

ABSTRACT
Peripheral muscle dysfunction is an important complication in patients with chronic obstructive pulmonary disease (COPD). The objective of this study was to explore the relationship between the levels of peroxisome proliferator-activated receptor α (PPARα) mRNA expression and the respiratory function and ultrastructure of mitochondria in the vastus lateralis of patients with COPD. Vastus lateralis biopsies were performed on 14 patients with COPD and 6 control subjects with normal lung function. PPARα mRNA levels in the muscle tissue were detected by real-time PCR. A Clark oxygen electrode was used to assess mitochondrial respiratory function. Mitochondrial number, fractional area in skeletal muscle cross-sections, and Z-line width were observed via transmission electron microscopy. The PPARα mRNA expression was significantly lower in COPD patients with low body mass index (BMIL) than in both COPD patients with normal body mass index (BMIN) and controls. Mitochondrial respiratory function (assessed by respiratory control ratio) was impaired in COPD patients, particularly in BMIL. Compared with that in the control group, mitochondrial number and fractional area were lower in the BMIL group, but were maintained in the BMIN group. Further, the Z-line became narrow in the BMIL group. PPARα mRNA expression was positively related to mitochondrial respiratory function and volume density. In COPD patients with BMIN, mitochondria volume density was maintained, while respiratory function decreased, whereas both volume density and respiratory function decreased in COPD patients with BMIL. PPARα mRNA expression levels are associated with decreased mitochondrial respiratory function and volume density, which may contribute to muscle dysfunction in COPD patients.
KEYWORDS: mitochondrial respiratory function, muscle ultrastructure, PPARα, skeletal muscle atrophy
Introduction
Chronic obstructive pulmonary disease (COPD), a chronic lung disease characterized by irreversible airflow limitation, is a systemic disease involved in multi-organ damage.1 Skeletal muscle dysfunction (SMD), an important complication in COPD patients, results in decreased muscle strength/e ndurance and high likelihood of fatigue,2,3 which decreases the quality of life and leads to serious economic burdens to families and society. Additionally, SMD is an important indicator for predicting mortality, independent of lung function, in COPD patients.4 The mechanisms leading to SMD in COPD is complex and related to factors such as a sedentary lifestyle of patients, cellular hypoxia, low-grade systemic inflammation, and oxidative stress.5-7 These factors reduce oxidative metabolism in the skeletal muscle or lead to muscle fiber-type shift, resulting in imbalanced muscle protein degradation and synthesis, increase skeletal muscle cell apoptosis, and autophagy8 ultimately leading to muscle dysfunction.9,10,11
Mitochondria are organelles involved not only in energy production for skeletal muscle movement but also in reactive oxygen species (ROS) generation; mitochondria are also related to skeletal muscle cell apoptosis. Its respiratory function (an important indicator of mitochondrial electron transport and oxidative phosphorylation), number, and volume size (i.e. mitochondrial volume density) have a direct impact on muscle function. Recent studies suggested that mitochondrial dysfunction exists in patients with COPD.12-15 Several studies have found that peroxisome proliferator-activated receptors (PPARs, steroid hormone receptor and member of a nuclear receptor superfamily) regulate the oxidative metabolism of skeletal muscle and promote fiber type conversion by mediating the activity of peroxisome proliferator. Remels et al. (2007) found that PPAR expression is decreased in the peripheral skeletal muscle of COPD patients under low oxygen and systemic inflammatory conditions. PPARα (a PPAR subtype, expressed highly in skeletal muscle) is significantly decreased in COPD patients with low body weight, and PPARα mRNA levels are related to citrate synthase, a key enzyme of the citric acid cycle. However, the respiratory function of mitochondria in COPD patients has not been directly confirmed.16
There have been no studies of mitochondrial respiratory function combined with mitochondrial ultrastructural detection and PPARα mRNA expression levels in the peripheral skeletal muscle of COPD patients. Therefore, the aim of the current study is to explore the pathophysiological mechanism of SMD in COPD patients by observing the mitochondrial respiratory function and ultrastructure of peripheral skeletal muscle while simultaneously detecting PPARα mRNA expression levels.
Material and methods
Subjects
Twenty subjects were recruited from the First Affiliated Hospital of Kunming Medical University, Kunming, People's Republic of China, including 6 controls and 14 stable COPD patients (diagnostic criteria in accordance with the 2014 Global Initiative for Chronic Obstructive Lung Disease guideline). None of the patients needed to change their current medication (including inhaled long-acting β-agonists and corticosteroid). Exclusion criteria were concurrent diseases, such as cancer, asthma, bronchiectasis, heart failure, paraplegia, liver and kidney dysfunction, endocrine dysfunction, and systemic immune disease. Eight COPD patients were categorized into a normal body mass index group (BMIN group, BMI > 21 kg·m−2 and FFMI > 16 kg·m−2 in men or > 15 kg·m−2 in women) and the remaining 6 COPD patients were categorized into a low body mass index group (BMIL group, BMI ≤ 21 kg·m−2 and FFMI ≤ 16 kg·m−2 in men or ≤ 15 kg·m−2 in women). Six femoral fracture patients (6 months to 1 y after internal fixation) served as age-matched controls, and none of these patients presented significant comorbidities. Muscle samples were obtained when the internal fixation was extracted. All subjects provided written informed consent and the study was approved by the ethics committee of Kunming Medical University.
Lung function, blood gas analysis, and body composition
Pulmonary function tests were performed with a pneumatonometer (Jaeger, Germany) after a resting period of 15–30 min. Forced vital capacity (FVC), the percentage of forced expiratory volume in 1 s over the predicted value (FEV1/Pred%), and the percentage of forced expiratory volume in 1 s over expiratory vital capacity (FEV1/FVC%) were recorded. Height (m) and body weight (kg) were measured in the early morning to calculate body mass indexes (BMI). Fat mass (FM) and fat-free mass (FFM) were measured by bioelectrical impedance (Beurer, Ulm, Germany) to calculate the fat mass index (FMI) and fat-free mass index (FFMI). Blood was collected from the radial artery over 15–30 min and blood gas analysis was performed (Premier 3000, Instrumentation Laboratory, Bedford, MA, USA), arterial oxygen tension (PaO2), and arterial carbon dioxide tension (PaCO2) were recorded.
Collection and processing of muscle tissue
Open biopsy was used to obtain 0.6 g vastus lateralis. The samples were placed on a gauze and arranged along the direction of the muscle fiber. Next, they were quickly divided into 3 parts. Of these, one part (∼400 mg) was placed into cold separation liquid containing 0.21 M mannitol, 0.07 M sucrose, 0.2 mM EDTA-Na2, and 5 mM Tris-HCl and sent to the laboratory for analysis. Separation of mitochondria was performed within 3 h of biopsy. A ∼50 mg part was placed in an RNase -free Eppendorf tube and sent to the laboratory for storage at −70°C for quantitative detection of PPARα mRNA. The final ∼50 mg part was placed into a 2 mL vial with electron microscopy fixative solution (2–3% glutaraldehyde diluted with 0.1 mM Millonig phosphate buffer [pH 7.2–7.4]). The vial was stored in the dark at 4°C. Detection was performed within 4 weeks of biopsy.
Separation of mitochondria and respiratory function tests
A tissue mitochondrial isolation kit (Beyotime, Jiangsu, China) was used to extract muscle mitochondria by refrigerated centrifugation. Mitochondrial protein content was determined by the Bradford method using Coomassie brilliant blue. A Clark oxygen electrode (Redmaple, Beijing, China) was used to determine mitochondrial respiratory function. Briefly, medium (2.0 mL 225 mM mannitol, 70 mM sucrose, 1 mM EDTA, 0.1% bovine serum albumin, and 10 mM KH2PO4, pH 7.4) was added to the reaction tank and preheated to 25°C, and then air was used for saturation. The oxygen electrode was inserted and the instrument was stabilized for 2 min. Extracted mitochondrial suspension (100 μL) was then added. After recording the baseline for 2 min, 20 μL exogenous substrate (0.2 mM disodium succinate) was added and state 4 (ST4) respiration occurred. After recording for ∼2 min, 10 μL 50 mM ADP was added and state 3 (ST3) respiration occurred. After the ADP was completely consumed, mitochondria reverted to ST4 respiration. The entire process was performed under magnetic stirring. Mitochondrial respiratory activity was calculated by oxygen consumption of one unit of mitochondrial protein in one unit of time (nmol·min−1·mg−1). Oxidative phosphorylation efficiency was assessed as the respiratory control ratio (RCR), presented as the ratio of oxygen consumption at ST3 and ST4.17
PPARα mRNA
Vastus lateralis (50 mg) sample was collected and preserved at −80°C. Total RNA extraction and cDNA synthesis were then performed within 1 month. Trizol reagent (Generay Biological Company, Shanghai, China) was used to extract total RNA from the sample and the RNA concentration was determined by spectrophotometry. Total RNA (2 μg) was used for reverse transcription of cDNA using a cDNA synthesis kit (Fermentas, Vilnius, Lithuania). The cDNA (3 μg) was amplified by real-time PCR using specific reagents (TOYOBO, Osaka, Japan) and a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The PPARα mRNA upstream primer was 5′-GGG ACA GAC TGA CAC CTT ACTT-3′ and the downstream primer was 5′-ATG GGA CCC TTA TCA ATC CTA-3′. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was also quantified to control the sample-to-sample differences in RNA concentration. The mRNA quantity was expressed as arbitrary units (Au).
Observation of muscle ultrastructure
Skeletal muscle tissue samples from all groups were cut into 1.0 × 1.0 × 1.0 mm pieces. Next, 2% glutaraldehyde/p hosphate-buffered saline fixative solution was used for post-fixation at 4°C for 1 h. Osmium tetroxide (1%, prepared in 0.1 mM phosphate-buffered saline) was used for fixation at 4°C for 2 h. Graded dehydration in ethanol, ethanol–acetone, and acetone was performed. After ethylene oxide replacement and saturation with 618 embedding solution and propylene oxide, vertical embedding was performed. Appropriate locations and fiberlongitudinal orientation were evaluated in the semi-thin sections from the central region of each sample, and an ultra-thin slicing machine (Ultracut R, Wetzlar, Germany) was used to prepare ultra-thin slices. The sections were placed on copper grids, and uranyl acetate and lead citrate electron staining was performed. The prepared grids were then observed by transmission electron microscopy (JEM-1011 (HC), Jeol, Tokyo, Japan) at 20,000 × magnification. Muscle mitochondria were identified by the mitochondrial electron density (black or dark gray), shape (oval or spherical), and subcellular location (both sides of the Z-line of muscle fibers). These digitized micrographs were analyzed by an interactive image analysis system (HMIAS-2000 high-resolution color medical image analysis system, Champion, Wuhan, China). Mitochondria were outlined in an overlaying bitmap, which was used to compute mitochondrial number and fractional area in a given muscle fiber area. Mitochondrial fractional area was computed as the percentile mitochondrial fractional area of total fiber area. Four to eight photographs were analyzed randomly so that at least 100 mitochondria per subject were examined. Subsequently, as a surrogate marker of muscle fiber typology, the average Z-line width was also determined for each micrograph.
Statistical analyses
The SPSS 11.5 statistical package was used for data processing (SPSS, Inc., Chicago, IL, USA). The data are presented as the mean ± SD. Comparison of means in 2 groups were done by independent sample t tests. One way analysis of variance was used for multiple mean comparisons. Comparison between groups was analyzed by Bonferroni t tests. The correlation analysis was conducted by the Pearson test. P < 0.05 was considered significant.
Results
General information and related clinical data
The age and height in the 3 groups showed no significant differences. As expected, FFMI was significantly lower in BMIL (13.49 ± 0.39 kg.m−2) than in both BMIN (17.09 ± 0.71 kg·m−2, P < 0.001) and controls (17.93 ± 1.61 kg·m−2, P < 0.001). No patients with BMIN showed abnormally low FFMI (< 16 kg·m−2); however, no difference in BMI or FFMI was found between the control and COPD BMIN groups. As expected, lung function in patients in the COPD group decreased significantly (P < 0.01), and PaO2 was decreased in COPD patients (P < 0.05) (Table 1).
Table 1.
General information of study subjects.
| COPD |
|||
|---|---|---|---|
| Control | BMIN | BMIL | |
| Gender (male/f emale) | 5/1 | 6/2 | 5/1 |
| Age (year) | 58.17 ± 5.34 | 62.75 ± 8.66 | 61.67 ± 9.72 |
| height (m) | 1.65 ± 0.03 | 1.67 ± 0.08 | 1.64 ± 0.04 |
| Body weight (kg) | 67.20 ± 4.48 | 64.78 ± 5.39 | 49.86 ± 2.86•,★ |
| Fat free body weight (kg) | 48.57 ± 3.70 | 47.59 ± 3.57 | 36.44 ± 2.68•,★ |
| BMI (kg·m−2) | 24.80 ± 1.84 | 23.26 ± 1.13 | 18.17 ± 0.40•,★ |
| FFMI (kg·m−2) | 17.93 ± 1.61 | 17.09 ± 0.71 | 13.49 ± 0.39•,★ |
| Smoking (pack-years) | 30 ± 16 | 27 ± 18 | 33 ± 13 |
| FEV1%pred | 102.0 ± 8.37 | 59.83 ± 11.52▴ | 37.75 ± 11.08•,F |
| FEV1/FVC% | 89.95 ± 4.83 | 66.12 ± 4.25▴ | 59.76 ± 3.43•,F |
| PaO2 (kpa) | 11.18 ± 1.74 | 9.51 ± 0.75△ | 8.88 ± 0.36○ |
| PaCO2 (kpa) | 5.30 ± 0.52 | 5.20 ± 0.81 | 5.78 ± 0.34 |
Data are presented as the mean ± SD, unless otherwise stated. COPD: chronic obstructive pulmonary disease; BMI: body mass index; FFMI: fat-free mass index; BMIN: normal body mass index; BMIL: low body mass index; FEV1: forced expiratory volume in one second; % pred: % predicted; FVC: forced vital capacity; PaO2: arterial oxygen tension; PaCO2: arterial carbon dioxide tension.
: P < 0.05,
: P < 0.01 versus controls;
: P < 0.05,
: P < 0.01 vs. controls;
: P < 0.05,
: P < 0.01 vs. BMIN.
Determination of mitochondrial respiratory function
ST3 showed significant differences between the healthy control group and COPD group (18.18 ± 2.65 vs. 13.23 ± 3.93, P = 0.002). After grouping of COPD patients, the difference between the BMIN and BMIL groups was significant (P < 0.01), but no difference was found between the control and BMIN groups (P = 0.43). Similarly, no significant difference in ST4 was found between the control and COPD groups. In the comparison of the respiratory control ratio, RCR (ST3 / ST4) among the 3 groups showed significant differences (P < 0.01).
Image analysis of muscle ultrastructure
Compared with the control group, the number of mitochondria in a unit of cross-sectional area and mean fractional area of mitochondria (volume density) in the COPD group decreased significantly (P < 0.01). After grouping of COPD patients, the volume density of mitochondria in the BMIN group showed a declining trend but was not statistically different compared with the control group. In the BMIL group, the volume density of mitochondria significantly decreased. No difference in Z-line width (representing muscle fiber type) was found between the BMIN and control groups. However, Z-line width between the control and BMIL groups was significantly different (P = 0.024), suggesting that type IIx fibers increased in the BMIL group. Specific data are presented in Table 2.
Table 2.
Results of mitochondrial respiratory function and image analysis of muscle ultrastructure.
| COPD |
||||
|---|---|---|---|---|
| Control | Total | BMIN | BMIL | |
| ST3 (nmol.min−1.mg−1) | 18.18 ± 2.65 | 12.23 ± 3.93 | 15.10 ± 2.36 | 8.40 ± 1.49•,★ |
| ST4 (nmol.min−1.mg−1) | 3.53 ± 0.29 | 3.43 ± 0.56 | 3.79 ± 0.36 | 2.94 ± 0.38F |
| RCR (ST3/S T4) | 5.13 ± 0.35 | 3.50 ± 0.71 | 3.97 ± 0.51▴ | 2.86 ± 0.32•,★ |
| Number of mitochondria in unit area (n/u −2) | 0.52 ± 0.06 | 0.38 ± 0.08 | 0.43 ± 0.08 | 0.32 ± 0.07• |
| Mean fractional area of mitochondria% | 4.31 ± 0.76 | 3.18 ± 0.75 | 3.71 ± 0.47 | 2.5 ± 0.37•,★ |
| Z line width (nm) | 103.2 ± 6.42 | 93.57 ± 9.8 | 99.8 ± 11.34 | 87. 2 ± 8.79○ |
Data are presented as the mean ± SD, unless otherwise stated. BMIN: normal body mass index group; BMIL: low body mass index group; ST3: state 3 respiration; ST4: state 4 respiration; RCR: respiratory control ratio.
: P < 0.01 vs. controls;
: P < 0.05,
: P < 0.01 vs. controls;
: P < 0.05,
: P < 0.01 vs. BMIN.
Expression of PPARα mRNA
In the control, BMIN, and BMIL groups, PPARα mRNA expression levels were 0.82 ± 0.10, 0.64 ± 0.09, and 0.41 ± 0.12, respectively. Compared to the control, the expression level of PPARα mRNA in BMIL was significantly decreased (P < 0.01). In the BMIN group, PPARα mRNA was decreased, but did not reach statistical significance (P = 0.12). After grouping of COPD patients, the difference in PPARα mRNA levels between the BMIN and BMIL groups was statistically significant (P = 0.02) (Fig. 1).
Figure 1.
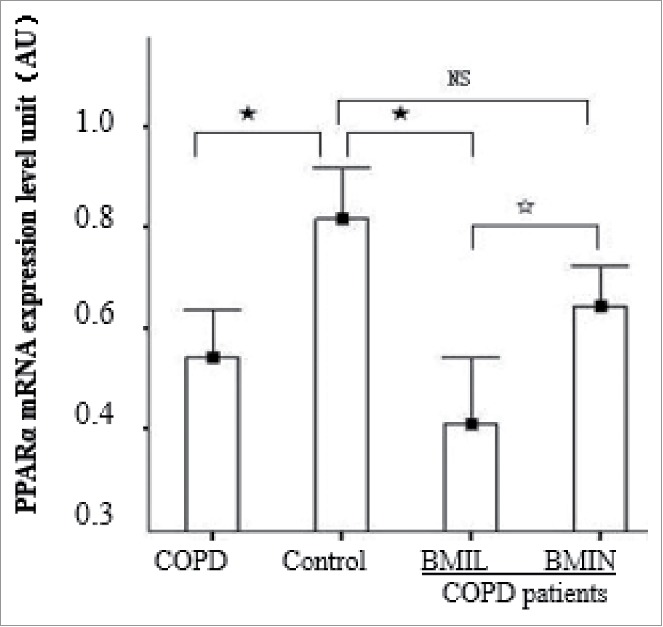
PPARα mRNA expression levels in each group. Control, control group; BMIN, COPD patients with normal body mass index; and BMIL, COPD patients with low body mass index. Longitudinal coordinates were PPARα/GAPDH mRNA; AU = arbitrary units. F: P < 0.05; ★: P < 0.01; NS: not statistically significant.
Correlations among data
The PPARα mRNA expression level was positively correlated with the RCR, Z-line width, number of mitochondria per unit area, and mean fractional area of mitochondria (Fig. 2). Oxygen partial pressure was positively correlated with the expression level of PPARα mRNA and Z-line width (r = 0.47 and 0.54; P = 0.04 and 0.01, respectively). Z-line width was positively correlated with the RCR and mean fractional area of mitochondria.
Figure 2.

* Healthy control group (control); ◇: COPD with BMIN; △: COPD with BMIL. a) Relationship between PPARα mRNA expression level and RCR: r = 0.71, P < 0.01. b) Relationship of PPARα mRNA expression and Z-line: r = 0.63, P < 0.01. c,d) PPARα mRNA expression and the number of mitochondria per unit area and mean fractional area of mitochondria; r values = 0.69 and 0.73, respectively, P < 0.01.
Discussion
In this study, we found that in normal weight COPD patients (assessed by FFMI and BMI), the mitochondrial electron transport chain and oxidative phosphorylation (assessed by respiratory control ratio, an important indicator of mitochondrial electron transport and oxidative phosphorylation) were abnormal, but the volume density of mitochondria was maintained. However, in low-weight COPD patients, both mitochondrial respiratory function and volume density were decreased. Thus, the mitochondrial electron transport chain and oxidative phosphorylation changed before mitochondrial volume density reduction and an early event of muscle dysfunction in COPD patients. PPARα mRNA expression also decreased in COPD patients and is related to mitochondrial respiratory function and the mean fractional area of mitochondria in muscle cross sections. Thus, decreased PPARα mRNA expression levels may contribute to the decrease in mitochondrial respiratory function and volume density.
In the mitochondria, energy for skeletal muscle movement and ROS are generated; this organelle is also related to the muscle fiber-type and oxidation capacity. The citric acid cycle, electron transport, and oxidative phosphorylation are also performed in mitochondria. RCR is an important indicator of mitochondrial electron transport and oxidative phosphorylation. Mitochondrial dysfunction is an important factor of skeletal muscle dysfunction in COPD patients.18-21 As described previously,7,20 the activities of citrate synthase and succinate dehydrogenase, which are key enzymes in the citric acid cycle, decrease in the skeletal muscle mitochondria of COPD patients. ROS production in the mitochondria increases, and electron transport chain and oxidative phosphorylation become abnormal because of the lack of oxygen and increased inflammation.15,20 In the present study, the respiratory function of mitochondria (RCR assessment) decreased significantly in COPD patients with low body weight. We directly evaluated mitochondrial volume density, which had not been previously examined. Additionally, there have been few studies on electron microscopic analysis of mitochondria in COPD skeletal muscle. A study by Gosker et al.18 showed that the mitochondrial volume density decreased significantly in COPD patients (FEV1%pred: 53.7 ± 22.9, BMI: 26.5 ± 5.7 kg·m−2, FFMI: 18.3 ± 2.5 kg·m−2). In this study, patients were stratified by BMI and FFMI, and we found that the mitochondrial volume density did not decrease significantly in COPD patients with normal weight (FEV1%pred: 59.83 ± 11.52, BMI: 23.26 ± 1.13 kg·m−2, FFMI: 17.09 ± 0.71 kg·m−2). However, in COPD patients with low-weight (FEV1%pred: 37.75 ± 11.08, BMI: 18.17 ± 0.40 kg·m−2, FFMI: 13.49 ± 0.39 kg·m−2), the mitochondrial volume density decreased significantly. This has not been previously reported. Moreover, we found that mitochondrial respiratory function was decreased in all COPD patients. This is the first study in which mitochondrial ultrastructure and respiratory function were determined simultaneously while stratifying BMI and FFMI in the vastus lateralis muscle of COPD patients.
We also detected a positive correlation between PPARα mRNA gene expression level and volume density of mitochondria in the peripheral skeletal muscle of patients with COPD, which has not been reported previously. Some studies showed that PPARs is a potential therapeutic target for the treatment of skeletal muscle dysfunction.22,23 PPARs are members of the nuclear receptor superfamily and are divided into 3 types: PPARα, PPARδ, and PPARγ. PPARα is highly expressed in skeletal muscles.16,23 PPARs have powerful anti-inflammatory, antioxidant, and oxidative metabolism regulatory activities and are associated with muscle fiber type.22-24 First, the chief function of PPARα in the skeletal muscle is to regulate genetic transcription of fatty acid β-oxidation enzymes, thus increasing the assimilation of fatty acids by muscle tissue and decreasing the assimilation of glucose to reduce the production of lactic acid and muscle fatigue. Second, PPARα directly prevents ROS gene expression. It has been shown that PPARα agonists directly decrease oxidative stress. Third, PPARα inhibits nuclear factor-kB-driven transcription of inflammatory cytokines, thus indirectly reducing inflammatory responses and oxidative stress.23,24 PPARα can also alter ROS generation and oxidation of fatty acids by adjusting the UCP3 gene.25,26 However, the expression of PPARα was inhibited by cellular hypoxia and the sedentary lifestyle of the patients.27 Recent evidence suggests that mitochondrial biosynthesis is related to the peroxisome proliferator γ-activated receptor coactivator-1α (PGC-1α) signaling cascade.28 PPARα has strong regulatory functions in muscle metabolism and affects the PGC-1α signaling pathway, indicating a relationship with the biosynthesis of mitochondria.24
Adaptation of mitochondria to movement is referred to as “mitochondrial biogenesis,” which consists of 2 related changes: mitochondrial content increase in unit muscle tissue and changes in mitochondrial composition, particularly the protein/l ipid ratio. The physiologic role of mitochondrial biosynthesis is to change the metabolism and induce the muscles to use lipids rather than carbohydrates for metabolism, thus reducing lactic acid production and delaying fatigue; moreover, the biosynthesis of mitochondria is related to muscle fiber-type. We found that Z-line width decreased in patients with low FFMI. A study by Sjostrom et al.29 found a relationship between Z-line width and muscle fiber-type, and an 83% accuracy rate can be achieved using the following criteria: type I: 128 ± 10 nm; type IIA: 104 ± 8.5 nm; type IIx: 88 ± 9.1 nm. Thus, type I and IIx fibers can be identified accurately, and type II fiber significantly increased in low-weight COPD patients via glycolysis. Further, muscle fiber-type was positively related to mitochondrial respiratory function, number of mitochondria, and unit cross-sectional area of muscle. Martin Picard et al.30 demonstrated that the mitochondrial respiratory function of patients with moderate to severe COPD is normal when mitochondrial volume was taken into account by normalizing respiration per unit of citrate synthase activity, and these differences may be attributable to greater type II fiber expression in COPD patients. However, according to previous studies,7,20 citrate synthase activity is decreased in COPD patients, and thus it may be a reasonable indicator of mitochondrial volume. Further studies are needed to determine the exact mechanisms behind these findings.
Because this was an invasive study, the sample size and content were small and the mitochondrial membrane potential was not measured. This measurement can indirectly reflect proton leakage and ROS production levels. Additional indicators, such as mitochondrial ROS, key enzymes in the citric acid cycle, and oxidative phosphorylation were not evaluated. In addition, it is well known that the daily activity and senescence are important influencing factors of degeneration process of muscle biogenesis. Although no significant difference in the age was found between the control and COPD groups, the lung function in patients in the COPD group decreased significantly than the control group. The patients who have lower FEV1 and PaO2 are more likely to sedentary, thus affecting muscle function. This needs to be noticed in further studies.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Liu Hua from the First Affiliated Hospital of Kunming Medical University, Son Jinglun from the electron microscope facility of Kunming Medical University, and Wang Hua from the Department of Pathology, Kunming Medical University.
Funding
This study was supported by a grant from the Yunnan Provincial Science and Technology Department (2008CD011).
References
- [1].Choudhury G, Rabinovich R, MacNee W. Comorbidities and systemic effects of chronic obstructive pulmonary disease. Clinics Chest Med 2014; 35:101-30; PMID:24507840; https://doi.org/ 10.1016/j.ccm.2013.10.007 [DOI] [PubMed] [Google Scholar]
- [2].Hussain SN, Sandri M. Role of autophagy in COPD skeletal muscle dysfunction. J Applied Physiol 2013; 114:1273-81; PMID:23085958; https://doi.org/ 10.1152/japplphysiol.00893.2012 [DOI] [PubMed] [Google Scholar]
- [3].Maltais F, Decramer M, Casaburi R, Barreiro E, Burelle Y, Debigare R, Dekhuijzen PN, Franssen F, Gayan-Ramirez G, Gea J, et al.. An official American Thoracic Society/E uropean respiratory society statement: update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am J Respiratory Critical Care Med 2014; 189:e15-62; PMID:24787074; https://doi.org/ 10.1164/rccm.201402-0373ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Swallow EB, Reyes D, Hopkinson NS, Man WD, Porcher R, Cetti EJ, Moore AJ, Moxham J, Polkey MI. Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax 2007; 62:115-20; PMID:17090575; https://doi.org/ 10.1136/thx.2006.062026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gea J, Agusti A, Roca J. Pathophysiology of muscle dysfunction in COPD. J Applied Physiol 2013; 114:1222-34; PMID:23519228; https://doi.org/ 10.1152/japplphysiol.00981.2012 [DOI] [PubMed] [Google Scholar]
- [6].Barreiro E, Gea J. Respiratory and Limb muscle dysfunction in COPD. Copd 2015; 12:413-26; PMID:25438125; https://doi.org/ 10.3109/15412555.2014.974737 [DOI] [PubMed] [Google Scholar]
- [7].Wust RC, Degens H. Factors contributing to muscle wasting and dysfunction in COPD patients. Int J Chronic Obstructive Pulmonary Dis 2007; 2:289-300; PMID:18229567 [PMC free article] [PubMed] [Google Scholar]
- [8].Guo Y, Gosker HR, Schols AM, Kapchinsky S, Bourbeau J, Sandri M, Jagoe RT, Debigare R, Maltais F, Taivassalo T, et al.. Autophagy in locomotor muscles of patients with chronic obstructive pulmonary disease. Am J Respiratory Critical Care Med 2013; 188:1313-20; PMID:24228729; https://doi.org/ 10.1164/rccm.201304-0732OC [DOI] [PubMed] [Google Scholar]
- [9].Barreiro E, Gea J. Molecular and biological pathways of skeletal muscle dysfunction in chronic obstructive pulmonary disease. Chronic Respiratory Dis 2016; 13:297-311; PMID:27056059; https://doi.org/ 10.1177/1479972316642366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Langen RC, Gosker HR, Remels AH, Schols AM. Triggers and mechanisms of skeletal muscle wasting in chronic obstructive pulmonary disease. Int J Biochem Cell Biol 2013; 45:2245-56; https://doi.org/ 10.1016/j.biocel.2013.06.015 [DOI] [PubMed] [Google Scholar]
- [11].Sandri M. Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol 2013; 45:2121-9; https://doi.org/ 10.1016/j.biocel.2013.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hara H, Araya J, Ito S, Kobayashi K, Takasaka N, Yoshii Y, Wakui H, Kojima J, Shimizu K, Numata T, et al.. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am J Physiol Lung Cell Mol Physiol 2013; 305:L737-46; PMID:24056969; https://doi.org/ 10.1152/ajplung.00146.2013 [DOI] [PubMed] [Google Scholar]
- [13].Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, Dijk F, Kalicharan D, Kelders M, Gosker HR, et al.. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respiratory Res 2013; 14:97; PMID:24088173; https://doi.org/ 10.1186/1465-9921-14-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Puente-Maestu L, Lazaro A, Tejedor A, Camano S, Fuentes M, Cuervo M, Navarro BO, Agusti A. Effects of exercise on mitochondrial DNA content in skeletal muscle of patients with COPD. Thorax 2011; 66:121-7; PMID:21097816; https://doi.org/ 10.1136/thx.2010.153031 [DOI] [PubMed] [Google Scholar]
- [15].Puente-Maestu L, Tejedor A, Lazaro A, de Miguel J, Alvarez-Sala L, Gonzalez-Aragoneses F, Simon C, Agusti A. Site of mitochondrial reactive oxygen species production in skeletal muscle of chronic obstructive pulmonary disease and its relationship with exercise oxidative stress. Am J Respiratory Cell Mol Biol 2012; 47:358-62; PMID:22493009; https://doi.org/ 10.1165/rcmb.2011-0382OC [DOI] [PubMed] [Google Scholar]
- [16].Remels AH, Schrauwen P, Broekhuizen R, Willems J, Kersten S, Gosker HR, Schols AM. Peroxisome proliferator-activated receptor expression is reduced in skeletal muscle in COPD. European Respiratory J 2007; 30:245-52; PMID:17459894; https://doi.org/ 10.1183/09031936.00144106 [DOI] [PubMed] [Google Scholar]
- [17].Rabinovich RA, Bastos R, Ardite E, Llinas L, Orozco-Levi M, Gea J, Vilaro J, Barbera JA, Rodriguez-Roisin R, Fernandez-Checa JC, et al.. Mitochondrial dysfunction in COPD patients with low body mass index. European Respiratory J 2007; 29:643-50; PMID:17182653; https://doi.org/ 10.1183/09031936.00086306 [DOI] [PubMed] [Google Scholar]
- [18].Gosker HR, Hesselink MK, Duimel H, Ward KA, Schols AM. Reduced mitochondrial density in the vastus lateralis muscle of patients with COPD. Eur Respir J 2007; 30:73-9; PMID:17428811; https://doi.org/ 10.1183/09031936.00146906 [DOI] [PubMed] [Google Scholar]
- [19].Meyer A, Zoll J, Charles AL, Charloux A, de Blay F, Diemunsch P, Sibilia J, Piquard F, Geny B. Skeletal muscle mitochondrial dysfunction during chronic obstructive pulmonary disease: central actor and therapeutic target. Exp Physiol 2013; 98:1063-78; PMID:23377494; https://doi.org/ 10.1113/expphysiol.2012.069468 [DOI] [PubMed] [Google Scholar]
- [20].Puente-Maestu L, Perez-Parra J, Godoy R, Moreno N, Tejedor A, Gonzalez-Aragoneses F, Bravo JL, Alvarez FV, Camano S, Agusti A. Abnormal mitochondrial function in locomotor and respiratory muscles of COPD patients. European Respiratory J 2009; 33:1045-52; PMID:19129279; https://doi.org/ 10.1183/09031936.00112408 [DOI] [PubMed] [Google Scholar]
- [21].Puente-Maestu L, Lazaro A, Humanes B. Metabolic derangements in COPD muscle dysfunction. J Applied Physiol 2013; 114:1282-90; PMID:23288549; https://doi.org/ 10.1152/japplphysiol.00815.2012 [DOI] [PubMed] [Google Scholar]
- [22].Belvisi MG, Hele DJ. Peroxisome proliferator-activated receptors as novel targets in lung disease. Chest 2008; 134:152-7; PMID:18628217 [DOI] [PubMed] [Google Scholar]
- [23].Remels AH, Gosker HR, Schrauwen P, Langen RC, Schols AM. Peroxisome proliferator-activated receptors: a therapeutic target in COPD? European Respiratory J 2008; 31:502-8; PMID:18310397; https://doi.org/ 10.1183/09031936.00068207 [DOI] [PubMed] [Google Scholar]
- [24].Kramer DK, Ahlsen M, Norrbom J, Jansson E, Hjeltnes N, Gustafsson T, Krook A. Human skeletal muscle fibre type variations correlate with PPAR alpha, PPAR delta and PGC-1 alpha mRNA. Acta Physiologica 2006; 188:207-16; PMID:17054660; https://doi.org/ 10.1111/j.1748-1716.2006.01620.x [DOI] [PubMed] [Google Scholar]
- [25].Busiello RA, Savarese S, Lombardi A. Mitochondrial uncoupling proteins and energy metabolism. Frontiers Physiol 2015; 6:36; PMID:25713540; https://doi.org/ 10.3389/fphys.2015.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ramsden DB, Ho PW, Ho JW, Liu HF, So DH, Tse HM, Chan KH, Ho SL. Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behavior 2012; 2:468-78; PMID:22950050; https://doi.org/ 10.1002/brb3.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol 2001; 166:7543-8; https://doi.org/ 10.4049/jimmunol.166.12.7543 [DOI] [PubMed] [Google Scholar]
- [28].Remels AH, Gosker HR, Langen RC, Polkey M, Sliwinski P, Galdiz J, van den Borst B, Pansters NA, Schols AM. Classical NF-kappaB activation impairs skeletal muscle oxidative phenotype by reducing IKK-alpha expression. Biochim Et Biophys Acta 2014; 1842:175-85; PMID:24215713; https://doi.org/ 10.1016/j.bbadis.2013.11.001 [DOI] [PubMed] [Google Scholar]
- [29].Sjostrom M, Angquist KA, Bylund AC, Friden J, Gustavsson L, Schersten T. Morphometric analyses of human muscle fiber types. Muscle Nerve 1982; 5:538-53; PMID:6292711; https://doi.org/ 10.1002/mus.880050708 [DOI] [PubMed] [Google Scholar]
- [30].Picard M, Godin R, Sinnreich M, Baril J, Bourbeau J, Perrault H, Taivassalo T, Burelle Y. The mitochondrial phenotype of peripheral muscle in chronic obstructive pulmonary disease: disuse or dysfunction? Am J Respir Crit Care Med 2008; 178:1040-7; PMID:18755922; https://doi.org/ 10.1164/rccm.200807-1005OC [DOI] [PubMed] [Google Scholar]