

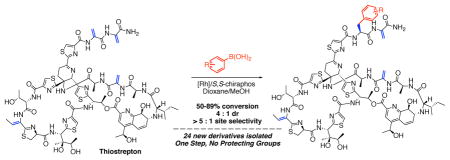
Abstract
The synthesis of complex, biologically active molecules by catalyst-controlled, selective functionalization of complex molecules is an emerging capability. We describe the application of Rh-catalyzed conjugate arylation to the modification of thiostrepton, a complex molecule with potent antibacterial properties for which few analogs are known. By this approach, we achieve the site- and stereoselective functionalization of one subterminal dehydroalanine residue (Dha16) present in thiostrepton. The broad scope of this method enabled the preparation and isolation of 24 new analogs of thiostrepton, the biological testing of which revealed that the antimicrobial activity of thiostrepton tolerates the alternation of Dha16 to a range of amino acids. Further analysis of this Rh-catalyzed process revealed that use of sodium or potassium salts was crucial for achieving high stereoselectivity. The catalyst system was studied further by application to the synthesis of amino esters and amides from dehydroalanine monomers, a process which was found to occur with up to 93 : 7 er under conditions milder than those previously reported for analogous reactions. Furthermore, the addition of the same sodium and potassium salts as applied in the case of thiostrepton lead to a nearly full reversal of the enantioselectivity of the reaction. As such, this study of site-selective catalysis in a complex molecular setting also delivered synergistic insights in the arena of enantioselective catalysis. In addition, these studies greatly expand the number of known thiostrepton analogs obtained by any method and reveal a high level of functional group tolerance for metal-catalyzed, site-selective modifications of highly complex natural products.
Graphical Abstract
Introduction
Catalyst-controlled, selective functionalization of highly complex natural products is an emerging approach to generate new compounds with potential applications as pharmaceuticals or tools to interrogate the biological function of the parent molecule.1 Although efficient in principle, this approach necessarily confronts a series of challenges, including (a) the compatibility of the reagents and reaction conditions with a highly functionalized substrate and (b) the site- and stereoselectivity with which reactions occur.1 Nonetheless, a growing number of reports have demonstrated the potential to achieve catalyst-controlled, site- and, when applicable, stereoselective functionalizations of several functional groups present in biologically relevant compounds.2
Thiostrepton (1, Fig. 1) is a canonical, complex, and highly potent antibiotic containing multiple, potentially reactive functional groups.3 Despite being readily available by fermentation, thiostrepton has seen limited use as a pharmaceutical due to poor pharmacological properties. Since its initial discovery in 1955,4 the biosynthesis5 and chemical synthesis6–8 of thiostrepton have been described, but only a few derivatives have been reported.9–11 The complexity of 1, exemplified in part by its three dehydroalanines, one dehydrothreonine, four thiazoles, one thiazoline, three isolated hydroxyl groups, a diol, an amine, an imine, and a highly-substituted pyridine, may contribute to the paucity of synthetic, semisynthetic, or biosynthetic derivatives that have been prepared and studied. Consequently, thiostrepton is a scientifically heuristic platform for the investigation of late-stage, site-, and stereoselective functionalization.
Figure 1.

The structure of thiostrepton 1 (top) and the suitability of dehydroalanine (Dha) for late stage chemical editing by Rh-catalyzed conjugate addition (bottom). The Dha residues in thiostrepton are shown in blue and are numbered in accordance with the peptide precursor. Strategies to incorporate Dha from amino acids are also shown.
The three dehydroalanine motifs within 1 are intriguing sites for selective functionalization (Fig. 1). Dehydroalanines are found in numerous medicinally important compounds including lantibiotics, thiopeptides, and other highly modified peptides of microbial origin.12,13
Moreover, in addition to their natural occurrence, dehydroalanines can be incorporated into complex molecules and proteins by β-elimination of serine-OH or cysteine-SH functional groups.14 Unlike the canonical twenty amino acids, dehydroalanine is electrophilic, making it suitable for selective functionalization even within the complex environment of a natural product, peptide, or protein.15,16 As such, a strategy for catalytic, nucleophilic addition to dehydroalanines in a complex environment would enable post-synthetic, catalyst-controlled chemical editing of many naturally-occurring compounds (Fig. 1).
With this objective, we aimed to apply Rh-catalyzed conjugate addition to functionalize selectively the dehydroalanines present in thiostrepton. While more commonly employed to create a β-stereocenter from an unsaturated carbonyl, in a few cases stereoselective, Rh-catalyzed conjugate addition to a dehydroalanine motif has been achieved, resulting in the formation of an α-stereocenter by asymmetric protonation of a Rh-enolate.17–23 Although reported catalytic methods require high reaction temperatures and specific substrates and reagents to achieve high yields and enantioselectivities, we hypothesized that further development of this reaction could make it suitable for both site- and diastereoselective editing of complex molecules containing dehydroalanines. Herein, we report a rhodium catalyst that operates under mild conditions and that controls both the site and the diastereoselectivity of the conjugate arylation of diverse boronic acids to thiostrepton. Furthermore, the application of these same reaction conditions to the asymmetric synthesis of protected amino acids from dehydroalanine monomers also led to the collateral discovery of a rare example of enantio-divergence owing to an achiral additive.
Results and Discussion
Site-Selective Arylation of Thiostrepton
To develop a Rh-catalyzed conjugate addition of boronic acids to thiostrepton, we began with reactions conditions typical for Rh-catalyzed conjugate addition reactions. We found that the reaction between 4-methoxyphenyl boronic acid and thiostrepton 1 at 50 °C using [Rh(nbd)2]BF4/rac-BINAP as catalyst led to the formation of two classes of products: products 2A/2A’ and 2B/2B’, resulting from conjugate additions to either Dha16 or Dha17, respectively, and products 3 and 3’, arising from a documented, base-mediated cleavage of Dha16 and/or Dha17 from thiostrepton (Fig. 2a).9 Using either KOH or Et3N as base, the major product observed is compound 3 (37–43% conversion by UPLC, Fig. 2b, entries 1–2). Smaller and roughly equal amounts of 2A and 2B also form, each as a mixture of two diastereomers. The site isomers 2A/2A’ and 2B/2B’ were assigned by 1D and 2D NMR analysis, which included a consideration of the chemical shifts of the signals from the carbonyl carbons in the remaining Dha residue of the product, as well as the correlations observed in 1H–13C HMBC spectra (see section III of the Supporting Information for a detailed explanation). Due to the significant amount of base-mediated cleavage that occurred under the initial reaction conditions, we investigated alternative bases that were either sterically hindered amines or non-nucleophilic inorganic bases. We found that K2CO3 delivers both higher conversion to 2A/2A’ and 2B/2B’ (16% total) and lower conversion to product 3 (25%) than were observed in the analogous reactions containing either KOH or Et3N as base (Fig. 2b, entry 3). Moreover, these catalytic conditions lead to 3 : 1 site-selectivity favoring functionalization of the subterminal Dha16 over that of terminal Dha17. This selectivity is alternate to that reported for un-catalyzed conjugate additions of thiols to thiostrepton, which are reported to occur at the terminal alkene.10 To our knowledge, there are no previously methods known to functionalize selectively the subterminal Dha (Dha16) of thiostrepton. When the reaction in the presence of K2CO3 is conducted at room temperature, the ratio of conjugate addition products to cleavage products further increases to approximately 1 : 1.5 (Fig. 2b, entry 4), although the overall conversion decreases slightly. The catalyst derived from the enantiomerically pure ligand S,S-BINAP affords more of the conjugate addition products than are produced by the racemic ligand, but overall, the yields of all reactions conducted using BINAP were low (Fig. 2b, entry 5).
Figure 2.

Conjugate additions of boronic acids to thiostrepton. a. Scheme depicting the products 2A, 2B, and 3 observed in the reaction using 4-OMe-phenyl boronic acid. Products 2A/2A’ and 2B/2B’ are pairs of diastereomers. Products 3 and 3’ arise from base mediated cleavage of the Dha residues. b. Tabulated results of the reaction depicted in Fig. 2a under varied conditions. Reaction temperatures at or above 80 °C resulted in significant decomposition of the starting material without product formation. Conversions to particular products were determined by UPLC using uncorrected areas. Conversion to 2A and 2B are the sum of the areas due to the two diastereomers of each product. The two diastereomers of 2A can be separated by UPLC. The two diastereomers of 2B co-elute as a single peak. See Fig. 2c and Supporting Information for site assignment. c. Representative UPLC trace of the crude reaction mixture formed under the conditions described in Entry 9. d. Thiostrepton analogs prepared by Rh-catalyzed conjugate arylation. Reaction conditions are as described in Fig. 2a. Tabulated conversions describe the sum of the conversion to the monoarylation products with the site selectivity as listed (see sections III–IV of the Supporting Information for details). *Products arising from reaction at Dha16 and Dha17 could not be separated completely. e. Representative UPLC traces demonstrating the purity of the products obtained by preparative HPLC and submitted for biological testing (see Table 1).
To identify a more active and selective catalyst, we evaluated ~20 chiral and achiral phosphine and diene ligands in place of BINAP (representative examples; Fig. 2b, entries 6–9; see section VII of the Supporting Information for additional results). Catalysts derived from most of the ligands that were evaluated afford little to no conjugate addition products. Of note, while the aforementioned BINAP-based ligands form the conjugate addition product 2A with up to 3:1 site-selectivity, the analogous reaction conducted with DPPB as the ligand enables a reversal of site-selectivity; Product 2B forms as the major product (1 : 5, 2A : 2B), albeit with only 4% conversion (Fig. 2b, entry 6). In sharp contrast to reactions conducted with any other ligand, reactions conducted with S,S-Chiraphos (Fig. 2b, entries 8) occur with moderate conversion to the conjugate addition products (45%), 8 : 1 site-selectivity for subterminal Dha16 over Dha17 and 4 : 1 chemoselectivity for conjugate addition over tail cleavage in reactions conducted at room temperature. At 50 °C (Fig. 2b, entry 9), the conjugate addition reaction proceeds to 67% conversion without loss of site or chemoselectivity. In a single purification, the four monoarylation products of the reaction under these conditions were isolated in 61% combined yield, revealing a high correlation between the conversion determined by UPLC (67%) and the isolated yield (61%). The four individual monoarylation products were purified by reverse phase preparative HPLC (See section III of the Supporting Information).
With these reaction conditions in hand, we evaluated the scope of the reaction (Fig 2d). A wide tolerance for boronic acids was observed, with the site-selectivity for Dha16 maintained in all cases (see section IV of the Supporting Information for details). Electron-neutral and electron-poor boronic acids were generally found to be more reactive than was 4-OMe phenyl boronic acid. The reaction of 3-CF3-phenylboronic acid forms the conjugate addition products 5 in 92% conversion at room temperature with only 1.5 equiv. of boronic acid. The combined monoarylation products of this reaction were obtained in 70% isolated yield. Ortho-, meta-, and para-substituted arylboronic acids were tolerated in the reaction, which was also suitable for the incorporation of heteroarenes into the thiostrepton scaffold. Finally, the dansylamide fluorophore can be incorporated into thiostrepton using this conjugate addition strategy, forming compound 14. Although initial experiments were performed at high catalyst loading, we evaluated the reaction of electron neutral (4-F) and electron poor (3-CF3) phenylboronic acids at lower catalyst loading and found that with these substrates, the reactions proceed to high conversion (75% and 78%, respectively) at 50 °C using only 6.25 mol% catalyst. Together, these results demonstrate the possibility to generate a wide range of derivatives of a complex molecule by Rh-catalyzed conjugate addition. However, little to no diastereoselectivity was observed, with all products reported in Fig. 2 forming as an approximately equal mixture of two diastereomers. Intrinsically, there remains value in this type of unselective outcome, because the individual diastereomers of the products formed from 11 of the 12 boronic acids shown in Fig. 2d were separable, enabling testing of their biological properties (vide infra).
Stereoselective Arylation of Thiostrepton
Having achieved catalyst-controlled site-selectivity, we turned our attention to modifying the reaction conditions to achieve catalyst-controlled stereoselectivity. This goal confronts at least two significant challenges. First, although the targeted transformation is diastereoselective, the reaction occurs on a flexible region of the molecule that lacks proximal stereo-centers. Thus, substrate-controlled diastereoselectivity is unlikely, and indeed under the conditions employed in Fig. 3, all products formed in ~1:1 dr. The second issue concerns catalyst control. Related precedents involving enantioselective conjugate addition of boronic acids to simple dehydroalanine substrates identify asymmetric protonation of an intermediate Rh-enolate as the enantio-determining step.17,19,24 Solutions to the general problem of enantioselective protonation of Rh-enolates rely on stoichiometric, acidic additives (such as substituted phenols or phthalamides) in place of protic solvents to achieve high enantioselectivities.17 These approaches have been applied specifically to the synthesis of protected amino acids from dehydroalanines, and high enantioselectivities and yields have been achieved. However, in addition to the use of stoichiometric organic additives, specialized ligands (difluorophos or bulky bidentate phosphites), and rarer aryl trifluoroborates, the reactions require high temperatures (>100 °C). This in particular implied that a direct implementation of literature conditions was not possible for the complex structure of 1.
Figure 3.

Reaction conditions for diastereoselective conjugate addition of boronic acids to thiostrepton. a. Model reaction used to develop a diastereoselective process. b. Outcome of the model reaction conducted under varied reaction conditions. c. HPLC traces of crude reactions conducted under aqueous conditions (described in Fig. 2) and methanolic conditions (Fig. 3b, entry 9). d. Outcome of diastereoselective reactions using varied boronic acid substrates. Reactions were conducted under conditions shown in Fig. 3b, entry 9. UPLC traces of crude reactions are shown to exemplify the change in diastereoselectivity in the presence of each ligand. Conversions are based on integration of the UPLC traces using uncorrected areas. Stereochemistry is depicted based on analogy to the stereochemical outcome of reactions of Dha monomers conducted under comparable conditions (vide infra).
We sought to define reaction conditions that would lead to stereoselectivity through evaluation of the reaction between thiostrepton and 4-F-phenylboronic acid in the absence of water and in the presence of various alternative proton sources (Fig. 3a). We found that only trace product forms in reactions containing either 2-methoxyphenol or phthalamide as acids (Fig. 3b, entries 1–4). In contrast, using isopropanol (10 vol %) in place of water, the conjugate addition products form with 6:1 site-selectivity (4A: 4B), with the major site isomer 4A forming in 4:1 dr (Fig. 3b, entry 5). However, only 11% conversion to conjugate addition products was observed, and further efforts to improve the conversion by modifying other reaction parameters were not successful. Using methanol in place of isopropanol (Fig. 3b, entries 7), we found that the conversion improves to 62% with a modest decrease in diastereoselectivity (3 : 1 dr) compared to that observed when using isopropanol (4 : 1 dr).
Given this improvement in conversion when using MeOH, we re-evaluated the effect of the base on the diastereoselectivity of the reaction under methanolic conditions (Fig. 3b, entries 6–10), and we found that only sodium and potassium bases afford diastereoselectivity, while reactions containing amine, cesium, and rubidium bases form the product with ~1 : 1 dr. Sodium carbonate was identified as the most suitable base for the reaction (Fig. 3b, entry 9); in the presence of 0.4 equiv. of Na2CO3, the reaction occurs with 84% conversion, 9:1 site selectivity, and 4: 1 diastereoselectivity for the formation of the major site isomer (Fig. 3b–c). Although the assignment of the absolute stereochemistry of 4A and 4A’ (and the other analogs that were prepared) is challenging and remains an ongoing effort in our laboratory, the stereochemistry depicted in Figure 3 is tentatively assigned on the basis of preliminary studies that established the absolute stereochemical outcome of reactions of dehydroalanine monomers conducted under the same catalytic conditions (vide infra).
Under the same conditions, we evaluated the selectivity of conjugate additions reactions between thiostrepton and other boronic acids (Fig. 3d and section IV of the Supporting Information). These studies revealed that electron-rich and electron-poor, ortho-, meta-, and para-substituted, and heteroaromatic boronic acids all react with comparable levels of diastereoselectivity. In contrast, reactions catalyzed by the opposite enantiomer of the catalyst (R,R-chiraphos) occur with conversions that are 2–3 times lower and with negligible diastereoselectivity, suggesting a mismatched effect between this enantiomer of the catalyst and the chiral structure of thiostrepton. However, we found that reactions containing the pseudoenantiomeric ligand R,R-catASium D proceeded in higher conversions and with higher selectivities than the analogous reactions containing R,R-Chiraphos; reactions containing R,R-catASium D formed the opposite diastereomer of the products compared to those formed in reactions containing S,S-chiraphos (Fig. 3d). The least reactive heteroaryl boronic acids did not react in the presence of catalysts formed from R,R-catASium D. The UPLC traces of crude reactions shown in Figure 3d exemplify catalyst-controlled selection for the different thiostrepton diastereomers formed during conjugate arylation.
To probe the effect of the base on the diastereoselectivity, we conducted the reaction to form thiostrepton analog 4 in the presence of either NaCl or KCl salts in places of their analogous carbonate bases, and we found that these salts effected the reaction with comparable levels of diastereoselectivity to that achieved with the carbonate bases, albeit with somewhat lower yields (Fig. 3b, entries 10–11). This suggests that the sodium or potassium cations may play a role in organizing the substrate and/or the catalyst to favor the formation of one diastereomer.
Implications for Enantioselective Catalysis
Having observed this effect in the case of a diastereoselective reaction of a complex molecule, we hypothesized that it could also be influential in enantioselective reactions of simpler substrates. We thus explored analogous conjugate addition reactions using protected Dha monomers in place of thiostrepton. The effect of the salt was found to be substantial (Fig. 4a). The omission of the alkali bases or salts from the reaction lead to nearly a full reversal of the enantioselectivity of the reaction. The addition of 1 equiv. of Na2CO3 to the reaction lead to the formation of the product with 98% conversion and 91:9 er in favor of the (S)-enantiomer (Fig 4b, entry 3). The reaction conducted in the absence of any additive occurred with 83:17 er in favor of the (R)-enantiomer (Fig 4b, entry 6). The reversal of selectivity was found to be conserved across several boronic acid and dehydroalanine substrates (Fig. 4c.)
Figure 4.

Outcome of conjugate addition reactions between protected Dha monomers and aryl boronic acids. Absolute configurations are assigned by comparison to literature data or by analogy (See Supporting Information for details) a. Observation of reversal of enantioselectivity in reactions conducted with and without additive. b. Analysis of the influence of varied cation sources on the outcome of the conjugate addition reaction. c. Demonstration of reversal of enantioselectivity in the presence of varied substrates. d. Synthesis of protected amino esters and amino amides using the Rh-catalyzed conjugate addition methods shown in Fig. 4a. Reactions were conducted with [Rh(cod)2]BF4 in place of [Rh(nbd)2]BF4.
This surprising reversal of enantioselectivity sheds light on the challenge to develop reactions relying on asymmetric protonation of Rh-enolates and complements prior work that found potassium aryl-trifluoroborates, rather than boronic acids, to be required for high enantioselectivity in analogous reactions.19,25 Ongoing studies in our laboratory seek to understand further these observations.
In addition to offering insight into the diastereoselectivity of reactions of thiostrepton, we were intrigued by the high level of enantioselectivity achieved in this reaction, given the limited number of catalyst systems known for enantioselective conjugate addition of boronic acids. A limited optimization of the reaction conditions revealed that [Rh(cod)2]BF4 afforded slightly higher selectivity than [Rh(nbd)2]BF4 (93 : 7 versus 91 : 9 er) in the model reaction between 4-F phenylboronic acid and Ac-Dha-OMe. Using these conditions, we prepared a series of additional amino ester and amino amide compounds (Fig. 4d).
Biological Activity of Thiostrepton Analogs
The aggregate of these studies enabled preparative semi-synthesis of 24 thiostrepton derivatives, including the first derivatives that are selectively functionalized at subterminal Dha16. This set of thiostrepton analogs significantly expands the number of known analogs of this highly complex natural product. Nineteen of the analogs were evaluated as antimicrobial agents against nine resistant and non-resistant organisms (see Supporting Information section XIII for complete data set). The majority of the analogs that were tested retained levels of activity that were within an order of magnitude of that of thiostrepton (Table 1). Many analogs retained activity that was within a factor of two of the level observed for thiostrepton (bolded entries, Table 1), with 2A’ having the highest activity. The data also show that the stereochemistry of the newly formed amino acids does affect the activity, with the magnitude of the difference in activity between the two diastereomers typically found to be 2–4 fold. The least active analogs were found to be those for which the Dha was modified by a very bulky or hydrophobic amino acid (such as 8A/A’ and 14A/A’). Together, these results show that conjugate arylation of Dha 16 may be a suitable strategy for modification of thiostrepton in order to improve the pharmacological properties of the molecule without inherent loss of activity.
Table 1.
Biological Activity of Analogs of Thiostrepton (See Supporting Information for details and additional data)
| Compound (Ar substituent) | MIC (ug/mL) for a range of organisms (VSE = Vancomycin susceptible, VRE = vancomycin resistant) | |||||
|---|---|---|---|---|---|---|
| S. aureus MRSA | E. faecalis VSE | E. faecalis VRE | E. faecium VRE | S. pneumoniae PSSP | S. pyogenes | |
| Thiostrepton 1 | 0.25 | 0.5 | 0.25 | 0.12 | 0.002 | 0.002 |
| Vancomycin | 1 | 2 | >32 | >32 | 0.25 | 0.5 |
| 2A (4-OMe) | 1 | 1 | 1 | 1 | 0.008 | 0.004 |
| 2A’ (4-OMe) | 0.5 | 1 | 0.5 | 0.25 | 0.002 | 0.002 |
| 4A (4-F) | 1 | 1 | 1 | 0.5 | 0.008 | 0.008 |
| 4A’ (4-F) | 2 | 2 | 1 | 1 | 0.015 | 0.008 |
| 5A (3-CF3) | 2 | 1 | 1 | 1 | 0.008 | 0.004 |
| 5A’ (3-CF3) | 2 | 1 | 1 | 0.5 | 0.008 | 0.004 |
| 6A (4-Cl) | 2 | 1 | 1 | 1 | 0.015 | 0.004 |
| 6A’ (4-Cl) | 2 | 2 | 1 | 1 | 0.008 | 0.004 |
| 7A (4-Me) | 2 | 1 | 0.5 | 0.5 | 0.004 | 0.002 |
| 8A (4-′Bu) | 4 | 2 | 2 | 2 | 0.008 | 0.008 |
| 8A’ (4-′Bu) | 8 | 4 | 2 | 2 | 0.03 | 0.015 |
| 9A (4-OCF3) | 2 | 1 | 2 | 1 | 0.008 | 0.008 |
| 10A (4-Morph.) | 4 | 2 | 2 | 1 | 0.015 | 0.015 |
| 10A’ (4-Morph.) | 2 | 1 | 1 | 0.5 | 0.008 | 0.004 |
| 11A (Benzofuran) | 2 | 2 | 1 | 1 | 0.008 | 0.004 |
| 11A’ (Benzofuran) | 4 | 2 | 4 | 2 | 0.015 | 0.015 |
| 12A (Dibenzofuran) | 2 | 2 | 2 | 1 | 0.008 | 0.008 |
| 14A (Dansyl) | 32 | 4 | 8 | 2 | 0.03 | 0.03 |
| 14A’ (Dansyl) | 64 | 4 | 4 | 2 | 0.15 | 0.15 |
Conclusions
We have developed a high yielding, and site- and diastereoselective conjugate addition reaction suitable for functionalization of the subterminal dehydroalanine residue of thiostrepton. In doing so, we have demonstrated the utility of this reaction to enable post-synthetic conversion of a dehydroalanine residue to a wide range of unnatural amino acids in a highly complex molecule; from the single, fully unprotected thiostrepton substrate, twenty-four new derivatives of this antibiotic were prepared by this approach. Moreover, we found that the simple reaction conditions developed for thiostrepton were also suitable for enantioselective conjugate addition to dehydroalanine monomers, providing access to a range of unnatural amino acid derivatives in high enantioselectivity. Thus, these synergistic studies of site-selective catalysis in a complex molecular setting also delivered new fundamental insights in the arena of enantioselective catalysis. In addition to providing access to the new compounds described herein, this method should enable the preparation of analogs of many other biologically relevant molecules containing one or more dehydroalanine motifs. They also exemplify the suitability of metal catalyzed asymmetric catalysts to address problems in the tailoring of high complexity natural products. Yet, many challenges remain. These include extending our findings to the comprehensive functionalization of alternate Dha sites, the dehyrothreonine site, if not all other loci of unsaturation in the structure.
Supplementary Material
Acknowledgments
Funding Sources
We are grateful to the National Institute of General Medical Sciences of the NIH (GM-068649) for support.
We are grateful to the U.S. National Institutes of Health for support of this research.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website. Experimental methods, characterization of all compounds, and copies of spectra can be found in the Supporting Information.
References
- 1.(a) Hartwig JF. Acc Chem Res. 2017;50:549–555. doi: 10.1021/acs.accounts.6b00546. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shugrue CR, Miller S. J Chem Rev. 2017 doi: 10.1021/acs.chemrev.7b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For representative specific examples, see: Lewis CA, Miller SJ. Angew Chem Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490.Kille S, Zilly FE, Acevedo JP, Reetz MT. Nat Chem. 2011;3:738–743. doi: 10.1038/nchem.1113.Pathak TP, Miller SJ. J Am Chem Soc. 2012;134:6120–6123. doi: 10.1021/ja301566t.Lee D, Williamson CL, Chan L, Taylor MS. J Am Chem Soc. 2012;134:8260–8267. doi: 10.1021/ja302549c.Pathak TP, Miller SJ. J Am Chem Soc. 2013;135:8415–8422. doi: 10.1021/ja4038998.Han S, Miller SJ. J Am Chem Soc. 2013;135:12414–12421. doi: 10.1021/ja406067v.Rosen BR, Simke LR, Thuy-Boun PS, Dixon DD, Yu J-Q, Baran PS. Angew Chem, Int Ed. 2013;52:7317–7320. doi: 10.1002/anie.201303838.Sun X, Lee H, Lee S, Tan KL. Nat Chem. 2013;5:790–795. doi: 10.1038/nchem.1726.He J, Hamann LG, Davies HML, Beckwith REJ. Nat Commun. 2015;6:5943. doi: 10.1038/ncomms6943.Yoganathan S, Miller SJ. J Med Chem. 2015;58:2367–2377. doi: 10.1021/jm501872s.Payne JT, Poor CB, Lewis JC. Angew Chem Int Ed. 2015;54:4226–4230. doi: 10.1002/anie.201411901.Tay JH, Arguelles AJ, DeMars MD, II, Zimmerman PM, Sherman DH, Nagorny P. J Am Chem Soc. 2017;139:8570–8578. doi: 10.1021/jacs.7b03198.
- 3.Chiu ML, Folcher M, Griffin P, Holt T, Klatt T, Thompson CJ. Biochemistry. 1996;35:2332–2341. doi: 10.1021/bi952073e. [DOI] [PubMed] [Google Scholar]
- 4.Donovick R, Pagano JF, Stout HA, Weinstein MJ. Antibiot Annu. 1955;3:554–559. [PubMed] [Google Scholar]
- 5.Kelly WL, Pan L, Li C. J Am Chem Soc. 2009;131:4327–4334. doi: 10.1021/ja807890a. [DOI] [PubMed] [Google Scholar]
- 6.Nicolaou KC. Angew Chem Int Ed. 2012;51:12414–12436. doi: 10.1002/anie.201205576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicolaou KC, Safina BS, Zak M, Estrada AA, Lee SH. Angew Chem Int Ed. 2004;116:5197–5202. [Google Scholar]
- 8.Nicolaou KC, Zak M, Safina BS, Lee SH, Estrada AA. Angew Chem Int Ed. 2004;43:5092–5097. doi: 10.1002/anie.200461341. [DOI] [PubMed] [Google Scholar]
- 9.Jonker HRA, Baumann S, Wolf A, Schoof S, Hiller F, Schulte KW, Kirschner KN, Schwalbe H, Arndt HD. Angew Chem Int Ed. 2011;50:3308–3312. doi: 10.1002/anie.201003582. [DOI] [PubMed] [Google Scholar]
- 10.Myers CL, Hang PC, Ng G, Yuen J, Honek JF. Bioorg Med Chem. 2010;18:4231–4237. doi: 10.1016/j.bmc.2010.04.098. [DOI] [PubMed] [Google Scholar]
- 11.Gruppi F, Xu X, Zhang B, Tang JA, Jerschow A, Canary JW. Angew Chem Int Ed. 2012;51:11787–11790. doi: 10.1002/anie.201204403. [DOI] [PubMed] [Google Scholar]
- 12.Siodłak D. Amino Acids. 2015;47:1–17. doi: 10.1007/s00726-014-1846-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gober JG, Ghodge SV, Bogart JW, Wever WJ, Watkins RR, Brustad EM, Bowers AA. ACS Chem Biol. 2017;12:1726–1731. doi: 10.1021/acschembio.7b00358. [DOI] [PubMed] [Google Scholar]
- 14.Chalker JM, Gunnoo SB, Boutureira O, Gerstberger SC, Fernández-González M, Bernardes GJL, Griffin L, Hailu H, Schofield CJ, Davis BG. Chem Sci. 2011;2:1666–1676. [Google Scholar]
- 15.Wright TH, Ben J, Bower Chalker JM, Bernardes GJL, Wiewiora R, Ng W-L, Raj R, Faulkner S, Vallée MRJ, Phanumartwiwath A, Coleman OD, Thézénas M-L, Khan M, Galan SRG, Lercher L, Schombs MW, Gerstberger S, Palm-Espling ME, Baldwin AJ, Kessler BM, Claridge TDW, Mohammed S, Davis BG. Science. 2016;354:aag1465-1–aag1465-11. doi: 10.1126/science.aag1465. [DOI] [PubMed] [Google Scholar]
- 16.deGruyter JN, Malins LR, Baran PS. Biochemistry. 2017;56:3863–3873. doi: 10.1021/acs.biochem.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phelan JP, Ellman JA. Beilstein J Org Chem. 2016;12:1203–1228. doi: 10.3762/bjoc.12.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hargrave JD, Bish G, Köhn GK, Frost CG. Org Biomol Chem. 2010;8:5120–5125. doi: 10.1039/c0ob00158a. [DOI] [PubMed] [Google Scholar]
- 19.Navarre L, Martinez R, Genet JP, Darses S. J Am Chem Soc. 2008;130:6159–6169. doi: 10.1021/ja710691p. [DOI] [PubMed] [Google Scholar]
- 20.Tian P, Dong HQ, Lin GQ. ACS Catal. 2012;2:95–119. [Google Scholar]
- 21.Sakai Masaaki, Hiroyuki Hayashi A, Miyaura N. Organometallics. 1997;16:4229–4231. [Google Scholar]
- 22.Navarre L, Darses S, Genet JP. Angew Chem Int Ed. 2004;43:719–723. doi: 10.1002/anie.200352518. [DOI] [PubMed] [Google Scholar]
- 23.Edwards HJ, Hargrave JD, Penrose SD, Frost CG. Chem Soc Rev. 2010;39:2093–2105. doi: 10.1039/b919762c. [DOI] [PubMed] [Google Scholar]
- 24.Luo Y, Carnell AJ, Lam HW. Angew Chem Int Ed. 2012;51:6762–6766. doi: 10.1002/anie.201202136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.