

ABSTRACT
Liver cancer is the sixth most prevalent cancer, and the third most frequent cause of cancer-related deaths. Circular RNAs (circRNAs), a kind of special endogenous ncRNAs, have been coming back to the forefront of cancer genomics research. In this study, we used a systems biology approach to construct and analyze the circRNA molecular regulatory networks in the context of liver cancer. We detected a total of 127 differentially expressed circRNAs and 3,235 differentially expressed mRNAs. We selected the top-5 upregulated circRNAs to construct a circRNA-miRNA-mRNA network. We enriched the pathways and gene ontology items and determined their participation in cancer-related pathways such as p53 signaling pathway and pathways involved in angiogenesis and cell cycle. Quantitative real-time PCR was performed to verify the top-five circRNAs. ROC analysis showed circZFR, circFUT8, circIPO11 could significantly distinguish the cancer samples, with an AUC of 0.7069, 0.7575, and 0.7103, respectively. Our results suggest the circRNA-miRNA-mRNA network may help us further understand the molecular mechanisms of tumor progression in liver cancer, and reveal novel biomarkers and therapeutic targets.
KEYWORDS: CircRNAs, circRNA-miRNA-mRNA, expression profile, liver cancer
Introduction
Liver cancer is the sixth most prevalent cancer and the third most frequent cause of cancer-related deaths.1-4 More than 700,000 cases of liver cancer were diagnosed in 2008.5 According to World Health Organization (WHO), liver cancer accounted for 9% of all cancer-related deaths worldwide in 2012.6 Although the survival rate of patients with liver cancer has improved, most patients who undergo surgical resection experience metatases and recurrence within 5 years.7-9 It is thus necessary to identify molecular targets and novel pathways that underlie tumorigenesis and progression of liver cancer.
Circular RNAs (circRNAs), are kinds of special endogenous ncRNAs that are predominantly generated by a process called ‘back-splicing’, followed by formation of covalently-closed continuous loops.10-13 Thanks to the technological breakthroughs in high-throughput deep sequencing, single-stranded circular RNAs are no longer perceived as splicing errors and are gradually assumed as the center stage in cancer genomics research.14 Because of the lack of free ends, circRNAs are resistant to exonucleases, which help these circRNAs escape the normal RNA turnover. These features confer numerous potential functions to circRNAs, such as transcription regulators15 or as competing endogenous RNAs to bind miRNAs (RNA sponges) or as RNA binding proteins (protein sponges), which helps these circRNAs modulate their local free concentration.16-20 These findings indicate the potential regulatory role of circRNAs in biologic development, and in pathogenesis and progression of diseases. Recent research has revealed aberrant expression of several circRNAs in colorectal cancer (CRC),21-23 gastric cancer (GC)24-26 and Alzheimer disease (AD);27,28 however, the expression profiles of circRNAs in liver cancer tissues are not well characterized.29-31 In the present study, we identified the differential expression profile of mRNA and circRNA in 3 pairs of liver cancer clinical specimens with use of circRNA and mRNA microarray detection. We detected a total of 127 circRNAs (DE-circRNAs) and 3235 mRNAs (DE-mRNAs) with significant differential expression. Further, we constructed a ‘cancer-related circular RNA molecular regulatory network’. We not only delineated the associated pathways and gene ontology items, but also performed a comprehensive functional network analysis. Our analysis revealed that 5 circRNAs could participate in pathways involved in cancer, the p53 signaling pathway, and in pathways involved in angiogenesis and cell cycle. The significant differential expressions of representative circRNAs were further confirmed using qRT-RCR.
Results
circRNA and mRNA expression profiles and coding genes-mediated pathway and GO analysis
We conducted Arraystar Human circRNA Array and Arraystar Human mRNA Array to analyze the circRNA and mRNA profiles in 3 pairs of liver cancer clinical tissues. We detected 127 (113 upregulated, 14 downregulated) circRNAs (DE-circRNAs) and 3,235 (1923 upregulated, 1312 downregulated) mRNAs (DE-mRNAs) with significant differential expressions (over 2-fold change; Fig. 1A and 1B, Table 1). Among these circRNAs, upregulated circRNAs were more common than downregulated circRNAs. The distribution of the circRNAs on the human chromosomes is depicted in Fig. 1C.
Figure 1.
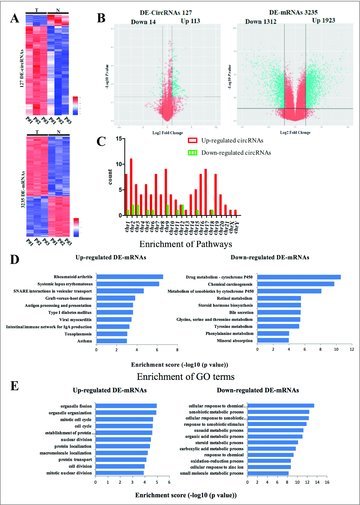
Differentially expressed circRNAs and mRNAs in 3 pairs of liver cancer patient tissues. (A) Heat maps of aberrantly expressed circRNAs and mRNAs. A total of 127 circRNAs and 3235 mRNAs showed significantly aberrant expression (≥ 2-fold change); P < 0.05 and FDR < 0.05. Each column represents one sample, and each row indicates a transcript. T: tumor tissues. N: normal tissues. (B) Volcano plots of aberrantly expressed circRNAs and mRNAs. The vertical green lines correspond to 2-fold upregulation and downregulation, respectively; the horizontal green line indicates a P-value of 0.05. The red points in the plots represent the significantly differentially expressed genes(C) Enrichment analysis of pathways term for DE-mRNAs. Pathway analysis was predominantly based on the KEGG database. The vertical axis represents the pathway category and the horizontal axis represents the -log10 (p value) of these significant pathways. (D) Enrichment analysis of GO term for DE-mRNAs. The vertical axis represents the biologic procession (BP) term and the horizontal axis represents the -log10 (p value) of these significant GO-BP term. (E) The distribution of differentially expressed circRNAs in human chromosomes.
We used KEGG pathway to enrich the function of 3235 DE-mRNAs [Fig. 1D]. We found that the upregulated mRNAs were enriched in immune system such as systemic lupus erythematosus, intestinal immune network for IgA production, while the downregulated mRNAs enrich in various metabolism. We also performed Gene Ontology analysis for the DE-mRNAs involved in biologic processes [Fig. 1E]. The upregulated genes are involved in organelle fission, cell cycle and cell division, while downregulated genes are involved in response to stimulus and metabolic process.
Construction of the circRNA-miRNA-mRNA interaction network
All the differentially expressed circRNAs were predicted according to the complementary miRNA matching sequence. A total of 369 miRNAs could be combined with circRNAs. An entire network of circRNA-miRNA interaction was delineated using Cytoscape [Fig. 2A]. The data displayed each circRNA and its potential complementary binding miRNAs [Supplementary Fig. 1].
Figure 2.
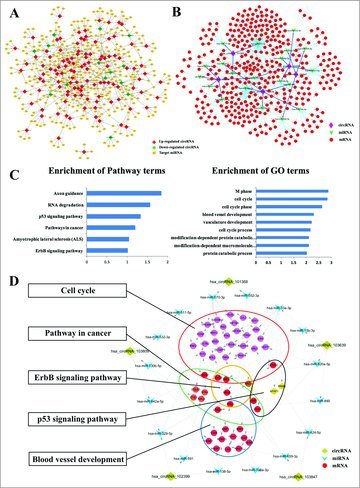
Construction of the circRNAs-miRNAs-mRNAs ceRNA regulatory networks. (A) The entire network of circRNA-miRNA interaction entire network of circRNA-miRNA interaction. (B) The predicted top-5 circRNAs targeted circRNA-miRNA-mRNA network based on sequence-pairing prediction. (C) Enrichment analysis of pathways term and GO term for mRNAs. (D) The key function module circRNA-miRNA-mRNA network.
To study the significant function of circRNAs, we choose top-5 upregulated circRNAs as the research objects to construct circRNA-miRNA-mRNA network. The microRNA/mRNA interaction was predicted with the miRNA database and then compared with the DE-mRNAs. An entire network of circRNA-miRNA-mRNAs interaction was delineated using Cytoscape [Fig. 2B].
We performed the co-expressed mRNAs associated pathways and gene ontology items enrichment analysis [Fig. 2C]. We found that the mRNAs participated in cancer-related pathways, such as the p53 signaling pathway, and pathways involved in angiogenesis and cell cycle. We chose these nodes to construct the key function module [Fig. 2D].
Validation for the differential expression level of circRNAs
To verify the accuracy of the prediction results, we determined the expression levels of the top-5 circRNAs in 40 paired clinical samples of liver cancer tissues and their paired adjacent normal liver tissues by qRT-PCR (Table 2). β-actin was used as the internal standard. We found that the expression levels of circZFR, circFUT8 and circIPO11 in liver cancer tissues were significantly higher than those in the corresponding non-tumorous tissues [Fig. 3]. The findings were consistent with those of microarray analysis.
Figure 3.
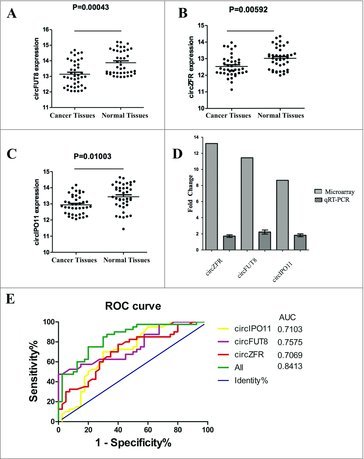
Validation for the expression of significant transcripts by quantitative RT-PCR and ROC analysis of the circRNAs. (A) The expression levels of circZFR are significantly higher than those in corresponding nontumorous tissues. P < 0.001. (B) The expression levels of circFUT8 are significantly higher than those in corresponding nontumorous tissues. P < 0.01. (C) The expression levels of circIPO11 are significantly higher than those in corresponding nontumorous tissues. P < 0.05. (D) The fold change of the circZFR, circFUT8 and circIPO11 expression levels by microarray and qRT-PCR. (E) ROC curve analysis of circZFR, circFUT8 and circIPO11 for discriminative ability between liver cancer cases and normal controls.
Representation of the data using an ROC plot showed circZFR, circFUT8, circIPO11 could significantly distinguish the cancer samples, with an AUC of 0.7069, 0.7575, and 0.7103, respectively (Fig. 3E)
Discussion
Thanks to the technological breakthroughs in high-throughput deep sequencing, single-stranded circular RNAs are no longer perceived as splicing errors and have evoked a renewed interest in cancer genomics research.14 Recent studies have shown that circRNAs may modulate cancer progression. Further, aberrant expression of circRNAs has been linked to the malignant cancer phenotypes.
To probe the molecular regulatory mechanisms of liver cancer, we explored the genome-wide expression profiles of circRNAs in 3 liver cancer specimens and their matched adjacent normal liver tissues using microarray. We identified 127 DE-circRNAs in liver cancer tissues. In the expression signatures, more than 15 circRNAs were upregulated (> 5-fold change), which suggests a role of these DE-circRNAs in the pathogenesis and progression of liver cancer. In particular, circFUT8 (hsa_circRNA_101368, hsa_circ_0003028), circZFR (hsa_circRNA_103809, hsa_circ_10072088), and circIPO11 (hsa_circRNA_103847, hsa_circ_0007915) were confirmed to be significantly dysregulated in liver cancer tissues by qRT-PCR.
A recent study identified 400 circRNAs in human cell free saliva.25 The study showed that hundreds of circRNAs had higher expressions than those of homologous linear transcripts in blood.32 Therefore, circRNAs could embody disease information that cannot be determined by conventional RNA assay, which suggests that the circRNAs are promising biomarkers for clinical application in the future.32 Our findings indicate that circFUT8, circZFR, circIPO11 represent potentially valuable diagnostic biomarkers of liver cancer. However, most differentially expressed circRNAs have not been studied yet.
Recent evidences have demonstrated that by sequestering the miRNAs, circular RNAs play a crucial role in fine tuning the miRNA-mediated regulation of gene expression. It is proposed that most targeted mRNAs would be completely suppressed when the expression of miRNAs vastly exceeds that of the ceRNAs. On the contrary, when the quantity of ceRNAs surpasses that of their miRNAs, the competition would reduce due to the limited availability of miRNAs.33,34 A wide body of evidence suggests a significant role of dysregulation of miRNA expression in the development of various human cancers.35,36 We constructed an entire network of circRNA-miRNA interaction. A total of 369 miRNAs could be combined with circRNAs.
Recent studies have shown that some diseases are related with miRNAs, which indicates that circular RNAs are important for disease regulation.37 For example, the circular RNA ciRS-7/CRD1as contains multiple, tandem miR-7 binding sites, and, thereby, acts as an endogenous miRNA “sponge” to adsorb, and hence quench, normal miR-7 functions.27 It has been demonstrated that miR-7 involves numerous pathways, and that miR-7 could directly down-regulate several oncogenes including EGFR, Raf1, Pak1, Ack1, IGF1R, PIK3CD and mTOR.38
Therefore, we speculated that circFUT8 might competitively bind with miR-570–3p, miR-17–3p, and miR-552–3p, and abrogate the inhibitory effect on the associated target genes. miR-570–3p is related to disease stage and colorectal cancer-specific mortality,39 miR-17–3p could synergistically induce the development of hepatocellular carcinoma40 and miR-552–3p were effective in inhibiting cell growth.41
The circRNA circZFR identified in this study was found to potentially interact with miR-511–5p, miR-130b-5p, miR-642a-5p, miR-532–3p and miR-329–5p. The circRNA circIPO11 was found to interact with miR-659–3p, miR-424–5p, and miR-106a-3p.
Li et al.38 reported a correlation between circRNAs and cancer miRNAs and proposed that the circRNA-miRNA axes might participate in cancer-related pathways, which is consistent with our findings.
Finally, as multiple circRNAs may contain binding sites for microRNAs, and those miRNAs in turn can target multiple genes, we considered a “Hub-gene model” for interaction networks among circRNAs, miRNAs and mRNA. From the constructed key function module, we found that the miR-570–3p interacted with most nodes. Therefore, the miR-570–3p can become our mechanism research focus in the future. The circRNAs profiled in this study have not been studied till date.
Accordingly, we propose the hypothesis that the mRNA-microRNA-circRNA axis may be the possible molecular regulatory mechanisms of liver cancer. Further biologic research on circRNAs and other associated functions and mechanisms will be performed in our following study.
In summary, the present study identified the association of circFUT8, circZFR and circIOP11 with liver cancer. While we speculate that a high level of circFUT8, circZFR and circIOP11 expression in liver cancer tissues is possibly correlated with tumor progression, the detailed molecular mechanisms by which these circRNA contribute to liver cancer proliferation, invasion, and metastasis require further research. Nonetheless, our research characterizes the circRNAs expression profiles and networks in liver cancer.
Material and methods
Source of specimens
Written consent was obtained from the patients before surgery. The study protocol was approved by the Institutional Review Board for the use of human subjects at China-Japan Union hospital of Jilin University, Changchun, China. A total of 30 pairs tissues are recruited from China-Japan Union hospital of Jilin University. None of the patients had received preoperative radiotherapy, chemotherapy or other cancer treatment. All the postoperative pathological diagnoses were done by 2 independent pathologists. All specimens were stored in liquid nitrogen immediately after surgical resection, and were transferred to the freezer at -80°C.
Circular RNA array analysis
Total RNA from each sample was quantified using the NanoDrop ND-1000. The sample preparation and microarray hybridization were performed based on the Arraystar's standard protocols. Briefly, total RNA from each sample was amplified and transcribed into fluorescent cRNA using random primer according to Arraystar's Super RNA Labeling protocol (Arraystar Inc.). The labeled cRNAs were hybridized onto the Arraystar Human circRNA Array (8 × 15K, Arraystar). After having washed the slides, the arrays were scanned by the Agilent Scanner G2505C.
Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images. Quantile normalization and subsequent data processing were performed using the R software package. Differentially expressed circRNAs with statistical significance between 2 groups were identified through Volcano Plot filtering. Differentially expressed circRNAs between 2 samples were identified through Fold Change filtering. Hierarchical Clustering was performed to show the distinguishable circRNAs expression pattern among samples.
GO and KEGG pathway analysis of differential expression genes
To unravel the biologic function of the differential expression genes, GO function and KEGG pathway enrichment analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID).42,43
Annotation for circRNA-miRNA interaction
The circRNA-microRNA interaction was predicted with Arraystar's home-made miRNA target prediction software based on TargetScan44 and miRanda,45 and the differentially expressed circRNAs within all the comparisons were annotated in detail with the circRNA-miRNA interaction information.
Annotation for miRNA-mRNA interaction
The microRNA-mRNA interaction was predicted with TargetScan (http://www.targetscan.org/), miRanda(http://www.microrna.org/), miRDB(http://www.mirdb.org/) that match the seed region of human miRNA sequences as obtained from miRBase (http://www.mirbase.org/), and then compared with the DE-mRNAs, Cytoscape (http://www.cytoscape.org/) was applied to build a circRNA–miRNA–mRNA interaction.
Quantitative real-time PCR
Total RNA was isolated from tissue and cell specimens using Trizol reagent (Invitrogen), RNA concentration was then measured using the Epoch Multi-volume Spectrophotometer System (BioTek, Vermont, USA) and these RNA samples werereversely transcribed into cDNA using a PrimeScript RT reagent Kit (TaKaRa Otsu, Shiga, Japan) and qPCR was then performed using SYBR Premix Ex Taq (TaKaRa) .qPCR was performed in Bio- Rad CFX96 system and the data were analyzed using the 2−ΔΔCT method. Each experiment was repeated at least 3 times.
The sequences of the 5 circRNAs were acquired from the database "CircInteractome" (http://circinteractome.nia.nih.gov/). β-actin ( Actin β , ACTB) , a housekeeping gene, was used as a control. Primers were synthesized by Sangon Biotech (Shanghai, China), with the following sequences: The primers for β-actin are forward: 5′- CTGGAACGGTGAAGGTGACA -3′ and reverse: 5′- AAGGGACTTCCTGTAACAATGCA -3′. The primers for hsa_circRNA_103809 are forward: 5′- TTTCCAAGCTGGCCCTTACG -3′ and reverse: 5′- ACAAACTGTCTGAAACGAG -3′. The primers for hsa_circRNA_101368 are forward: 5′- CCGAGAACTGTCCAAGATT -3′ and reverse: 5′- TCCTCCTGGTGATATGTAG -3′. The primers for hsa_circRNA_102399 are forward: 5′- CGACCTGTTTGAGAGTGGGAA -3′ and reverse: 5′- CAGTTCTGCATGGAGCGGTT -3′. The primers for hsa_circRNA_103639 are forward: 5′- TTACTCTACAGGAGTGGTTA -3′ and reverse: 5′- GCTTATTAAGGGTGTGGTAG -3′. The primers for hsa_circRNA_103847 are forward: 5′- CCAGACAGTGGCCTGAACTAA -3′ and reverse: 5′- AGTTGGTGATGAGCCCTGC -3′.
Statistical analysis
All statistical analyses were performed using SPSS version 18.0 (WPSS Ltd, Surrey, UK) and GraphPad Prism 6 software (GraphPad Software, Inc. La Jolla, CA, USA). The Receiver Operating Characteristic (ROC) curve and binary logistic regression analysis were used to analyze the sensitivity, specificity and area under the curve (AUC) with 95% CI .p value < 0.05 was considered to be statistically significant.
Table 2.
Clinicopathological characteristics of the study samples.
| Clinical parameters | Number of Case |
|---|---|
| Gender | |
| Male | 27 |
| Female | 13 |
| Age (52.6 Average) | |
| >50 | 29 |
| <50 | 11 |
| Differentiation status | |
| Poorly differentiated | 9 |
| Moderately differentiated | 27 |
| Highly differentiated | 4 |
Table 1.
The top 5 significantly differentially expressed circRNAs.
| CircRNAs | Fold Change | Regulation | Type | Chrom | Strand | Gene Symbol |
|---|---|---|---|---|---|---|
| hsa_circRNA_103809 | 13.22 | UP | exonic | chr5 | − | ZFR |
| hsa_circRNA_101368 | 11.44 | UP | exonic | chr14 | + | FUT8 |
| hsa_circRNA_102399 | 10.58 | UP | exonic | chr19 | + | CNN2 |
| hsa_circRNA_103639 | 10.37 | UP | exonic | chr4 | + | DCUN1D4 |
| hsa_circRNA_103847 | 8.65 | UP | exonic | chr5 | + | IPO11 |
| hsa_circRNA_001547 | 4.48 | DOWN | intronic | chr9 | − | BICD2 |
| hsa_circRNA_400010 | 3.54 | DOWN | intronic | chr1 | + | NASP |
| hsa_circRNA_000639 | 3.53 | DOWN | intronic | chr18 | + | SETBP1 |
| hsa_circRNA_100759 | 3.27 | DOWN | exonic | chr11 | − | DENND5A |
| hsa_circRNA_102950 | 3.11 | DOWN | exonic | chr2 | + | AGAP1 |
Supplementary Material
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China (#81602594 and #81472662), Foundation of Jilin Province Education Department (#2016453), Innovation fund of Jilin University (#JCKY-QKJC25), and Project 2016039 was supported by the Graduate Innovation fund of Jilin University.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65:87–108;http://dx.doi.org/10.3322/caac.21262 [DOI] [PubMed] [Google Scholar]
- [2].Waziry R, Grebely J, Amin J, Alavi M, Hajarizadeh B, George J, Matthews GV, Law M, Dore GJ. Trends in hepatocellular carcinoma among people with HBV or HCV notification in Australia (2000-2014). J Hepatol 2016; 65:1086–93; PMID:27569777; http://dx.doi.org/10.1016/j.jhep.2016.08.010 [DOI] [PubMed] [Google Scholar]
- [3].Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012; 379:1245–55; PMID:22353262; http://dx.doi.org/10.1016/S0140-6736(11)61347-0 [DOI] [PubMed] [Google Scholar]
- [4].Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017; 67:7–30; http://dx.doi.org/10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- [5].Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010; 127:2893–917; PMID:21351269; http://dx.doi.org/10.1002/ijc.25516 [DOI] [PubMed] [Google Scholar]
- [6].Stewart BW, Wild C. International Agency for Research on Cancer, World Health Organization. World cancer report 2014. Lyon, France Geneva, Switzerland: International Agency for Research on Cancer WHO Press, 2014 [Google Scholar]
- [7].Zhang Y, Li T, Guo P, Kang J, Wei Q, Jia X, Zhao W, Huai W, Qiu Y, Sun L, et al.. MiR-424-5p reversed epithelial-mesenchymal transition of anchorage-independent HCC cells by directly targeting ICAT and suppressed HCC progression. Sci Rep 2014; 4:6248; PMID:25175916; http://dx.doi.org/10.1038/srep06248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chan KM, Kuo CF, Hsu JT, Chiou MJ, Wang YC, Wu TH, Lee CF, Wu TJ, Chou HS, Lee WC. Metformin confers risk reduction for developing hepatocellular carcinoma recurrence after liver resection. Liver Int 2017; 37:434–41; PMID:27775209; http://dx.doi.org/10.1111/liv.13280 [DOI] [PubMed] [Google Scholar]
- [9].Wu TJ, Chang SS, Li CW, Hsu YH, Chen TC, Lee WC, Yeh CT, Hung MC. Severe Hepatitis Promotes Hepatocellular Carcinoma Recurrence via NF-kappaB Pathway-Mediated Epithelial-Mesenchymal Transition after Resection. Clin Cancer Res 2016; 22:1800–12; PMID:26655845; http://dx.doi.org/10.1158/1078-0432.CCR-15-0780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilusz JE, Sharp PA. Molecular biology. A circuitous route to noncoding RNA. Science 2013; 340:440–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen LL, Yang L. Regulation of circRNA biogenesis. RNA Biol 2015; 12:381–8; PMID:25746834; http://dx.doi.org/10.1080/15476286.2015.1020271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lasda E, Parker R. Circular RNAs: diversity of form and function. RNA 2014; 20:1829–42; PMID:25404635; http://dx.doi.org/10.1261/rna.047126.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vicens Q, Westhof E. Biogenesis of Circular RNAs. Cell 2014; 159:13–4; PMID:25259915; http://dx.doi.org/10.1016/j.cell.2014.09.005 [DOI] [PubMed] [Google Scholar]
- [14].Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol 2014; 32:453–61; PMID:24811520; http://dx.doi.org/10.1038/nbt.2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013; 19:141–57; PMID:23249747; http://dx.doi.org/10.1261/rna.035667.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature 2013; 495:384–8; PMID:23446346; http://dx.doi.org/10.1038/nature11993 [DOI] [PubMed] [Google Scholar]
- [17].Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al.. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013; 495:333–8; PMID:23446348; http://dx.doi.org/10.1038/nature11928 [DOI] [PubMed] [Google Scholar]
- [18].Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 2014; 15:409; PMID:25070500; http://dx.doi.org/10.1186/s13059-014-0409-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 2011; 30:4414–22; PMID:21964070; http://dx.doi.org/10.1038/emboj.2011.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014; 56:55–66; PMID:25242144; http://dx.doi.org/10.1016/j.molcel.2014.08.019 [DOI] [PubMed] [Google Scholar]
- [21].Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, Mesteri I, Grunt TW, Zeillinger R, Pils D. Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 2015; 5:8057; PMID:25624062; http://dx.doi.org/10.1038/srep08057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang X, Zhang Y, Huang L, Zhang J, Pan F, Li B, Yan Y, Jia B, Liu H, Li S, et al.. Decreased expression of hsa_circ_001988 in colorectal cancer and its clinical significances. Int J Clin Exp Pathol 2015; 8:16020–5; PMID:26884878 [PMC free article] [PubMed] [Google Scholar]
- [23].Xie H, Ren X, Xin S, Lan X, Lu G, Lin Y, Yang S, Zeng Z, Liao W, Ding YQ, et al.. Emerging roles of circRNA_001569 targeting miR-145 in the proliferation and invasion of colorectal cancer. Oncotarget 2016; 7:26680–91; PMID:27058418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, Celniker SE, Graveley BR, Lai EC. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 2014; 9:1966–80; PMID:25544350; http://dx.doi.org/10.1016/j.celrep.2014.10.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, Wong DT, Xiao X. The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin Chem 2015; 61:221–30; PMID:25376581; http://dx.doi.org/10.1373/clinchem.2014.230433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li P, Chen S, Chen H, Mo X, Li T, Shao Y, Xiao B, Guo J. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta 2015; 444:132–6; PMID:25689795; http://dx.doi.org/10.1016/j.cca.2015.02.018 [DOI] [PubMed] [Google Scholar]
- [27].Lukiw WJ. Circular RNA (circRNA) in Alzheimer's disease (AD). Front Genet 2013; 4:307; PMID:24427167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhao Y, Alexandrov PN, Jaber V, Lukiw WJ. Deficiency in the Ubiquitin Conjugating Enzyme UBE2A in Alzheimer's Disease (AD) is Linked to Deficits in a Natural Circular miRNA-7 Sponge (circRNA; ciRS-7). Genes (Basel) 2016; 7:pii: E116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Qin M, Liu G, Huo X, Tao X, Sun X, Ge Z, Yang J, Fan J, Liu L, Qin W. Hsa_circ_0001649: A circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark 2016; 16:161–9; PMID:26600397; http://dx.doi.org/10.3233/CBM-150552 [DOI] [PubMed] [Google Scholar]
- [30].Shang X, Li G, Liu H, Li T, Liu J, Zhao Q, Wang C. Comprehensive Circular RNA Profiling Reveals That hsa_circ_0005075, a New Circular RNA Biomarker, Is Involved in Hepatocellular Crcinoma Development. Medicine (Baltimore) 2016; 95:e3811; PMID:27258521; http://dx.doi.org/10.1097/MD.0000000000003811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fu L, Yao T, Chen Q, Mo X, Hu Y, Guo J. Screening differential circular RNA expression profiles reveals hsa_circ_0004018 is associated with hepatocellular carcinoma. Oncotarget. 2017; Apr 6; PMID: 28430660; https://doi.org/10.18632/oncotarget.16881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Memczak S, Papavasileiou P, Peters O, Rajewsky N. Identification and Characterization of Circular RNAs As a New Class of Putative Biomarkers in Human Blood. PLoS One 2015; 10:e0141214; PMID:26485708; http://dx.doi.org/10.1371/journal.pone.0141214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 2011; 146:353–8; PMID:21802130; http://dx.doi.org/10.1016/j.cell.2011.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat Genet 2011; 43:854–9; PMID:21857679; http://dx.doi.org/10.1038/ng.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al.. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 2006; 103:2257–61; PMID:16461460; http://dx.doi.org/10.1073/pnas.0510565103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bandyopadhyay S, Mitra R, Maulik U, Zhang MQ. Development of the human cancer microRNA network. Silence 2010; 1:6; PMID:20226080; http://dx.doi.org/10.1186/1758-907X-1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ghosal S, Das S, Sen R, Basak P, Chakrabarti J. Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet 2013; 4:283; PMID:24339831; http://dx.doi.org/10.3389/fgene.2013.00283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li J, Yang J, Zhou P, Le Y, Zhou C, Wang S, Xu D, Lin HK, Gong Z. Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am J Cancer Res 2015; 5:472–80; PMID:25973291 [PMC free article] [PubMed] [Google Scholar]
- [39].Slattery ML, Herrick JS, Mullany LE, Valeri N, Stevens J, Caan BJ, Samowitz W, Wolff RK. An evaluation and replication of miRNAs with disease stage and colorectal cancer-specific mortality. Int J Cancer 2015; 137:428–38; PMID:25484364; http://dx.doi.org/10.1002/ijc.29384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shan SW, Fang L, Shatseva T, Rutnam ZJ, Yang X, Du W, Lu WY, Xuan JW, Deng Z, Yang BB. Mature miR-17-5p and passenger miR-17-3p induce hepatocellular carcinoma by targeting PTEN, GalNT7 and vimentin in different signal pathways. J Cell Sci 2013; 126:1517–30; PMID:23418359; http://dx.doi.org/10.1242/jcs.122895 [DOI] [PubMed] [Google Scholar]
- [41].Choi YC, Yoon S, Byun Y, Lee G, Kee H, Jeong Y, Yoon J, Baek K. MicroRNA library screening identifies growth-suppressive microRNAs that regulate genes involved in cell cycle progression and apoptosis. Exp Cell Res 2015; 339:320–32; PMID:26485640; http://dx.doi.org/10.1016/j.yexcr.2015.10.012 [DOI] [PubMed] [Google Scholar]
- [42].Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1–13; PMID:19033363; http://dx.doi.org/10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44–57; PMID:19131956; http://dx.doi.org/10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- [44].Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol 2003; 5:R1; PMID:14709173; http://dx.doi.org/10.1186/gb-2003-5-1-r1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 2012; 13:271–82; PMID:22411466 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.