

Abstract
Tetramethylpyrazine (TMP) was originally isolated from a traditional Chinese herbal medicine, Ligusticum chuanxiong. In the present study, TMP exhibits potent antitumor activities in vitro. However, the molecular mechanisms remain to be defined. Hence, this study aims to investigate the antiproliferative and apoptotic effects of TMP on HepG2 and elucidate the underlying mechanisms. Analyses using Cell Counting Kit-8 and real-time cell analyzer indicated that TMP significantly inhibited HepG2 cell proliferation. We also observed that TMP induced cell cycle arrest at the G0/G1 checkpoint and apoptosis, using flow cytometry and high-content screening. Furthermore, our results predicted that TMP could directly decrease mitochondrial membrane potential (Δψm), increase the release of cytochrome c, and increase caspase activation, indicating that mitochondrial pathway apoptosis could be the mechanism for TMP within HepG2 cells. Moreover, TMP altered expression of p53 and the Bcl-2/Bax protein ratio, which revealed that TMP induced cell cycle arrest and caspase-dependent mitochondrial apoptosis in HepG2 cells in vitro. These studies provided mechanistic insights into the antitumor properties of TMP, which may be explored as a potential option for treatment of hepatocellular carcinoma.
Keywords: tetramethylpyrazine (TMP), hepatocellular carcinoma, mitochondrial pathway apoptosis, p53, cytochrome c release, caspase activation
Introduction
Hepatocellular carcinoma (HCC) is one of the most common and malignant diseases in the world. Its incidence has been increasing globally, and more than 70% of all newly diagnosed liver cancers occur in Eastern Asia and Central Africa.1,2 The main cause of HCC is the cirrhotic liver, which is associated with chronic hepatitis B virus (HBV) or hepatitis C virus infections and the consumption of alcohol. Dysregulation of cellular proliferation and apoptosis are frequent events related to the malignant phenotype and poor responsiveness of HCC toward chemotherapy.3 Therefore, developing effective pharmacological therapy for such dysregulation is a valid target for HCC clinical treatment.
Cell proliferation depends on the rates of both cell division and cell death. Tumors frequently have decreased cell death as a primary mode of increased cell proliferation.4 Genetic changes resulting in loss of programmed cell death are likely to be critical components of tumorigenesis. Many of the gene products that appear to control apoptotic tendencies are regulators of cell cycle progression. Two apoptotic pathways, the mitochondrial-dependent intrinsic pathway and the death receptor–mediated extrinsic pathway, have been elucidated.5,6 Furthermore, the tumor suppressor p53 initiates various cellular responses that can lead to cell cycle arrest and apoptosis, which also plays a role in the mitochondrial apoptosis pathway because its activation can directly induce Bax expression.7,8 Their roles in HepG2 apoptosis remain to be defined, and they could be potential targets for drug-induced HepG2 cell cycle arrest and apoptosis implicated in antitumor therapy.
Ligusticum chuanxiong Hort is a plant classified in the family Apiaceae. It was known as a herb that promotes the circulation of blood in traditional Chinese medicine. Its main active constituent is tetramethylpyrazine (TMP), which can ameliorate microcirculation and also exhibits antioxidant activity and calcium antagonism. Recent studies suggest that TMP exhibits therapeutic promise for hepatic fibrosis and potent antitumor activities.9,10 However, the underlying molecular mechanisms remain to be defined. In the present study, our results demonstrated that TMP induced cell cycle arrest at the G0/G1 checkpoint and promoted mitochondrial apoptosis dependent on caspase activation in HepG2 cells. These effects were mediated by activation of p53 and upregulation of the Bax/Bcl-2 protein ratio. These discoveries provide novel insights into TMP induction of HepG2 cell cycle arrest and apoptosis and its anti–liver cancer properties.
Materials and Methods
Reagents and Antibodies
TMP was purchased from National Institutes for Food and Drug Control (Beijing, China). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories Science and Technology (Kamimashiki gun Kumamoto, Japan). The primary antibodies against cleaved-caspase-3, cleaved-caspase-9, Bax, Bcl-2, p53, and cytochrome c (cyt c) were purchased from Cell Signaling Technology (Danvers, MA). Cellomics Multiparameter Cytotoxicity 3 Kit and Hoechst 33342 staining were purchased from Thermo Fisher Scientific (Massachusetts, USA). Annexin V-FITC/PI kit and propidium iodide (PI) were purchased from Invitrogen (California, USA).
Cell Culture and Preparation of TMP
HepG2 cell line was purchased from Shanghai Institute of Biochemistry and Cell Biology. Cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY) and supplemented with 10% heat-inactivated fetal bovine serum (Sijiqing Biotechnology Co, Hangzhou, China), 100 U/mL penicillin, and 100 µg/mL streptomycin (Hyclone, Thermo Fisher Scientific Inc, Massachusetts, USA) at 37°C with a humidified atmosphere of 5% CO2.
TMP was weighed and dissolved into dimethylsulfoxide (DMSO) as a 280 000 µmol/L stock solution; then diluted into 175, 350, 700, 1400, and 2800 µmol/L of serial solutions, respectively; and next sterilized through a 0.22-µm filter membrane for subsequent study.
Cell Viability Analysis
Cytotoxicity activity was assayed by CCK-8 as described.11,12 Briefly, cells were seeded into 96-well plates (Costar, Corning Inc, Corning, NY) at a density of 1 × 104 cells/well, cultured for 24 hours, and then divided into a control treated with DMSO (Beijing Solarbio Science and Technology Co, Ltd, Beijing, China) and those undergoing TMP treatments (the final sample solutions at 5 concentrations of 175, 350, 700, 1400, and 2800 µmol/L). After incubation for 48 hours, the cell viability was assayed by CCK-8. All experiments were performed at least 3 times. Data were calculated as the percentage of cell inhibition rate (%): [1 − (Sample solution absorbance value/Control absorbance value)] × 100%.
Real-Time Monitoring of Density-Dependent Growth
To monitor cellular adhesion and growth responses, we have exploited the system from Roche Applied Sciences, consisting of microtiter plates (E-plates) with integrated gold microarrays in the bottom of the wells for continuous and label-free measurements of cellular status in real-time by the Real-time Cell Analyzer (RTCA DP) instrument.13-15 In our study, cells were seeded in E-plates at a density of 1 × 105 cells/well. E-plates were then transferred to the RTCA-DP instrument for automated real-time monitoring at standard incubator conditions: quadruplet readout of the parameter “Cell Index” every 1 hour for the following 48 hours.
Flow Cytometer Analysis of Cell Cycle and Apoptosis
To determine cell cycle and apoptosis, PI cell cycle kit and Annexin V-FITC/PI staining assay were performed as previously described.16,17 The cells were seeded in 6-well plates at a density of 1 × 105 cells/well, incubated for 24 hours, and then exposed to TMP at a concentration of 700 µmol/L for 12, 24, and 48 hours, respectively. After treatment, the cells were stained with Annexin V-FITC/PI kit according to the manufacturer’s instructions and were suspended in a fluorochrome solution containing 5 µg/mL PI, 1 mg/mL sodium citrate, 0.1 mg/mL RNase, and 1% Triton X-100 and then analyzed with a flow cytometer (FACSCalibur, BD Instruments Inc, Franklin Lakes, NJ) and FlowJo 7.1.0 software (Tree Star, Ashland, OR). All experiments were performed in triplicate, and for each measurement, at least 20 000 cells were counted.
High-Content Screening (HCS) Analysis of Cell Apoptosis
HCS is a tool for generating data on multiple parameters in single cell as well as in populations of cells, which is used to measure the toxicity or proapoptotic effect of compounds on the cell.16,18 In this experiment, cells were incubated with TMP for 48 hours and stained with fluorescent dyes, including Hoechst 33342, Annexin V-FITC, and PI. Then, plates were sealed and run immediately on the HCS Assay Scan VTI HCS Reader to acquire images. Images were analyzed with HCS software.
HCS Analysis of Mitochondrial Function
Using HCS technology, TMP induction of HepG2 mitochondrial pathway apoptosis at different TMP doses was assayed by Assay Scan VTI HCS Reader with the Cellomics Multiparameter Cytotoxicity 3 Kit. The principle of the assay was that cells were labeled with fluorescent dyes that would indicate the cellular properties of interest, including nucleus, mitochondrial membrane potential (Δψm) and cyt c. All procedures were performed according to the manufacturer’s instructions.19-21 Afterward, plates were sealed and run immediately on the Assay Scan VTI HCS Reader to acquire images. Images were analyzed with HCS software.
Simple Western Analysis
Simple Western analyses were performed according to the ProteinSimple user manual.22-24 In brief, cell lysate samples were mixed with a master mix to a final concentration of sample buffer, fluorescent molecular weight markers, and dithiothreitol and then heated at 95°C for 5 minutes. Next, the samples, blocking reagent, primary antibodies, chemiluminescent substrate, and separation and stacking matrices were also dispensed to designated wells in a 384-well plate. After plate loading, the separation electrophoresis and immunodetection steps took place in the capillary system and were fully automated. The collected signal intensity was subsequently processed, converted to an electropherogram, and integrated using Compass software (ProteinSimple).
Statistical Analysis
All experimental values were presented as means ± SD. Data were analyzed using SPSS 15.0 software. The significance of difference was determined by 1-way ANOVA with the least-significant difference tests, and Students’s t-test was used for comparing the 2 treatments, as needed. Values of P < .05 were considered to be statistically significant.
Results
Effects of TMP on Viability of HepG2 Cells
To investigate the effect of TMP on the survival of HepG2 cells, a wide range of doses of TMP, from 175 to 2800 µmol/L, were incubated with HepG2 cells for 48 hours. Cell viability was determined by CCK-8 assay. As shown in Figure 1A, TMP significantly increased HepG2 cell inhibition in a dose-dependent manner (P < .01) compared with controls. Moreover, we further characterized the TMP-incubated HepG2 cell growth rate using the real-time cell analysis system, which allows continuous data recording over a period of several days (Figures 1B and 1C). In our experiment, measurements on untreated and TMP-stimulated cells demonstrated that the proliferation rate of TMP-treated cells was remarkably reduced in a dose- and time-dependent manner (P < .01).
Figure 1.
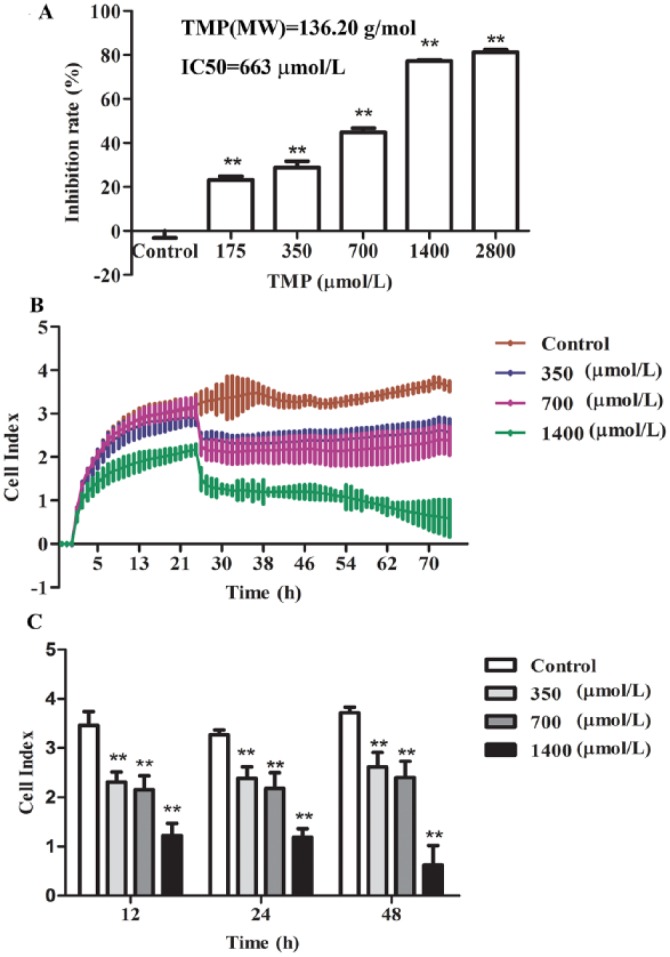
The effects of tetramethylpyrazine (TMP) on HepG2 cell viability and real-time monitoring of cellular proliferation. A. The HepG2 cells were treated with TMP at concentrations of 175, 350, 700, 1400, and 2800 µmol/L for 48 hours, and then, cell viability was assessed using the Cell Counting Kit-8 assay. B. Cells were seeded in an E-plate and then monitored for 72 hours with the real-time cell analyzer instrument. C. The proliferation of TMP-treated cells for 12, 24, and 48 hours, respectively. Values are expressed as mean ± SD from 3 independent experiments, *P < .05, **P < .01 compared with control treatment.
Effects of TMP on HepG2 Cell Cycle and Apoptosis
Flow cytometric analysis of HepG2 cells stained with PI showed a significant increase in G0/G1 when TMP was induced for 12 hours and subG1 phase when TMP was induced form 12 to 48 hours (P < .01; Figures 2A and 2B). These results demonstrated that TMP could arrest HepG2 cells at the G0/G1 phase and induce cell apoptosis. Subsequently, Annexin V-FITC/PI staining was used to quantitatively determine the percentage of cells that were actively undergoing apoptosis. Cells were incubated with TMP for 12, 24, and 48 hours, respectively; stained with Annexin V-FITC/PI; and analyzed by flow cytometry. As shown in Figures 2C and 2D, compared with controls, the number of apoptotic cells significantly increased in the TMP-treated cells in a time-dependent manner (P < .01). Additional evidence for TMP induction of HepG2 apoptosis was provided by Hoechst staining and Annexin V-FITC/PI, as analyzed by HCS (Figures 3A-3D). Data analyzed by HCS showed that compared with control treatment, the nuclear size became smaller (P < .05) and both the Annexin V-FITC and PI fluorescence intensity significantly increased (P < .01) in TMP-treated cells. Collectively, these data indicated that TMP could induce HepG2 cell cycle arrest and apoptosis.
Figure 2.

The effects of tetramethylpyrazine (TMP) on HepG2 cell cycle and apoptosis using flow cytometry. Cells were treated with TMP at a concentration of 700 µmol/L for 12, 24, and 48 hours, respectively. (A) Cell cycle distribution and (B) cell number percentage in each phase (subG1, G0/G1, S, and G2/M) were detected and calculated. (C) Images and (D) quantification of apoptotic cells were analyzed and expressed. Data are presented as mean ± SD from triplicate samples. *P < .05, **P < .01 compared with the control group.
Figure 3.

The effects of tetramethylpyrazine (TMP) on HepG2 cell apoptosis using high-content screening (HCS) analysis. (A) HepG2 cells were stained by Hoechst 33342 and Annexin V-FITC/PI (propidium iodide) fluorescent dyes, and (B) the nucleus size, (C) Annexin V-FITC fluorescence, and (D) PI fluorescence were evaluated by HCS. Data are presented as mean ± SD from triplicate samples. *P < .05, **P < .01 compared with the control group.
Effects of TMP on HepG2 Cell Mitochondrial Function and Caspase Activation
To further elucidate the mechanism of cell apoptosis, we next explored the pathway by which TMP promoted HepG2 apoptosis. Mitochondria play a pivotal role as the gatekeepers of apoptosis.25 During apoptosis, several key events occur in mitochondria, including a decrease in Δψm and increase in the release of cyt c.26,27 Using HCS technology, nuclear, Δψm and the release of cyt c from mitochondrial were costained with fluorescent dyes, and these images were taken by the HCS assay scan VTI Reader. Data derived from these images were analyzed using HCS software. The results (Figures 4A-4D) suggested that the mitochondrial pathway is involved in TMP-induced HepG2 apoptosis, and compared with control treatment, after cells were exposed to TMP for 48 hours, the fluorescent intensity associated with size of the nucleus and Δψm significantly decreased (P < .05), whereas that associated with the release of cyt c from mitochondria markedly increased (P < .01).
Figure 4.

The mitochondrial function induced by tetramethylpyrazine (TMP) in apoptotic HepG2 cells. Cells were treated with TMP for 48 hours at a concentration of 700 µmol/L and then, respectively stained with the Hoechst 33342, cytochrome c, and mitochondrial membrane potential (Δψm). The release of cytochrome c from mitochondria to cytosol is shown in image (A) and quantification of the average cytochrome c intensity in (B). The cell Δψm is shown in image (C) and quantification of the average Δψm intensity in (D). Data are presented as mean ± SD from triplicate samples. *P < .05 and **P < .01 compared with the control group.
Next, despite varying conditions under which apoptosis can occur, caspase activation is a universal event, and for that reason, it is considered an apoptotic marker. In the current study, we demonstrated that TMP significantly increased cleaved-caspase-3 (P < .01) and cleaved-caspase-9 (P < .01) compared with the control group (Figures 5A-5E). Our results indicate that TMP could induce HepG2 cell mitochondrial-mediated and caspase-dependent apoptosis.
Figure 5.

The activation of caspases induced by tetramethylpyrazine (TMP) in apoptotic HepG2 cells. HepG2 cells were incubated with TMP for 48 hours at a concentration of 700 µmol/L, and then, cleaved-caspase-3, cleaved-caspase-9, and β-actin were assessed via simple Western blotting. (A-C) The electropherogram, (D) pseudo-gel, and (E) quantification of simple Western blotting were measured using antibody for blotting. Data are presented as mean ± SD from triplicate samples. *P < .05, **P < .01 compared with the control group.
Effects of TMP on p53 Signaling and Bcl-2/Bax Protein Ratio
Some cell survival pathways control foundational cellular functons such as proliferation, cell cycle, and apoptosis and can be affected by many types of cellular stimuli or toxic insults.28 We investigated the signaling pathway by which TMP induced HepG2 cell cycle arrest and apoptosis by simple Western analysis (Figures 6A-6E). Because the Bax/Bcl-2 ratio is critical for regulating the release of cytocrome c from mitochondria, we investigated the potential involvement of the Bcl-2 family of proteins in the process of TMP-mediated hepatocyte apoptosis. Our subsequent Western blot assays demonstrated that compared with control treatment, TMP-treated cells showed an obvious decrease in Bcl-2/Bax protein ratio (P < .01; Figures 6A-6F). Results in Figures 6C to 6F show that the upregulation of p53 was detected in TMP-treated cells. Thus, these data suggest that TMP could induce G0/G1 cell cycle arrest and destroy mitochondrial function via activation of p53 and upregulation of the Bcl-2/Bax protein ratio.
Figure 6.

The p53 signaling and Bax/Bcl-2 protein ratio mediates tetramethylpyrazine (TMP)-induced HepG2 apoptosis. HepG2 cells were incubated with TMP for 48 hours at a concentration of 700 µmol/L, and then Bax, Bcl-2, p53, and β-actin were assessed via simple Western blotting (A-D). The electropherogram (E) pseudo-gel and (F) quantification of simple Western blotting were measured using antibody for blotting. Data are presented as mean ± SD from triplicate samples. *P < .05, **P < .01 compared with the control group.
Discussion
HCC is most common in Asian countries, where the HBV is endemic.29 Induction of cell apoptosis has been proposed to be a potential strategy for treatment of HCC.30,31 Previous studies demonstrated that TMP could protect the liver and reverse tumor multidrug resistance.9,10 Our present study explored the TMP effects on HepG2 cell proliferation and apoptosis and underlying mechanisms in vitro and further predicted the potential molecular target for TMP actions.
To clarify the effect of TMP on regulating HepG2 cellular proliferation, we initially examined the dose-response effect of TMP on cell viability. The results obtained by the CCK-8 assay and real-time cell analysis system demonstrated that TMP markedly inhibited HepG2 cell proliferation in a dose- and time-dependent manner.
Cell cycle arrest in cancer cells has been known to be a major indicator for anticancer effects; the loss of cell cycle control has been implicated in tumor development and proliferation.32 Subsequently, we performed flow cytometry to examine the effect of TMP on the cell cycle. Our data suggested that TMP induced a significant cell-cycle arrest in the G0/G1 phase during cell cycle progression and apoptosis. As a mechanism for cell death, apoptosis plays important roles in many normal processes, ranging from fetal development to adult tissue homeostasis.33 Many tumors have been associated with the inhibition of apoptosis. Apoptotic induction has been a new target for innovative mechanism-based drug discovery.34 Hence, to further confirm whether TMP can induce cell apoptosis, we used the Annexin V-FITC/PI kit for flow cytometry and HCS, respectively. These results indicated that TMP could induce HepG2 cell cycle arrest and apoptosis.
Apoptosis can be initiated via 2 alternative signaling pathways: the mitochondrion-mediated intrinsic apoptotic pathway and the death receptor–mediated extrinsic apoptotic pathway.35,36 Mitochondria play critical roles in the regulation of various apoptotic processes, including drug-induced apoptosis.37 On induction of apoptosis, activation of the ion channel complex triggers mitochondrial membrane permeabilization that finally results in an increased permeability of the outer mitochondrial membrane, which permits the release of mitochondrial proapoptotic proteins, such as cyt c, into the cytoplasm.38,39 In our study, we assessed the effect of TMP on cell nuclear size, Δψm, and the release of cyt c from mitochondria using HCS analysis. Strikingly, these results confirmed that TMP induced HepG2 cell apoptosis via the cyt c–dependent mitochondrial pathway.
A further characteristic of apoptosis is the activation of caspase family members. It is now well established that in most cell types, once cyt c is released into the cytosol, it triggers caspase-9 activation, results in the cleavage of caspase-3, and ultimately leads to the activation of the execution phase of apoptosis.40 In this study, as expected, we found that TMP significantly increased cleaved-caspase-3 and cleaved-caspase-9 activation. These observations suggested that TMP induced HepG2 cell apoptosis by activating caspases.
Bcl-2 family members are critical regulators of the mitochondrial pathway.41,42 Death-promoting members of the Bcl-2 family, such as Bax, play key roles in the chemical-induced release of cyt c, thereby activating caspases and the mitochondrial apoptotic pathway.43,44 To determine which apoptosis-related proteins are regulated by TMP, the expression of Bax and Bcl-2 protein were measured by Western blotting analysis. In this study, we demonstrated that TMP downregulated the Bcl-2/Bax protein ratio in HepG2 cells, inducing the release of cyt c and activating caspase-3-dependent mitochondrial apoptosis. The p53 gene is an essential component of the pathway leading to 2 different critical functions: G1 growth arrest and apoptosis. Also, the p53-dependent expression of Bax gene triggers apoptosis as a facile cytotoxic response to chemotherapeutic agents.45,46 Our mechanistic investigations uncovered an activation of p53 signaling responsible for TMP-induced cell cycle arrest and mitochondrial apoptosis in HepG2 cells.
Conclusion
In conclusion, our results demonstrated that TMP induced HepG2 cell G0/G1 arrest and stimulated mitochondrial apoptosis dependent on cyt c–mediated caspase cascades in vitro. Moreover, the above results demonstrated that the activation of p53 and downregulation of the Bcl-2/Bax protein ratio by TMP resulted in HepG2 cell cycle arrest and apoptosis. Molecular mechanisms indicated that p53 could be the target molecule for TMP within HepG2. Our results provide new insight into the mechanisms underlying the anti–liver cancer effect of TMP and its implications for anti-HCC therapy.
Footnotes
Authors’ Note: LB and XY contributed equally to the work.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from National Natural Sciences Foundation of China (81273638, 81503486) and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). In addition, this program was supported by Natural Science Research Project of Jiangsu Provincial Education Department (14KJD360004).
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90. [DOI] [PubMed] [Google Scholar]
- 2. Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893-1907. [DOI] [PubMed] [Google Scholar]
- 3. Carbajo-Pescador S, Steinmetz C, Kashyap A, et al. Melatonin induces transcriptional regulation of Bim by FoxO3a in HepG2 cells. Br J Cancer. 2013;108:442-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316-323. [DOI] [PubMed] [Google Scholar]
- 5. Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770-776. [DOI] [PubMed] [Google Scholar]
- 6. Kuranaga E. Caspase signaling in animal development. Dev Growth Differ. 2011;53:137-148. [DOI] [PubMed] [Google Scholar]
- 7. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413-431. [DOI] [PubMed] [Google Scholar]
- 8. Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13-20. [DOI] [PubMed] [Google Scholar]
- 9. Zhang F, Kong DS, Zhang ZL, et al. Tetramethylpyrazine induces G0/G1 cell cycle arrest and stimulates mitochondrial-mediated and caspase-dependent apoptosis through modulating ERK/p53 signaling in hepatic stellate cells in vitro. Apoptosis. 2013;18:135-149. [DOI] [PubMed] [Google Scholar]
- 10. Zhang YX, Liu XM, Zuo T, Liu Y, Zhang JH. Tetramethylpyrazine reverses multidrug resistance in breast cancer cells through regulating the expression and function of P-glycoprotein. Med Oncol. 2012;29:534-538. [DOI] [PubMed] [Google Scholar]
- 11. Gao M, Zhang WC, Liu QS, Hu JJ, Liu GT, Du GH. Pinocembrin prevents glutamate-induced apoptosis in SH-SY5Y neuronal cells via decrease of bax/bcl-2 ratio. Eur J Pharmacol. 2008;591:73-79. [DOI] [PubMed] [Google Scholar]
- 12. Kei T, Shuuji M, Shin Y, Tadayoshi S, Nobuhiko T, Michihiko I. Tumor necrosis factor-related apoptosis-inducing ligand 1 (TRAIL1) enhances the transition of red blood cells from the larval to adult type during metamorphosis in Xenopus. Blood. 2010;115:850-859. [DOI] [PubMed] [Google Scholar]
- 13. Hellevik T, Pettersen I, Berg V, et al. Cancer-associated fibroblasts from human NSCLC survive ablative doses of radiation but their invasive capacity is reduced. Radiat Oncol. 2012;7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei PL, Kuo LJ, Wang W, et al. Silencing of glucose-regulated protein 78(GRP78) enhances cell migration through the upregulation of vimentin on hepatocellular carcinoma cells. Ann Surg Oncol. 2012;19:S572-S579. [DOI] [PubMed] [Google Scholar]
- 15. Hendrik U, Susanne S, Stephanie G, Frank G, Fred F. Differential roles of Src in transforming growth factor-β regulation of growth arrest, epithelial-to-mesenchymal transition and cell migration in pancreatic ductal adenocarcinoma cells. Int J Oncol. 2011;38:797-805. [DOI] [PubMed] [Google Scholar]
- 16. Xiang SL, Sun ZH, He QZ, Yan F, Wang Y, Zhang J. Aspirin inhibits ErbB2 to induce apoptosis in cervical cancer cells. Med Oncol. 2010;27:379-387. [DOI] [PubMed] [Google Scholar]
- 17. Lv L, Zhou ZX, Huang XZ, et al. Inhibition of peptidyl-prolyl cis/trans isomerase Pin1 induces cell cycle arrest and apoptosis in vascular smooth muscle cells. Apoptosis. 2010;15:41-54. [DOI] [PubMed] [Google Scholar]
- 18. Sun S, Du GJ, Qi LW, Williams S, Wang CZ, Yuan CS. Hydrophobic constituents and their potential anticancer activities from Devil’s Club Oplopanax horridus. J Ethnopharmacol. 2010;132:280-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu JJ, Diaz D, O Brien PJ. Applications of cytotoxicity assays and pre-lethal mechanistic assays for assessment of human hepatotoxicity potential. Chem Biol Interact. 2004;150:115-128. [DOI] [PubMed] [Google Scholar]
- 20. Dambach DM, Andrews BA, Moulin F. New technologies and screening strategies for hepatotoxicity: use of in vitro methods. Toxicol Pathol. 2005;33:17-26. [DOI] [PubMed] [Google Scholar]
- 21. Giuliano KA, Haskins JR, Taylor DL. Advances in high content screening for drug discovery. Assay Drug Dev Technol. 2003;1:565-577. [DOI] [PubMed] [Google Scholar]
- 22. Liu SB, Sardi S, Sonom B, et al. The application of a novel nanovolume capillary electrophoresis-based protein analysis system in personalized and translational medicine research. J Bioanal Biomed. 2013;S3:1-10. [Google Scholar]
- 23. Chen JQ, Heldman MR, Herrmann MA, et al. Absolute quantitation of endogenous proteins with precision and accuracy using a capillary Western system. Anal Biochem. 2013;442:97-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rustandi RR, Loughney JW, Hamm M, et al. Qualitative and quantitative evaluation of simon, a new CE-based automated Western blot system as applied to vaccine development. Electrophoresis. 2012;33:2790-2797. [DOI] [PubMed] [Google Scholar]
- 25. Wang C, Youle RJ. The role of mitochondria in apoptosis. Ann Rev Genet. 2009;43:95-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691-699. [DOI] [PubMed] [Google Scholar]
- 27. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maddika S, Ande SR, Panigrahi S, et al. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat. 2007;10:13-29. [DOI] [PubMed] [Google Scholar]
- 29. Bosch FX, Ribes J, Cleries R, Diaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis. 2005;9:191-211. [DOI] [PubMed] [Google Scholar]
- 30. Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513-519. [DOI] [PubMed] [Google Scholar]
- 31. Schulze-Bergkamen H, Krammer PH. Apoptosis in cancer-implications for therapy. Semin Oncol. 2004;31:90-119. [DOI] [PubMed] [Google Scholar]
- 32. Liu Y, Cao Y, Zhang W, et al. A small molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2012;11:1672-1682. [DOI] [PubMed] [Google Scholar]
- 33. Frankfurt OS, Krishan A. Apoptosis-based drug screening and detection of selective toxicity to cancer cells. Anticancer Drugs. 2003;14:555-561. [DOI] [PubMed] [Google Scholar]
- 34. Ye M, Liu JK, Lu ZX, et al. Grifolin, a potential antitumor natural product from the mushroom Albatrellus confluens, inhibits tumor cell growth by inducing apoptosis in vitro. FEBS Lett. 2005;579:3437-3443. [DOI] [PubMed] [Google Scholar]
- 35. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305-1308. [DOI] [PubMed] [Google Scholar]
- 36. Green DR. Apoptotic pathways: the roads to run. Cell. 1998;94:695-698. [DOI] [PubMed] [Google Scholar]
- 37. Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513-519. [DOI] [PubMed] [Google Scholar]
- 38. Mayer B, Rainer O. Mitochondrial regulation of apoptosis. News Physiol Sci. 2003;18:89-94. [DOI] [PubMed] [Google Scholar]
- 39. Zamzani N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev. 2001;2:67-71. [DOI] [PubMed] [Google Scholar]
- 40. Jiang XJ, Wang XD. Cytochrome c-mediated apoptosis. Ann Rev Biochem. 2004;73:87-106. [DOI] [PubMed] [Google Scholar]
- 41. Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun. 2003;304:437-444. [DOI] [PubMed] [Google Scholar]
- 43. Rosse T, Olivier R, Monney L, et al. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496-499. [DOI] [PubMed] [Google Scholar]
- 44. Zhang A, Wang Y, Xie HY, Zheng S. Calcitriol inhibits hepatocyte apoptosis in rat allograft by regulating apoptosis-associated genes. Int Immunopharmacol. 2007;7:1122-1128. [DOI] [PubMed] [Google Scholar]
- 45. Hagopian GS, Mills GB, Khokhar AR, Bast RC, Jr, Siddik ZH. Expression of p53 in cisplatin-resistant ovarian cancer cell lines: modulation with the novel platinum analogue (1R, 2R-diaminocyclohexane) (trans-diacetato) (dichloro)-platinum (IV). Cancer Res. 1999;5:655-663. [PubMed] [Google Scholar]
- 46. Prokop A, Wieder T, Sturm I, et al. Relapse in childhood acute lymphoblastic leukemia is associated with a decrease of the Bax/Bcl-2 ratio and loss of spontaneous caspase-3 processing in vivo. Leukemia. 2000;14:1606-1613. [DOI] [PubMed] [Google Scholar]