPK/CAM, GST, and LTP, which are potentially involved in embryogenic callus formation. Additionally, 35 of them overlapped in all 3 comparisons of IME vs. ME.
LTP is related to embryogenic callus development through the regulation of the embryogenic callus formation pathway [59], and Ca2+ has been suggested to play an intermediary role during plant embryogenesis [50, 51]. GST was previously shown to be induced by various stresses involved in the SE through complex interactions with other transcriptional regulators and auxin metabolism [54, 64]. Germins and GLPs are thought to play a significant role in stress and in somatic embryogenesis in wheat and Germins had oxalate oxidase activity [52, 53], 6 differently expressed genes related to GLPs (4) and oxalate oxidase (2) were identified in IME vs. ME comparisons (Additional file 11 b-c), while LEA, ABA and ethylene are stress-related substances for the acquisition of embryogenic competence by plant cells [55, 56]. Many genes thatrespond to various abiotic stresses have previously been found to be induced by auxin [4]. For example, Hsps in carrot somatic cells was found to be an auxin-responsive gene during somatic embryo development [60].
Auxin as a plant growth hormoneis considered to be essential for the initiation of cell division and differentiation [65] before it can express embryogenic competence [66], as well as in the initiation of embryogenesis from somatic tissues [67]. Auxin accumulation was previously detected in developing somatic embryos in Arabidopsis and carrot [68, 69], and the endogenous auxin concentration reached a peak at the embryogenic callus stage during somatic embryogenesis [40]. In sunflower, an endogenous auxin pulse was believed to be involved in the induction of somatic embryogenesis [70]. Exogenous auxin application and endogenous auxin content were both determining factors during the induction phase. The level of free and conjugated IAA was previously found to be highly regulated with embryonic callus formation in wheat [26]. In this study, KEGG analysis of DEGs in IME vs. ME comparison demonstrated that many genes were involved in plant hormone signal transduction with down-regulation during the process of embryogenic callus formation (Fig. 8a). Research of SE identification and characterization indicated that stress-related genes and proteins were associated with SE in stress-induced acquisition [71]. Thus the 346 DEGs of SSEGs, especially the 35 overlapping DEGs, might be involved in embryogenic callus formation.
TFs are important factors involved in plant development as well as in regeneration process. A series of studies on SE development revealed that complex transcription regulation networks existed in cell differentiation, maintaining embryogenic competency, embryogenic patterning, meristem maintenance, as well as roles in stress and hormone-mediated signaling [40, 44]. In this study, we identified 271 differentially expressed TFs in the comparisons of IME vs. ME, 10 of which were simultaneously differentially expressed in all 3 comparisons. Of the 271 TFs, bHLH, AP2/ERF, b-ZIP, and WRKY were highly represented.
Zinc finger family proteins have been proven to be involved in stress, the regulation of plant cell death and callus differentiation [57, 58]. BohLOL1, encoding an LSD1-like zinc finger protein in bamboo and participating in growth regulation and response to biotic stress, was up-regulated by auxins, cytokinins, and hydrogen peroxide in in vitro culture [72]. PEI1, encoding a protein containing a Cys3-His zinc finger domain, is an embryo-specific transcription factor that plays an important role during Arabidopsis embryogenesis [73]. The over expression of OsLSD1, a rice zinc finger protein, accelerated callus differentiation in transformed rice tissues [58]. In the present study, the up-regulated unigenes that encode the zinc finger family protein in the IME vs. ME comparisons were 15/18, 8/18, and 0/3 at 3, 6, and 15 DC, respectively (Additional file 8). The ratio of the up-regulated DEGs was higher at the phase of embryogenic callus initiation than at the development and completed phase, indicating that they have a regulatory role in wheat embryogenic callus formation.
A member of the AP2/ERF domain family BABY BOOM (BBM) was identified as a marker of SE in cell cultures of Brassica napus (B. napus L.).The ectopic expression of BBM can lead to somatic embryogenesis in Arabidopsis and B. napus [21, 74], while over-expression can induce embryo formation and enhance regeneration ability in tobacco [75], sweet pepper [76], and in cacao [77]. The A. thaliana EMBRYOMAKER (EMK), another member of the AP2/ERF domain family, plays a redundant role in the maintenance of embryonic cell identity. Ectopic expression of AtEMK can promote the initiation of somatic embryos from cotyledons [78, 79] revealed that AP2/ERF expression was related to the dramatic regulation of somatic embryogenesis in Hevea. In the present study, there were 20 AP2/ERF genes differentially expressed in the comparisons of IME vs. ME at 3 stages (Additional file 8), with 2 genes (comp126788_c0, comp125594_c0) being differentially expressed in IME vs. ME and in the comparisons between stages in IME, but not in comparison between stages in ME. Thus these AP2/ERF genes may be related to embryogenic callus formation in wheat.
Members of bHLH are involved in growth, developmental, and abiotic stress responses [80], axillary meristem formation [81]. They also participate in epidermal cell type specification in Arabidopsis [82], and are involved in brassinosteroid and abscisic acid signaling in rice and Arabidopsis [83]. BIM1, a bHLH protein controlling Arabidopsis embryonic patterning and interacting with the auxin and BR signaling pathways [84], which interacts with other proteins and TFs such as the key regulator MYB families that involved in stress response and regulated embryogenesis pathways [85]. In this study, the ratio of up-regulated unigenes of bHLH in IME vs. ME comparisons was 12/19, 3/18, and 2/11 at 3, 6, and 15d, respectively, suggesting that a complex regulation exists in embryogenic callus formation in wheat. Additionally, the majority of MYB genes was up-regulated in embryogenic initiation and showed a relatively low expression level during somatic embryo development, indicating that the members of these 2 families play important roles in embryogenic callus formation.
The WRKY TFs have been shown to be involved in the response to biotic and/or abiotic stresses [86], and were found to be up-regulated during SE in plants. The expression of the member genes in this family was shown to be associated with embryogenic callus formation [87]. In this study, the DEGs at 3 DC possessed the highest numbers of WRKY than other stages, with the ratio of up-regulated unigenes in IME vs. ME comparisons being 14/15, 2/6, and 5/5 at 3, 6, and 15d, respectively.
B3 domain transcription factors in Arabidopisis (LEC2, FUS3 and ABI3) encode regulatory proteins involved in embryogenesis and the induction of somatic embryo development [16]. LEC1 (LEAFY COTYLEDON1), a CCAAT box-binding factor, along with LEC2 and FUS3, are essential for plant embryo development. For instance, LEC2 promotes embryogenic induction in the somatic tissues of Arabidopsis [88]. Furthermore, cacaoTcLEC2, a functional ortholog of AtLEC2, is involved in similar genetic regulatory networks during cacao somatic embryogenesis [89]. In this study, 4, 1, and 1 B3 TFs were differentially expressed in IME vs. ME at 3, 6 and 15d, with all of them being up-regulated. Three of them were differentially expressed in the comparison between stages in IME and/or ME.
The MADS-domain transcriptional regulator AGAMOUS-LIKE15 (AGL15) had been reported to enhance somatic embryo development in Arabidopsis and soybean when ectopically expressed [90]. Soybean orthologs of the Arabidopsis MADS box genes, AGAMOUS-Like15 (GmAGL15) and GmAGL18, constitutively expressed increased embryogenic competence of explants. ABI3 and FUSCA3 were found to be directly up-regulated by GmAGL15 [22]. WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators, which was highly expressed during early somatic embryo development [91]. In the present study, MADS and wux genes were differentially expressed at 3d and 6d. Histone H3.3 participates in the epigenetic transmission of active chromatin states in animal and Histone H3 showed appreciable levels in the somatic embryos at all stages of somatic embryo development in Alfalfa [92, 93], 15 Histone DEG was identified with 3 of H3 in IME vs. ME comparisons (Additional file 11c).
Somatic embryogenesis is a complex process. TF with auxin and stress treatment were essential for the acquisition of embryogenic competence by carrot somatic cells [5]. In this study, many stress factors including oxidative, heat and salt stress, were differentially expressed in the comparisons. Thus, the Aux, TF, and stress related genes regulated the embryogenic callus formation. Except for SSEGs and TFs, there were 39 genes that were present in all 3 comparisons of IME vs. ME and were involved in GO biological process of response to stimulus, in multi-organism process, reproductive process and reproduction, function as expansin, cellulose synthase, mobile element transfer protein, multicopper oxidase, ATPase family associated with various cellular activities (AAA), glutaredoxin, ethylene insensitive 3, senescence-associated protein, universal stress protein family and pathogenesis-related protein/dehydrase and lipid transport, and were perhaps related toembryogenic callus formation.
The 7 unknown DEGs presented in three of the IME vs. ME comparisons and in the comparisons between stages in IMEs but not in MEs, function as xyloglucan endotransglucosylase/hydrolase (XTH) protein 22, xyloglucan endo-transglycosylase (XET) C-terminus, root cap, cortical cell-delineating protein, U-box domain/E3 ubiquitin-protein ligase A. thaliana, glycerol-3-phosphate acyltransferase 2 A. thaliana, gag-polypeptide of LTR copia-type/ reverse transcriptase Integrase core domain (Table 6). XTH, a cell wall-modifying enzyme, can act as cell wall-loosening enzymes [94], and may possess XET or endohydrolase activities [95], indicating their special roles in embryogenic callus formation.
Conclusion
In summary, the combination of tissue culture of wheat embryos and RNA-seq approaches by global gene expression patterns during embryogenic callus formation were performed to investigate the regulatory genes in embryogenic callus formation. Expression and function analysis indicated that the DEGs of 271 TFs (10 overlapped in all 3 comparisons of IME vs. ME), 346 SSEGs (35 overlapped in all 3 comparisons of IME vs. ME), 39 BPGs, and 7 unknown unigenes that overlapped in IME vs. ME and in comparisons between stages in IMEs, but not in any one of comparison in ME, might play an important role in embryogenic callus formation in wheat. The present comparative profiling provided new insights into the molecular mechanisms for the regulation of embryogenic callus formation processes. However, as molecular markers are required to follow specific events during embryogenic callus formation, further experiments are required to evaluate the interaction between stress and auxin signaling during embryogenic callus development.
Additional files
Primers used in this study for qRT-PCR. (XLSX 14 kb)
Lengths distribution of all assembled unigenes (XLSX 11 kb)
Functional annotation of unigenes against Nr, Pfam, KEGG, COG, GO Swissprot databases. (XLS 20569 kb)
Gene ontology (GO) classification, COG classification and KEGG classification of unigenes. a. The GO terms are summarized into three main categories: biological process, cellular component and molecular function. b. COG classification of the unigenes. c. KEGG classification of 5933 KO annotated unigenes. (XLSX 111 kb)
Expression of genes in the 6 libraries in wheat callus based on the results of RNA-Seq. (XLSX 6574 kb)
Information of DEGs in the comparisons of IME vs. ME. (XLSX 397 kb)
Information of DEGs in the comparisons between stages in IMEs and MEs. (XLSX 319 kb)
Functional annotation of all DEG against Nr, Pfam, Swissprot, KOG, KEGG, GO and COG databases. (XLSX 1510 kb)
KEGG enrichment analysis of all DEGs. (XLSX 65 kb)
List of all TFs and expression of DEGs TFs in the 6 libraries. (XLSX 93 kb)
Expression and annotation of differentially expressed aux and SSEGs IME vs. ME. (XLSX 86 kb)
KEGG pathways of plant hormone signal transduction in comparisons of IME vs. ME and between stages. (PDF 122 kb)
Acknowledgements
This project was funded by the National Key Research and Development Program (2016YFD0101802), Henan Major Science and Technology Projects (161100110500) and Henan Science and Technology Innovation Outstanding Youth Funding (174100510001) of China.
Availability of data and materials
The sequencing raw data of this article have been deposited as BioProject ID [PRJNA353135] in the NCBI SRA under the accession number [SRP093588]. All the supporting data are included in Additional files.
Abbreviations
- 3 DC (6 DC; 15 DC)
cultured for 3 d (6 d; 15 d)
- ABA
Abscisic acid
- ABI3
ABA insensitive 3
- AGL
AGAMOUS-like
- ARF
Auxin-responsive factor
- BBM
Baby boom
- BPGs
bio-processes-involved genes
- CDPK
Calcium-dependent protein kinase
- COG
Cluster of orthologous groups
- DEGs
Differentially expressed genes
- ERF
Ethylene-responsive transcription factor
- GLP
Germin like protein
- GO
Gene ontology
- GST
Glutathione S-transferase
- HSP
Heat-shock protein
- IAA
Indole-3-aceticacid
- IMEs
Immature embryos
- KEGG
Kyoto encyclopedia of genes and genomes
- LEA
Late embryogenesis abundant proteins
- LEC
Leafy cotyledon
- LTP
lipidtransfer protein
- MEs
Mature embryos
- RPKM
Reads per kilobase of exon model per million mapped reads
- SE
Somatic embryogenesis
- SSEGs
Stress and somatic embryogenesis related genes
- TF
Transcription factor
- XET
Xyloglucan endo-transglycosylase
- XTH
Xyloglucan endo-transglucosylase/hydrolase protein
Authors’ contributions
DC conceived and designed the study. ZC and FC performed bioinformatics analysis and wrote the manuscript. JC, JS, ZD, XY, YW and XZ cultivated the wheat plantlets, extracted RNA and participated in the bioinformatics analysis. HX and FC critically reviewed the manuscript, and provided guidance. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12870-017-1204-2) contains supplementary material, which is available to authorized users.
Contributor Information
Zongli Chu, Email: chuzl10000@126.com.
Junying Chen, Email: chenjunying3978@126.com.
Junyan Sun, Email: xysjy66@163.com.
Zhongdong Dong, Email: dongzhongdong@163.com.
Xia Yang, Email: tianshiyangxia@163.com.
Ying Wang, Email: 18238839189@163.com.
Haixia Xu, Email: xuhaixia@sohu.com.
Xiaoke Zhang, Email: zhangxiaoke66@126.com.
Feng Chen, Phone: +86-0371-63558537, Email: chf0088@163.com.
Dangqun Cui, Phone: +86-0371-63558537, Email: cdq62@sohu.com.
References
- 1.Dunwell JM. Transgenic approaches to crop improvement. Journal of experimental botany. 2000;51:Spec No:487–Spec No:496. doi: 10.1093/jexbot/51.suppl_1.487. [DOI] [PubMed] [Google Scholar]
- 2.Zimmerman JL. Somatic embryogenesis: a model for early development in higher plants. Plant Cell. 1993;5(10):1411–1423. doi: 10.1105/tpc.5.10.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang L, Y-h L, Bian L, H-l S. Somatic embryogenesis and plantlet regeneration from mature zygotic embryos of Manchurian ash (Fraxinus mandshurica Rupr.) Plant Cell, Tissue and Organ Culture (PCTOC) 2013;115(2):115–125. doi: 10.1007/s11240-013-0345-8. [DOI] [Google Scholar]
- 4.Jin F, Hu L, Yuan D, Xu J, Gao W, He L, Yang X, Zhang X. Comparative transcriptome analysis between somatic embryos (SEs) and zygotic embryos in cotton: evidence for stress response functions in SE development. Plant Biotechnol J. 2014;12(2):161–173. doi: 10.1111/pbi.12123. [DOI] [PubMed] [Google Scholar]
- 5.Kikuchi A, Sanuki N, Higashi K, Koshiba T, Kamada H. Abscisic acid and stress treatment are essential for the acquisition of embryogenic competence by carrot somatic cells. Planta. 2006;223(4):637–645. doi: 10.1007/s00425-005-0114-y. [DOI] [PubMed] [Google Scholar]
- 6.Tyankova ND, Zagorska NA. Genetic control of in vitro response in wheat (Triticum Aestivum L.) In Vitro Cellular & Developmental Biology - Plant. 2001;37(5):524–530. doi: 10.1007/s11627-001-0091-1. [DOI] [Google Scholar]
- 7.Hoenemann C, Richardt S, Kruger K, Zimmer AD, Hohe A, Rensing SA. Large impact of the apoplast on somatic embryogenesis in Cyclamen Persicum offers possibilities for improved developmental control in vitro. BMC Plant Biol. 2010;10:77. doi: 10.1186/1471-2229-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin HC, Morcillo F, Dussert S, Tranchant-Dubreuil C, Tregear JW, Tranbarger TJ. Transcriptome analysis during somatic embryogenesis of the tropical monocot Elaeis Guineensis: evidence for conserved gene functions in early development. Plant Mol Biol. 2009;70(1–2):173–192. doi: 10.1007/s11103-009-9464-3. [DOI] [PubMed] [Google Scholar]
- 9.Zeng F, Zhang X, Zhu L, Tu L, Guo X, Nie Y. Isolation and characterization of genes associated to cotton somatic embryogenesis by suppression subtractive hybridization and macroarray. Plant Mol Biol. 2006;60(2):167–183. doi: 10.1007/s11103-005-3381-x. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Li F, Zhang C, Liu C, Zhang X. Differential gene expression of cotton cultivar CCRI24 during somatic embryogenesis. J Plant Physiol. 2009;166(12):1275–1283. doi: 10.1016/j.jplph.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Vale H r, Vielle-Calzada J-P, Hartog EDLS MV, Boutilier K, Grossniklaus U, SCd V. The Arabidopsis somatic embryogenesis receptor kinase 1 gene is expressed in developing ovules and embryos and enhances embryogenic competence in culture. Plant Physiol. 2001;127(3):803–816. doi: 10.1104/pp.010324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thibaud-Nissen F, Shealy RT, Khanna A, Vodkin LO. Clustering of microarray data reveals transcript patterns associated with somatic embryogenesis in soybean. Plant Physiol. 2003;132(1):118–136. doi: 10.1104/pp.103.019968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lucau-Danila A, Laborde L, Legrand S, Huot L, Hot D, Lemoine Y, Hilbert JL, Hawkins S, Quillet MC, Hendriks T, et al. Identification of novel genes potentially involved in somatic embryogenesis in chicory (Cichorium Intybus L) BMC Plant Biol. 2010;10:122. doi: 10.1186/1471-2229-10-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lotan T, Ohto M-a, Yee KM, West MAL, Lo R, Kwong RW, Yamagishi K, Fischer RL, Goldberg RB, Harada JJ. Arabidopsis LEAFY COTYLEDON1 Is Sufficient To induce embryo development in vegetative cells. Cell. 1998;93(7):1195–1205. doi: 10.1016/S0092-8674(00)81463-4. [DOI] [PubMed] [Google Scholar]
- 15.Ledwoń A, Gaj MD. LEAFY COTYLEDON1, FUSCA3 expression and auxin treatment in relation to somatic embryogenesis induction in Arabidopsis. Plant Growth Regul. 2011;65(1):157–167. doi: 10.1007/s10725-011-9585-y. [DOI] [Google Scholar]
- 16.Gaj MD, Zhang S, Harada JJ, Lemaux PG. Leafy cotyledon genes are essential for induction of somatic embryogenesis of Arabidopsis. Planta. 2005;222(6):977–988. doi: 10.1007/s00425-005-0041-y. [DOI] [PubMed] [Google Scholar]
- 17.Stone SL, Kwong LW, Yee KM, Pelletier J, Lepiniec L, Fischer RL, Goldberg RB, Harada JJ. LEAFY COTYLEDON2 encodes a B3 domain transcription factor that induces embryo development. Proc Natl Acad Sci. 2001;98(20):11806–11811. doi: 10.1073/pnas.201413498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCarty DR, Hattori T, Carson CB, Vasil V, Lazar M, Vasil IK. The Viviparous-1 developmental gene of maize encodes a novel transcriptional activator. Cell. 1991;66(5):895–905. doi: 10.1016/0092-8674(91)90436-3. [DOI] [PubMed] [Google Scholar]
- 19.Piyatrakul P, Putranto R-A, Martin F, Rio M, Dessailly F, Leclercq J, Dufayard J-F, Lardet L, Montoro P. Some ethylene biosynthesis and AP2/ERF genes reveal a specific pattern of expression during somatic embryogenesis in Hevea Brasiliensis. BMC Plant Biol. 2012;12:244. doi: 10.1186/1471-2229-12-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kulinska-Lukaszek K, Tobojka M, Adamiok A, Kurczynska EU. Expression of the BBM gene during somatic embryogenesis of Arabidopsis Thaliana. Biol Plant. 2012;56(2):389–394. doi: 10.1007/s10535-012-0105-3. [DOI] [Google Scholar]
- 21.Boutilier K. Ectopic expression of BABY BOOM triggers a conversion from vegetative to embryonic growth. The Plant Cell Online. 2002;14(8):1737–1749. doi: 10.1105/tpc.001941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Q, Perry SE. Alterations in the transcriptome of soybean in response to enhanced somatic embryogenesis promoted by orthologs of Agamous-like15 and Agamous-like18. Plant Physiol. 2014;164(3):1365–1377. doi: 10.1104/pp.113.234062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.YH S, Zhao XY, Liu YB, Zhang CL, O'Neill SD, Zhang XS, Auxin-induced WUS. Expression is essential for embryonic stem cell renewal during somatic embryogenesis in Arabidopsis. Plant J. 2009;59(3):448–460. doi: 10.1111/j.1365-313X.2009.03880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delporte F, Pretova A, du Jardin P, Watillon B. Morpho-histology and genotype dependence of in vitro morphogenesis in mature embryo cultures of wheat. Protoplasma. 2014;251(6):1455–1470. doi: 10.1007/s00709-014-0647-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delporte F, Mostade O, Jacquemin JM. Plant regeneration through callus initiation fromthin mature embryo fragments of wheat. Plant Cell Tissue Organ Cult. 2001;67:73–80. doi: 10.1023/A:1011697316212. [DOI] [Google Scholar]
- 26.Singla B, Tyagi AK, Khurana JP, Khurana P. Analysis of expression profile of selected genes expressed during auxin-induced somatic embryogenesis in leaf base system of wheat (Triticum Aestivum) and their possible interactions. Plant Mol Biol. 2007;65(5):677–692. doi: 10.1007/s11103-007-9234-z. [DOI] [PubMed] [Google Scholar]
- 27.Delporte F, Muhovski Y, Pretova A, Watillon B. Analysis of expression profiles of selected genes associated with the regenerative property and the receptivity to gene transfer during somatic embryogenesis in Triticum Aestivum L. Mol Biol Rep. 2013;40(10):5883–5906. doi: 10.1007/s11033-013-2696-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mardis ER, Next-generation DNA. Sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 29.Schuster SC. Next-generation sequencing transforms today’s biology. Nat Methods. 2008;5(1):16–18. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- 30.Shendure J, Ji H, Next-generation DNA. sequencing. Nat Biotechnol. 2008;26(10):1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 31.Fan Z, Li J, Li X, Wu B, Wang J, Liu Z, Yin H. Genome-wide transcriptome profiling provides insights into floral bud development of summer-flowering Camellia Azalea. Sci Rep. 2015;5:9729. doi: 10.1038/srep09729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsanakas GF, Manioudak ME, Economou AS, Kalaitzis P. De novo transcriptome analysis of petal senescence in Gardenia Jasminoides Ellis. BMC Genomics. 2014;15:554. doi: 10.1186/1471-2164-15-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Zhang W, Zhang L, Li N, Peng J, Wang Y, Zhong C, Yang Y, Sun S, Liang S, et al. Transcriptomic insights into antagonistic effects of gibberellin and abscisic acid on petal growth in Gerbera Hybrida. Front Plant Sci. 2015;6:168. doi: 10.3389/fpls.2015.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu W, Li R, Zhang N, Ma F, Jiao Y, Wang Z. Transcriptome profiling of Vitis Amurensis, an extremely cold-tolerant Chinese wild Vitis species, reveals candidate genes and events that potentially connected to cold stress. Plant Mol Biol. 2014;86(4–5):527–541. doi: 10.1007/s11103-014-0245-2. [DOI] [PubMed] [Google Scholar]
- 35.Peng Z, He S, Gong W, Sun J, Pan Z, Xu F, Lu Y, Du X. Comprehensive analysis of differentially expressed genes and transcriptional regulation induced by salt stress in two contrasting cotton genotypes. BMC Genomics. 2014;15:760. doi: 10.1186/1471-2164-15-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhai L, Xu L, Wang Y, Huang D, Yu R, Limera C, Gong Y, Liu L. Genome-wide identification of embryogenesis-associated microRNAs in radish (Raphanus Sativus L.) by high-throughput sequencing. Plant Mol Biol Report. 2014;32(4):900–915. doi: 10.1007/s11105-014-0700-x. [DOI] [Google Scholar]
- 37.Qi B, Yang Y, Yin Y, Xu M, Li H. De novo sequencing, assembly, and analysis of the Taxodium ‘Zhongshansa’ roots and shoots transcriptome in response to short-term waterlogging. BMC Plant Biol. 2014;14:201. doi: 10.1186/s12870-014-0201-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H, Yang Y, Wang C, Liu M, Li H, Fu Y, Wang Y, Nie Y, Liu X, Ji W. Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genomics. 2014;15:898. doi: 10.1186/1471-2164-15-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oono Y, Kobayashi F, Kawahara Y, Yazawa T, Handa H, Itoh T, Matsumoto T. Characterisation of the wheat (Triticum Aestivum L.) transcriptome by de novo assembly for the discovery of phosphate starvation-responsive genes: gene expression in pi-stressed wheat. BMC Genomics. 2013;14:77. doi: 10.1186/1471-2164-14-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang X, Zhang X, Yuan D, Jin F, YZJ X. Transcript profiling reveals complex auxin signalling pathway and transcription regulation involved in dedifferentiation and redifferentiation during somatic embryogenesis in cotton. BMC Plant Biol. 2012;12:110. doi: 10.1186/1471-2229-12-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Q, Zhang S, Wang J. Transcriptome analysis of callus from Picea Balfouriana. BMC Genomics. 2014;15:553. doi: 10.1186/1471-2164-15-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi X, Zhang C, Liu Q, Zhe Z, Bo Z, Bao M. De novo comparative transcriptome analysis provides new insights into sucrose induced somatic embryogenesis in camphor tree ( Cinnamomum camphor a L) BMC Genomics. 2016;17(1):1–18. doi: 10.1186/s12864-015-2357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salvo SAGD, Hirsch CN, Buell CR, Kaeppler SM, Kaeppler HF. Whole transcriptome profiling of maize during early somatic embryogenesis reveals altered expression of stress factors and embryogenesis-related genes. PLoS One. 2014;9:10. doi: 10.1371/journal.pone.0111407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang X, Chen J, Bao Y, Liu L, Jiang H, An X, Dai L, Wang B, Peng D. Transcript profiling reveals auxin and cytokinin signaling pathways and transcription regulation during in vitro organogenesis of ramie (Boehmeria Nivea L. gaud) PLoS One. 2014;9(11):e113768. doi: 10.1371/journal.pone.0113768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenchley R, Spannagl M, Pfeifer M, Barker GL, D'Amore R, Allen AM, McKenzie N, Kramer M, Kerhornou A, Bolser D, et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature. 2012;491(7426):705–710. doi: 10.1038/nature11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 48.Guo AY, Chen X, Gao G, Zhang H, Zhu QH, Liu XC, Zhong YF, Gu X, He K, Luo J. PlantTFDB: a comprehensive plant transcription factor database. Nucleic Acids Res. 2008;36(Database issue):966–969. doi: 10.1093/nar/gkm841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25 [DOI] [PubMed]
- 50.Yang X, Zhang X. Regulation of somatic embryogenesis in higher plants. Crit Rev Plant Sci. 2010;29(1):36–57. doi: 10.1080/07352680903436291. [DOI] [Google Scholar]
- 51.Kiselev KV, Shumakova OA, Manyakhin AY, Mazeika AN. Influence of calcium influx induced by the calcium ionophore, A23187, on resveratrol content and the expression of CDPK and STS genes in the cell cultures of Vitis Amurensis. Plant Growth Regul. 2012;68(3):371–381. doi: 10.1007/s10725-012-9725-z. [DOI] [Google Scholar]
- 52.Caliskan M, Turet M, Cuming AC. Formation of wheat (Triticum Aestivum L.) embryogenic callus involves peroxide-generating germin-like oxalate oxidase. Planta. 2004;219(1):132–140. doi: 10.1007/s00425-003-1199-9. [DOI] [PubMed] [Google Scholar]
- 53.Patnaik D, Khurana P. Germins and germin like proteins: an overview. Indian J Exp Biol. 2001;39(3):191–200. [PubMed] [Google Scholar]
- 54.Galland R, Randoux B, Vasseur J, Hilbert JL. A glutathione S-transferase cDNA identified by mRNA differential display is upregulated during somatic embryogenesis in Cichorium. Biochim Biophys Acta. 2002;1522(3):212–216. doi: 10.1016/S0167-4781(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 55.Hundertmark M, Hincha DK. LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis Thaliana. BMC Genomics. 2008;9(1):118. doi: 10.1186/1471-2164-9-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kikawada T, Nakahara Y, Kanamori Y, Iwata K, Watanabe M, McGee B, Tunnacliffe A, Okuda T. Dehydration-induced expression of LEA proteins in an anhydrobiotic chironomid. Biochem Biophys Res Comm. 2006;348 [DOI] [PubMed]
- 57.Dietrich RA, Richberg MH, Schmidt R, Dean C, Dangl JL. A novel zinc finger protein is encoded by the Arabidopsis LSD1 gene and functions as a negative regulator of plant cell death. Cell. 1997;88(5):685–694. doi: 10.1016/S0092-8674(00)81911-X. [DOI] [PubMed] [Google Scholar]
- 58.Wang L, Pei ZY, He C. OsLSD1, a rice zinc finger protein, regulates programmed cell death and callus differentiation. Mol Plant-Microbe Interact. 2005;18(5):375–384. doi: 10.1094/MPMI-18-0375. [DOI] [PubMed] [Google Scholar]
- 59.Sterk P, Booij H, Schellekens GA, Van KA, De Vries SC. Cell-specific expression of the carrot EP2 lipid transfer protein gene. Plant Cell. 1991;3(9):907–921. doi: 10.1105/tpc.3.9.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kitamiya E, Suzuki S, Sano T, Nagata T. Isolation of two genes that were induced upon the initiation of somatic embryogenesis on carrot hypocotyls by high concentrations of 2,4-D. Plant Cell Rep. 2000;19(6):551–557. doi: 10.1007/s002990050772. [DOI] [PubMed] [Google Scholar]
- 61.Lyndon RF. Hormonal regulation of plant growth and development. PLoS Biol. 2004;2(9):1270–1273. doi: 10.1371/journal.pbio.0020311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elbl P, Lira BS, Andrade SCS, Jo L, Santos ALWD, Coutinho LL, Floh EIS, Rossi M: Comparative transcriptome analysis of early somatic embryo formation and seed development in Brazilian pine, Araucaria angustifolia (Bertol.) Kuntze. Plant Cell Tissue & Organ Culture 2015;120(3):903–915
- 63.Grafi G, Chalifa-Caspi V, Nagar T, Plaschkes I, Barak S, Ransbotyn V. Plant response to stress meets dedifferentiation. Planta. 2011;233(3):433–438. doi: 10.1007/s00425-011-1366-3. [DOI] [PubMed] [Google Scholar]
- 64.Qin D, Wu H, Peng H, Yao Y, Ni Z, Li Z, Zhou C, Sun Q. Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum Aestivum L.) by using wheat genome Array. BMC Genomics. 2008;9(1):1–19. doi: 10.1186/1471-2164-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao Z, Andersen SU, Ljung K, Dolezal K, Miotk A, Schultheiss SJ, Lohmann JU. Hormonal control of the shoot stem-cell niche. Nature. 2010;465(7301):1089–1092. doi: 10.1038/nature09126. [DOI] [PubMed] [Google Scholar]
- 66.Su YH, Zhang XS. Auxin gradients trigger de novo formation of stem cells during somatic embryogenesis. Plant Signal Behav. 2014;4(7):574–576. doi: 10.4161/psb.4.7.8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fehér A, Pasternak T, Dudits D. Transition of somatic plant cells to an embryogenic state. Plant Cell Tissue Organ Cult. 2003;74(3):201–228. doi: 10.1023/A:1024033216561. [DOI] [Google Scholar]
- 68.Michalczuk L, Cooke TJ, Cohen JD. Auxin levels at different stages of carrot somatic embryogenesis. Phytochemistry. 1992;31(92):1097–1103. doi: 10.1016/0031-9422(92)80241-6. [DOI] [Google Scholar]
- 69.Kurczynska EU, Gaj MD, Ujczak A, Mazur E. Histological analysis of direct somatic embryogenesis in Arabidopsis Thaliana (L.) Heynh. Planta. 2007;226(3):619–628. doi: 10.1007/s00425-007-0510-6. [DOI] [PubMed] [Google Scholar]
- 70.Thomas C, Bronner R, Molinier J, Prinsen E, van Onckelen H, Hahne G. Immuno-cytochemical localization of indole-3-acetic acid during induction of somatic embryogenesis in cultured sunflower embryos. Planta. 2002;215(4):577–583. doi: 10.1007/s00425-002-0791-8. [DOI] [PubMed] [Google Scholar]
- 71.Omid K, Abbas S. The molecular basis for stress-induced acquisition of somatic embryogenesis. Mol Biol Rep. 2010;37(5):2493–2507. doi: 10.1007/s11033-009-9764-3. [DOI] [PubMed] [Google Scholar]
- 72.Yeh SH, Lin CS, FH W, Wang AY. Analysis of the expression of BohLOL1, which encodes an LSD1-like zinc finger protein in Bambusa Oldhamii. Planta. 2011;234(6):1179–1189. doi: 10.1007/s00425-011-1467-z. [DOI] [PubMed] [Google Scholar]
- 73.Li Z, Thomas TL. PEI1, an embryo-specific zinc finger protein gene required for heart-stage embryo formation in Arabidopsis. Plant Cell. 1998;10:383–398. doi: 10.1105/tpc.10.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.El Ouakfaoui S, Schnell J, Abdeen A, Colville A, Labbe H, Han S, Baum B, Laberge S, Miki B. Control of somatic embryogenesis and embryo development by AP2 transcription factors. Plant Mol Biol. 2010;74(4–5):313–326. doi: 10.1007/s11103-010-9674-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Srinivasan C, Liu Z, Heidmann I, Supena ED, Fukuoka H, Joosen R, Lambalk J, Angenent G, Scorza R, Custers JB, et al. Heterologous expression of the BABY BOOM AP2/ERF transcription factor enhances the regeneration capacity of tobacco (Nicotiana Tabacum L) Planta. 2007;225(2):341–351. doi: 10.1007/s00425-006-0358-1. [DOI] [PubMed] [Google Scholar]
- 76.Heidmann I, de Lange B, Lambalk J, Angenent GC, Boutilier K. Efficient sweet pepper transformation mediated by the BABY BOOM transcription factor. Plant Cell Rep. 2011;30(6):1107–1115. doi: 10.1007/s00299-011-1018-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Florez SL, Erwin RL, Maximova SN, Guiltinan MJ, Curtis WR. Enhanced somatic embryogenesis in Theobroma Cacao using the homologous BABY BOOM transcription factor. BMC Plant Biol. 2015;15:121. doi: 10.1186/s12870-015-0479-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsuwamoto R, Yokoi S, Takahata Y, Arabidopsis EMBRYOMAKER. Encoding an AP2 domain transcription factor plays a key role in developmental change from vegetative to embryonic phase. Plant Mol Biol. 2010;73(4–5):481–492. doi: 10.1007/s11103-010-9634-3. [DOI] [PubMed] [Google Scholar]
- 79.Piyatrakul P, Putranto RA, Martin F, Rio M, Dessailly F, Leclercq J, Dufayard JF, Lardet L, Montoro P. Some ethylene biosynthesis and AP2/ERF genes reveal a specific pattern of expression during somatic embryogenesis in Hevea Brasiliensis. BMC Plant Biol. 2012;12:244. doi: 10.1186/1471-2229-12-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou J, Li F, Wang JL, Ma Y, Chong K, YY X. Basic helix-loop-helix transcription factor from wild rice (OrbHLH2) improves tolerance to salt- and osmotic stress in Arabidopsis. J Plant Physiol. 2009;166(12):1296–1306. doi: 10.1016/j.jplph.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 81.Yang F, Wang Q, Schmitz G, Muller D, Theres K. The bHLH protein ROX acts in concert with RAX1 and LAS to modulate axillary meristem formation in Arabidopsis. Plant J. 2012;71(1):61–70. doi: 10.1111/j.1365-313X.2012.04970.x. [DOI] [PubMed] [Google Scholar]
- 82.Zhao H, Li X, Ma L. Basic helix-loop-helix transcription factors and epidermal cell fate determination in Arabidopsis. Plant Signal Behav. 2012;7(12):1556–1560. doi: 10.4161/psb.22404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang LY, Bai MY, Wu J, Zhu JY, Wang H, Zhang Z, Wang W, Sun Y, Zhao J, Sun X, et al. Antagonistic HLH/bHLH transcription factors mediate brassinosteroid regulation of cell elongation and plant development in rice and Arabidopsis. Plant Cell. 2009;21(12):3767–3780. doi: 10.1105/tpc.109.070441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chandler JW, Cole M, Flier A, Werr W. BIM1, a bHLH protein involved in brassinosteroid signalling, controls Arabidopsis embryonic patterning via interaction with DORNROSCHEN and DORNROSCHEN-LIKE. Plant Mol Biol. 2009;69(1–2):57–68. doi: 10.1007/s11103-008-9405-6. [DOI] [PubMed] [Google Scholar]
- 85.Wang X, Niu Q-W, Teng C, Li C, Mu J, Chua N-H, Zuo J. Overexpression of PGA37/MYB118 and MYB115 promotes vegetative-to-embryonic transition in Arabidopsis. Cell Res. 2009;19(2):224–235. doi: 10.1038/cr.2008.276. [DOI] [PubMed] [Google Scholar]
- 86.Tripathi P, Rabara RC, Rushton PJ. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta. 2014;239(2):255–266. doi: 10.1007/s00425-013-1985-y. [DOI] [PubMed] [Google Scholar]
- 87.Lagace M, Matton DP. Characterization of a WRKY transcription factor expressed in late torpedo-stage embryos of Solanum Chacoense. Planta. 2004;219(1):185–189. doi: 10.1007/s00425-004-1253-2. [DOI] [PubMed] [Google Scholar]
- 88.Wojcikowska B, Jaskola K, Gasiorek P, Meus M, Nowak K, Gaj MD. LEAFY COTYLEDON2 (LEC2) promotes embryogenic induction in somatic tissues of Arabidopsis, via YUCCA-mediated auxin biosynthesis. Planta. 2013;238(3):425–440. doi: 10.1007/s00425-013-1892-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Clemens A, Maximova SN, Guiltinan MJ. The Theobroma cacao B3 domain transcription factor TcLEC2 plays a duel role in control of embryo development and maturation. 2014;14(1):1-16. [DOI] [PMC free article] [PubMed]
- 90.Thakare D, Tang W, Hill K, Perry SE. The MADS-domain transcriptional regulator AGAMOUS-LIKE15 promotes somatic embryo development in Arabidopsis and soybean. Plant Physiol. 2008;146(4):1663–1672. doi: 10.1104/pp.108.115832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leibfried A, To JP. Busch W, Stehling S, Kehle A, Demar M, Kieber JJ, Lohmann JU. WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators. Nature. 2005;438(7071):1172–1175. doi: 10.1038/nature04270. [DOI] [PubMed] [Google Scholar]
- 92.Kapros T, Dudits D. Differential expression of histone H3 gene variants during cell cycle and somatic embryogenesis in alfalfa. Plant Physiol. 1992;98(2):621. doi: 10.1104/pp.98.2.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Orsi GA, Couble P, Loppin B. Epigenetic and replacement roles of histone variant H3.3 in reproduction and development. Int J Dev Biol. 2009;53(3):231–243. doi: 10.1387/ijdb.082653go. [DOI] [PubMed] [Google Scholar]
- 94.Van Sandt VS, Suslov D, Verbelen JP, Vissenberg K. Xyloglucan Endotransglucosylase activity loosens a plant Cell Wall. Ann Bot. 2008;100(7):1467–1473. doi: 10.1093/aob/mcm248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saladié M, Rose JKC, Cosgrove DJ, Catalá C. Characterization of a new xyloglucan endotransglucosylase/hydrolase (XTH) from ripening tomato fruit and implications for the diverse modes of enzymic action. Plant J. 2006;47(2):282–295. doi: 10.1111/j.1365-313X.2006.02784.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in this study for qRT-PCR. (XLSX 14 kb)
Lengths distribution of all assembled unigenes (XLSX 11 kb)
Functional annotation of unigenes against Nr, Pfam, KEGG, COG, GO Swissprot databases. (XLS 20569 kb)
Gene ontology (GO) classification, COG classification and KEGG classification of unigenes. a. The GO terms are summarized into three main categories: biological process, cellular component and molecular function. b. COG classification of the unigenes. c. KEGG classification of 5933 KO annotated unigenes. (XLSX 111 kb)
Expression of genes in the 6 libraries in wheat callus based on the results of RNA-Seq. (XLSX 6574 kb)
Information of DEGs in the comparisons of IME vs. ME. (XLSX 397 kb)
Information of DEGs in the comparisons between stages in IMEs and MEs. (XLSX 319 kb)
Functional annotation of all DEG against Nr, Pfam, Swissprot, KOG, KEGG, GO and COG databases. (XLSX 1510 kb)
KEGG enrichment analysis of all DEGs. (XLSX 65 kb)
List of all TFs and expression of DEGs TFs in the 6 libraries. (XLSX 93 kb)
Expression and annotation of differentially expressed aux and SSEGs IME vs. ME. (XLSX 86 kb)
KEGG pathways of plant hormone signal transduction in comparisons of IME vs. ME and between stages. (PDF 122 kb)
Data Availability Statement
The sequencing raw data of this article have been deposited as BioProject ID [PRJNA353135] in the NCBI SRA under the accession number [SRP093588]. All the supporting data are included in Additional files.