

Abstract
Alkbh1 is a mammalian homolog of the Escherichia coli DNA repair enzyme AlkB, an Fe(II) and 2-oxoglutarate dependent dioxygenase that removes alkyl lesions from DNA bases. The human homolog ALKBH1 has been associated with six different enzymatic activities including DNA or tRNA hydroxylation, cleavage at abasic (AP) sites in DNA, as well as demethylation of histones. The reported cellular roles of this protein reflect the diverse enzymatic activities and include direct DNA repair, tRNA modification, and histone modification. We demonstrate that ALKBH1 produced in mammalian cells (ALKBH1293) is similar to the protein produced in bacteria (ALKBH1Ec) with regard to its m6A demethylase and AP lyase activities. In addition, we find that ALKBH1293 forms a covalent adduct with the 5′ product of the lyase product in a manner analogous to ALKBH1Ec. Localization and subcellular fractionation studies with the endogenous protein in two human cell strains confirm that ALKBH1 is primarily in the mitochondria. Two strains of CRISPR/Cas9-created ALKBH1-deficient HEK293 cells showed increases in mtDNA copy number and mitochondrial dysfunction as revealed by growth measurements and citrate synthase activity assays.
1. Introduction
AlkB, an Escherichia coli protein that catalyzes DNA repair, is an Fe(II)/2-oxoglutarate (2OG)-dependent dioxygenase. This enzyme hydroxylates 1-methyladenine (m1A) and 3-methylcytosine (m3C), releasing formaldehyde to restore the native bases [1, 2]. Mammals possess nine AlkB homologs (Alkbh1 – Alkbh8 and FTO) [3–5], but their biological functions are diverse (for references to individual proteins see [6, 7]). Alkbh2 repairs methylated DNA bases in vivo. Alkbh3 demethylates these lesions in single-stranded (ss) DNA, but additionally acts on 6-methyladenine (m6A) in tRNA. Alkbh5 and FTO also act on m6A, but using mRNA with effects on gene expression. In vivo, Alkbh5 affects mouse fertility while FTO helps determine body mass. Alkbh8 acts on tRNA by modifying 5-methyluridine in the anticodon wobble position, important in regulating translation. Alkbh4 functions in actin demethylation and plays a role in cell division. Alkbh7 localizes to the mitochondria and plays roles in programmed necrosis and fatty acid oxidation; its absence yields obese mice. Alkbh6 has no known function or substrate. Lastly, Alkbh1 (closest in sequence to AlkB) has a remarkably wide range of cellular roles and enzymatic activities.
Alkbh1 was first reported to have weak m3C demethylase activity using ssDNA [8]. Human ALKBH1 possesses m6A demethylation activity, implying a role in epigenetic gene silencing [9]. Demethylation of m1A at A58 in tRNAiMet suggests the enzyme regulates translation [10]. In mitochondria, ALKBH1 hydroxylates 5-methylcytosine (m5C) in tRNAiMet to generate 5-formylcytosine (f5C) [11], increasing translation. ALKBH1 also converts m5C to f5C in cytoplasmic tRNALeu and mt-tRNAiMet [12]. The protein also possesses apurinic/apyrimidinic (AP) lyase activity, cleaving DNA at abasic sites by a β-elimination mechanism. Surprisingly, ALKBH1 is a single-turnover lyase because it forms a covalent adduct with the 5′-product [13–15]. Lastly, ALKBH1 was reported to be a histone H2A dioxygenase involved in neural development [16]. In agreement with these diverse roles and enzymatic activities, ALKBH1 is in three compartments of the cell – the cytoplasm, the nucleus and the mitochondria [8, 16–18]. Furthermore, two murine studies showed Alkbh1−/− mice are born at a lower than expected Mendelian ratio, the number of male pups is greater than female pups, and the Alkbh1-deficient animals are smaller than their littermates [19, 20]. In addition, one early study reported that ALKBH1 plays a role in placental trophoblast lineage differentiation [17].
Most prior studies of ALKBH1 used protein generated in recombinant E. coli cells (ALKBH1Ec), raising the question of which functions occur in mammalian cells. Here, we show that ALKBH1 produced in mammalian cells (ALKBH1293) catalyzes demethylation of m6A in ssDNA and β-elimination at AP sites. We demonstrate that ALKBH1293 covalently attaches to the 5′ product of the AP lyase reaction, analogous to ALKBH1Ec. Furthermore, we show ALKBH1 is most prominent in the mitochondria and provide evidence that ALKBH1-deficient HEK293 cells exhibit mitochondrial dysfunction.
2. Materials and methods
2. 1. Cloning
ALKBH1 was PCR amplified from pBAR67 while introducing flanking BamHI and NotI restriction sites. The PCR fragment was cloned into pGEM t-easy and after partial digestion with BamHI and NotI the appropriate fragment was inserted into pLH203 (from Kefei Yu), generating pABH62 that encodes a maltose-binding protein (MBP) tagged-ALKBH1 fusion protein.
We created a genomic block fragment (gBlock, IDT DNA) containing the EcoRI-NcoI-FLAG-linker-HA sequence followed by the ALKBH1 sequence to nt 231, located in a NsiI site. The gBlock was cloned into pCR2.1 and the desired fragment was subcloned into pABH44 [20] using the EcoRI and NsiI sites. The fragment encoding FLAG-HA-tagged ALKBH1 was subcloned into pCDNA6 using EcoRI and XhoI restriction sites, creating pABH59.
2.2. Generation of ALKBH1-deficient HEK293 cells
ALKBH1-deficient human cells were generated using CRISPR/Cas9-mediated gene targeting in HEK293 cells [21]. Q5 site-directed mutagenesis was carried out using primers 5′-ACAGATCTTGGGTCTCTTCCGGTGTTTCGTCCTTTCC-3′ and 5′-GTTTTAGAGCTAGAAATAGCAAGTT-3′ along with the Cas9 targeting vector pAK1 (from Kefei Yu) to create pTAM2 containing a gRNA specific for exon 3 of ALKBH1. To generate ALKBH1-deficient HEK293 cells, pTAM2 (4 μg) was cotransfected with hCas9 (2 μg) and pSUPER-puro (0.5 μg, Oligoengine), puromycin-resistant clones were isolated, and ALKBH1 expression was examined by immunoblotting. We analyzed ALKBH1-deficient clones by PCR-amplifying a fragment in exon 3, and demonstrated the two alleles contained frameshift insertions or deletions. To create ALKBH1-deficient clones that express Flag-HA ALKBH1, clone 3–36 was transfected with pABH59 using fugene6 (Promega) and selected for blasticidin resistance (5 μg/ml, Gibco, Fisher Scientific). Single clones were isolated and ALKBH1 expression was verified by immunoblotting.
2.3. Mammalian cell culture
HEK293 and HEK293T cells were cultured at 37 °C with 5% CO2 in Dulbecco Modified Eagle Medium (DMEM) with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine, 0.1 mM non-essential amino acids, and 1 mM sodium pyruvate. To measure growth, cells growing on DMEM containing 4.5 g/l glucose were harvested, counted, and seeded at 20,000 cells per well in 24-well plates containing DMEM with 4.5 g/l glucose, 1 g/l glucose, or 1 g/l galactose. Relative proliferation was assessed by adding 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 1 mg/ml) and measuring absorbance at 570 nm [22].
2.4. Protein production and purification
HEK293T cells were plated and transiently transfected with 10 μg of plasmid and polyethylenimine (1 mg/ml, Polysciences) at 3 μl/μg of DNA per 100 mm dish. Cells were grown for 72 h, and cell lysates were prepared by resuspending in 4 ml buffer (20 mM Tris, 0.2 M NaCl, pH 7.5), sonicating, and centrifuging (12,000 rpm, 30 min, 4 °C). Cell-free lysates were loaded onto amylose resin (1×5 cm), washed, and MBP-ALKBH1293 was eluted (20 mM Tris, pH 7.5, 0.1 M NaCl, and 10 mM maltose). Purified protein was exchanged into 20 mM Tris, pH 8, with 1 mM EDTA (TE) buffer using a PD-10 disposable column (GE Healthcare Life Sciences) and concentrated using an Amicon centrifugal filter (Millipore). When indicated, the MBP-tag was cleaved with tobacco etch virus (TEV) protease. ALKBH1Ec was produced in E. coli Codon Plus RIPL cells harboring pBAR67 and purified as described previously [13, 14].
2.5. ALKBH1 enzyme activity and adduct formation assays
The m6A demethylase, AP lyase, and adduct formation assays were performed as previously described [14, 15, 20]. For m6A demethylase assays, MBP-ALKBH1293 (5 μM) was incubated with 5′-FAM-labeled oligo_m6A1 (1 μM, 5′-AACTTCGTGCAGGCATGGG(m6A)TCT-TGTCTACT-3′, Sigma-Aldrich), 1 mM Fe(NH4)2SO4, 1 mM L-ascorbic acid, and 1 mM 2OG in 50 mM bis-Tris (pH 7) at 37 °C for 1 h, annealed to 1.1 equivalents of oligo_m6A2 (5′-AGTAGACAAGATCCCATGCCTGCACGAAGTT-3′), and digested with DpnII (0.5 U/μl) for 1 h at 37 °C. We analyzed samples by polyacrylamide gel electrophoresis (PAGE) and visualized product formation using a fluorescence scanner. TEV protease cleavage produced ALKBH1293. For assays of AP lyase or adduct formation, 5′- or 3′-FAM-labeled oligo_AP1 (10 μM, 5′-AACTTCGTGCAGGCATGGTAG(dU)TTGTCTACT-3′, IDT DNA) was incubated with uracil DNA glycosylase (2 U) to create the AP site. ALKBH1Ec, MBP-ALKBH1293, and ALKBH1293 were incubated with the 3′-FAM-labeled oligo_AP1 (1 μM) in 20 mM Tris, pH 8, containing 1 mM EDTA, and incubated for 1 h at 37 °C. Reactions were quenched with 10 mM methoxyamine, products were analyzed by PAGE, and adduct formation was quantified by sodium dodecyl sulfate (SDS)-PAGE. For assessing adduct formation, ALKBH1Ec and MBP-ALKBH1293 (5 μM) were incubated with 5′-FAM labeled oligo_AP1 (1 μM) at 37 °C for 1 h and analyzed by SDS-PAGE.
2.6. Quantitative end-point PCR (qPCR)
We carried out qPCR as described [23, 24]. Genomic DNA was isolated using the DNeasy blood & tissue kit (Qiagen) with samples eluted in 100 μl. DNA concentration was determined using Quantifluor (Promega), samples were diluted to 10 ng/μl, and 3 ng/μl samples were used in the qPCR. All PCR analyses used 1 U/reaction of GoTaq polymerase and 10 mM dNTP in 1x GoGreen colorless buffer (Promega). mtDNA was amplified using primer 14620 (5′-CCCCACAAACCCCATTACTAAACCCA-3′, 300 nM) and primer 14841 (5′-TTTCATCATGCGGAGATGTTGGATGG-3′, 300 nM) by initial denaturation at 93 °C (2 min) then 19 cycles at 93 °C (30 s), annealing at 57 °C (25 s), elongation at 72 °C (45 s), and a final step at 72 °C (7 min). For each DNA sample, we used 7.5 and 15 ng DNA and measured the amplified products using Quantifluor. Results were considered to be in the exponential range for 40–60 % relative amplification. Nuclear DNA was amplified with the γ-globin primer set (Clontech) using primers GH20 and PC04. The same PCR components and parameters were utilized for nuclear DNA amplification, but with 15 and 30 ng DNA for each sample and increasing to 24 cycles.
2.7. Immunoblotting
Following SDS-PAGE proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon-P, Millipore Sigma). The membranes were incubated with monoclonal anti-hALKBH1 (Abcam), anti-GAPDH antibody (Genetex), or polyclonal anti-Cox4 (Genetex), either at 4 °C overnight or at room temperature for 1 h in 3% non-fat dry milk in phosphate buffered saline (PBS) containing 0.1% Tween 20 (PBST), washed, incubated with rabbit anti-mouse IgG peroxidase antibody (Millipore Sigma) or goat anti-rabbit IgG peroxidase antibody (Millipore Sigma), washed, and developed using Clarity ECL™ Western blotting substrate (Bio-Rad).
2.8. Immunocytochemistry and subcellular fractionation
Cells were incubated on glass chamber slides for 24 h, treated with MitoTracker Red CMXRos (Invitrogen) for 30 min at 37 °C, washed with PBS, fixed in 4% paraformaldehyde (1 h at room temperature), washed, incubated with 50 mM NH4Cl in PBS (15 min at room temperature), permeabilized (0.1% NP-40 in buffer A, 0.25% gelatin, and 0.01% saponin in PBS) for 1 h, and washed again. Cells were blocked with PBS containing 1% bovine serum albumin (BSA) and 10% normal goat serum (Invitrogen) for 1 h, incubated with anti-ALKBH1 antibody in PBS containing 1% BSA (1:500) overnight at 4 °C, washed with PBS for 10 min, and incubated with goat anti-mouse IgG Alexa Fluor 488 (Invitrogen) for 1 – 2 h in the dark. The sample was incubated with 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI) stain and washed, anti-fade was added, and the slides were dried overnight at 4 °C. Mitochondria were isolated using the Qproteome Mitochondria Isolation kit (Qiagen).
2.9. CS activity assays
Cells were grown in medium containing either 4.5 g/l glucose, 1 g/l glucose, or 1 g/l galactose for one passage before plating 1×106 cells, incubated for 3 d (4.5 g/l glucose) or 6 d (1 g/l glucose and galactose), and harvested. Cell lysates were prepared in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate) containing protease inhibitor (Roche), incubated (37 °C, 5 min), and centrifuged (17,000 g, 30 min, 4 °C). CS assays (Sigma technical bulletin) used 40–80 μg of protein in each reaction and were monitored by using a Shimadzu spectrophotometer.
3. Results
3.1. MBP-ALKBH1293 has m6A demethylase and AP lyase activities similar to ALKBH1Ec
We previously showed that His-tagged ALKBH1Ec possessed low m6A demethylase activity using ssDNA and cleaved 3′ of abasic sites in ssDNA and double-stranded (ds) DNA via β-elimination [13–15]. Here, we tested whether ALKBH1293 also exhibits these activities. Purified MBP-ALKBH1293 exhibited one band at the expected size (85 kDa) and two smaller signals (Fig. 1a) that, according to anti-ALKBH1 antibody cross-reactivity (not shown), are degradation products.
Fig. 1. Enzymatic activities of mammalian cell-produced ALKBH1.
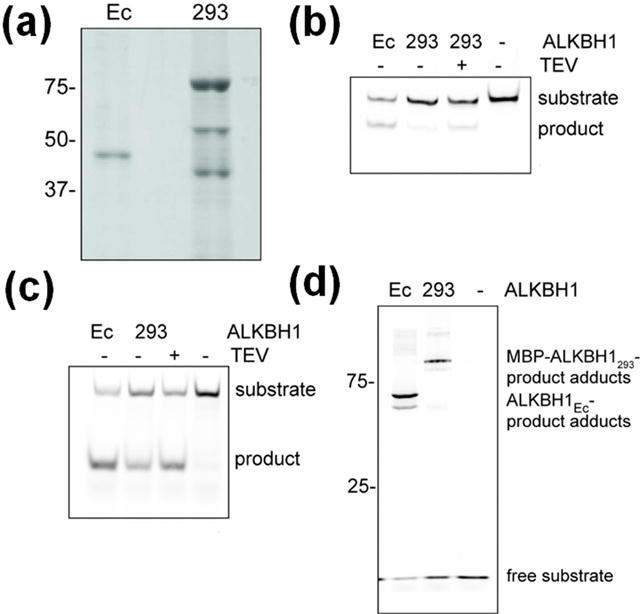
(a) Purified MBP-ALKBH1293 analyzed by SDS-PAGE, with purified ALKBH1Ec used as a control. (b) The m6A demethylase activities of ALKBH1Ec, MBP-ALKBH1293, and ALKBH1293 determined by incubating with m6A-containing oligonucleotide, annealing to the complementary oligonucleotide, digesting with DpnII, and analyzing by PAGE and a fluorescence scanner. (c) The AP lyase activity of ALKBH1Ec, MBP-ALKBH1293, and ALKBH1293 determined by incubating with a 3′-fluorescently-labeled oligonucleotide and analyzing as in panel b. (d) Adduct formation associated with AP lyase activity of ALKBH1Ec and MBP-ALKBH1293 examined using a 5′-fluorescently-labeled oligonucleotide and analyzed as for panel b.
To examine the m6A demethylase activity of MBP-ALKBH1293, the protein was incubated with a methylated oligonucleotide in the presence of ferrous iron and 2OG. The MBP-ALKBH1293 hydroxylated m6A in ssDNA leading to demethylation, but it generated less than 5% of the product made by ALKBH1Ec (Fig. 1b). To test whether the fused MBP domain was responsible for the low activity, the MBP-tag was cleaved using TEV protease. Product formation increased approximately 3-fold to ~15% of that made by ALKBH1Ec, indicating the MBP domain impairs DNA hydroxylation (Fig. 1b).
To test the AP lyase activity of MBP-ALKBH1293, enzyme was incubated with a 3′-fluorescently-labeled oligonucleotide containing one AP site and DNA cleavage was assessed by PAGE. MBP-ALKBH1293 cleaved the AP-containing DNA (Fig. 1c), but the activity was slightly lower (57 ± 10%) than that of ALKBH1Ec. After removing the MBP-tag, AP lyase activity increased (to 70 ± 21% of ALKBH1Ec) showing that the MBP tag interferes with this activity (Fig. 1c). Thus, ALKBH1293 catalyzes the β-elimination reaction at a level similar to ALKBH1Ec.
3.2. The MBP-ALKBH1293 forms an adduct with the α,β-unsaturated aldehyde from AP cleavage
Unusual for an AP lyase, ALKBH1Ec is a single-turnover enzyme that forms an adduct with the α,β-unsaturated aldehyde 5′ product of the β-elimination reaction [14, 15, 20]. We tested whether ALKBH1293 catalyzes the same covalent attachment reaction. When MBP-ALKBH1293 was incubated with 5′-fluorescently-labeled oligonucleotide with an AP site, adduct formation occurred at levels similar to those formed by ALKBH1Ec (Fig. 1d), suggesting that covalent adduct generation is inherent to ALKBH1 and may have a biological function.
3.3. ALKBH1 localizes to the mitochondria in HEK293 cells
We re-examined the subcellular localization of ALKBH1, focusing on the endogenous protein in different mammalian cell strains. Prior studies have shown ALKBH1 expression in multiple cellular compartments [8, 11, 16, 18], consistent with roles in nuclear DNA repair, cytoplasmic translation through tRNA demethylation, and mitochondrial protein synthesis via hydroxylation of mt-tRNA [8–12], but these studies relied heavily on tagged proteins and HeLa cells. Immunostaining of ALKBH1 in murine Alkbh1−/− fibroblasts expressing human ALKBH1 revealed colocalization with mitotracker, consistent with a predominantly mitochondrial location (Fig. 2a). To corroborate these findings, we performed cell fractionation with MSU1.1 and HEK293 human cells. In both cells, ALKBH1 is predominantly found in the mitochondria (Fig. 2b–d). These data confirm that endogenous ALKBH1 localizes to the mitochondria, consistent with some previous studies [8, 11].
Fig. 2. Subcellular localization of human ALKBH1.
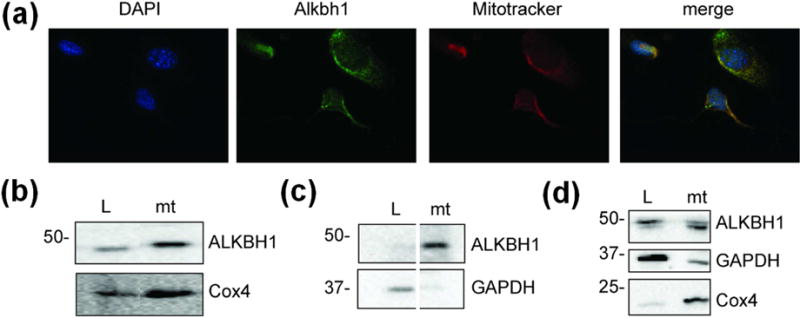
(a) Immunocytochemistry with murine Alkbh1−/− fibroblasts expressing ALKBH1. The DAPI-stained DNA is blue, ALKBH1-bound FITC-conjugated antibody is green, and the mitotracker-localized mitochondria are red. (b) Immunoblot using anti-ALKBH1 and anti-Cox4 antibodies with subcellular fractions from MSU1.1 cells. (c) Immunoblot using anti-ALKBH1 and anti-GAPDH antibodies as in panel b, to confirm the separation of cytoplasmic and mitochondrial proteins (samples are from nonadjacent lanes of the same gel). (d) Subcellular localization of ALKBH1 in HEK293T cells. L, Whole-cell lysate; mt, mitochondria.
3.4. ALKBH1 is required for normal cell proliferation
Since deficits in mitochondrial metabolism can have profound effects on cell proliferation, we tested growth effects of ALKBH1-deficiency. We created an ALKBH1-deficient HEK293 strain using CRISPR/Cas9 technology, targeting exon 3 as in an Alkbh1−/− mouse [20]. We screened clones by immunoblotting (Fig. 3a), confirmed targets by sequencing, and selected two clones (3–13 and 3–36) for further studies.
Fig. 3. Representative growth curves of ALKBH1-producing and ALKBH1-deficient HEK293 cells.
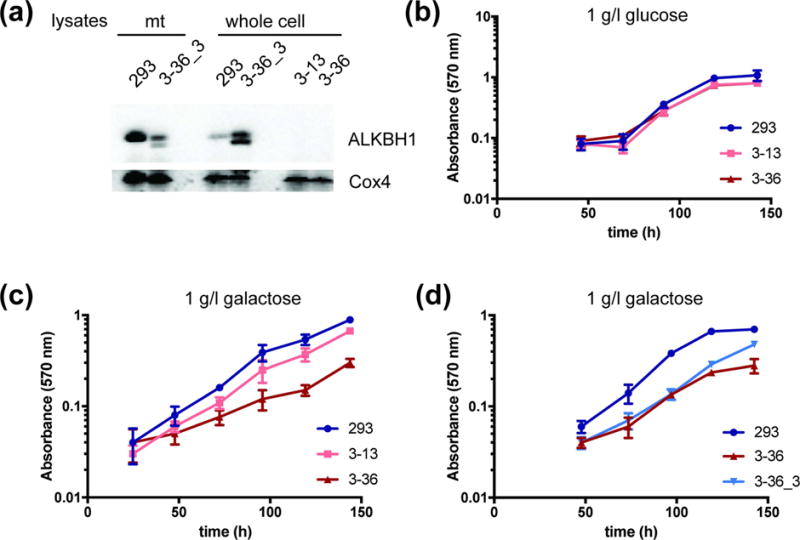
(a) ALKBH1 expression in mitochondria and whole-cell lysates of HEK293, ALKBH1-deficient (3–13, 3–36), or Flag-HA-tagged ALKBH1 producing (3–36_3) strains. ALKBH1 expression (endogenous and flag-tagged) was verified in single clones using anti-ALKBH1 antibody, with anti-Cox4 antibody as a loading control. (b) Proliferation of HEK293 cells and two ALKBH1-deficient clones (3–13 and 3–36) in medium containing 1 g/l glucose, measured by MTT staining. (c) Growth of WT and two ALKBH1-deficient strains on 1 g/l galactose, as in panel b. (d) Growth of WT, 3–36 and 3–36_3 cells expressing Flag-HA-ALKBH1 strains on 1 g/l galactose, as in panel b. Data are the mean of four replicates and error bars indicate SD.
Growth rates for the two ALKBH1-deficient HEK293 cell clones in standard media (4.5 g/l glucose) were not different from those producing the protein, consistent with our previous studies of Alkbh−/− murine fibroblasts which had no proliferation defects. Because high glucose concentrations can mask mitochondrial deficiency, we tested for growth on more limiting glucose (1 g/l) and galactose (1 g/l). Galactose accentuates growth deficits from mitochondrial defects by channeling ATP production through oxidative phosphorylation, thus reducing the energy yield from glycolysis [12]. We saw no differences in proliferation between ALKBH1-proficient and -deficient cells in low glucose media (Fig. 3b), whereas both ALKBH1-deficient cells strains grew slower than wild-type (WT) cells on galactose (Fig. 3c). These data are consistent with defective mitochondrial function in cells lacking ALKBH1 and confirm results by others [12]. We corroborated this conclusion by rescuing the proliferation deficit via expressing ALKBH1 in an ALKBH1-deficient strain (Fig. 3d).
3.5. ALKBH1-deficient cells exhibit mitochondrial dysfunction
As further evidence of a mitochondrial defect, we investigated whether ALKBH1-deficient cells grown on 4.5 g/l glucose, 1 g/l glucose, or 1 g/l galactose exhibit differences in activities for CS. Activity measurements with lysates from ALKBH1-deficient cells cultured in 4.5 g/l glucose showed a slight decrease of CS activity compared to lysates from WT cells, whereas the reduction was more pronounced in cells grown on 1 g/l glucose or 1 g/l galactose (Fig. 4a). The deficits in CS activity were reversed by stable expression of FLAG-ALKBH1, with levels equal to or greater than in WT HEK293 cells. We conclude that ALKBH1 deficiency in HEK293 cells results in partially dysfunctional mitochondria, confirming findings of our proliferation studies with 1 g/l galactose.
Fig. 4. CS activity and mtDNA copy number in ALKBH1-producing and ALKBH1-deficient clones.
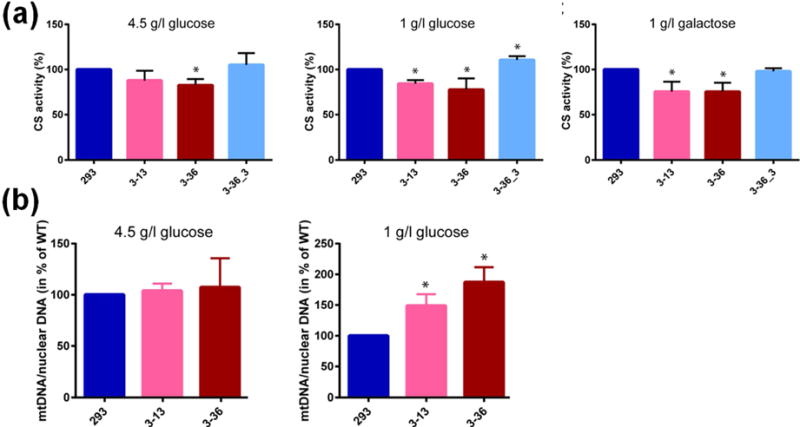
(a) CS activity was measured using lysates from ALKBH1-WT (293), ALKBH1-deficient strains (3–13, 3–36), and Flag-HA-ALKBH1-expressing complemented strain (3–36_3) grown in the media indicated. Data are the mean of three independent experiments and error bars indicate SD. *, P < 0.05 using the Student’s t-test. In the middle panel, the CS activity of strain 3–36_3 was increased over the WT cells in a statistically significant manner. (b) The mtDNA copy numbers of ALKBH1-producing and ALKBH1-deficient clones were calculated by end-point qPCR for cells grown 3 d on 4.5 g/l glucose or 6 d on 1 g/l glucose. DNA concentrations were determined as described [23, 24]. Data are the mean of three independent experiments and error bars indicate SD. *, P < 0.05 according to the Student’s t-test.
3.6. mtDNA copy numbers are increased in ALKBH1-deficient cells
We tested whether ALKBH1 deficiency also affects mtDNA copy number by quantitatively amplifying a small mtDNA fragment and a small nuclear DNA fragment using end-point qPCR and calculating the ratio of these signals [23, 24]. The ratios of WT cells compared to those of the two ALKBH1-deficient clones grown on 4.5 g/l and 1 g/l glucose demonstrate the mtDNA copy number increases in a statistically significant manner in ALKBH1-deficient cells (Fig. 4b). These data demonstrate a mitochondrial function for ALKBH1 and indicate that loss of ALKBH1 results in mitochondrial dysfunction.
4. Discussion
We have shown that ALKBH1 exhibits m6A demethylase and AP lyase activities along with the ability to form a covalent adduct with AP-containing DNA when produced in a human cell line, similar to that produced in E. coli [13–15]. The enzyme activity levels of ALKBH1293 are similar to those of ALKBH1Ec, so heterologously-produced enzyme is useful for further biochemical characterization. Our ALKBH1293 results support findings from ALKBH1Ec variants that the m6A demethylase and AP lyase activities are located in different regions of the protein and utilize distinct residues [15]; i.e., the MBP domain more greatly affects the demethylase over the lyase activity and product formation increased differently after MBP-tag removal.
We found ALKBH1 localizes to the mitochondria and ALKBH1-deficient cell strains are affected in mitochondrial function. HEK293 cells lacking ALKBH1 have a proliferation deficit when cultured in galactose; a phenotype that is reversed by expressing ALKBH1 in these strains. Moreover, we found lower CS activities from ALKBH1-deficient cells grown on either 1 g/l glucose or 1 g/l galactose compared to the WT strain, with production of Flag-HA-tagged ALKBH1 restoring the activities to WT levels. These data support an emerging consensus: ALKBH1 functions in the mitochondria and the lack of ALKBH1 results in clear deficits in mitochondrial metabolism. Our results agree with the work of others [12] who found slow growth of ALKBH1-deficient HEK293 cells (targeting exon 4) versus WT cells (on media containing either glucose or galactose) and decreased levels of oxidative phosphorylation. Together, these data provide evidence that ALKBH1 has an important role in mitochondrial function. Still lacking, however, is a clear understanding of how ALKBH1 deficiency impairs mitochondrial metabolism and cell proliferation. ALKBH1 is unlikely to act as an m6A demethylase in the organelle because this epigenetic marker is not present in mtDNA [9]. ALKBH1’s lyase activity could be crucial for mtDNA repair, but Alkbh1−/− murine fibroblasts are not more susceptible to oxidative or alkylation damage [20]. Further studies will need to specifically address the ALKBH1 function in the mitochondria.
Supplementary Material
Highlights.
Mammalian produced ALKBH1 (ALKBH1293) possesses m6A demethylase and AP lyase activity.
ALKBH1293 forms an adduct with the 5′-product of the AP lyase reaction.
ALKBH1 localizes to the mitochondria in MSU1.1 and HEK293 cells.
ALKBH1-deficient cells exhibit mitochondrial dysfunction.
Acknowledgments
We thank Kefei Yu for plasmids and advice. This work was initiated with support from the National Institutes of Health (GM063582 to R.P.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Falnes PØ, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 2.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 3.Kurowski MA, Bhagwat AS, Papaj G, Bujnicki JM. Phylogenomic identification of five new human homologues of the DNA repair enzyme AlkB. BMC Genomics. 2003;4:48. doi: 10.1186/1471-2164-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez-Pulido L, Andrade-Navarro MA. The FTO (fat mass and obesity associated) gene codes for a novel member of the non-heme dioxygenase superfamily. BMC Biochem. 2007;8:23. doi: 10.1186/1471-2091-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O’Rahilly S, Schofield CJ. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM. The AlkB family of Fe(II)/α-ketoglutarate dependent dioxygenases: Repairing nucleic acid alkylation damage and beyond. J Biol Chem. 2015;290:20734–20742. doi: 10.1074/jbc.R115.656462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Müller TA, Hausinger RP. AlkB and its homologues - DNA repair and beyond. In: Schofield CJ, Hausinger RP, editors. 2-Oxoglutarate-Dependent Oxygenases. Royal Society of Chemistry; Cambridge, UK: 2015. pp. 246–262. [Google Scholar]
- 8.Westbye MP, Feyzi E, Aas PA, Vågbo CB, Talstad VA, Kavli B, Hagen L, Sundheim O, Akbari M, Liabakk NB, Slupphaug G, Otterlei M, Krokan HE. Human AlkB homolog 1 is a mitochondrial protein that demethylates 3-methylcytosine in DNA and RNA. J Biol Chem. 2008;283:25046–25056. doi: 10.1074/jbc.M803776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, Liu Y, Byrum SD, Mackintosh SG, Zhong M, Tackett A, Wang G, Hon LS, Fang G, Swenberg JA, Xia AZ. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature. 2016;532:329–333. doi: 10.1038/nature17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu F, Clark WC, Luo G, Wang X, Fu Y, Wei J, Wang X, Hao Z, Dai Q, Zheng G, Ma H, Han D, Evans M, Klungland A, Pan T, He C. ALKBH1-mediated tRNA demethylation regulates translation. Cell. 2016;167:816–828. doi: 10.1016/j.cell.2016.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haag S, Sloan KE, Ranjan N, Warda AS, Kretschmer J, Blessing C, Hübner B, Seikowski J, Dennierlein S, Rehling P, Rodnina MV, Höbartner C, Bohnsack MT. NSUN3 and ABH1 modify the wobble position of mt-tRNAMet to expand codon recognition in mitochondrial translation. EMBO J. 2016;35:2104–2119. doi: 10.15252/embj.201694885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawarada L, Suzuki T, Ohira T, Hirata S, Myauchi K, Suzuki T. ALKBH1 is an RNA dioxygenase responsible for cytoplasmic and mitochondrial tRNA modifications. Nucleic Acids Res. 2017;45:7401–7415. doi: 10.1093/nar/gkx354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Müller TA, Andrzejak MM, Hausinger RP. A covalent protein-DNA 5′-product adduct is generated following AP lyase activity of human ALKBH1 (AlkB homologue 1) Biochem J. 2013;452:509–518. doi: 10.1042/BJ20121908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Müller TA, Meek K, Hausinger RP. Human AlkB homologue 1 (ABH1) exhibits DNA lyase activity at abasic sites. DNA Repair. 2010;9:58–65. doi: 10.1016/j.dnarep.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Müller TA, Tobar M, Perian MN, Hausinger RP. Biochemical characterization of AP lyase and m6A demethylase activities of human AlkB homologue 1 (ALKBH1) Biochemistry. 2017;56:1899–1910. doi: 10.1021/acs.biochem.7b00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ougland R, Lando D, Jonson I, Dahl JA, Moen MN, Nordstrand LM, Rognes T, Lee JT, Klungland A, Kouzarides T, Larsen E. ALKBH1 is a histone H2A dioxygenase involved in neural differentiation. Stem Cells. 2012;30:2672–2682. doi: 10.1002/stem.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan Z, Sikandar S, Witherspoon M, Dizon D, Nguyen T, Benirschke K, Wiley C, Vrana P, Lipkin SM. Impaired placental trophoblast lineage differentiation in Alkbh1(−/−) mice. Developmental Dynamics. 2008;237:316–327. doi: 10.1002/dvdy.21418. [DOI] [PubMed] [Google Scholar]
- 18.Tsujikawa K, Koike K, Kitae K, Shinkawa A, Arima H, Suzuki T, Tsuchiya M, Makino Y, Furukawa T, Konishi N, Yamamoto H. Expression and sub-cellular localization of human ABH family molecules. Journal of cellular and molecular medicine. 2007;11:1105–1116. doi: 10.1111/j.1582-4934.2007.00094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nordstrand LM, Svärd J, Larsen E, Nilsen A, Ougland R, Furu K, Lien GF, Rognes T, Namekawa SH, Lee JT, Klungland A. Mice lacking Alkbh1 display sex-ratio distortion and unilateral eye defects. PLoS One. 2010;5:e13827. doi: 10.1371/journal.pone.0013827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Müller TA, Yu K, Hausinger RP, Meek K. ALKBH1 is dispensable for abasic site cleavage during base excision repair and class switch recombination. PLoS One. 2013;8:e67403. doi: 10.1371/journal.pone.0067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norviille JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy S, de Melo AJ, Xu Y, Tadi SK, Negrel A, Hendrickson E, Modesti M, Meek K. XRCC4/XLF interaction is variably required for DNA repair and is not required for ligase IV stimulation. Molec Cell Biol. 2015;35:3017–3028. doi: 10.1128/MCB.01503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furda AM, Bess AS, Meyer JN, Van Houten B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR, Meth. Molec Biol. 2012;920:111–132. doi: 10.1007/978-1-61779-998-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Meth Molec Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.