

Abstract
Coxsackievirus B virus infections are suspected environmental triggers of Type 1 diabetes (T1D) and macrophage anti-viral responses may provide a link to virus-induced T1D. We previously demonstrated an important role for NADPH oxidase (NOX)-derived superoxide production during T1D pathogenesis, as NOX-deficient Non-Obese Diabetic (NOD) mice (NOD.Ncf1m1J) were protected against T1D, due, in part, to impaired pro-inflammatory Toll-like receptor (TLR) signaling in NOD.Ncf1m1J macrophages. Therefore, we hypothesized that loss of NOX-derived superoxide would dampen diabetogenic anti-viral macrophage responses and protect from virus-induced diabetes. Upon infection with a suspected diabetogenic virus, Coxsackievirus B3 (CB3), NOD.Ncf1m1J mice remained resistant to virus-induced autoimmune diabetes. A concomitant decrease in circulating inflammatory chemokines, blunted anti-viral gene signature within the pancreas, and reduced pro-inflammatory M1 macrophage responses were observed. Importantly, exogenous superoxide addition to CB3-infected NOD.Ncf1m1J bone marrow-derived macrophages rescued the inflammatory anti-viral M1 macrophage response, revealing the redox-dependent mechanisms of STAT1 signaling and dsRNA viral sensors in macrophages. We report that superoxide production following CB3 infection may exacerbate pancreatic β-cell destruction in T1D by influencing pro-inflammatory M1 macrophage responses, and mechanistically linking oxidative stress, inflammation, and diabetogenic virus infections.
Introduction
Viral infections have long been suspected environmental instigators of Type 1 diabetes (T1D) (1, 2). Cytomegalovirus, Epstein-Barr virus, Rubella, Mumps, Rotavirus, retroviruses, and Coxsackieviruses have been associated with T1D (1, 3). Studies of Coxsackievirus infection in human cadaveric donors (4, 5) and the Non-obese diabetic (NOD) mouse model of T1D have provided extensive knowledge of how viruses may trigger T1D (4–9). Specifically, these ssRNA viruses have a tropism for the pancreas, and all six serotypes (CB1-6) successfully infect islets (10, 11). Histological analysis of pancreatic islets from recent-onset T1D patients revealed the presence of a Coxsackievirus infection (4, 12) which in some cases, was β-cell-specific (5, 12). Pre-diabetic NOD mice infected with Coxsackievirus B3 (CB3) or B4 (CB4) displayed accelerated T1D progression (6, 11), which was partly due to the uptake of infected β-cells by antigen-presenting cells and bystander activation of autoreactive T cells (8, 9). Furthermore, this acceleration required established insulitis (7, 13), suggesting that Coxsackievirus infections impact disease onset through exacerbating inflammation and breaking peripheral tolerance.
T1D is an organ-specific autoimmune disease consisting of chronic production of pro-inflammatory cytokines (TNF-α, IFN-γ, IL-1β), Type I interferons (IFN-α/β), and reactive oxygen species (ROS) synthesis that drives autoimmune responses and β-cell destruction (14). Classically-activated M1 macrophages are a major source of ROS and pro-inflammatory cytokines partly due to STAT1 activation (15). Importantly, macrophages are absolutely essential in the pathogenesis of T1D (16, 17). Within the NOD mouse, they display hyperactivation of the NF-κB signaling pathway (18) leading to aberrant production of inflammatory cytokines and chemokines to facilitate β-cell cytotoxicity (19), but can also function as antigen-presenting cells to activate autoreactive T cells (20). Macrophages can sufficiently transfer the diabetogenicity of CB4 infection, as adoptive transfer of macrophages from CB4-infected NOD.scid mice was able to trigger diabetes onset in T cell receptor-transgenic NOD.BDC-2.5 mice (9).
T1D is a relapsing and remitting disease exhibiting hallmarks of chronic oxidative stress and inflammation that contribute to autoimmune dysregulation (21). Oxidative stress is intimately linked to inflammatory responses, and free radical production functions as a crucial third signal for efficient activation of T cells (22–25). Our lab has established the importance of oxidative stress in T1D, as the absence of NADPH oxidase (NOX)-derived superoxide, conferred by a mutation (Ncf1m1J) on the essential p47phox subunit of the NOX complex, delays T1D in NOD.Ncf1m1J mice (16, 22). We have recently shown that loss of superoxide causes an inherent skewing of macrophages from an M1 to an M2 phenotype during spontaneous and adoptive transfer of T1D (26). Importantly, we also reported that NOX-deficient macrophages display diminished TLR3-dependent inflammatory responses following stimulation with the viral dsRNA mimic, poly(I:C) (27). Therefore, shifting macrophage differentiation away from an inflammatory M1 phenotype within the NOD mouse can significantly change disease outcome.
Since virus-induced T1D is a consequence of heightened innate immune anti-viral responses, NOD pro-inflammatory M1 macrophage differentiation and immune responses to poly(I:C) is dependent on NOX-derived superoxide, and T1D-resistant NOD.Ncf1m1J mice lack the ability to generate NOX-derived superoxide, we propose that the commonality of virus infections and induction of autoimmune destruction of pancreatic β-cells is the generation of oxidative stress. However, the role for free radicals in anti-viral responses in T1D remains unknown. Macrophages play critical roles in both T1D and in combating Coxsackievirus infection, and they express one of the highest levels of NOX2. Altogether, we hypothesized that NOX-derived superoxide production during Coxsackievirus infection would potentiate pro-inflammatory M1 macrophage anti-viral responses and aid in triggering autoimmunity.
Materials and Methods
Mice
NOD/ShiLtJ and NOD.Ncf1m1J mice were bred and housed under pathogen free conditions at the Research Support Building animal facility at the University of Alabama at Birmingham. Female and male mice between 8 and 16 weeks of age were used. Mice received standard chow and acidified water weekly except for NOD.Ncf1m1J, which received 80 mg of Trimethoprim/Sulfamethoxazole (Hi-Tech Pharmacal) in drinking water. All animal studies were performed in accordance with the UAB Institutional Animal Use and Care Committee in compliance with the laws of the United States of America.
Bone marrow-derived macrophage (BM-MΦ) generation, virus infection, and xanthine oxidase (XO) addition
NOD and NOD.Ncf1m1J BM-MΦ were generated as previously described (27). Coxsackievirus B3 (CB3) (Woodruff variant) was graciously provided by Dr. Marc Horwitz (University of British Columbia) and maintained as described (6). To provide exogenous superoxide, culture media was supplemented with 1mU/mL of XO (Sigma) as published (28). In vitro infections were performed at a multiplicity of infection (MOI) range of 10–50.
In vivo infections, diabetes incidence, and viral plaque assays
NOD and NOD.Ncf1m1J mice were infected by intraperitoneal injection of 100 PFU CB3 in HBSS, or HBSS alone as control. For diabetes incidence studies, female mice were infected or uninfected at 12 weeks of age, and monitored several times weekly for diabetes by glucosuria (Diastix). Mice were considered diabetic following 2 consecutive blood glucose readings ≥300 mg/dL. To determine viral titers, whole pancreata were harvested, weighed, homogenized in HeLa growth media (DMEM supplemented with 10% heat-inactivated FBS, 4mM L-glutamine, 1mM sodium pyruvate, 100 units/mL penicillin-streptomycin), frozen and thawed 3 times, and used for plaque assays on HeLa cells (29).
Quantitative real-time PCR, Western blotting and ELISAs
For RNA analysis, BM-MΦ were harvested using TRIzol (Invitrogen) and pancreata were initially harvested with RNAlater (Ambion), then lysed in TRIzol using the TissueLyser II (Qiagen). mRNA was isolated by the RNeasy kit (Qiagen) and reverse transcribed into cDNA using Superscript III (Invitrogen). TaqMan gene expression was analyzed with the following primers (Applied Biosystems): Emr1 (Mm00802529_m1), Tlr3 (Mm00628112_m1), Ifih1 (Mm00459183), Ddx58 (Mm01216853_m1), Tnf (Mm00443258_m1), Ifnb1 (Mm00439552_s1), Isg15 (Mm01705338_s1), and Gapdh (Mm99999915_g1). Relative gene expression levels were calculated using the 2(−ΔΔCt) method, normalized to Gapdh, and wild-type unstimulated levels set to 1. For Western blotting, 25μg of whole cell lysates were separated, transferred to nitrocellulose membrane (Millipore), and probed with antibodies against P-STAT1 (Y701), STAT1, MDA5, RIG-I (Cell Signaling), or β-actin (Sigma-Aldrich) as described (27), then visualized on an Odyssey CLx Imager with Image Studio v4.0 software using LI-COR secondary anti-rabbit or anti-mouse antibodies conjugated to either IRDye 680RD or IRDye 800CW. TNF-α, CXCL10, CCL5 (R&D Systems), and IFN-α/IFN-β (PBL Assay Science) was quantified by ELISAs.
Flow cytometric analysis
Pancreatic cells were isolated by collagenase digestion and macrophages were identified by surface staining with anti-F4/80 (clone BM8, eBioscience) and anti-CD11b (clone M1/70, BD Biosciences) (30). Intracellular staining was performed with anti-TNF-α (clone MP6-XT22, R&D Biosystems) and the appropriate isotype control (R&D Biosystems). Samples were collected (400,000 events) on an Attune NxT flow cytometer (Thermo Scientific) and analyzed with FlowJo version 10.0.7 Software (Treestar Incorporated) following the gating scheme (Suppl. Fig. 1).
Immuno-spin trapping and immunofluorescence
NOD and NOD.Ncf1m1J BM-MΦ were cultured on chamber slides (Lab-Tek) and infected with 10 MOI CB3. For immuno-spin trapping, macromolecular-centered free radicals were detected with 1 mmol/L 5,5-dimethyl-1-pyrroline N-oxide (DMPO; Dojindo), then stained for DMPO adducts by immunofluorescence, as described (28). P-STAT1 (Y701) was detected with FITC-conjugated donkey anti-rabbit IgG (H+L) secondary antibody (1:500). Macrophages were stained with DAPI and anti-F4/80 surface staining. Images were obtained with an Olympus IX81 Inverted Microscope at a 40X objective and analyzed with cellSens Dimension imaging software version 1.12. To quantitate fluorescence intensity, 3–6 images were obtained for each treatment group, collected at the same exposure time, adjusted to the same intensity level for standardization, and measured using ImageJ Software (NIH), as previously described (28).
Statistical analysis
Data were analyzed using GraphPad Prism Version 5.0 statistical software. The difference between mean values and standard error of the mean was assessed using the 2-tailed Student’s t test, with p < 0.05 considered significant. Descriptive statistics that were calculated for serum IFN-α/IFN-β levels included means and standard deviations. The exact Wilcoxon rank-sum test was used, statistical tests were two-sided and performed using a significance level of 5% with SAS software, version 9.4 (SAS Institute).
Results
CB3 infection of bone marrow-derived macrophages elicits NOX-derived superoxide production
Previous studies from our lab and others have underscored the importance of oxidative stress and free radical production during T1D progression in human patients and NOD mice (16, 22, 31, 32). Macrophages are major producers of NOX-derived superoxide, essential immune cells for T1D pathogenesis, and critical for the clearance of Coxsackievirus infection (33). To establish if diabetogenic CB3 infection (11) can induce NOX-derived superoxide production by macrophages, we performed in vitro CB3 infections of both NOD and NOD.Ncf1m1J BM-MΦ and measured macromolecular-centered free radical adduct production by immuno spin-trapping (Fig. 1, Suppl. Fig. 2). As early as 4 hours post-infection, NOD macrophages elicited an oxidative burst as shown by the appearance of green fluorescent puncta (Fig. 1A). Importantly, there was a decrease (p<0.0001) in DMPO-adducts detected by immuno-spin trapping within CB3-infected NOD.Ncf1m1J BM-MΦ cultures (Fig. 1B). These results demonstrate that upon Coxsackievirus infection, NOX-derived superoxide is induced by macrophages.
FIGURE 1. CB3-induced oxidative burst by NOD macrophages is ablated in the absence of NOX-derived superoxide.
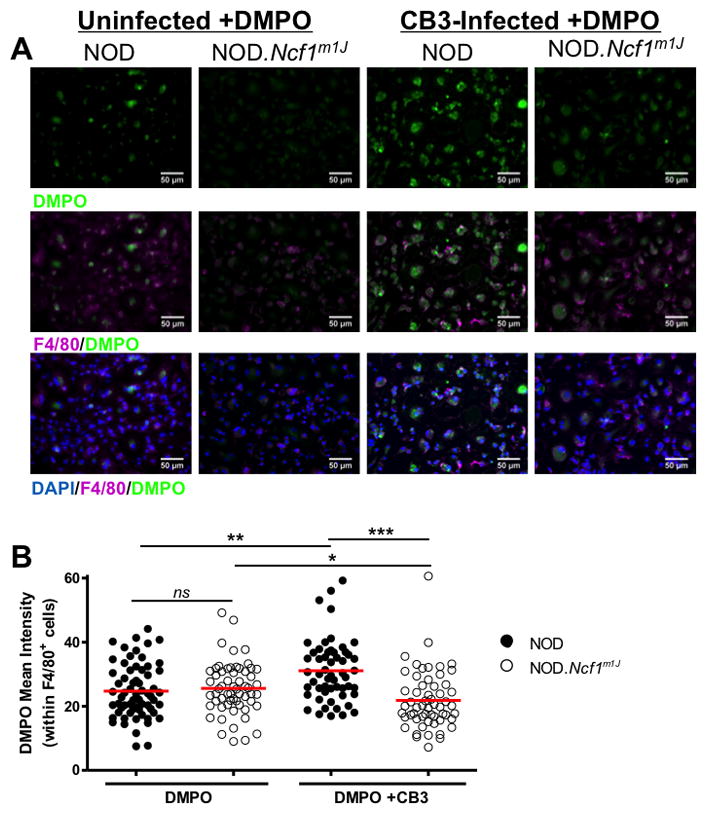
NOD and NOD.Ncf1m1J BM-MΦ were cultured on chamber slides in the presence or absence of DMPO and 10 MOI CB3 infection for 4 hours. Cells were identified by DAPI nuclear stain (blue) and anti-F4/80 macrophage marker staining (purple), and free radical formation of DMPO adducts was detected by anti-DMPO (green) (A). Mean fluorescence intensity of DMPO adducts were quantified using ImageJ analysis software and normalized to F4/80 expression intensity (B). Each dot represents anti-DMPO fluorescence on a single F4/80+ cell with the following total cells quantified: NOD (n=64), NOD.Ncf1m1J (n=59), NOD+CB3 (n=55), NOD.Ncf1m1J+CB3 (n=57). Results are representative of 2 independent experiments. ***p<0.0001; **p<0.01; *p<0.05
Superoxide deficiency protects NOD.Ncf1m1J mice from CB3-induced diabetes without compromising pancreatic viral clearance
In the absence of NOX-derived superoxide production, NOD.Ncf1m1J mice exhibit a delay in spontaneous and adoptive transfer of T1D (16, 22). Given that NOX-derived superoxide is produced by macrophages in response to CB3 infection (Fig. 1), we sought to determine if NOD.Ncf1m1J mice exhibit a delay in CB3-induced T1D. Twelve week-old, pre-diabetic female NOD and NOD.Ncf1m1J mice were infected with 100 PFU of CB3, then observed for onset of T1D in comparison with HBSS-treated mice (Fig. 2A). NOD mice showed acceleration in diabetes onset as early as 2 weeks and culminating at 5 weeks post-infection (p=0.0176) compared to HBSS controls (Fig. 2A). Acceleration of T1D was transient, as a cohort of CB3-infected NOD mice failed to succumb to autoimmune diabetes following 5 weeks post-infection. NOD.Ncf1m1J mice were significantly protected against spontaneous T1D (p<0.0001), CB3-accelerated T1D at 5 weeks post-infection (p=0.0371), and at the end of the study (p<0.05) compared to HBSS-treated NOD mice. These results show that superoxide production by the immune system plays a critical role in how viral infections trigger autoimmunity in NOD mice.
FIGURE 2. NOD.Ncf1m1J mice are significantly protected against CB3-induced acceleration of autoimmune diabetes without compromising viral clearance.
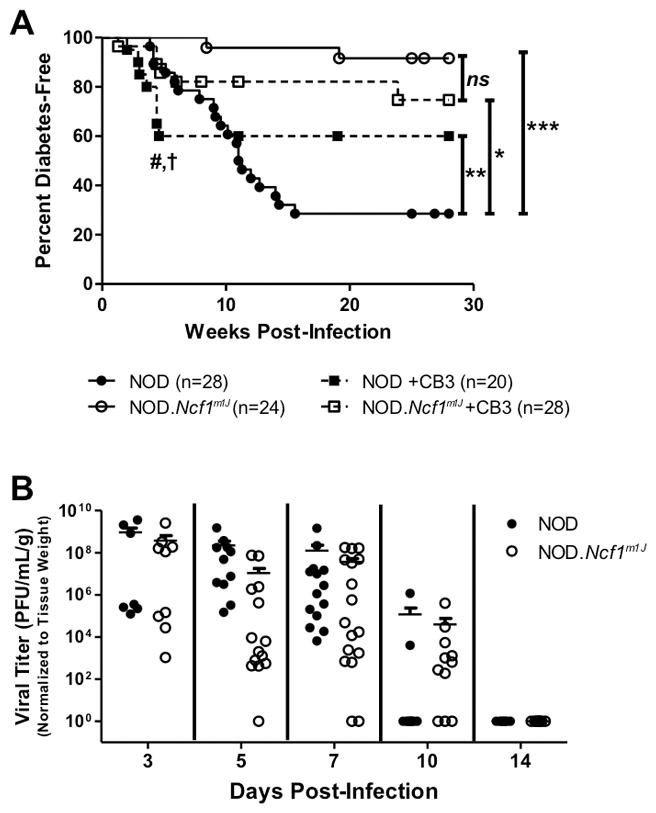
Kaplan Meier survival curve of twelve week-old NOD and NOD.Ncf1m1J mice infected with 100 PFU CB3 i.p. (n=20 and 28, respectively), or uninfected (n=28 and 24, respectively) (A). # symbol indicates significant (p=0.0176) differences between NOD and NOD+CB3 groups, and † symbol indicates significant (p=0.0371) differences between NOD and NOD.Ncf1m1J CB3-infected groups at 5 weeks post-infection; **p<0.01 between NOD and NOD+CB3 curves when analyzed between 10–28 weeks post-infection. Viral titer of CB3 in the pancreata of NOD and NOD.Ncf1m1J mice on days 3 (n=11 and 14), 5 (n=11 and 14), 7 (n=14 and 17), 10 (n=10 and 11) and 14 (n=7 and 8, respectively) post-infection (B). Titers were normalized to mg of pancreatic tissue. Each dot represents an individual mouse. ***p<0.0001; **p<0.01; *p<0.05; ns, not significant
NOX-derived superoxide is essential for eradicating bacterial and fungal infections (34). While the contribution of superoxide during viral clearance is not fully established, studies have suggested that lack of dietary antioxidants can enhance Coxsackievirus infection (35). To determine if the loss of superoxide production impairs the clearance of CB3 infection, viral titers were measured from the pancreata of NOD and NOD.Ncf1m1J at 3, 5, 7, 10 and 14 days post-infection (Fig. 2B). Surprisingly, even in the absence of NOX-derived superoxide, NOD.Ncf1m1J pancreata harbored similar viral titers as compared to NOD pancreata and CB3 was efficiently cleared from NOD and NOD.Ncf1m1J pancreata by 2 weeks post-infection (Fig 2B). Therefore, superoxide deficiency does not restrict the infectivity of CB3 within the pancreas, nor does it hinder immune clearance of CB3.
Circulating pro-inflammatory chemokines and Type I interferons are decreased in CB3-infected NOD.Ncf1m1J mice
CXCL10, an inflammatory chemokine, is important in the acute immune response against Coxsackievirus infection (36) and expressed within the islets of T1D patients compared to healthy controls (37). Additionally, protective Ccl5 SNPs in T1D human studies are correlated with decreased serum-specific CCL5 production (38). To determine if superoxide deficiency caused diminutions in the chemokine response to CB3 infection, we compared levels of CXCL10 and CCL5 in the sera of NOD and NOD.Ncf1m1J mice following infection (Fig. 3). CXCL10 was detected as early as day 3 and remained elevated in contrast to CB3-infected NOD.Ncf1m1J mice through day 7 post-infection (Fig. 3A). There was a 4.3- (p=0.0219) and 2-fold (p=0.0483) reduction of CXCL10 in the sera of NOD.Ncf1m1J mice at 5 and 7 days post-infection, respectively (Fig. 3A). CCL5 levels followed similar kinetics throughout infection, and in contrast to NOD, CCL5 levels were decreased 3.2- (p=0.0268) and 3.7-fold (p=0.0087) in the sera of CB3-infected NOD.Ncf1m1J mice at 5 and 7 days post-infection, respectively (Fig. 3B). The marked decreases of serum CXCL10 and CCL5 during viral infection in NOD.Ncf1m1J mice demonstrates an important role for NOX-derived superoxide in regulating chemokine production.
FIGURE 3. NOD.Ncf1m1J mice have decreased circulating inflammatory chemokines and Type I interferons throughout CB3 infection.
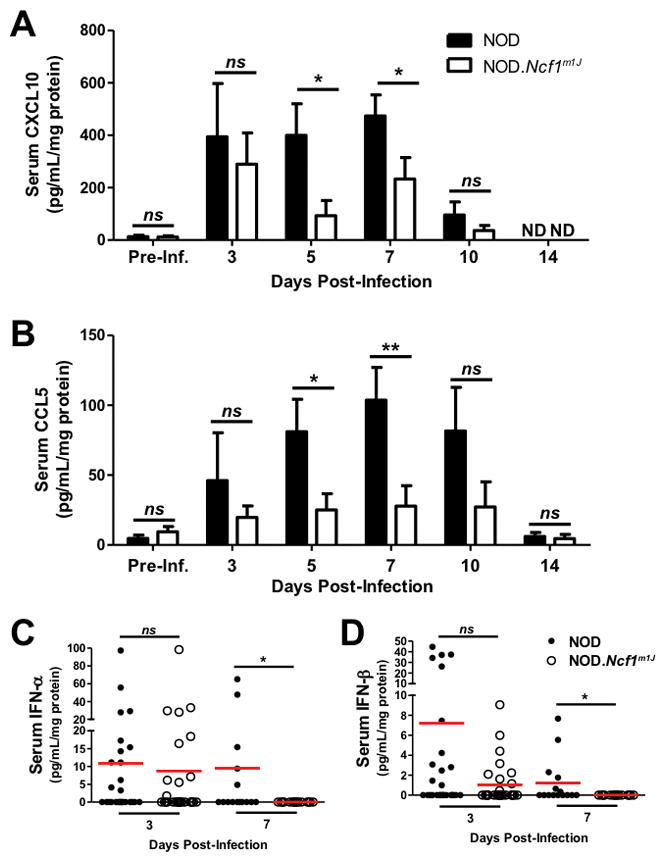
Levels of circulating CXCL10 (A), CCL5 (B), IFN-α (C), and IFN-β (D) were measured from the serum of each individual mouse by ELISA, compared with pre-infection levels, and then normalized to serum protein concentration as determined by BCA protein assay. Bars for (A) and (B) represent the average readings of all mice in each group from Fig. 2B. For (C) and (D), dots represent individual mice (NOD day 3=27; NOD.Ncf1m1J day 3=28; NOD day 7=16; NOD.Ncf1m1J day 7=16). **p<0.01; *p<0.05
To determine if Type I interferon synthesis is compromised in CB3-infected NOD and NOD.Ncf1m1J mice, we assessed levels of IFN-α and IFN-β in the serum of mice at days 3 and 7 post-infection. At 3 days post-infection, NOD.Ncf1m1J mice displayed similar levels of IFN-α and IFN-β production, as compared with NOD mice (Fig. 3C). However, by 7 days post-infection, IFN-α and IFN-β were still detectable in the NOD serum, but completely absent in the NOD.Ncf1m1J serum (p=0.018 and p=0.007, respectively) (Fig. 3D). Thus, the synthesis of Type I IFN at 3 days post-infection may facilitate viral clearance, while the extended superoxide-dependent Type I IFN responses detected in NOD serum at day 7 may confer susceptibility to CB3-accelerated T1D.
Pro-inflammatory anti-viral mRNA accumulation is ablated in the pancreata of CB3-infected NOD.Ncf1m1J mice
With the confirmed loss of an oxidative burst by NOD.Ncf1m1J macrophages upon CB3 infection (Fig. 1) and decreased circulating CXCL10 and CCL5 levels (Fig. 3), we hypothesized that the delay in CB3-accelerated T1D in NOD.Ncf1m1J mice (Fig. 2A) was partly due to the absence of synergistic effects of free radicals on pro-inflammatory macrophage anti-viral responses. To address this, we analyzed innate immune anti-viral responses within the pancreas of uninfected and CB3-infected NOD and NOD.Ncf1m1J mice by qRT-PCR (Fig. 4). By day 7 post-infection, an influx of macrophages was detected in the pancreata of both NOD and NOD.Ncf1m1J mice as indicated by a 10.6- and 6.8-fold increase, respectively, in the transcript expression of Emr1, which encodes for the macrophage marker F4/80. Interestingly, analysis of the transcription factor, Stat1, involved in both Type 1 IFN signaling and inflammatory M1 macrophage differentiation, was decreased 2.5-fold (p=0.0012) in CB3-infected NOD.Ncf1m1J pancreata compared to NOD. In addition to decreased Stat1, mRNA accumulation of inflammatory response gene Tnf was reduced by 4-fold (p=0.0472). Anti-viral mRNA accumulation was also severely attenuated within NOX-deficient pancreata, with a 8.3-fold (p=0.0104) reduction in the expression of Ifnb1, and a 4.1-fold (p=0.0041) decrease in expression of the IFN-stimulated gene, Isg15 (Fig. 4). Altogether, these results demonstrate that NOX-derived superoxide and the activation of redox-dependent signaling pathways enhance the innate immune anti-viral response against CB3 infection.
FIGURE 4. Expression of innate anti-viral response genes upon CB3 infection is severely hindered in NOD.Ncf1m1J pancreata.
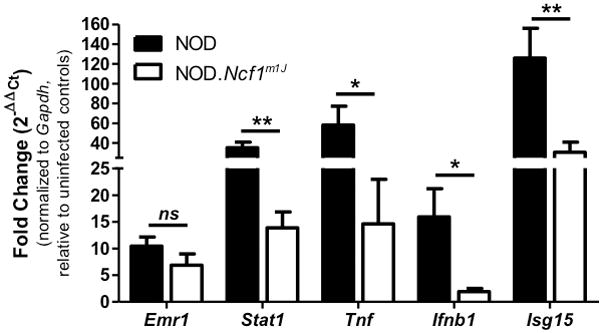
mRNA fold change of Emr1, Stat1, Tnf, Ifnb1, and Isg15 within NOD and NOD.Ncf1m1J pancreata at day 7 post 100 PFU CB3 infection. Data shown are representative results from 23 individual mice. **p<0.01; *p<0.05; ns, not significant
Pancreas-infiltrating macrophages from CB3-infected NOD.Ncf1m1J mice are less inflammatory
To investigate the impact of superoxide deficiency on macrophage responses to viral infection, the pancreata from HBSS-treated or CB3-infected NOD and NOD.Ncf1m1J mice were immunophenotyped for infiltrating macrophages (F4/80+ CD11b+) by flow cytometry (Fig. 5, Supp. Fig. 1). Despite the dampened serum levels of CXCL10 and CCL5 in CB3-infected NOD.Ncf1m1J mice (Fig. 3), and decreased (p=0.03) total number of infiltrating cells within the NOD.Ncf1m1J pancreas (22.5x106 ±3.4x106 (NOD.Ncf1m1J) vs. 32.7x106 ±3.0x106 (NOD)) (Fig. 5A), macrophage recruitment into the pancreas was similar between NOD and NOD.Ncf1m1J mice by both percentage (22.9±3.4 (NOD) vs. 20.6±4.6 (NOD.Ncf1m1J)) and cell counts (8.6x106 ±1.9x106 (NOD) vs. 6.2x106 ±2.0x106 (NOD.Ncf1m1J)) at 7 days post-infection (Fig. 5A). To determine if the loss of NOX-derived superoxide hampers pro-inflammatory TNF-α synthesis of pancreas-infiltrating macrophages, intracellular cytokine staining was performed and revealed a decrease in the percentage (1.9-fold, p=0.0293), count (2.3-fold, p=0.0335), and geometric mean fluorescence intensity (gMFI) (2.4-fold, p=0.0064) compared to CB3-infected NOD mice (Fig. 5B, D). Macrophages recovered from the pancreas at day 7 post-CB3 infection were in vitro challenged with LPS for 4 hours. Interestingly, NOD.Ncf1m1J macrophages still displayed decreased intracellular TNF-α synthesis compared to NOD, by percentage (2.1-fold, p=0.0206), total number (2.9-fold, p=0.0136), and gMFI (3.0-fold, p=0.0255) (Fig. 5C, D). Therefore, our data demonstrates the importance of NOX-derived superoxide to mature and enhance pro-inflammatory M1 macrophage responses in the pancreas following CB3-infection.
FIGURE 5. CB3-infected NOD.Ncf1m1J mice have a reduced amount of TNF-α-producing pancreas-infiltrating macrophages.
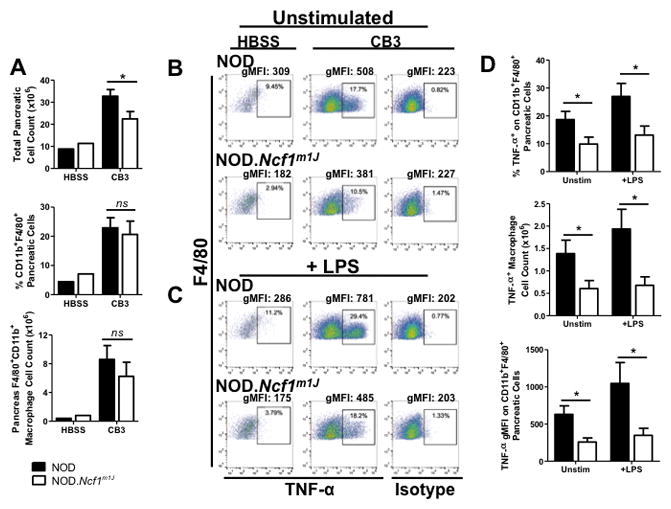
Macrophages within the pancreata of NOD and NOD.Ncf1m1J mice infected with 100 PFU CB3 or HBSS-treated were analyzed by flow cytometry. Analysis of cell counts in infected pancreata was performed for total cells and CD11b+, F4/80+ macrophages (A). Dot plots of TNF-α by F4/80 following a 4 hour unstimulated (B) or LPS-stimulated (C) culture, with events pre-gated on the CD11b+ F4/80+ population. Compiled data (D) show overall results of percentage, total macrophage count and geometric mean fluorescence intensity (gMFI). Data shown are representative results from 4 independent experiments with a total of 16 individual mice per group. *p<0.05; ns, not significant
NOD.Ncf1m1J macrophages display a reduced anti-viral response upon CB3 infection
To determine the redox-regulated mechanisms involved in macrophage anti-viral responses, NOD and NOD.Ncf1m1J BM-MΦ were infected with CB3 and induction of pro-inflammatory innate immune responses were assessed. First, to confirm the in vivo phenotype of a decreased anti-viral signature within NOD.Ncf1m1J pancreata (Fig. 4), mRNA accumulation of pro-inflammatory and Type-1 IFN response genes was assessed (Fig. 6A). While infection of NOD BM-MΦ with CB3 induced robust inflammatory responses, NOD.Ncf1m1J BM-MΦ displayed reductions in Tnf (1.4-fold, p=0.0001), Cxcl10 (2-fold, p < 0.0001), and Ccl5 (1.5-fold, p=0.0023) transcripts, in addition to attenuated Ifnb1 (1.9-fold, p=0.0106) and Isg15 (1.4-fold, p=0.0059) Type 1 IFN responses. Additionally, superoxide-deficiency induced significant diminutions in the viral RNA sensors, Tlr3 (1.3-fold, p=0.0026) and Ddx58 (1.5-fold, p=0.0095), while Ifih1 remained unaffected (Fig. 6A). To corroborate the observed decrease in mRNA accumulation, ELISAs of cytokine and chemokine production were analyzed within culture supernatants. Compared to NOD, production of TNF-α (Fig. 6B), CXCL10 (Fig. 6C), CCL5 (Fig. 6D), and IFN-β (Fig. 6E) by NOD.Ncf1m1J BM-MΦ were diminished 2-fold (p<0.0001), 1.4-fold (p=0.0001), 2-fold (p=0.0009), and 11-fold (p<0.0001), respectively. These results support our previous reports that inflammatory macrophage responses are diminished in the absence of superoxide production (26, 27) and more importantly, provide evidence that redox status can influence macrophage anti-viral responses.
FIGURE 6. NOD.Ncf1m1J BM-MΦ display hindered inflammatory and anti-viral responses upon CB3 virus infection.
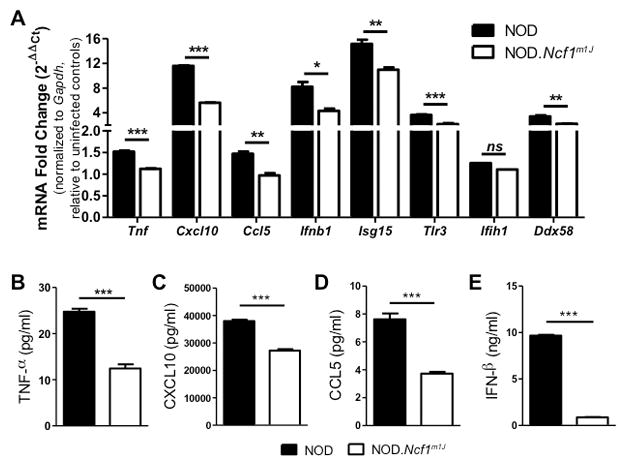
NOD and NOD.Ncf1m1J BM-MΦ were infected with 10–25 MOI CB3. Quantitative RT-PCR analysis was performed for Tnf, Cxcl10 Ccl5, Ifnb1, Isg15, Tlr3, Ifih1 and Ddx58 after 6 hours of infection (A). Production of TNF-α (B), CXCL10 (C), CCL5 (D), and IFN-β (E) were measured 24 hours post-infection via ELISA. Data shown represent average of 3 independent experiments performed in triplicate. ND, not detected; ***p<0.0001; **p<0.01; *p<0.05; ns, not significant
Addition of exogenous superoxide restores anti-viral RNA sensing and M1 macrophage differentiation in NOD.Ncf1m1J macrophages
To determine whether anti-viral responses in macrophages were redox-regulated, exogenous superoxide was added to CB3-infected NOD and NOD.Ncf1m1J BM-MΦs via xanthine oxidase (XO), an enzyme that produces superoxide as a byproduct during the production of uric acid from xanthine. Western blot analysis of cellular lysate at 24 hours post-infection revealed that MDA5, a viral dsRNA sensor critical for anti-viral responses, was dampened 2.6-fold (p=0.0024) in CB3-infected NOD.Ncf1m1J BM-MΦ compared to NOD (Fig. 7A, B). With the addition of XO to NOD.Ncf1m1J BM-MΦ, expression of MDA5 increased by 1.6-fold, restoring to near NOD wild-type levels (Fig. 7A, B). Interestingly, even without CB3 challenge, XO addition enhanced MDA5 basal expression by more than 15-fold in both NOD (p=0.0403) and NOD.Ncf1m1J (p=0.0495) BM-MΦ (Fig, 7A, B). RIG-I, another cytosolic viral RNA sensor, was upregulated by over 2-fold in NOD (p=0.0188) and NOD.Ncf1m1J (p=0.0067) BM-MΦ with XO alone (Fig. 7C, D). Compared to NOD BM-MΦ, RIG-I expression in CB3-infected NOD.Ncf1m1J BM-MΦ was decreased by 1.6-fold (p=0.0011) and addition of exogenous superoxide restored RIG-I protein levels by 1.3-fold back to wild-type NOD levels (Fig. 7C, D). Together, these results suggest that NOX-derived superoxide enhances the sensitivity of macrophages to detect and respond to viral infections.
FIGURE 7. Addition of exogenous superoxide restores viral RNA sensor levels and STAT1 activation.
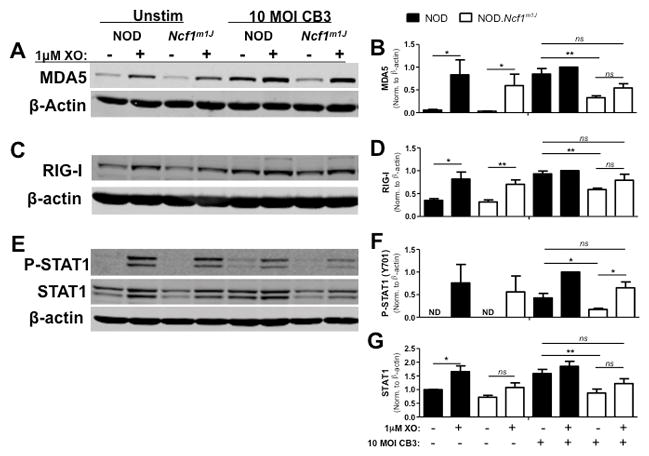
NOD and NOD.Ncf1m1J BM-MΦ were infected with 10 MOI CB3 and co-treated with 1mU/mL XO for 6–24 hours. Whole cell lysates were probed by Western blot for MDA5 (A) and RIG-I (C) at 24 hours, and P-STAT1 (Y701) and STAT1 (E) at 6 hours. Blots were then probed with β-actin as a loading control. Densitometry for MDA5 (B), RIG-I (D), P-STAT1 (Y701) (F) and STAT1 (G) was calculated by normalizing to β-actin and setting expression relative to the NOD CB3+XO treatment group. Western blot images are representative of, and densitometry plots are compiled from, at least 3 independent experiments. **p<0.01; *p<0.05; ns, not significant
Since the absence of NOX-derived superoxide compromised M1 macrophage differentiation during spontaneous and adoptive transfer of T1D in NOD.Ncf1m1J mice (26), we determined if CB3-infected NOD.Ncf1m1J macrophages displayed decreases in STAT1 signaling, a transcription factor involved in inflammatory M1 differentiation and Type-1 IFN signaling. By 6 hours, the addition of XO alone induced expression of P-STAT1 (Y701) in NOD and NOD.Ncf1m1J macrophages (Fig. 7E, F). Importantly, upon CB3 infection there was a 2.5-fold decrease (p=0.0481) in P-STAT1 (Y701) in NOD.Ncf1m1J macrophages, which was rescued with a 3.8-fold increase (p=0.0108) upon addition of XO (Fig. 7F). Interestingly, STAT1 protein expression may also be redox-regulated, as NOD.Ncf1m1J macrophages displayed a 1.8-fold decrease (p=0.0092) in total STAT1 levels upon CB3 infection (Fig. 7E, G).
To further assess STAT1 activation and M1 differentiation, nuclear translocation of P-STAT1 was analyzed by immunofluorescence staining (Fig. 8). Six hours post-infection, NOD and NOD.Ncf1m1J BM-MΦ were co-stained for P-STAT1 (Y701), F4/80, and DAPI. Nuclear expression of P-STAT1 (Y701) was readily detected upon CB3-infection in F4/80+ NOD macrophages, and was significantly decreased (p<0.0001) in CB3-infected NOD.Ncf1m1J macrophages (Fig. 8A, B). Similar to the immunoblot analysis, treatment with XO alone elicited nuclear translocation of P-STAT1 (Y701) for NOD and NOD.Ncf1m1J macrophages (p<0.0001 for both), and XO addition rescued the NOD.Ncf1m1J phenotype upon CB3 infection (Fig. 8A, B). Therefore, superoxide production can enhance M1 macrophage differentiation by increasing STAT1 phosphorylation and nuclear translocation following diabetogenic CB3 viral infections.
FIGURE 8. Loss of NOX-derived superoxide decreases nuclear localization of P-STAT1 (Y701) in CB3-infected NOD.Ncf1m1J BM-MΦ.
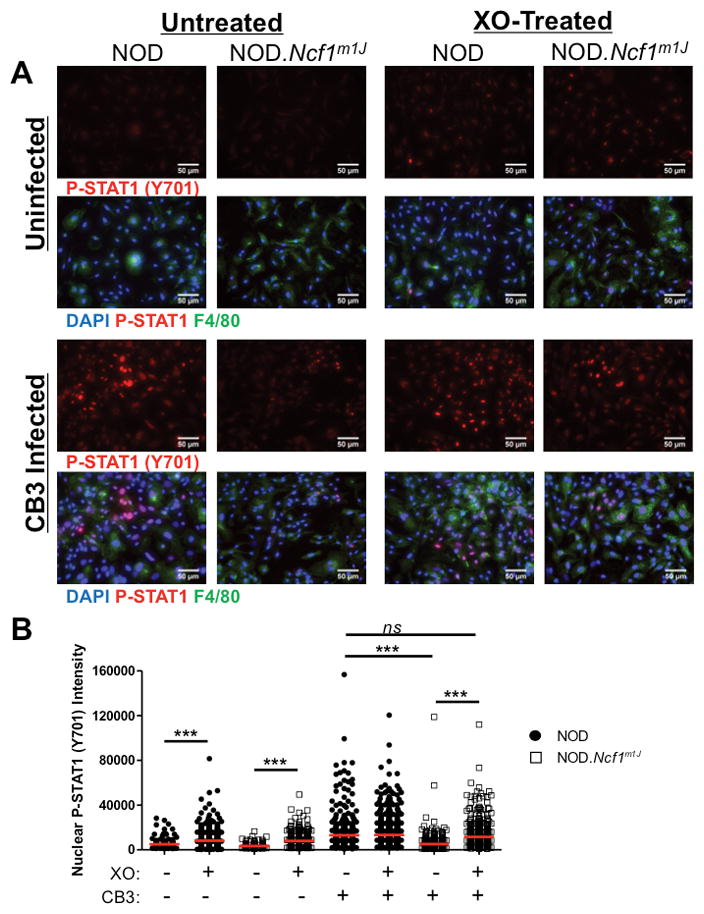
NOD and NOD.Ncf1m1J BM-MΦ were cultured on chamber slides then infected with 10 MOI CB3 in the presence or absence of XO, for 6 hours for P-STAT1 (Y701) expression (A, red stain) and co-stained with anti-F4/80 macrophage marker (A, green stain) and DAPI nuclear stain (A, blue). Mean fluorescence intensity of P-STAT1 (Y701) within DAPI-positive nuclei (B) was quantified using ImageJ analysis software. Each dot represents P-STAT1 (Y701) fluorescence within an individual nucleus. Data was compiled from at least 3 separate 40X images per well with the following total number of events: NOD (n=179), NOD+XO (n=455), NOD.Ncf1m1J (n=227), NOD.Ncf1m1J+XO (n=329), NOD+CB3 (n=374), NOD+XO+CB3 (n=729), NOD.Ncf1m1J+CB3 (n=519), NOD.Ncf1m1J+XO+CB3 (n=604). ***p<0.0001; ns, not significant
Discussion
T1D is a chronic inflammatory autoimmune disease, and studies in both the NOD mouse model and human patients have shown that oxidative stress coincides with the inflammatory progression of T1D (32). Free radicals can influence innate and adaptive immune cell activation and effector responses in T1D (23–26). Recently, our lab demonstrated that NOX-derived superoxide within the islet microenvironment can influence macrophage differentiation and progression to T1D (26). Islet-resident NOD macrophages progressively displayed a pro-inflammatory M1 phenotype during spontaneous T1D, while NOD.Ncf1m1J islet-resident macrophages were skewed towards a non-inflammatory M2 phenotype and fostered a protective microenvironment (26). Given the prominent role of macrophages in driving autoimmunity (16, 17), we hypothesized that environmental cues, such as viral infections, may trigger diabetogenic pro-inflammatory macrophages responses that are regulated by the NOX complex.
Our results demonstrate that innate immune signaling by macrophages following CB3 infection is redox-regulated and provides an essential link with diabetogenic viruses, oxidative stress, and the activation of autoimmunity in T1D. We provide evidence that superoxide-deficient NOD.Ncf1m1J mice exhibited a significant delay in CB3-accelerated T1D in contrast to NOD mice. Interestingly, we also observed that not all CB3-infected NOD mice became diabetic and corroborated a previous report by Serreze, et al using a CB4-accelerated T1D model (13). The discrepancy in acceleration and protection from Coxsackievirus-induced T1D may be due to the degree of insulitis within NOD mice prior to CB3 infection, as infections of NOD mice with low levels of insulitis exhibit a delay in T1D (39), while infections of NOD mice with established insulitis exhibit a break in tolerance and progression to T1D (11, 13). Given the range of virulence, persistence and cellular specificity across variants within both CB3 (11) and CB4 strains (10), it is also plausible that the CB3 variant used in our studies causes an acute infection that is rapidly cleared prior to efficiently generating a sustained level of oxidative stress and inflammation necessary to activate all effector T cells in the pancreas, especially of those mice with lower insulitis at the time of infection. Our data support this possibility, as CB3 viral titer was not detected from the pancreata of NOD and NOD.Ncf1m1J mice at 2 weeks post-infection, suggesting that this strain confers an acute, rather than chronic infection. However, understanding how oxidative stress is involved in persistent Coxsackievirus infections could provide even further insight into how viruses can trigger T1D in humans.
Interestingly, even in the absence of superoxide, the anti-viral response of NOD.Ncf1m1J mice was sufficient for viral clearance, but unable to trigger T1D. The pro-inflammatory innate immune response by NOD.Ncf1m1J macrophages following CB3 infection was not completely ablated, but was at sufficient levels to contribute towards viral clearance. This may be particularly true for NOD macrophages, as they are inherently hyperinflammatory and in the absence of superoxide synthesis, NOD.Ncf1m1J macrophages have an anti-viral response that is still capable of CB3 clearance, but not sufficient to accelerate autoimmune diabetes. Our data assessing serum Type 1 IFN responses supports this conclusion, as the early day 3 Type I IFN responses remained intact, and that a prolonged response was seen at day 7 in T1D-susceptible NOD, but not NOD.Ncf1m1J serum. Our results showing similar IFN-α levels between NOD and NOD.Ncf1m1J serum also aligns with results from a study by Lincez, et al, showing that IFN-α responses are associated with Coxsackie B virus clearance (40). Additionally, the loss of NOX-derived superoxide may not compromise the anti-viral response of other immune cells necessary for viral clearance including natural killer cells, CD8 T cells, and B cells. Future studies will determine if NOD.Ncf1m1J CD8 T cells display increases in cytolytic function and/or if B cells produce an enhanced humoral response following CB3 infection.
One mechanism of protection against virus-induced T1D afforded by the Ncf1m1J mutation was due to dampened inflammatory macrophage responses. Activated NOD macrophages exhibit a hyper-inflammatory phenotype (18), which may also constitute an exacerbated anti-viral response during diabetogenic viral infections. The role of innate immune responses have garnered great interest recently, as anti-viral response signatures can be detected in T1D-susceptible individuals prior to autoantibody seroconversion (41, 42). TNF-α, IFN-β, CXCL10, and CCL5 were highly produced following CB3-infection of NOD macrophages, but were significantly reduced in NOD.Ncf1m1J macrophages. Pancreas-infiltrating CD11b+ F4/80+ NOD.Ncf1m1J macrophages were less inflammatory as shown by decreased TNF-α production in contrast to NOD mice. Further corroborating the dampened inflammatory response of CB3-infected NOD.Ncf1m1J macrophages, a decrease in anti-viral signaling pathway activation was also observed. CB3 infection of NOD macrophages resulted in the upregulation of the viral RNA sensors, MDA5 and RIG-I that was dampened in NOD.Ncf1m1J macrophages. Importantly, exogenous addition of superoxide enhanced the expression of STAT1, MDA5, and RIG-I in CB3-infected NOD.Ncf1m1J macrophages to wild-type levels. Therefore, these results support the hypothesis that oxidative stress can enhance pro-inflammatory anti-viral responses following diabetogenic viral infections in the NOD mouse and ultimately, contribute to viral-induced T1D. Similar results were also observed with RIG-I and MDA5 expression in nasal epithelial cells following Influenza A Virus infection, as Duox2-derived hydrogen peroxide (H2O2) could induce RIG-I and MDA5 transcription (43). The novel findings in our study provide further mechanistic data of the connections with redox biology, dysregulated autoimmune responses in T1D, and how viral infections can exacerbate those interactions.
Our current study demonstrates that STAT1 expression and phosphorylation in macrophages are redox-dependent upon CB3 infection, as exogenous superoxide restored STAT1 activation in NOD.Ncf1m1J macrophages. The effect of free radicals on STAT1 may be direct and/or indirect via the upstream JAK1, JAK2, or TYK2 kinases. In support of the latter, H2O2 can induce P-STAT1 (Y701) expression in glia by a JAK2-dependent mechanism (44). Additionally, oxidation of several protein tyrosine phosphatase family members exacerbated STAT1 signaling in insulitic pre-diabetic NOD pancreata (45). Therefore, these results warrant further investigation into the redox-dependent mechanism of STAT1 activation in pro-inflammatory M1 macrophage differentiation. Interestingly, while superoxide-deficiency during the natural progression of T1D resulted in a skewed anti-inflammatory M2 macrophage phenotype (26), mRNA accumulation of M2-related markers including Cd206 and Stat6 were undetected in CB3-infected NOD.Ncf1m1J pancreata or in vitro-challenged macrophages (data not shown). Altogether, blocking the redox-dependent cues necessary for M1 macrophage differentiation is sufficient to delay CB3-accelerated T1D.
Supplementary Material
Acknowledgments
This work was supported by an NIH/NIDDK R01 award (DK099550) and an American Diabetes Association Career Development Award (7-12-CD-11) and the NIH NIAID (5T32AI007051-35) Immunologic Diseases and Basic Immunology T32 training grant (ARB and LEP). The following core facilities were used to generate data for the manuscript: Animal Resources Program (G20RR025858, Sam Cartner, DVM, PhD) and the Comprehensive Arthritis, Musculoskeletal, and Autoimmunity Center: Epitope Recognition Immunoreagent Core (P30 AR48311, Mary Ann Accavitti-Loper, PhD).
The authors are grateful to Jessie Barra and Drs. Jon Piganelli, Sasanka Ramanadham, and Ruth McDowell for critical reading of the manuscript. We are also appreciative of the biostatistical analyses performed by Dr. Robert Oster from the Division of Preventive Medicine at UAB.
The abbreviations used are
- T1D
Type 1 diabetes
- NOD
Non-Obese Diabetic
- NOX
NADPH oxidase
- Ncf1
neutrophil cytosolic factor 1
- ROS
reactive oxygen species
- TLR
Toll-like receptor
- STAT
signal transducer and activator of transcription
- MDA5
melanoma differentiation-associated protein 5
- RIG-I
retinoic acid-inducible protein I
- XO
xanthine oxidase
- DMPO
5,5-dimethyl-1-pyrroline-N-oxide
Footnotes
Author Contributions
No conflicts of interest relevant to this research article were reported. A.R.B. researched data, wrote the manuscript, and reviewed/edited manuscript. S.D. researched data and reviewed/edited manuscript. L.E.P. researched data and reviewed/edited manuscript. Z.K. researched data. H.M.T. researched data, wrote the manuscript, and reviewed/edited manuscript. Dr. Hubert M. Tse is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Jun HS, Yoon JW. A new look at viruses in type 1 diabetes. Diabetes Metab Res Rev. 2003;19:8–31. doi: 10.1002/dmrr.337. [DOI] [PubMed] [Google Scholar]
- 2.Hyoty H. Enterovirus infections and type 1 diabetes. Annals of medicine. 2002;34:138–147. [PubMed] [Google Scholar]
- 3.Schneider DA, von Herrath MG. Potential viral pathogenic mechanism in human type 1 diabetes. Diabetologia. 2014;57:2009–2018. doi: 10.1007/s00125-014-3340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krogvold L, Edwin B, Buanes T, Frisk G, Skog O, Anagandula M, Korsgren O, Undlien D, Eike MC, Richardson SJ, Leete P, Morgan NG, Oikarinen S, Oikarinen M, Laiho JE, Hyoty H, Ludvigsson J, Hanssen KF, Dahl-Jorgensen K. Detection of a low-grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes. 2015;64:1682–1687. doi: 10.2337/db14-1370. [DOI] [PubMed] [Google Scholar]
- 5.Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, Mosca F, Boggi U, Muda AO, Del Prato S, Elliott JF, Covacci A, Rappuoli R, Roep BO, Marchetti P. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5115–5120. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nature medicine. 1998;4:781–785. doi: 10.1038/nm0798-781. [DOI] [PubMed] [Google Scholar]
- 7.Horwitz MS, Fine C, Ilic A, Sarvetnick N. Requirements for viral-mediated autoimmune diabetes: beta-cell damage and immune infiltration. J Autoimmun. 2001;16:211–217. doi: 10.1006/jaut.2000.0486. [DOI] [PubMed] [Google Scholar]
- 8.Horwitz MS, Ilic A, Fine C, Rodriguez E, Sarvetnick N. Presented antigen from damaged pancreatic beta cells activates autoreactive T cells in virus-mediated autoimmune diabetes. The Journal of clinical investigation. 2002;109:79–87. doi: 10.1172/JCI11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horwitz MS, Ilic A, Fine C, Balasa B, Sarvetnick N. Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells. Clin Immunol. 2004;110:134–144. doi: 10.1016/j.clim.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Frisk G, Diderholm H. Tissue culture of isolated human pancreatic islets infected with different strains of coxsackievirus B4: assessment of virus replication and effects on islet morphology and insulin release. International journal of experimental diabetes research. 2000;1:165–175. doi: 10.1155/EDR.2000.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. Coxsackievirus B3 infection and type 1 diabetes development in NOD mice: insulitis determines susceptibility of pancreatic islets to virus infection. Virology. 2004;329:381–394. doi: 10.1016/j.virol.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 12.Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52:1143–1151. doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- 13.Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes. 2000;49:708–711. doi: 10.2337/diabetes.49.5.708. [DOI] [PubMed] [Google Scholar]
- 14.Lightfoot YL, Chen J, Mathews CE. Immune-mediated beta-cell death in type 1 diabetes: lessons from human beta-cell lines. European journal of clinical investigation. 2012;42:1244–1251. doi: 10.1111/j.1365-2362.2012.02711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thayer TC, Delano M, Liu C, Chen J, Padgett LE, Tse HM, Annamali M, Piganelli JD, Moldawer LL, Mathews CE. Superoxide production by macrophages and T cells is critical for the induction of autoreactivity and type 1 diabetes. Diabetes. 2011;60:2144–2151. doi: 10.2337/db10-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jun HS, Yoon CS, Zbytnuik L, van Rooijen N, Yoon JW. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. The Journal of experimental medicine. 1999;189:347–358. doi: 10.1084/jem.189.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sen P, Bhattacharyya S, Wallet M, Wong CP, Poligone B, Sen M, Baldwin AS, Jr, Tisch R. NF-kappa B hyperactivation has differential effects on the APC function of nonobese diabetic mouse macrophages. J Immunol. 2003;170:1770–1780. doi: 10.4049/jimmunol.170.4.1770. [DOI] [PubMed] [Google Scholar]
- 19.Arnush M, Scarim AL, Heitmeier MR, Kelly CB, Corbett JA. Potential role of resident islet macrophage activation in the initiation of autoimmune diabetes. J Immunol. 1998;160:2684–2691. [PubMed] [Google Scholar]
- 20.Jun HS, Santamaria P, Lim HW, Zhang ML, Yoon JW. Absolute requirement of macrophages for the development and activation of beta-cell cytotoxic CD8+ T-cells in T-cell receptor transgenic NOD mice. Diabetes. 1999;48:34–42. doi: 10.2337/diabetes.48.1.34. [DOI] [PubMed] [Google Scholar]
- 21.von Herrath M, Sanda S, Herold K. Type 1 diabetes as a relapsing-remitting disease? Nature reviews Immunology. 2007;7:988–994. doi: 10.1038/nri2192. [DOI] [PubMed] [Google Scholar]
- 22.Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, Mathews CE. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol. 2010;185:5247–5258. doi: 10.4049/jimmunol.1001472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tse HM, Milton MJ, Schreiner S, Profozich JL, Trucco M, Piganelli JD. Disruption of innate-mediated proinflammatory cytokine and reactive oxygen species third signal leads to antigen-specific hyporesponsiveness. J Immunol. 2007;178:908–917. doi: 10.4049/jimmunol.178.2.908. [DOI] [PubMed] [Google Scholar]
- 24.Sklavos MM, Tse HM, Piganelli JD. Redox modulation inhibits CD8 T cell effector function. Free radical biology & medicine. 2008;45:1477–1486. doi: 10.1016/j.freeradbiomed.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 25.Padgett LE, Tse HM. NADPH Oxidase-Derived Superoxide Provides a Third Signal for CD4 T Cell Effector Responses. Journal of immunology. 2016;197:1733–1742. doi: 10.4049/jimmunol.1502581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Padgett LE, Burg AR, Lei W, Tse HM. Loss of NADPH oxidase-derived superoxide skews macrophage phenotypes to delay type 1 diabetes. Diabetes. 2015;64:937–946. doi: 10.2337/db14-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seleme MC, Lei W, Burg AR, Goh KY, Metz A, Steele C, Tse HM. Dysregulated TLR3-dependent signaling and innate immune activation in superoxide-deficient macrophages from nonobese diabetic mice. Free Radic Biol Med. 2012;52:2047–2056. doi: 10.1016/j.freeradbiomed.2012.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Padgett LE, Anderson B, Liu C, Ganini D, Mason RP, Piganelli JD, Mathews CE, Tse HM. Loss of NOX-Derived Superoxide Exacerbates Diabetogenic CD4 T-Cell Effector Responses in Type 1 Diabetes. Diabetes. 2015;64:4171–4183. doi: 10.2337/db15-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horwitz MS, Krahl T, Fine C, Lee J, Sarvetnick N. Protection from lethal coxsackievirus-induced pancreatitis by expression of gamma interferon. J Virol. 1999;73:1756–1766. doi: 10.1128/jvi.73.3.1756-1766.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cantor J, Haskins K. Recruitment and activation of macrophages by pathogenic CD4 T cells in type 1 diabetes: evidence for involvement of CCR8 and CCL1. J Immunol. 2007;179:5760–5767. doi: 10.4049/jimmunol.179.9.5760. [DOI] [PubMed] [Google Scholar]
- 31.Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Annals of the New York Academy of Sciences. 2013;1281:16–35. doi: 10.1111/j.1749-6632.2012.06826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gil-del Valle L, de la CML, Toledo A, Vilaro N, Tapanes R, Otero MA. Altered redox status in patients with diabetes mellitus type I. Pharmacological research : the official journal of the Italian Pharmacological Society. 2005;51:375–380. doi: 10.1016/j.phrs.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Richer MJ, Lavallee DJ, Shanina I, Horwitz MS. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PloS one. 2009;4:e4127. doi: 10.1371/journal.pone.0004127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Segal BH, Grimm MJ, Khan AN, Han W, Blackwell TS. Regulation of innate immunity by NADPH oxidase. Free radical biology & medicine. 2012;53:72–80. doi: 10.1016/j.freeradbiomed.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beck MA, Williams-Toone D, Levander OA. Coxsackievirus B3-resistant mice become susceptible in Se/vitamin E deficiency. Free radical biology & medicine. 2003;34:1263–1270. doi: 10.1016/s0891-5849(03)00101-1. [DOI] [PubMed] [Google Scholar]
- 36.Antonelli A, Fallahi P, Ferrari SM, Pupilli C, d’Annunzio G, Lorini R, Vanelli M, Ferrannini E. Serum Th1 (CXCL10) and Th2 (CCL2) chemokine levels in children with newly diagnosed Type 1 diabetes: a longitudinal study. Diabetic medicine : a journal of the British Diabetic Association. 2008;25:1349–1353. doi: 10.1111/j.1464-5491.2008.02577.x. [DOI] [PubMed] [Google Scholar]
- 37.Roep BO, Kleijwegt FS, van Halteren AG, Bonato V, Boggi U, Vendrame F, Marchetti P, Dotta F. Islet inflammation and CXCL10 in recent-onset type 1 diabetes. Clinical and experimental immunology. 2010;159:338–343. doi: 10.1111/j.1365-2249.2009.04087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhernakova A, Alizadeh BZ, Eerligh P, Hanifi-Moghaddam P, Schloot NC, Diosdado B, Wijmenga C, Roep BO, Koeleman BP. Genetic variants of RANTES are associated with serum RANTES level and protection for type 1 diabetes. Genes Immun. 2006;7:544–549. doi: 10.1038/sj.gene.6364326. [DOI] [PubMed] [Google Scholar]
- 39.Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, Lane PH, Romero JR, Leser JS. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol. 2002;76:12097–12111. doi: 10.1128/JVI.76.23.12097-12111.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lincez PJ, Shanina I, Horwitz MS. Reduced expression of the MDA5 Gene IFIH1 prevents autoimmune diabetes. Diabetes. 2015;64:2184–2193. doi: 10.2337/db14-1223. [DOI] [PubMed] [Google Scholar]
- 41.Ferreira RC, Guo H, Coulson RM, Smyth DJ, Pekalski ML, Burren OS, Cutler AJ, Doecke JD, Flint S, McKinney EF, Lyons PA, Smith KG, Achenbach P, Beyerlein A, Dunger DB, Wicker LS, Todd JA, Bonifacio E, Wallace C, Ziegler AG. A type I interferon transcriptional signature precedes autoimmunity in children genetically at-risk of type 1 diabetes. Diabetes. 2014 doi: 10.2337/db13-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kallionpaa H, Elo LL, Laajala E, Mykkanen J, Ricano-Ponce I, Vaarma M, Laajala TD, Hyoty H, Ilonen J, Veijola R, Simell T, Wijmenga C, Knip M, Lahdesmaki H, Simell O, Lahesmaa R. Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes. 2014 doi: 10.2337/db13-1775. [DOI] [PubMed] [Google Scholar]
- 43.Kim HJ, Kim CH, Kim MJ, Ryu JH, Seong SY, Kim S, Lim SJ, Holtzman MJ, Yoon JH. The Induction of Pattern-Recognition Receptor Expression against Influenza A Virus through Duox2-Derived Reactive Oxygen Species in Nasal Mucosa. Am J Respir Cell Mol Biol. 2015;53:525–535. doi: 10.1165/rcmb.2014-0334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorina R, Sanfeliu C, Galito A, Messeguer A, Planas AM. Exposure of glia to pro-oxidant agents revealed selective Stat1 activation by H2O2 and Jak2-independent antioxidant features of the Jak2 inhibitor AG490. Glia. 2007;55:1313–1324. doi: 10.1002/glia.20542. [DOI] [PubMed] [Google Scholar]
- 45.Stanley WJ, Litwak SA, Quah HS, Tan SM, Kay TW, Tiganis T, de Haan JB, Thomas HE, Gurzov EN. Inactivation of Protein Tyrosine Phosphatases Enhances Interferon Signaling in Pancreatic Islets. Diabetes. 2015;64:2489–2496. doi: 10.2337/db14-1575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.