

Abstract
Purpose of review
We review the genetics of the autosomal dominant, multi-system disorder, Alagille syndrome and provide a summary on how current functional models and emerging biotechnologies are equipped to guide scientists towards novel therapies. The importance of haploinsufficiency as a disease mechanism will be underscored throughout this discussion.
Recent findings
Alagille syndrome, a human disorder affecting the liver, heart, vasculature, kidney, and other systems, is caused by mutations in the Notch signaling pathway ligand, Jagged1 (JAG1) or the receptor, NOTCH2. Current advances in animal modeling, in vitro cell culture, and human induced pluripotent stem cells, provide new opportunities in which to study disease mechanisms and manifestations.
Summary
We anticipate that the availability of innovative functional models will allow scientists to test new gene therapies or small molecule treatments in physiologically-relevant systems. With these advances, we look forward to the development of new methods to help Alagille syndrome patients.
Keywords: Alagille syndrome, haploinsufficiency, gene therapy, liver disease, Jagged1, Notch2
Introduction
Alagille syndrome (ALGS) is an autosomal dominant disease with an estimated incidence of 1:30,000 to 1:50,000 live births, and is characterized by 5 main clinical features including: cholestasis with bile duct paucity, cardiac defects, posterior embryotoxon, characteristic facies, and butterfly vertebrae [1, 2]. Affected individuals present with at least 3 of these 5 features, although additional clinical manifestations, such as renal, bone, or vascular anomalies, have also been noted [3, 4]. Given this wide range of phenotypes, patients undergo multidisciplinary treatment via a team of clinical specialists. Despite our extensive understanding of the disease characteristics, there is no curative treatment for ALGS. Medications, including ursodeoxycholic acid, cholestyramine, rifampin, naltrexone, and more recently, sertraline, have been used to treat the symptoms of pruritus and xanthomas that commonly accompany the cholestatic feature of the disease [2, 5]. More invasive interventions, such as biliary diversion and liver transplantation, are sometimes required for treatment of refractory pruritus or progressive liver disease [6–8]. Complex cardiac disease is treated surgically when symptoms indicate, and less serious manifestations are followed as dictated by the cardiac lesion and severity [4].
The genetics of ALGS are well-defined and mutations in the Jagged1 (JAG1) gene are found in close to 95% of affected individuals, with most mutation types leading to a non-functional protein. This data suggests that the disease mechanism occurs through haploinsufficiency. Since the original identification of the genetic etiology of ALGS 20 years ago, a strong focus has been to fully characterize the genetic presentation of the disease and its associated molecular pathway in order to understand disease pathogenesis. During this time, and particularly with more recent scientific advances, a wide range of animal and in vitro disease models have been identified and optimized to guide studies towards a therapeutic objective. Of particular interest with these studies, and what will be the focus of this review, is the associated liver disease since it is an important and ongoing cause of morbidity and mortality in many ALGS patients. Currently, the models, reagents, and technology exist such that the field is poised to ask and answer therapeutically-relevant questions that have the potential to enhance the treatment options available for affected individuals. We will discuss what is known about ALGS genetics and disease mechanisms and review current functional models in order to understand possible novel therapeutic options in the setting of advancing molecular technology.
Genetics of ALGS
Causative genes and phenotypic variability
ALGS is caused by mutations in genes that impair Notch signaling, a highly conserved pathway that is fundamental to the development of multiple organ systems, which explains the pleiotropic nature of the disease. JAG1 is one of 5 known Notch signaling ligands (JAG1, JAG2, DLL1, DLL3, and DLL4), and it is expressed as a cell surface protein with extracellular, intracellular, and transmembrane domains. There are 4 Notch receptors in humans (NOTCH1-4), which are also cell surface proteins composed of two peptides (an extracellular and an intracellular peptide) that are covalently linked. Through a paracrine signaling mechanism, Notch ligands bind Notch receptors on adjacent cells and initiate a cascade of events that includes proteolytic cleavage of the Notch intracellular domain (NICD) and subsequent translocation of the NICD to the nucleus where it directly regulates transcription of target genes. For a detailed understanding of the important molecular events involved in Notch signaling, we recommend referring to one of the many review articles that have been published on this topic [9–11].
In ALGS, 94–95% of patients have a heterozygous mutation in the Notch ligand, JAG1 (Figure 1A), and 1–2% of patients have a heterozygous mutation in the Notch receptor, NOTCH2 [12–15, 2]. The vast majority of JAG1 mutations are truncating mutations (nonsense or frameshift) or whole/partial gene deletions, which can be located across the extracellular domain of the protein. It is suggested that ALGS is caused by haploinsufficiency of JAG1 on the basis that individuals with whole gene deletions can have identical phenotypes to those with intragenic mutations [16, 1]. A number of missense mutations have also been studied, and in most cases, it has been shown that these proteins are not properly trafficked to the cell surface and thus unable to physiologically interact with NOTCH2, further supporting a model of haploinsufficiency [17–20]. A dominant negative mechanism was proposed by Boyer-Di Ponio et. al. based on the observation that mutant JAG1 protein was observed in cell culture media and was able to compete with immobilized protein to inhibit Notch signaling when cells were transfected with either a missense or nonsense JAG1 cDNA [21]. However, the relevance of this cell culture model to the clinical disease has not been established. The numerous studies that show trafficking and protein folding defects of mutant proteins, as well as the observation of similar phenotypes for individuals with missense mutations and those with full gene deletions, support a more likely disease mechanism of haploinsufficiency and gene dosage [22–24, 16, 20, 17–19]. The mechanism of action of the smaller number of NOTCH2 mutations is not as clear, although it has been shown that they have impaired signaling ability [25].
Figure 1.
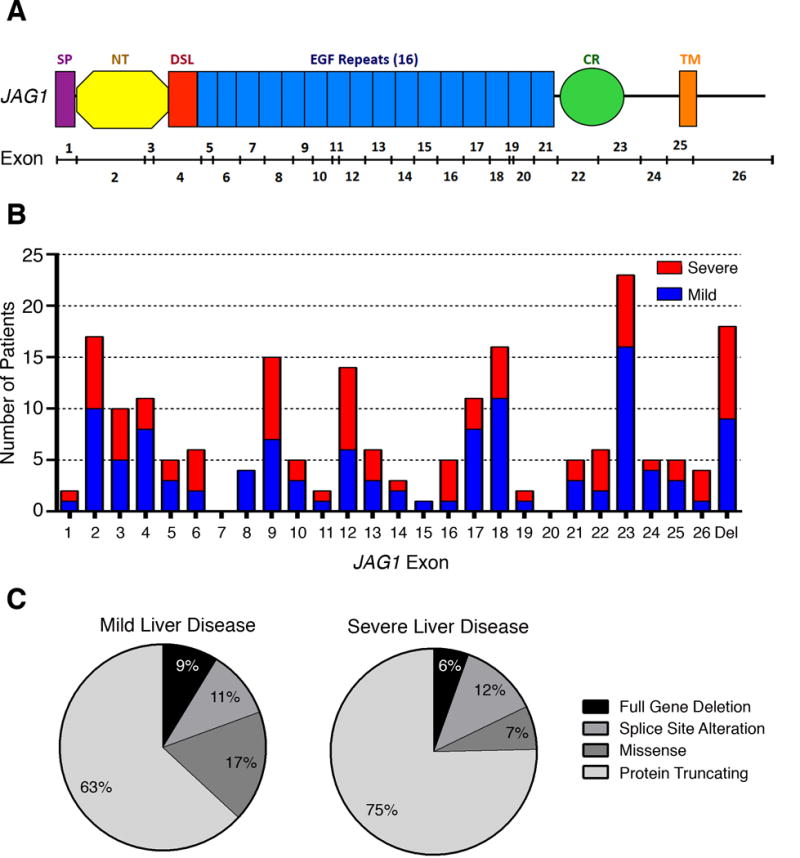
JAG1 mutation type or location does not predict liver disease severity. (A) Schematic of JAG1 protein. Protein domains are listed above the schematic and include: SP-signal peptide, DSL-Delta/Serate/Lag-2, EGF-epidermal growth factor-like, CR-cysteine-rich, TM-transmembrane. Exons are indicated below the protein schematic. (B) Exonic distribution of mutations in 201 patients stratified as having mild (blue) or severe (red) liver disease. Mutations are evenly dispersed across JAG1 and there is no apparent genotype-phenotype correlation by location. Del-gene deletion (C) Mutation type in mild (n=103) and severe (n=73) patients. Both affected populations have a similar distribution of mutation types. Liver disease stratification and data include patients from the GWAS published by Tsai and Gilbert et. al.1
1. Tsai, E.A. et al. THBS2 Is a Candidate Modifier of Liver Disease Severity in Alagille Syndrome. Cell Mol Gastroenterol Hepatol 2, 663–675 e2 (2016).
Although the majority of patients have de novo JAG1 mutations, up to 40% of affected individuals inherit a mutation from a parent ([23, 16] and Spinner et al. unpublished data), many of whom are seemingly unaffected or only mildly affected. The extreme phenotypic variability that is seen among families harboring the same mutation is a hallmark feature of ALGS [26–29]. Variable expressivity has been observed for mutations found in both JAG1 and NOTCH2 [30, 25]. This type of variability has particularly been associated with liver disease (Figure 1B,C), with the clinical manifestations ranging from mild biochemical abnormalities to severe cholestasis, and roughly 15% of ALGS patients will ultimately require a liver transplant [29]. Multiple studies have sought to identify predictive factors that will allow clinicians to distinguish who will likely advance to end-stage liver disease and require a transplant, but these studies have met with varying success [31–33]. Although it was found that liver disease status under 5 years of age was not a stable predictor of long-term disease progression, a new prognostic method, which analyzes serum total bilirubin, liver biopsy fibrosis, and xanthomas, has shown some benefit in helping to predict the likelihood for future liver transplant [31, 32].
Candidate modifiers of liver disease severity
In the absence of an identified environmental factor that could influence disease severity or a single biomarker that can successfully predict liver disease variability, studies have been conducted to question whether a second genetic or epigenetic modifier may affect disease severity and progression. Multiple reports have interrogated the role that post-translational modification of the JAG1 or NOTCH2 proteins may have on modifying the disease phenotype. Of particular interest is the function of glycosyltransferases, which can alter receptor-ligand affinity by facilitating O-fucosylation or O-glycosylation of EGF-like repeats, which are functional motifs that are present in both JAG1 and NOTCH2 proteins [34–36].
Using mouse models, Ryan et. al. showed a role for a family of glycosyltransferases, the Fringe genes, in bile duct proliferation by analyzing mice that were haploinsufficient for both Jag1 and each of 3 related Fringe genes [34]. These mice showed differences in postnatal bile duct growth that suggested a possible modifier role for this gene family in ALGS. Similarly, a separate study investigating the liver-specific glycosyltransferase, Rumi (the mouse homolog of the human gene POGLUT1), showed that haploinsufficiency of both Jag1 and Rumi resulted in an increase of bile duct paucity and a decrease in the total number of biliary cells in the periportal regions [36]. Importantly, since both Jag1 and Rumi heterozygote animals had no phenotype alone in this model (a new heterozygous model of Jag1 does have an ALGS-like phenotype and will be further discussed below [35]), the authors concluded that the liver disease induced by the double heterozygotes showed dosage-sensitivity of Rumi to Jag1, such that when JAG1 is limiting, downstream signaling becomes highly sensitive to Rumi-specific O-glycosylation. Building on this finding, a subsequent study using a more refined Jag1 mouse model of ALGS confirmed a role for Rumi in altering the liver phenotype, but remarkably observed the opposite effect. In this mouse model, decreasing levels of Rumi were found to oppose JAG1, suggesting that a reduction of Rumi was protective of liver function [35]. These observations propose that Rumi functions to glycosylate the extracellular domain of JAG1, which further abrogates JAG1 function. Therapeutically, the liver-specific expression of Rumi offers an advantageous way to target glycosylation events only in the tissue of interest, which would be ideal for the development of potential treatments. Although these studies do not conclusively identify glycosyltransferases as genetic modifiers of ALGS, they do provide evidence warranting future study to accurately pinpoint this post-translational effect on downstream Notch signaling.
Work by our group has also attempted to identify genetic modifiers of liver disease severity in ALGS. We performed a genome-wide association study (GWAS) after stratifying ALGS patients with known JAG1 mutations into 2 groups based on whether they had a mild or severe presentation of liver disease in an effort to uncover a putative risk allele [33]. We identified a candidate locus upstream of the THROMBOSPONDIN 2 (THBS2) gene, which encodes an extracellular matrix protein that we show to be expressed in bile ducts and periportal regions of the mouse liver. We further demonstrated that THBS2 protein both interacts with NOTCH2 and can inhibit JAG1-NOTCH2 binding, supporting a role for THBS2 as a candidate modifier of liver disease severity in patients who have a JAG1 mutation. This research is ongoing, and we are currently pursuing animal models to further define a possible functional role for THBS2 in ALGS.
Functional modeling of Alagille syndrome
Haploinsufficiency
Haploinsufficiency is a disease mechanism that arises when loss of a single allele of a gene results in insufficient protein product to support downstream function. This is distinct from other types of autosomal dominance, such as that induced by a dominant negative mechanism in which the mutant allele inhibits the proper function of the wild type allele. The mechanism by which haploinsufficiency induces a phenotype has been proposed to result from 2 scenarios that are eloquently described by Wilkie (1994) and include: 1) obstruction of a metabolic pathway via loss of an important, rate-limiting intermediary and 2) reduced dosage of a gene that is required to work at a specific threshold [37]. In its first description as a disease caused by mutations in JAG1, ALGS was hypothesized to result from haploinsufficiency arising from a reduction in gene dosage based on the observation that patients with gene deletions and patients with frameshift or missense mutations share a clinically-similar disease presentation [14]. This hypothesis has been subsequently supported by studies showing that missense mutations in JAG1 result in improper trafficking of the protein to the cell surface, and thus a reduction in gene dosage rather than aberrant expression of a mutant protein [17–20].
With a well-defined genetic etiology and disease mechanism of haploinsufficiency, models of ALGS are vital to advance scientific understanding of this disease towards clinical treatments. Below, we aim to provide a broad overview on the current state of both animal and in vitro cellular models of ALGS. We have referenced multiple recent reviews that elaborate many of these studies in more detail than we are able to here, and we encourage you to refer to these for additional information.
Animal models
The first mouse model of Jag1 was introduced only two years after mutations affecting Notch signaling were established as the underlying cause of Alagille syndrome [38]. As would be expected given the developmental importance of Notch signaling in multiple organ systems, Jag1 homozygous null mice died by embryonic day 10, as a consequence of vascular defects [38]. Disappointingly, the heterozygous Jag1 loss of function mice described by Xue et. al. did not exhibit any of the ALGS-associated phenotypes seen in human disease other than an eye dysmorphology [38]. However, when these heterozygous Jag1 null mice were crossed with mice that expressed one wild type and one hypomorphic Notch2 allele, the resulting progeny displayed phenotypic characteristics that are strongly associated with ALGS, including intrahepatic bile duct paucity, severe cardiac defects, and renal disease [39]. Although promising, the liver phenotype in these mice is more severe than is seen in human ALGS patients, with bile duct paucity arising at birth, and other features of ALGS are absent, including vertebral and craniofacial abnormalities. Moreover, these mice require mutations in both Jag1 and Notch2 to produce a phenotype, whereas the human disease only requires a single mutation in a single gene.
In the 2 decades that have ensued since these seminal papers, multiple groups have continued the quest to establish a mouse model of ALGS. These studies have included the generation of conditional knockout animals that have extended our understanding of early liver development and elucidated the specific cellular context that underlies the hepatic phenotype in ALGS, namely that JAG1 expression in the portal vein mesenchyme, but not in hepatoblasts, is required to avoid the associated liver disease [40, 41]. A comprehensive review of these and other fundamental Jag1 and Notch2 mouse models that have contributed to our understanding of ALGS was published in 2015 and will not be further discussed in this report [42].
More recently, and perhaps most excitingly, has been the first publication of a heterozygous Jag1 null mouse that appears to mimic the human disease and thus represent the first haploinsufficient mouse model of ALGS [35, 43]. This was accomplished by repeatedly backcrossing the Jag1 null mouse on a C57BL/6 background to eliminate genetic heterogeneity. Cytokeratin staining in livers of the resultant heterozygous animals show less organization of the ductal plate in late embryogenesis, and bile duct paucity at birth in concordance with a reduction in biliary epithelial cells and vascular smooth muscle cells perinatally [35]. An essential concept of haploinsufficiency is a dependency on gene dosage to effect phenotype. Thakurdas et. al. took their ALGS model a step further and eliminated one copy of the Rumi gene which, as discussed above, functions to post-translationally modify JAG1 by adding O-glucose to JAG1 EGF repeats, a process that has been previously identified to affect Notch signaling [36, 35]. Mice heterozygous for both Jag1 and Rumi were found to have reduced bile duct paucity, suggesting that loss of Rumi helps to rescue the hepatic phenotype in the Jag1 heterozygous animals [35]. Collectively, this report not only characterizes a heterozygous model of Jag1 that recapitulates the human ALGS phenotype, but also shows that the severity of the liver defect in these animals can be ameliorated by abrogating JAG1 glycosylation through a reduction in Rumi, providing a model that truly mimics the haploinsufficiency seen with the human disease.
In addition to mice, zebrafish have also proven valuable as animal models of ALGS. Due to gene duplication events, zebrafish express 3 different jagged genes, and work has shown that cooperative knockdown by morpholino injection targeting the jagged1b and jagged2 genes results in a highly disrupted intrahepatic biliary system with extreme bile duct paucity that phenocopies ALGS [44]. Importantly, Lorent et. al. were able to show that the severity of liver disease exhibited dose-dependency, and that the phenotype could be rescued by injection of full-length human JAG1 mRNA, again supporting a disease mechanism of haploinsufficiency of Jagged protein [44].
Induced pluripotent stem cell (iPSC) models
A prerequisite to the identification of effective therapeutics for any disease is the development of an in vitro assay to study patient-derived tissue samples. Differentiated somatic cells pose long-term culturing problems due to their inherent low proliferative capacity. Conditions have recently been described to achieve long-term culture of bi-potent liver progenitor cells acquired from liver tissue, which form organoids that are genetically-stable in vitro and appropriately express biliary markers [45]. Even more recently, a cocktail of small molecules has been published that have the potential to convert mature hepatocytes into bipotent chemically-induced liver progenitor cells with the capacity to differentiate into both mature hepatocytes and biliary epithelial cells [46]. Indeed, Katsuda et. al. were able to show that these cells are able to repopulate chronically-injured liver tissue [46]. Thus far, these experiments have been done only in rat and mouse, and it remains to be seen whether human hepatocytes are chemically-susceptible to this conversion. Although promising, the pathophysiological importance of these hepatic cell models is dampened slightly by both their static nature–they inherently allow only for the study of events downstream of differentiation and not key developmental time points that may be vital to disease initiation–and the inaccessible location of the liver, which requires invasive techniques for sample acquisition.
Since the discovery of the Yamanaka factors and their ability to induce pluripotency when overexpressed in human somatic cells in 2006, the emergent field of iPSC biology has become instrumental in facilitating advances in disease therapies [47, 48]. At the epicenter of iPSC clinical endeavors are hepatic cells since they offer a preclinical method for performing pharmacokinetics and toxicology studies. A review on modeling liver disease by iPSCs has recently been published and thoroughly describes the successes and future of hepatic iPSC models by focusing on 4 groups who have each made independent advances in modeling techniques, which will be briefly mentioned here [49].
Original reports characterizing protocols for the generation of cholangiocytes from human embryonic stem cells, iPSCs, and hepatoblasts (HepaRG cells) showed early success in obtaining these cells in monolayer [50, 51]. Notably, studies showed that these cells were functionally similar to cholangiocytes, could respond to calcium stimulation, and also were capable of forming duct-like structures and primary cilia [50, 51]. To build on this work, subsequent methods were focused on creating even more physiologically-relevant systems that use 3D culture conditions resulting in cells that exhibit fluid secretion and have been used to model ALGS [52, 53]. Of particular interest is the work done by Ogawa et. al., which capitalizes on the importance of Notch signaling in the developing liver via a protocol that requires co-culture of iPSCs with OP9 stromal cells, a cell type that expresses JAG1 [52]. The utilization of these cells provides an intriguing context within which to study the way that various JAG1 or NOTCH2 mutations impact cholangiocyte development, and it will be exciting to see the work that derives from those types of experiments. Cumulatively, the work described in the past 5 years by these groups certainly define advanced protocols that provide relevant cell types and assays that can be used to interrogate the behavior of these cells to therapeutic manipulation [49]
Clinical applications: towards a treatment
As mentioned in the introduction, current treatments for the liver phenotype of ALGS include medication to treat the sequelae of poor liver function (including pruritus) as well as more invasive options, such as liver transplant, for patients presenting with more severe forms of cholestatic disease. The fundamental importance of the Notch pathway throughout development and in the pathogenesis of many different diseases has already instigated the study of therapies that target and modify Notch signaling [54]. Here, we theorize on the future of ALGS therapeutics that could be envisioned given the current state of functional disease models and cell-based technologies.
Potential for iPSC therapies: transplantation
The emergence of in vitro models of iPSC-derived cholangiocytes provides an exciting and promising new direction towards ALGS therapeutics. At its core, ALGS is a cholangiopathy, and even patients who do not require a liver transplant are burdened with impaired liver function. iPSC technology provides a unique mechanism whereby a patient’s own cells can be corrected and reintroduced into a diseased liver, a process that eliminates the risk of rejection and immunosuppression that is frequently seen with liver transplantation. Investigations aimed to reduce barriers to this type of therapy, which include low engraftment frequency and long-term engraftment potential, have already shown promise [52, 51]. In describing their iPSC model, De Assuncao et. al. additionally showed that their differentiated human cholangiocytes were able to engraft in portal tracts of immune competent mice after retrograde intra-biliary infusion, and that these cells formed de novo duct-like structures by 3 days post engraftment [51]. Similarly, Ogawa et. al. also assayed the engraftment capacity of their differentiated cholangiocytes by implanting them with Matrigel into mammary fat pads of immunocompromised mice and assessed functionality by showing that these cells were capable of transporting a dye, rhodamine 123, into the lumen [52]. Although still in their early stages, these studies are beginning to investigate the feasibility of iPSC transplantation in the liver.
Equally important as the ability of iPSCs to engraft into liver tissue is their ability to introduce a genetically-corrected, healthy liver cell. At the current state of research, this concept is primarily hypothetical for ALGS, although a variety of possibilities to achieve this result can be postulated. Given a disease mechanism of haploinsufficiency, it can be hypothesized that increasing the expression levels of JAG1 in the diseased liver may overcome this deficiency by either 1) rescuing the mutant allele, or 2) overexpressing the wild type allele. As mentioned above, animal studies have shown that JAG1 expression is dispensable in hepatocytes and biliary epithelial cells, but necessary in the portal vein mesenchyme [40, 41], thus cell type-specificity is an important consideration when visualizing these types of experiments.
The discovery of clustered regularly interspaced short palindromic repeats (CRISPR)-mediated systems for gene expression modulation has been paradigm-shifting for the field of gene therapy [55–59]. Briefly, this 2-component system involves a guide RNA that site-specifically targets a gene of interest, a CRISPR, and a Cas9 enzyme that associates with a CRISPR and is able to nick DNA [59]. The result is that DNA replication can be rendered inactive via the nick, or that a new sequence can be inserted through homologous recombination with a CRISPR sequence. Gene editing by homologous recombination through either CRISPR- or zinc-finger nuclease-based systems provide a mechanism to site-specifically correct a mutation in vitro and repair both nonsense- and missense-mutant alleles [55–58]. Success using this type of technology in iPSCs has been seen with CFTR, the gene that is defective in cystic fibrosis [60]. Cultured iPSCs with defective CFTR have been shown to have restored function as assayed by reestablishment of chloride channel transport after zinc-finger nuclease-mediated editing of a mutated locus [60]. Although promising, these technologies are limited specifically to missense- and/or nonsense-mutant alleles (42% of reported JAG1 mutations). This technology would not be applicable to patients with full gene deletions (11%), frameshift mutations caused by insertions and deletions (36%) or splice site mutations (11%) (Spinner lab, unpublished data).
To circumvent this issue, upregulation of the wild type allele is an auspicious alternative. A modification of the CRISPR system allows for the CAS9 enzyme to be rendered inactive and tagged with transcriptional activators, so that rather than catalytically producing a nick, it becomes rewired to promote gene expression at locations that are specifically recognized by the associated CRISPR [56, 61, 58, 57]. Modification of this system, and other types of RNA activating technologies has resulted in similar techniques with varying advantages and disadvantages. For instance, Fimiani et. al. designed a synthetic ribonucleoprotein transcription factor that shows the same specificity as CRISPR systems, but with the advantage of a smaller size, which they suggest may help with delivery [62]. It is possible to envision utilizing these types of technologies in differentiated cholangiocytes from patient-derived iPSCs to study potential therapeutic options of upregulating the endogenous, wild type JAG1 allele. As techniques for generating these types of cells are already being optimized, it is likely that many of these experiments may already be underway.
Small molecule screening
Although genetic engineering and transplantation of iPSCs is an obvious therapeutic option for ALGS, iPSCs are also excellent model systems for candidate drug screening. The ability to tissue-specifically differentiate these cells provides an optimal method to screen small molecule libraries in the unique setting of individual patient samples with various genetic backgrounds, including different JAG1 or NOTCH2 mutations. These types of studies are currently ongoing for a variety of other diseases. Frontotemporal lobar degeneration occurs via haploinsufficiency of the GRN gene, which encodes the protein progranulin (PGRN), and a recent study to screen a library of small molecules identified a naturally-occurring disaccharide, trehalose, as a molecule capable of increasing endogenous PGRN levels [63]. This group confirmed their findings in both an animal model of the disease and in iPSCs that had innate GRN mutations. Similarly, Ogawa et. al., whose work on optimizing iPSC culture conditions for cholangiocytes was previously introduced, tested the ability of their system to function in drug-screening by using cells derived from patients with mutations in the gene CFTR, which is impaired in cystic fibrosis [52]. Here, they showed that a cocktail of molecules hypothesized to show efficacy as a possible therapy were in fact able to restore function using a fluid transport assay. Finally, Sampaziotis et. al., showed that in their hepatic iPSC model, they could block cleavage of NOTCH2 through the use of a known gamma-secretase inhibitor, and prevent downstream signaling, both confirming the physiologic relevancy of their model and setting the stage for screening of novel compounds [53]. The recent description of a haploinsufficient JAG1 mouse further provides a way to take these types of in vitro findings and test them directly in an animal model [35]. Although still in its early stages, the concept of utilizing small molecules to modulate JAG1-NOTCH2 signaling is no longer in its infancy, and the existence of physiologically-relevant in vitro systems in which to study these therapies provides an ideal model for the maturation of novel treatment modalities for ALGS.
Conclusions
In the 20 years since ALGS was first recognized as a haploinsufficient disease caused by impaired Notch signaling, the fields of genetics and biology have harmonized to produce a strong understanding of disease etiology. Although questions still remain regarding phenotypic disparities between patients, progress with animal and in vitro modeling and advances in biotechnology have provided scientists with resources to tackle these lingering problems. It is our hope that the possibilities offered by these scientific advances will lead to exciting, novel treatment therapies for patients with ALGS.
Acknowledgments
We would like to thank Ellen Tsai, Kathy Loomes, Marcella Devoto, Ian Krantz, David Piccoli, and Binita Kamath for continued collaboration. We are grateful to the many people who have contributed to our understanding of the genetics of Alagille syndrome including Ian Krantz, Ray Colliton, Jennifer Morrissette, Dan Warthen, Ryan McDaniell, Rob Bauer, Laura Leonard, Christopher Grochowski, and Ramakrishnan Rajagopalan. Parts of this work were supported by R01DK081702.
Footnotes
Conflicts of Interest: The authors disclose no conflicts
Human and Animal Rights: All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet. 2016;9:75–82. doi: 10.2147/TACG.S86420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spinner NB, Leonard LD, Krantz ID. Alagille Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al., editors. GeneReviews(R) Seattle (WA): 2013. [Google Scholar]
- 3.Kamath BM, Podkameni G, Hutchinson AL, Leonard LD, Gerfen J, Krantz ID, et al. Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A. 2012;158A(1):85–9. doi: 10.1002/ajmg.a.34369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251–7. doi: 10.1038/ejhg.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thebaut A, Habes D, Gottrand F, Rivet C, Cohen J, Debray D, et al. Sertraline as an Additional Treatment for Cholestatic Pruritus in Children. J Pediatr Gastroenterol Nutr. 2016 doi: 10.1097/MPG.0000000000001385. [DOI] [PubMed] [Google Scholar]
- 6.Emerick KM, Whitington PF. Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. Hepatology. 2002;35(6):1501–6. doi: 10.1053/jhep.2002.33332. [DOI] [PubMed] [Google Scholar]
- 7.Mattei P, von Allmen D, Piccoli D, Rand E. Relief of intractable pruritus in Alagille syndrome by partial external biliary diversion. J Pediatr Surg. 2006;41(1):104–7. doi: 10.1016/j.jpedsurg.2005.10.014. discussion -7. [DOI] [PubMed] [Google Scholar]
- 8.Pawlowska J, Socha P, Jankowska I. Factors affecting catch-up growth after liver transplantation in children with cholestatic liver diseases. Ann Transplant. 2010;15(1):72–6. [PubMed] [Google Scholar]
- 9.Grochowski CM, Loomes KM, Spinner NB. Jagged1 (JAG1): Structure, expression, and disease associations. Gene. 2016;576(1 Pt 3):381–4. doi: 10.1016/j.gene.2015.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. Semin Cell Dev Biol. 2012;23(4):450–7. doi: 10.1016/j.semcdb.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17(11):722–35. doi: 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- 12.Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet. 1997;34(2):152–7. doi: 10.1136/jmg.34.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16(3):243–51. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 14.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16(3):235–42. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 15.McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79(1):169–73. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in alagille syndrome. Hum Mutat. 2001;17(1):18–33. doi: 10.1002/1098-1004(2001)17:1<18::AID-HUMU3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 17.Bauer RC, Laney AO, Smith R, Gerfen J, Morrissette JJ, Woyciechowski S, et al. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum Mutat. 2010;31(5):594–601. doi: 10.1002/humu.21231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu F, Morrissette JJ, Spinner NB. Conditional JAG1 mutation shows the developing heart is more sensitive than developing liver to JAG1 dosage. Am J Hum Genet. 2003;72(4):1065–70. doi: 10.1086/374386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrissette JD, Colliton RP, Spinner NB. Defective intracellular transport and processing of JAG1 missense mutations in Alagille syndrome. Hum Mol Genet. 2001;10(4):405–13. doi: 10.1093/hmg/10.4.405. [DOI] [PubMed] [Google Scholar]
- 20.Tada M, Itoh S, Ishii-Watabe A, Suzuki T, Kawasaki N. Functional analysis of the Notch ligand Jagged1 missense mutant proteins underlying Alagille syndrome. FEBS J. 2012;279(12):2096–107. doi: 10.1111/j.1742-4658.2012.08595.x. [DOI] [PubMed] [Google Scholar]
- 21.Boyer-Di Ponio J, Wright-Crosnier C, Groyer-Picard MT, Driancourt C, Beau I, Hadchouel M, et al. Biological function of mutant forms of JAGGED1 proteins in Alagille syndrome: inhibitory effect on Notch signaling. Hum Mol Genet. 2007;16(22):2683–92. doi: 10.1093/hmg/ddm222. [DOI] [PubMed] [Google Scholar]
- 22.Crosnier C, Driancourt C, Raynaud N, Dhorne-Pollet S, Pollet N, Bernard O, et al. Mutations in JAGGED1 gene are predominantly sporadic in Alagille syndrome. Gastroenterology. 1999;116(5):1141–8. doi: 10.1016/s0016-5085(99)70017-x. [DOI] [PubMed] [Google Scholar]
- 23.Krantz ID, Colliton RP, Genin A, Rand EB, Li L, Piccoli DA, et al. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet. 1998;62(6):1361–9. doi: 10.1086/301875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McElhinney DB, Krantz ID, Bason L, Piccoli DA, Emerick KM, Spinner NB, et al. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106(20):2567–74. doi: 10.1161/01.cir.0000037221.45902.69. [DOI] [PubMed] [Google Scholar]
- 25.Kamath BM, Bauer RC, Loomes KM, Chao G, Gerfen J, Hutchinson A, et al. NOTCH2 mutations in Alagille syndrome. J Med Genet. 2012;49(2):138–44. doi: 10.1136/jmedgenet-2011-100544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dhorne-Pollet S, Deleuze JF, Hadchouel M, Bonaiti-Pellie C. Segregation analysis of Alagille syndrome. J Med Genet. 1994;31(6):453–7. doi: 10.1136/jmg.31.6.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elmslie FV, Vivian AJ, Gardiner H, Hall C, Mowat AP, Winter RM. Alagille syndrome: family studies. J Med Genet. 1995;32(4):264–8. doi: 10.1136/jmg.32.4.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shulman SA, Hyams JS, Gunta R, Greenstein RM, Cassidy SB. Arteriohepatic dysplasia (Alagille syndrome): extreme variability among affected family members. Am J Med Genet. 1984;19(2):325–32. doi: 10.1002/ajmg.1320190215. [DOI] [PubMed] [Google Scholar]
- 29.Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29(3):822–9. doi: 10.1002/hep.510290331. [DOI] [PubMed] [Google Scholar]
- 30.Guegan K, Stals K, Day M, Turnpenny P, Ellard S. JAG1 mutations are found in approximately one third of patients presenting with only one or two clinical features of Alagille syndrome. Clin Genet. 2012;82(1):33–40. doi: 10.1111/j.1399-0004.2011.01749.x. [DOI] [PubMed] [Google Scholar]
- 31.Kamath BM, Munoz PS, Bab N, Baker A, Chen Z, Spinner NB, et al. A longitudinal study to identify laboratory predictors of liver disease outcome in Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;50(5):526–30. doi: 10.1097/MPG.0b013e3181cea48d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mouzaki M, Bass LM, Sokol RJ, Piccoli DA, Quammie C, Loomes KM, et al. Early life predictive markers of liver disease outcome in an International, Multicentre Cohort of children with Alagille syndrome. Liver Int. 2016;36(5):755–60. doi: 10.1111/liv.12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai EA, Gilbert MA, Grochowski CM, Underkoffler LA, Meng H, Zhang X, et al. THBS2 Is a Candidate Modifier of Liver Disease Severity in Alagille Syndrome. Cell Mol Gastroenterol Hepatol. 2016;2(5):663–75 e2. doi: 10.1016/j.jcmgh.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryan MJ, Bales C, Nelson A, Gonzalez DM, Underkoffler L, Segalov M, et al. Bile duct proliferation in Jag1/fringe heterozygous mice identifies candidate modifiers of the Alagille syndrome hepatic phenotype. Hepatology. 2008;48(6):1989–97. doi: 10.1002/hep.22538. [DOI] [PubMed] [Google Scholar]
- 35•.Thakurdas SM, Lopez MF, Kakuda S, Fernandez-Valdivia R, Zarrin-Khameh N, Haltiwanger RS, et al. Jagged1 heterozygosity in mice results in a congenital cholangiopathy which is reversed by concomitant deletion of one copy of Poglut1 (Rumi) Hepatology. 2016;63(2):550–65. doi: 10.1002/hep.28024. First description of a haploinsufficient mouse model of Jag1 that mimics human ALGS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandez-Valdivia R, Takeuchi H, Samarghandi A, Lopez M, Leonardi J, Haltiwanger RS, et al. Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development. 2011;138(10):1925–34. doi: 10.1242/dev.060020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilkie AO. The molecular basis of genetic dominance. J Med Genet. 1994;31(2):89–98. doi: 10.1136/jmg.31.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, et al. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8(5):723–30. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- 39.McCright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development. 2002;129(4):1075–82. doi: 10.1242/dev.129.4.1075. [DOI] [PubMed] [Google Scholar]
- 40.Loomes KM, Russo P, Ryan M, Nelson A, Underkoffler L, Glover C, et al. Bile duct proliferation in liver-specific Jag1 conditional knockout mice: effects of gene dosage. Hepatology. 2007;45(2):323–30. doi: 10.1002/hep.21460. [DOI] [PubMed] [Google Scholar]
- 41.Hofmann JJ, Zovein AC, Koh H, Radtke F, Weinmaster G, Iruela-Arispe ML. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Development. 2010;137(23):4061–72. doi: 10.1242/dev.052118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geisler F, Strazzabosco M. Emerging roles of Notch signaling in liver disease. Hepatology. 2015;61(1):382–92. doi: 10.1002/hep.27268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huppert SS. A faithful JAGGED1 haploinsufficiency mouse model of arteriohepatic dysplasia (Alagille syndrome) after all. Hepatology. 2016;63(2):365–7. doi: 10.1002/hep.28338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lorent K, Yeo SY, Oda T, Chandrasekharappa S, Chitnis A, Matthews RP, et al. Inhibition of Jagged-mediated Notch signaling disrupts zebrafish biliary development and generates multi-organ defects compatible with an Alagille syndrome phenocopy. Development. 2004;131(22):5753–66. doi: 10.1242/dev.01411. [DOI] [PubMed] [Google Scholar]
- 45.Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160(1–2):299–312. doi: 10.1016/j.cell.2014.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katsuda T, Kawamata M, Hagiwara K, Takahashi RU, Yamamoto Y, Camargo FD, et al. Conversion of Terminally Committed Hepatocytes to Culturable Bipotent Progenitor Cells with Regenerative Capacity. Cell Stem Cell. 2017;20(1):41–55. doi: 10.1016/j.stem.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 48.Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115–30. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49•.Ghanekar A, Kamath BM. Cholangiocytes derived from induced pluripotent stem cells for disease modeling. Curr Opin Gastroenterol. 2016;32(3):210–5. doi: 10.1097/MOG.0000000000000260. Thorough review on recent iPSC culture models within the past 5 years. [DOI] [PubMed] [Google Scholar]
- 50.Dianat N, Dubois-Pot-Schneider H, Steichen C, Desterke C, Leclerc P, Raveux A, et al. Generation of functional cholangiocyte-like cells from human pluripotent stem cells and HepaRG cells. Hepatology. 2014;60(2):700–14. doi: 10.1002/hep.27165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Assuncao TM, Sun Y, Jalan-Sakrikar N, Drinane MC, Huang BQ, Li Y, et al. Development and characterization of human-induced pluripotent stem cell-derived cholangiocytes. Lab Invest. 2015;95(10):1218. doi: 10.1038/labinvest.2015.99. [DOI] [PubMed] [Google Scholar]
- 52•.Ogawa M, Ogawa S, Bear CE, Ahmadi S, Chin S, Li B, et al. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat Biotechnol. 2015;33(8):853–61. doi: 10.1038/nbt.3294. Important work describing a Notch-dependent 3D culture system for deriving cholangiocytes from iPSCs. [DOI] [PubMed] [Google Scholar]
- 53.Sampaziotis F, Cardoso de Brito M, Madrigal P, Bertero A, Saeb-Parsy K, Soares FA, et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat Biotechnol. 2015;33(8):845–52. doi: 10.1038/nbt.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov. 2014;13(5):357–78. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 55.Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23(10):1163–71. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–51. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10(10):977–9. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10(10):973–6. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crane AM, Kramer P, Bui JH, Chung WJ, Li XS, Gonzalez-Garay ML, et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports. 2015;4(4):569–77. doi: 10.1016/j.stemcr.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173–83. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fimiani C, Goina E, Mallamaci A. Upregulating endogenous genes by an RNA-programmable artificial transactivator. Nucleic Acids Res. 2015;43(16):7850–64. doi: 10.1093/nar/gkv682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holler CJ, Taylor G, McEachin ZT, Deng Q, Watkins WJ, Hudson K, et al. Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: a novel therapeutic lead to treat frontotemporal dementia. Mol Neurodegener. 2016;11(1):46. doi: 10.1186/s13024-016-0114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]