

Abstract
Most pharmaceutical formulations also include a certain amount of lubricant to improve their flowability and prevent their adhesion to the surfaces of processing equipment. Magnesium stearate is an additive that is most frequently used as a lubricant. Magnesium stearate is capable of forming films on other tablet excipients during prolonged mixing, leading to a prolonged drug liberation time, a decrease in hardness, and an increase in disintegration time. It is hydrophobic, and there are many reports in the literature concerning its adverse effect on dissolution rates.
The objective of this study was to evaluate the effects of two different concentrations of magnesium stearate on dissolution properties of ranitidine hydrochloride coated tablet formulations labeled to contain 150 mg. The uniformity content was also checked.
During the drug formulation development, several samples were designed for choice of the formulation. For this study, two formulations containing 0,77 and 1,1% of magnesium stearate added in the manufacture of cores were chosen. Fraction of ranitidine hydrochloride released in dissolution medium was calculated from calibration curves. The data were analyzed using pharmaco-peial test for similarity of dissolution profiles (f2 equation), previously proposed by Moore and Flanner.
Application of f2 equation showed differences in time-course of ranitidine hydrochloride dissolution properties. The obtained values indicate differences in drug release from analyzed ranitidine hydrochloride formulations and could cause differences in therapeutic response.
Keywords: magnesium stearate, dissolution properties^, equation, coated tablets, ranitidine hydrochloride
INTRODUCTION
Lubrication is a process of high importance in the pharmaceutical industry. Lubricants are added to tablet formulations for two reasons: (a) to prevent adhesion of granules to the tooling-anti-adherent; (b) to improve granule flow properties-glidant. As anti-adherents, they reduce the friction between the wall and granules as the tablet is formed and ejected (1,2). As glidants, they can enhance the blending of the active components and decrease processing problems and weight variability during compaction (3). There are numerous examples of the effects of lubrication on densification and compactabil-ity of mixtures (4, 5, 6), and also on tablet properties such as tensile strength, friability, disintegration (7, 8). Among lubricants, magnesium stearate is the most widely used one. Commonly used concentrations range between 0,25-5% (9). It appears in different crystal forms, shows different particle size and shape, and occurs in several hydrate forms (10,11,12). Its lubricating effectiveness was described by Delacourte et al. (13). Magnesium stearate is capable of forming films on other tablet excipients during prolonged mixing, leading to a prolonged drug liberation time, a decrease in hardness, and an increase in disintegration time. It is hydrophobic, and there are many reports in the literature concerning its adverse effect on dissolution rates. A tablet is compressed using high forces to form a solid compact of relatively low porosity, which enables it to endure subsequent handling (14, 15). Drug absorption from solid dosage forms after oral administration depends on the release of the drug substance from the drug product, the dissolution or solubi-lization of the drug under physiological conditions, and the permeability across the gastrointestinal tract. Because of the critical nature of the first two of these steps, in vitro dissolution may be relevant to the prediction of in vivo performance (16). The dissolution test facilitates assessment of the dissolution properties of the drug itself and thereby selection of the most appropriate excip-ients as well as optimization of proportions among them that result in the desired drug release behavior (17,18). The objective of this study was to evaluate the effects of two different concentrations of magnesium stearate on dissolution properties of ranitidine hydrochloride coated tablet formulations labeled to contain 150 mg. The uniformity content was also checked. Ranitidine hydrochloride is a histamine H2-receptor antagonist. It is widely prescribed in active duodenal ulcers, gastric ulcers, Zollinger-Ellison syndrome, gastroesophageal reflux disease, and erosive esophagitis. During the drug formulation development, several samples were designed for choice of the formulation. For this study, two formulations containing 0,77 and 1,1% of magnesium stearate added in the manufacture of cores were chosen.
MATERIALS AND METHODS
Reagents
The used reagents were all of analytical grade, unless otherwise stated. Ranitidine hydrochloride working standard, hydrochloric acid, ammonium acetate, acetonitrile were provided by Merck (Darmstadt, Germany).
Tablet ingredients
For the tablet formulations the following ingredients were used and provided by different producers: ra-nitidine hydrochloride, titan dioxide (Merck Darmstadt, Germany), magnesium stearate and ethanol (Riedel-de Haen, Seelze-Hanover, Germany), micro-crystalline cellulose, macrogol 6000 (Fluka, Buchs, Switzerland), maize starch, povidone, hydroxypro-pyl cellulose (Sigma-Aldrich, Steinheim, Germany).
Tablet preparation
The process started with wet granulation where ra-nitidine hydrochloride, maize starch and povidone were mixed and granulated with ethanol. The granulation was dried to the prescribed moisture content and sieved. Sieved magnesium stearate was added. This mixture was homogenized and compressed into cores. For the process of film coating, a homogenous dispersion (hydroxypropyl cellulose, titan dioxide, mac-rogol 6000, ethanol) was prepared and sprayed onto cores. Both formulations (“A” and “B”) were prepared under the same technological conditions. The only difference between them was the content of magnesium stearate added; 0,77 and 1,1% respectively.
Preparation of standard solutions
A standard curve of absorbance versus concentration was constructed using solutions of ranitidine hydro-chloride in the dissolution medium (artificial gastric juice, pH=1,2; without pepsine, previously degasified) ranging in concentration from 0,0059 to 0,0099 mg/ ml. Absorbance versus concentration plot was linear over this concentration range and was used to determine percent of drug dissolved in the dissolution experiments. UV absorbance of each standard solution was measured spectrophotometrically at 228 nm.
Dissolution test conditions and analysis procedure
The dissolution tests of ranitidine hydrochloride coated tablets (n=6) were performed using USP apparatus 2 (n=6), Van Kel VK 7010 dissolution tester, at a stirring speed of 50 rpm (Van Kel, Cary, NC, USA). The dissolution apparatus was maintained at 37°C throughout the experiment. Samples in the amount of 5 ml were withdrawn at 15 min intervals (15th, 30th and 45th min). Prior to use, the dissolution media were equilibrated at 37°C overnight to deaerate the medium so that bubble formation during the test, due to escape of dissolved gases, was minimized. Dissolution samples were collected for analysis and replaced with an equal volume of fresh dissolution fluid to maintain a constant total volume. These samples were filtered using a 0,45 μιη membrane filter (Sartorious GmbH, Goettingen, Germany). The dissolution apparatus was connected with UV/VIS spectrophotometer Shimadzu 1601 (Shimadzu, Kyoto, Japan). Determination of dissolution rates for the active ingredient in film tablets is carried out by the previously mentioned spectrophotometric method. All dissolution tests were performed in triplicate.
Applied method to compare dissolution profiles
In this study, model independent approach that compare the dissolution profiles of a pair of drug products were applied to the dissolution data. The data were analyzed using pharmacopeial test for similarity of dissolution profiles f equation), previously proposed by Moore and Flanner (19). This similarity factor has been adopted by the Center for Drug Evaluation and Research (FDA) and by Human Medicines Evaluation Unit of The European Agency for the Evaluation of Medicinal Products (EMEA), as a criterion for the assessment of the similarity between two in vitro dissolution profiles and is included in the “Guidance on Immediate Release Solid Oral Dosage Forms; Scale-up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Dissolution Testing; In Vivo Bioequivalence Documentation” (20), commonly called SUPAC IR, and in the “Note For Guidance on Quality of Modified Release Products: A. Oral Dosage Forms; B. Transdermal Dosage Forms; Section I (Quality)” (21). The similarity factor f2) as defined by FDA and EMEA is a logarithmic reciprocal square root transformation of one plus the mean squared (the average sum of squares) differences of drug percent dissolved between the test and the reference products:
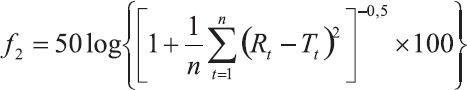
where n is the number of dissolution time points, Rt is the reference assay at time point t.
Content uniformity testing
The following experiment examined the content uniformity of the active ingredient in tablets and was carried out by high-performance liquid chromatography (HPLC) method. The system consisted of a pump, injection valve, autosampler and variable wavelength detector (Shimadzu, Kyoto, Japan). The mobile phase consisted of (70:30, v/v) mixture of 0,1 mol/l ammonium acetate solution and acetonitrile. The flow rate was 1,2 ml/min, the injection volume 20 μ!, the column temperature 25°C and the detection wavelength 322 nm. LiChrospher RP18 column (250ΓηΓηχ4,01ηιηχ5μιη) was used throughout the experiments. Under the abovementioned conditions, the retention time of the analyte was ca. 6 min.
RESULTS AND DISCUSSION
The results of dissolution studies are summarized in Table 1, Table 2 and Figure 1, which show the fraction of the dissolved drug as a function of time. According to the USP (22), in vitro release of raniti-dine hydrochloride coated tablets fulfilled all requirements if the dissolution of each of the six coated tablets was not less than 80% of the declared contents for a period of 45 minutes. In our study, in vitro release of ranitidine hydrochloride from both formulations fulfilled these requirements and exhibited release profile: 102,05 and 96,87%, for “A” and “B” formulation.
TABLE 1.
Fraction of dissolved ranitidine hydrochloride from the ‘A’ formulation as a function of time.
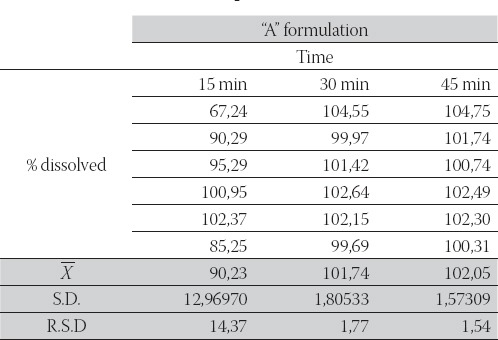
TABLE 2.
Fraction of dissolved ranitidine hydrochloride from the “B” formulation as a function of time.

FIGURE 1.
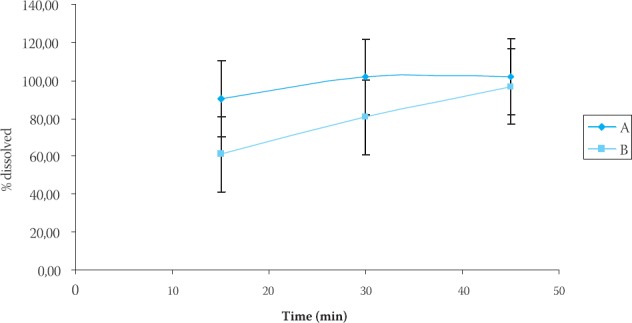
In vitro release of ranitidine hydrochloride from a coated tablets (’K and “B” formulation)
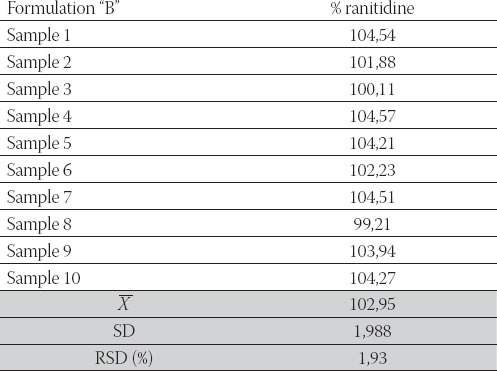
Fractions of ranitidine hydrochloride released in dissolution medium were calculated from calibration curves. The data were analyzed using pharmacopeial test for similarity of dissolution profiles (f2 equation), previously proposed by Moore and Flanner. The f value between 50 and 100 suggests that the dissolution profiles are similar. The value of 100 suggests that the test and reference dissolution profiles are identical. The smaller the value, the larger the dissimilarity between dissolution profiles. Fractions of the released ranitidine hydrochloride were compared using this value. After interpolating data for dissolution profiles for “A” and “B” formulation we obtained valuef2= 41,33. This value indicates differences in drug release from analyzed ranitidine hydrochloride coated tablets. We can hypothesize that magnesium stearate added in “B” formulation (concentration 1,1%), being hydro-phobic in nature, forms a stronger film at the surface of other excipients or the API (ranitidine hydrochloride). In that manner, it enables the dissolution medium to remain on the surface of the particle causing slower wettabilty, and dissolution rate as well. Determination of the content uniformity of ranitidine in our formulations was carried out by HPLC method. The procedure was performed on ten tablets separately. According to the USP (22), the content uniformity of ra-nitidine expressed as % of the declared content should be within the limits of 85-115% and relative standard deviation (R.S.D.) should be equal or smaller than 6%. The results of content uniformity studies are summarized in Table 3 and Table 4 which show the percentage of drug present in each tablet (n=10), standard deviation (S.D.) and relative standard deviation (R.S.D.) as well, for each formulation. The results of the content uniformity analysis of our tablet formulations were 102,35 ±2,21% and 102,95±1,93% for formulation A and B, respectively, which fulfills pharmacopoeial requirements.
TABLE 3.
Content uniformity of ranitidine expressed as % of declared content-Formulation “A”
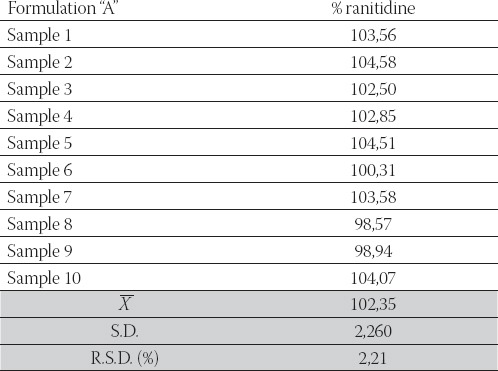
TABLE 4.
Content uniformity of ranitidine expressed as % of declared content-Formulation “B”
CONCLUSION
◊ According to the results obtained in this study, we can conclude that our “A” and “B” formulations satisfied pharma-copeial requirements concerning dissolution rate and content uniformity.
◊ Application off equation shows differences in time-course of ranitidine hydrochloride dissolution, and indicates change in dissolution properties. With the advent of international harmonization of scientific protocols and implementation of SUPAC guidelines including site-to site manufacturing conditions, such changes have important regulatory implications.
◊ The obtained values indicate differences in drug release from analyzed ranitidine hydrochloride formulations and may cause differences in therapeutic response.
◊ We can hypothesize that magnesium stearate added in “B” formulation (concentration 1,1%), being hydrophobic in nature, forms a stronger film at the surface of other excipients or the API (ranitidine hydrochloride). In that manner, it enables the dissolution medium to remain on the surface of the particle causing slower wettabilty as well as dissolution rate.
◊ Application off equation showed differences in time-course of ranitidine hydrochloride dissolution. With the advent of international harmonization of scientific protocols and implementation of SUPAC guidelines including site-to site manufacturing conditions, such process comparisons have important regulatory implications.
◊ However, more data on in vivo drug absorption profiles will provide additional information needed to complete profile characterization of the investigated formulations.
REFERENCES
- 1.Moodya G, Rubinstein M.H, Fitz Simmons R.A. 1984. Tablet lubricants. I. Theory and modes of action. Int. J. Pharm. 1984;9:75–184. [Google Scholar]
- 2.Mehrotra A, Llusa M, Faqih A, Levin M, Muzzio F. J. Influence of shear intensity and total shear on properties of blends and tablets of lactose and cellulose lubricated with magnesium stearate. Int. J. Pharm. 2007;336:284–291. doi: 10.1016/j.ijpharm.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 3.Mackin L, Sartnurak S, Thomas I, Moore S. The impact of low levels of amorphous material (<5%) on the blending characteristics of a direct compression formulation. Int. J. Pharm. 2002;23:213–226. doi: 10.1016/s0378-5173(01)00880-8. [DOI] [PubMed] [Google Scholar]
- 4.Vromans H, Lerk C.F. Densification properties and compatibility of mixtures of pharmaceutical excipients with and without magnesium stearate. Int. J. Pharm. 1988;46:183–192. [Google Scholar]
- 5.van Veen B, Bolhuis G.K, Wu Y.S, Zuurman K, Frijlink H.W. Compaction mechanism and tablet strength of unlubricated and lubricated (silicified) microcrystalline cellulose. Eur. J. Pharm. Biopharm. 2005;59:133–138. doi: 10.1016/j.ejpb.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Ebba F, Piccerelle P, Prinderre P, Opota D, Joachim J. Stress relaxation studies of granules as a function of different lubricants. Eur. J. Pharm. Biopharm. 2001;52:211–220. doi: 10.1016/s0939-6411(01)00171-0. [DOI] [PubMed] [Google Scholar]
- 7.Johansson M.E. Granular magnesium stearate as a lubricant in tablet formulations. Int. J. Pharm. 1984;21:307–315. [Google Scholar]
- 8.de Lourdes GaróSerra M, Robles L.V. Compatibility of agglomerated mixtures of calcium carbonate and microcrystalline cellulose. Int J Pharm. 2003;258:153–163. doi: 10.1016/s0378-5173(03)00185-6. [DOI] [PubMed] [Google Scholar]
- 9.Shangraw R.F, Demarest D.A. A survey of current industrial practices in the formulation and manufacture of tablets and capsules. Pharm. Technol. 1993;17(1):32–44. [Google Scholar]
- 10.Ertel K.D, Carstensen J.T. Chemical physical, and lubricant properties of magnesium stearate. J. Pharm. Sci. 1988;77:625–629. doi: 10.1002/jps.2600770715. [DOI] [PubMed] [Google Scholar]
- 11.Steffens K.J, Koglin J. The magnesium stearate problem III. International Conference on Pharmaceutical Ingredients and Intermediates. 1992:118–125. [Google Scholar]
- 12.Rowe R, Sheskey P, Weller P. Handbook of pharmaceutical excipients. 4th ed. New York: APhA Publications; 2003. [Google Scholar]
- 13.Delacourte A, Guyot J.C, Colombo P, Catellani P.L. Effectiveness of lubricants and lubrication mechanism in tablet technology. Drug Dev. Ind. Pharm. 1995;21:2187–2199. [Google Scholar]
- 14.Roblot L, Puisieux F, Duchéne D. A. Study on lubrication by magnesium stearate. The influence of the proportion of lubricant and the mixing process on the tablet characteristics. Labo. Phar-ma. Probl. Tech. 1983;31:843–847. [Google Scholar]
- 15.Jarosz P.J, Parrott E.L. Effect of lubricants on tensile strengths of tablets. Drug Dev. Ind. Pharm. 1984;10:259–273. [Google Scholar]
- 16.Shah V.P, Lesko L.J, Fan J, Fleischer N, Handerson J, Malinowski H, Makary M, Ouderkirk L, Roy S, Sathe P, Singh G.J.P, Tillman L, Tsong Y, Williams R.L. FDA guidance for industry: dissolution testing of immediate release solid oral dosage forms. Dissolution Technol. 1997;4:15–22. [Google Scholar]
- 17.Amidon G.L, Lennernas H, Shah V.P, Crison J.R. A theoretical basis for a biopharmaceutical drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 18.Dressman J.B, Amidon G.L, Reppas C, Shah V.P. Dissolution as a prognostic tool for oral drug absorption: immediate release dosage forms. Pharm. Res. 1998;15:11–22. doi: 10.1023/a:1011984216775. [DOI] [PubMed] [Google Scholar]
- 19.Moore J.W, Flanner H.H. Mathematical comparison of dissolution profiles. Pharm. Tech. 1996;20:64–74. [Google Scholar]
- 20.Anonymous. Guidance on Immediate Release Solid Oral Dosage Forms; Scale-up and Postapproval Changes: Chemistry, Manufacturing, and Controls. In Vitro Dissolution Testing;In Vivo Bioequivalence Documentation, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) 1995 [Google Scholar]
- 21.Note For Guidance on Quality of Modified Release Products. A Oral Dosage Forms; B Transdermal Dosage Forms; Section I (Quality), European Agency for the Evaluation of Medicinal Products Human Medicines Evaluation Unit Committee for Proprietary Medicinal Products (CPMP) 1999 [Google Scholar]
- 22.United States Pharmacopoeia. National Formulary. USP 30th revision (November 2006) NF 25th edition. Rockville, MD: The United States Pharmacopoeial Convention, Inc; 2006. [(November 2006)]. [Google Scholar]