

Abstract
Background
Pulmonary arterial hypertension (PAH) is characterized by abnormal growth and enhanced glycolysis of pulmonary artery endothelial cells (PAECs). However, the mechanisms underlying alterations in energy production have not been identified.
Methods
Here, we examined the miRNA and proteomic profiles of blood outgrowth endothelial cells (BOECs) from patients with heritable PAH (HPAH) due to mutations in the bone morphogenetic protein receptor type 2 (BMPR2) gene and patients with idiopathic PAH (IPAH) to determine mechanisms underlying abnormal endothelial glycolysis. We hypothesized that in BOECs from PAH patients, the downregulation of miR-124, determined using a tiered systems biology approach, is responsible for increased expression of the splicing factor polypyrimidine-tract-binding protein (PTBP1), resulting in alternative splicing of pyruvate kinase muscle isoforms 1 and 2 (PKM1 and 2) and consequently, increased PKM2 expression. We questioned whether this alternative regulation plays a critical role in the hyperglycolytic phenotype of PAH endothelial cells.
Results
HPAH and IPAH BOECs recapitulated the metabolic abnormalities observed in PAECs from IPAH patients, confirming a switch from oxidative phosphorylation to aerobic glycolysis. Overexpression of miR-124, or siRNA silencing of PTPB1, restored normal proliferation and glycolysis in HPAH BOECs, corrected the dysregulation of glycolytic genes and lactate production, and partially restored mitochondrial respiration. BMPR2 knockdown in control BOECs reduced expression of miR-124, increased PTPB1, and enhanced glycolysis. Moreover, we observed reduced miR-124, increased PTPB1 and PKM2 expression and significant dysregulation of glycolytic genes in the rat SUGEN-hypoxia model of severe PAH, characterized by reduced BMPR2 expression and endothelial hyperproliferation, supporting the relevance of this mechanism in vivo.
Conclusions
Pulmonary vascular and circulating progenitor endothelial cells isolated from patients with PAH demonstrate downregulation of miR-124 leading to the metabolic and proliferative abnormalities in PAH ECs via PTPB1 and PKM1/PKM2. Therefore, the manipulation of this miRNA, or its targets, could represent a novel therapeutic approach for the treatment of PAH.
Keywords: metabolism, pulmonary hypertension, endothelial cell, endothelial progenitor cells, glycolysis
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a rare disease characterized by profound vascular remodelling in the small peripheral arteries of the lung, leading to a progressive increase in pulmonary vascular resistance1. The consequence of pulmonary vascular obliteration is right heart failure and increased mortality2. The classes of PAH include heritable PAH (HPAH), caused primarily by mutations in the gene encoding the bone morphogenetic protein receptor type 2 (BMPR2), and an idiopathic form (IPAH), which resembles the inherited disease both clinically and at a molecular level3. Endothelial cell (EC) dysfunction is considered to be a critical initiating factor in the pathobiology of PAH, manifested by increased susceptibility to apoptosis and heightened permeability, but also enhanced endothelial proliferation, contributing to the formation of plexiform lesions2. Recent studies have identified abnormalities in cellular bioenergetics in ECs isolated from patients with IPAH, associated with increased proliferation. Specifically, alterations in glucose uptake and utilization, accompanied by a reduction in mitochondrial oxidative phosphorylation, have been demonstrated, accompanied by similar findings in smooth muscle cells and fibroblasts from the hypertensive pulmonary vasculature4–7.
It is established that healthy ECs generate most of their energy from glycolysis8. It has been proposed that the reliance of ECs on glycolysis allows these cells to reduce oxygen consumption thereby increasing the oxygen availability to perivascular tissues9, and enabling ECs to adapt quickly to pro-angiogenic stimuli by providing more rapid ATP production than oxidative phosphorylation, when shifting from a quiescent state to proliferation and migration8. Pulmonary artery ECs (PAECs) from PAH patients exhibit a further shift to lactate synthesis and aerobic glycolysis, accompanied by reduced oxidative phosphorylation6, 7. This phenomenon, known as the “Warburg effect”, was originally observed in cancer cells and is considered necessary for the efficient allocation of nutrients between energy production and macromolecular biosynthesis in highly proliferative cells10.
Despite the introduction of several vasodilator treatments, PAH remains a life-limiting disease and existing therapies fail to target its underlying cellular and molecular abnormalities11. Thus, novel therapeutic approaches are urgently needed. Recent experimental approaches aimed at the normalization of glucose oxidation or the modulation of the balance between fatty acid oxidation and glucose oxidation have shown promise as therapies for PAH, confirming the importance of metabolic abnormalities in this disease12, 13. Restoring normal glycolysis in pulmonary vascular ECs might therefore represent a new and effective approach in the treatment of PAH.
MicroRNAs (miRNAs) are a class of small, non-coding RNAs that negatively regulate gene expression by targeting specific messenger RNAs (mRNAs) and inducing their degradation or translational repression. Several studies have confirmed the importance of miRNA-mediated gene regulation in cardiovascular development and pathology14 and in metabolic abnormalities15. MicroRNAs are also involved in the pathobiology of PAH16, with effects on cell proliferation and apoptosis17, 18 and mitochondrial metabolism19. Recently, a role for miR-124 was described in the proliferation of pulmonary artery smooth muscle cells (PASMCs) and fibroblasts (PAFs)4, 20, linking decreased expression of miR-124 with the highly proliferative, migratory, and inflammatory phenotype of hypertensive pulmonary artery adventitial fibroblasts20.
In the present study, consistent with previous reports in ECs derived from the hypertensive pulmonary artery6, 7, we confirmed the abnormal phenotype of blood outgrowth endothelial cells (BOECs) derived from PAH patients, associated with enhanced glycolysis and reduced mitochondrial oxidative phosphorylation. Employing unbiased miRNA and proteomic profiling, we identified miR-124 amongst the most downregulated miRNAs in BOECs from HPAH and IPAH patients, associated with increased levels of a target protein, the splicing factor polypyrimidine-tract-binding protein (PTBP1). We further show that upregulation of PTPB1 by decreased miR-124 expression, as reported in cancer cells21, contributes to the metabolic and proliferative abnormalities of PAH BOECs. The mechanism involved switching of expression between two pyruvate kinase muscle isoforms, PKM1, normally expressed in adult cells, and PKM2, associated with hyperproliferative and hyperglycolytic cells22. Our experiments demonstrate a link between BMPR2 dysregulation and the expression levels of miR-124, PTBP1 and downstream targets, reaffirming a central role for BMPR-II in the development of PAH. Dysregulation of miR-124 and PTBP1 was also observed in the rat SUGEN-hypoxia model of severe PAH, supporting a conserved mechanism for glycolysis regulation between patients and experimental models of PAH. Notably, in vitro manipulation of miR-124 or PTBP1 restored normal glycolysis in ECs, partially restored mitochondrial TCA cycle activity and corrected the hyperproliferative EC phenotype, suggesting that inhibition of this critical pathway could provide a promising new strategy in the treatment of PAH.
METHODS
A full description of the methods is presented in the supplementary material.
Cell culture
BOECs were isolated and cultured as previously described23. Human PAECs were purchased from Lonza, Workingham, United Kingdom. Cells were maintained in complete endothelial cell growth medium-2 (EGM-2MV) and were used at passages 4 to 7. Human PAECs were derived from patients with idiopathic PAH or unused donor lungs. Clinical information on PAH patients for PAEC and BOEC isolations is summarized in Suppl. Table 1.
Measurement of glycolytic flux
Glycolytic flux of cells was measured by monitoring the conversion of 5-3H-glucose to 3H2O as previously described7. Briefly, 2 × 105 cells/well (BOECs from HPAH or healthy controls, or control PAECs) were plated in a 48 well plate in normal medium (EGM-2 MV 10% FBS). 24 hours after seeding (or 48 to 72 hours post-transfection as indicated in figure legend), 5-3H-glucose (PerkinElmer Life Sciences Inc., Boston, MA, USA) was added to a final concentration of 0.5 μCi/well (0.0185 MBq/well). Samples were incubated for 2 hours at 37 °C in a humidified incubator under 5% CO2. Then, 200 μl/well of supernatant was collected and placed into glass vials containing hanging wells with filter paper soaked with H20. Vials were capped, sealed with rubber stoppers, and incubated for 2–3 days at 37 °C to reach equilibrium. During incubation, 3H2O generated by glycolysis diffused from the bottom of the glass vials to the filter paper carried by the hanging wells through evaporation, condensation and absorption. The filter paper was then transferred into scintillation vials containing 5 ml of scintillation liquid and counted in a scintillation counter. Appropriate 3H-glucose-only and H2O-only controls were included, enabling the calculation of H2O in each sample and thus the flux of glycolysis as described7.
Proliferation assays
BOECs were maintained in complete EGM-2MV. Cells were plated in 24-well plates at 10,000 cells per well, then transfected with a Silencer® for PTBP1, a miR-124 Pre-miR™ miRNA Precursor or a Pre-miR™ miRNA scramble negative control (Applied Biosystems, Foster City, CA, USA) as described in the Supplemental material. Cells were quiesced in serum-free medium for 4 hours and counted on days 0, 2, 5 and 7 using trypan blue exclusion.
SUGEN-hypoxia rat model
Male Sprague–Dawley rats (12 weeks old; 190–200 g) received a single injection of SUGEN-5416 on day 1 (20 mg/kg) and were then maintained in 10% O2 for 3 weeks. Rats were returned to normoxia for a further 5 weeks. Right ventricular systolic pressure (RVSP) and right ventricular hypertrophy (ratio of right ventricle [RV] weight to the left ventricle plus septal weight [LV+S] were measured as described previously24. Following sacrifice, lung tissue was harvested and frozen in liquid nitrogen. Protocols were conducted under the Animals (Scientific Procedures) Act 1986 (Amendment Regulations 2012) following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body.
Statistical Analysis
Values are expressed as fold change or mean ± SEM. Student t-test, 1-way ANOVA or 2-way ANOVA were used for statistical analysis, as specified in figure legends. Differences with p-values <0.05 were considered statistically significant. For qPCR, samples were tested in triplicate and data were analyzed using Bonferroni post-hoc test.
RESULTS
Mutation or deficiency of BMPR2 is associated with increased glycolysis in ECs
To compare the metabolic profiles of control and HPAH BOECs, conditioned media (n=4 of each) were collected and analysed on the Ultrahigh Performance Liquid Chromatography/Electrospray Ionization Tandem Mass Spectrometry Platform (Metabolon Inc.), as previously described25. Metabolic profiles of HPAH BOECs differed significantly from controls, as determined by multivariate analysis. In particular, hierarchical clustering analysis (Suppl. Figure 1A) and partial least square-discriminant analysis (Suppl. Figure 1B) showed distinct metabolic profiles between the two groups. Metabolites with the highest loading weights across principal component 1 (Suppl. Figure 1B), explaining ~25% of the total variance, included nucleotides, glycolytic products, amino acids and lipids (Suppl. Figure 1A). The metabolite showing the most statistically significant change (p=0.0001) was pyruvate (Suppl. Figure 1C), which was increased over 7.5-fold in HPAH cells compared with controls.
Consistent with the metabolomic findings, glycolysis measured by 3H-glucose metabolism, was significantly higher in BOECs from HPAH patients carrying a BMPR2 mutation (Figure 1A), and IPAH patients (Figure 1B) compared with cells derived from control individuals. Lactate concentrations in cell supernatants were also higher in HPAH (Figure 1C) and IPAH (Figure 1D). Since the expression level of BMPR2 is suppressed in pulmonary vascular cells of PAH patients with non-genetic forms of PAH26, we measured glycolysis in control BOECs and PAECs after silencing of BMPR2 with a short-interfering RNA (siRNA), or control siRNA. Seventy two hours after transfection, glycolytic flux was significantly increased in siBMPR2-treated BOECs (Figure 1E) and PAECs (Figure 1F).
Figure 1. Glycolysis is increased in BOECs from HPAH/IPAH patients and in siBMPR2-treated control ECs.
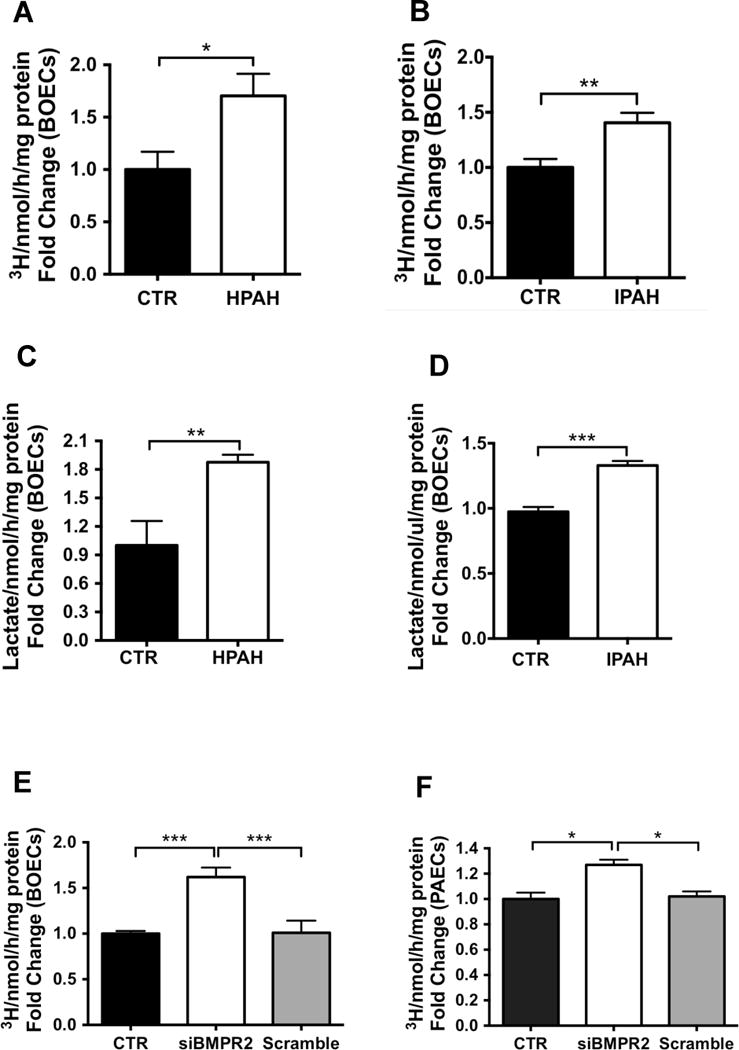
(A–B) Glycolytic flux in BMPR2 mutant HPAH BOECs (A, n=4) and IPAH BOECs (B, n=4) compared to control BOECs (n=3). (C–D) Lactate production was measured in HPAH BOECs (C) or IPAH BOECs (D) compared to control BOECs. (E–F) Glycolytic flux in control BOECs (E) or PAECs (F) transfected with siBMPR2 compared to transfection reagent alone (CTR) or scrambled siRNA. Sample were tested in triplicate. Data are presented as the mean ± S.E.M. of four biological replicates and analyzed using an unpaired t-test (A–D), or a 1-way ANOVA followed by Bonferroni post hoc test (E–F) (***p<0.001, **p<0.01, *p<0.05).
miRNA and proteomic screens identify an altered miR-124/PTBP1 axis in PAH BOECs
To elucidate the molecular mechanisms by which loss of BMPR2 leads to altered endothelial glycolysis, we employed an unbiased genome-wide screen to detect miRNAs dysregulated in BOECs derived from 4 HPAH, 3 IPAH and 4 control subjects (Suppl. Table 1). A total of 1066 miRNAs were measured by qPCR-array. 17 miRNAs were altered in the principal comparison of HPAH versus control subjects (from −2.8 to +2.0 fold change, unadjusted p<0.05; Figure 2A–B, Suppl. Table 2). Comparing miRNA levels in IPAH versus control BOECs revealed 34 altered miRNAs (from −3.2 to +3.6 fold change, Figure 2B, Suppl. Table 2). 4 miRNAs showed concordant changes in both HPAH and IPAH BOECs versus controls (Figure 2C). Of these, miR-124 exhibited the most pronounced and significant reduction (from −2.8 to −3.2 fold, p=0.006). Due to the modest sample size, these changes did not achieve statistical significance after a false discovery rate correction was applied (Figure 2C, Suppl. Table 2). However, several of the miRNAs identified were subsequently validated in larger sample sizes by quantitative PCR analysis (Suppl. Figure 2). Also the reduction in miR-124 was further confirmed in BOECs from larger groups of HPAH and IPAH patients (Suppl. Table 1) and using a different qPCR platform (Figure 2D–E). To determine whether BMPR-II is involved in the regulation of miR-124, we examined miR-124 expression after silencing of BMPR2 in control BOECs. miR-124 was significantly downregulated 24 and 48 hours following siRNA transfection, with miR-124 expression returning to baseline levels after 72 hours (Figure 2F).
Figure 2. miR-124 expression is reduced in PAH BOECs and in control cells following BMPR2 silencing.
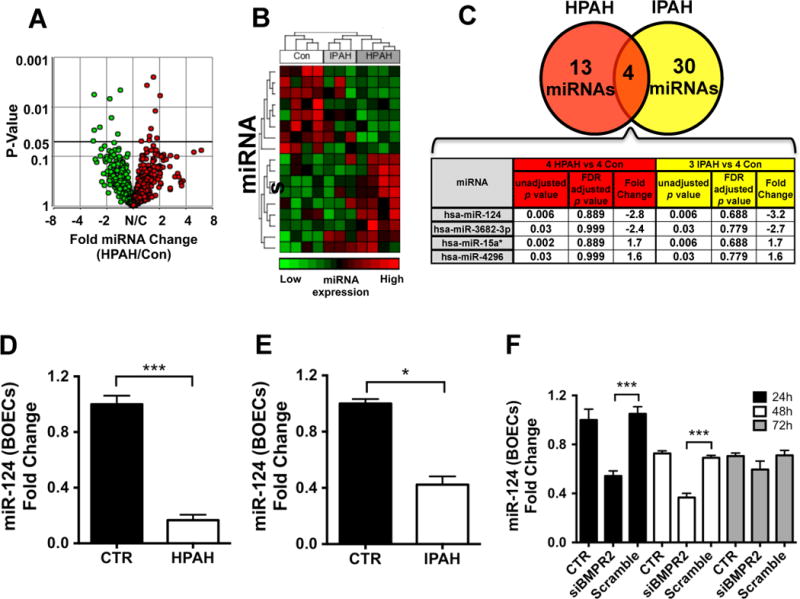
1066 miRNAs were screened by quantitative PCR-array in BOECs from 4 HPAH, 3 IPAH and 4 control subjects. (A) Volcano plot showing 17 miRNAs significantly altered (unadjusted p-value<0.05) between HPAH patients and control subjects. (B) Heat-map showing relative expression patterns for the miRNAs altered in HPAH and IPAH patients, and the hierarchical clustering across all subjects. (C) 4 miRNAs were concordantly dysregulated in both groups of patients. Unadjusted p-values, adjusted p-values (false discovery rate (FDR) adjusted p-value) and fold change (FC) are shown. Importantly, it should be noted that the miRNA candidates identified in this screen did not pass typical false discovery rate thresholds, and therefore were prioritized based on p-values unadjusted for multiple comparisons. miR-124 expression was validated using 7 HPAH, 5 IPAH and 8 control BOEC lines (D–E) to confirm the observations in the quantitative PCR-array. (F) Control BOECs were transfected with a siBMPR2 and tested for miR-124 expression 24, 48 and 72 hours post-transfection compared to transfection reagent alone (CTR) or scrambled siRNA (n=3). Data are presented as the mean ± S.E.M. Samples were tested in triplicate. Data were analyzed using an unpaired t-test (D–E) or a 1-way ANOVA followed by Bonferroni post-hoc test (F). (***p<0.001, *p<0.05).
In addition to miRNA profiling, we examined the proteome of HPAH BOECs compared to control cells using a highly quantitative multiplex proteomic approach based on labelling peptides from eight different samples with eight individual isobaric tags (iTRAQ), allowing simultaneous assessment of relative and absolute changes27 (Suppl. Tables 3 and 4). The ubiquitous RNA binding protein PTBP1, a known target of miR-12428, was among the most upregulated proteins (Suppl. Table 4). Although originally identified as a splicing factor, PTBP1 plays a central role in several cellular processes related to mRNA metabolism including polyadenylation, mRNA stability and translation initiation29. A comparison between the miRNA microarray and the proteomic study is provided in Suppl. Table 5.
miR-124 and PTBP1 are aberrantly expressed in PAH BOECs and are regulated by BMPR-II
The upregulation of PTBP1 in HPAH and IPAH BOECs was confirmed by qPCR and immunoblotting (Figure 3A–F). In addition, immunohistochemistry in human lung tissue confirmed the localization of PTBP1 in the pulmonary vascular endothelium, with positive staining also observed in the adventitia (Figure 3G). To determine whether PTBP1 is linked to BMPR-II expression, we examined PTBP1 expression following siRNA silencing of BMPR2 in control BOECs, and demonstrated increased expression of PTBP1 mRNA 72 hours post-transfection (Figure 3H). Similarly, increased PTBP1 protein levels were observed 72 hours post-transfection (Figure 3I–J).
Figure 3. PTBP1 expression is increased in PAH BOECs and in siBMPR2-treated control cells.
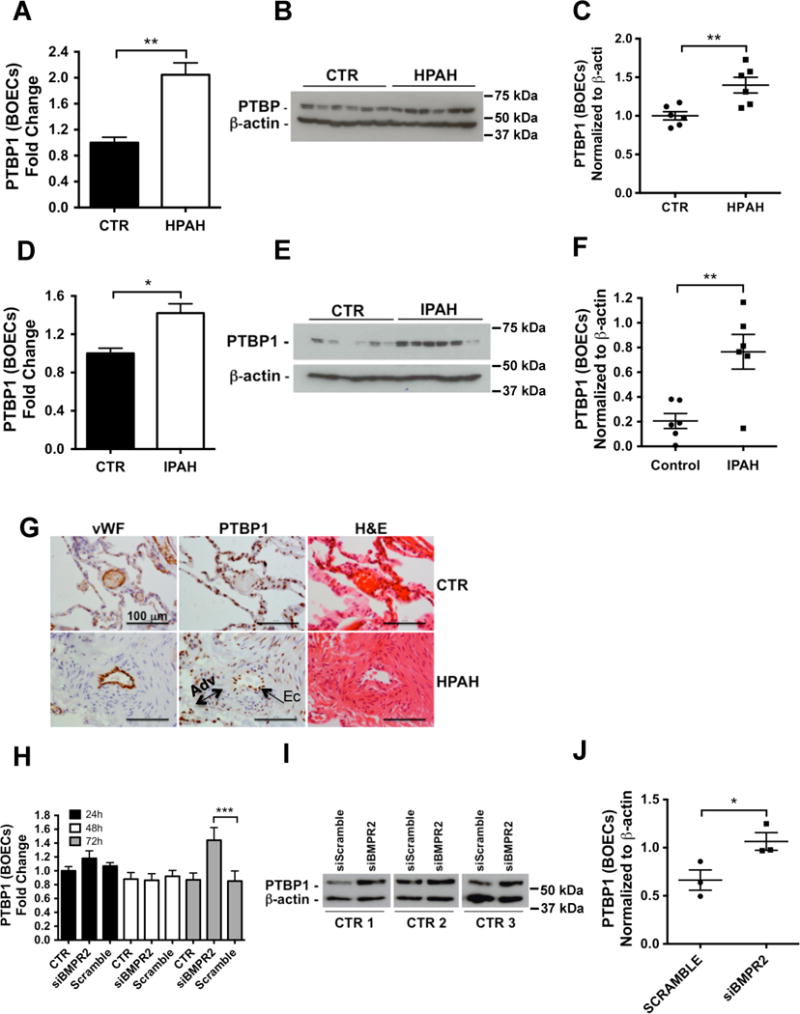
(A, D) qPCR analysis of PTBP1 was conducted in HPAH BOECs (A, n=7) or IPAH BOECs (D, n=5) versus control cells (n=8). (**p<0.01, *p<0.05). (B, E) PTBP1 protein expression was determined in whole cell lysates from 6 HPAH (B) and 6 IPAH (E) samples compared with 6 controls and normalized to β-actin expression. (C, F) Quantitative densitometry analysis of the immunoblots shown in B and E, respectively (**p<0.01). (G) PTBP1 localisation was confirmed by immunostaining in lung tissue from HPAH patients and non-affected controls. Scale bars: 100 μm. (H) PTBP1 gene expression was assessed by qPCR in control BOECs transfected with siBMPR2 or scrambled siRNA. Cells treated with the transfection reagent alone were used as a negative control (CTR). Total RNA was isolated 24, 48 and 72 hours post-transfection. Data were analyzed using a 1-way ANOVA followed by Bonferroni post hoc test. (n=3, ***p<0.001). (I–J) Immunoblot analysis of PTBP1 expression 72h hours after transfection with a siBMPR2 or scrambled siRNA. Non-transfected cells were used as a negative control (CTR). Expression was normalized to β-actin. (J) Quantitative densitometry analysis of the immunoblot (*p<0.05). Data were analyzed using an unpaired t-test.
Increased PTBP1 regulates the expression of the glycolytic factor, PKM2
PTBP1 is a critical splicing factor determining the relative expression of pyruvate kinase isoforms, PKM1 and PKM230, which are major regulators of glycolysis. Therefore, we determined the balance between PKM1/2 in ECs from PAH patients. Using qPCR we observed increased PKM2 expression in HPAH and IPAH BOECs compared with control cells (Figure 4A–B). Conversely, the expression of PKM1 was reduced in HPAH and IPAH BOECs (Suppl. Figure 3). Notably, the expression of the precursor mRNA coding for both PKM1 and PKM2 did not differ between the groups, consistent with post-transcriptional regulation by PTBP1 (Suppl. Figure 4A–B). Using confocal microscopy we confirmed that PKM2 protein levels were increased in PAH cells, mainly localized to the perinuclear area of the cell, where PKM2 is known to interact with elements of the cytoskeleton and other glycolytic enzymes to form the glycolytic enzyme complex31 (Figure 4C). To confirm a role for BMPR-II in the regulation of PKM2 we silenced BMPR2 in control BOECs and showed that PKM2 expression was significantly upregulated 72 hours post-transfection (Figure 4D). Again, we observed no change in the expression of the precursor mRNA for both PKM1 and PKM2 (Suppl. Figure 4C). We also measured the mRNA levels of two important glycolytic genes, monocarboxylate transporter 1 (MCT1) and lactate dehydrogenase A (LDHA)32, 33. MCT1, a transporter that catalyzes the transport of lactate and pyruvate across the plasma membrane, is also a predicted target of miR-12434. LDHA catalyses the final step of glycolysis, the inter-conversion of pyruvate and L-lactate. These experiments confirmed upregulation of MCT1 and LDHA in both HPAH and IPAH BOECs, and also in control BOECs 72 hours following BMPR2 siRNA knockdown (Figure 4E–J).
Figure 4. PKM2 levels are elevated in PAH BOECs and in siBMPR2 control BOECs.
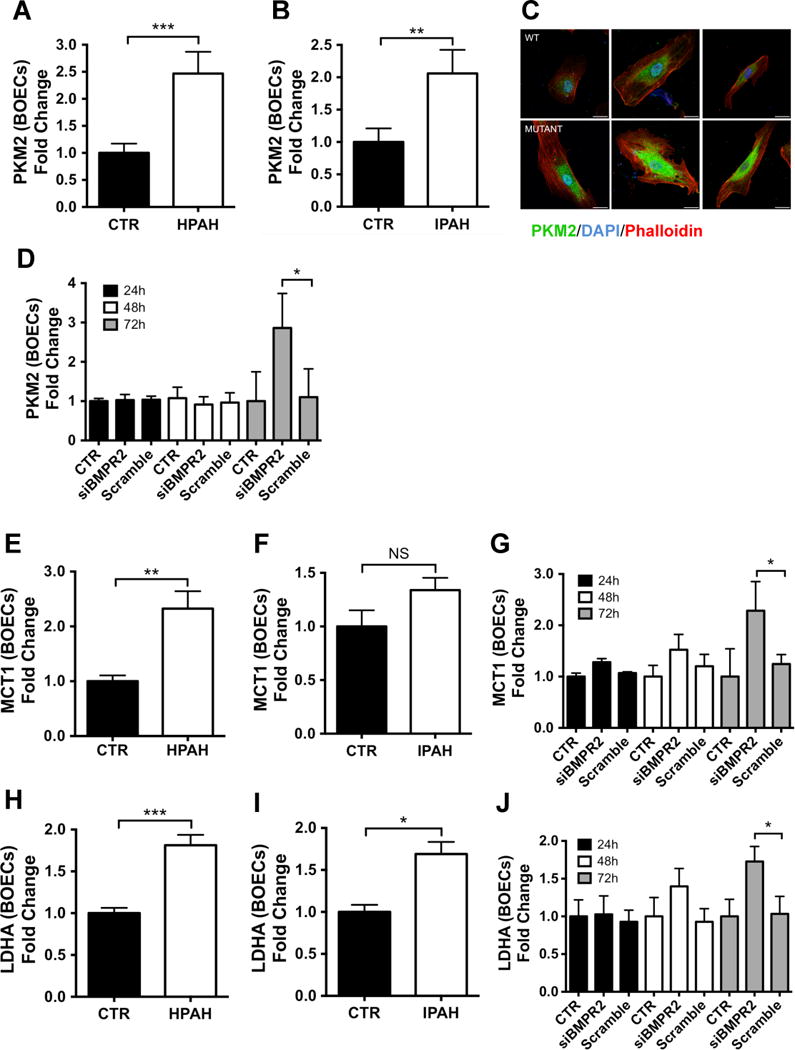
(A–B) The expression of PKM2 was assessed by qPCR in BOECs isolated from HPAH (A, n=7) or IPAH (B, n=5) patients versus controls (n=8). Data were analyzed using an unpaired t-test (***p<0.001, **p<0.01). (C) PKM2 localisation and expression was confirmed by immunofluorescence labeling of PKM2 in HPAH BOECs and control samples (n=3). PKM2 is stained in green; actin was counterstained using phalloidin (red) and nuclei counterstained using DAPI (blue). Scale bars: 25 μm. (D) PKM2 gene expression was assessed in control BOECs (n=3) transfected with siBMPR2 or scrambled siRNA control. Non-transfected cells were used as a negative control (CTR). Data were analyzed using a 1-way ANOVA followed by Bonferroni post hoc test (*p<0.05). (E, F, H, I) Glycolytic factors MCT1 and LDHA were analysed by qPCR in BOECs isolated from HPAH (E, H, n=7) and IPAH (F, I, n=5) versus controls (n=8). Data were analyzed using an unpaired t-test (***p<0.001, **p<0.01, *p<0.05). (G, J) siBMPR2-transfected control BOECs were analysed for MCT1 (G) and LDHA (J) expression levels compared to scrambled control. Cells treated with the transfection reagent alone were used as a negative control (CTR). Data were analyzed using a 1-way ANOVA followed by Bonferroni post hoc test (n=3, *p<0.05). Data are presented as the mean ± S.E.M. Samples were tested in triplicate.
Dysregulation of miR-124, PTBP1 and PKM2 in PAECs derived from PAH patients
Although we and others have previously confirmed that BOECs are highly representative of ECs23,35,36 and demonstrate the same abnormal cellular phenotypes reported in PAECs from PAH patients,23 we sought to confirm our observations in PAECs derived from IPAH patients (Suppl. Table 1). qPCR analysis confirmed the significant downregulation of miR-124 in PAH PAECs (Figure 5A), whereas immunoblotting confirmed increased expression of PTBP1 and PKM2 in IPAH samples (Figure 5C–E). Moreover, immunofluorescence staining of remodeled pulmonary arteries demonstrated high expression levels of PTBP1 in endothelial and adventitial cells, whereas PTBP1 was undetectable in normal donor arteries (Figure 5B).
Figure 5. miR-124, PTBP1 and PKM2 are dysregulated in IPAH PAECs.
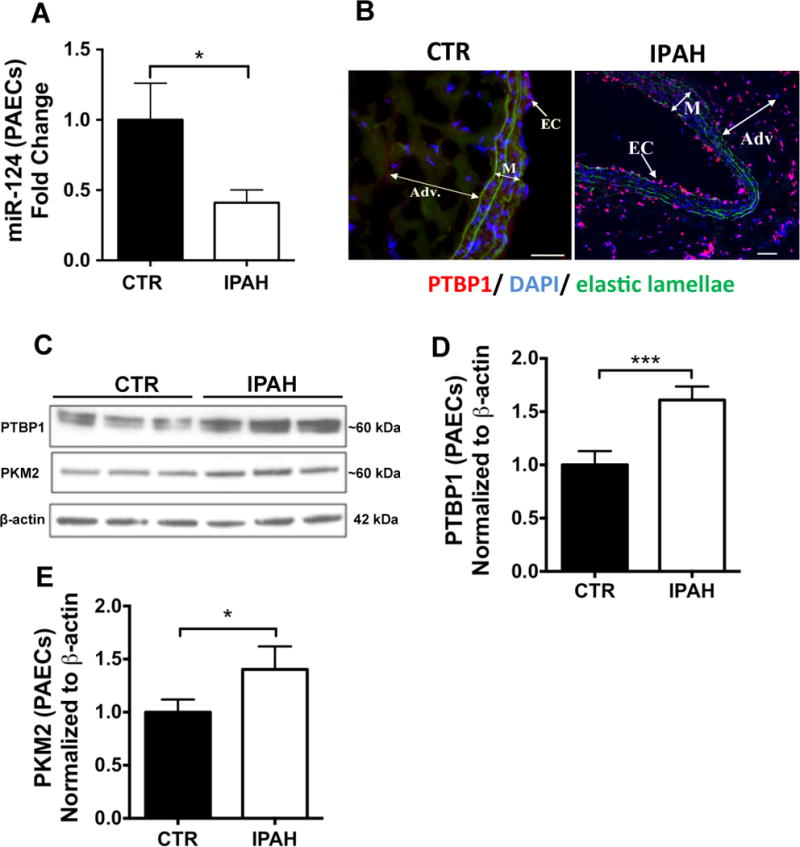
(A) miR-124 expression was analysed by qPCR in IPAH versus control PAECs (n=4, *p<0.05). Data are presented as the mean ± S.E.M. Sample were tested in triplicate. Data were analysed using an unpaired t-test. (B) Immunofluorescence staining demonstrated high expression levels of PTBP1 (red) in endothelial and adventitial cells (EC and Adv., respectively. M = media) in the pulmonary artery of IPAH patients, whereas it is undetectable in normal donor arteries. Scale bars: 100 μm. (C) Immunoblotting for PTBP1 and PKM2 protein expression in PAEC. (D–E) Quantitative densitometry analysis of PTBP1 (D) and PKM2 (E) protein expression. Immunoblotting was conducted upon whole cell lysates from PAECs from 3 IPAH patients compared with 3 controls and normalized to β-actin expression. Data were analysed using an unpaired t-test (***p<0.001, *p<0.05).
Supplementation of miR-124 in PAH BOECs normalises expression of glycolysis-related genes
We reasoned that restoration of miR-124 levels should normalise the expression of genes involved in the glycolytic program. Thus, we employed a miR-124-specific mimic to show that, 48 hours after miR-124 upregulation in HPAH BOECs, the expression of the glycolysis-related genes, LDHA, pyruvate dehydrogenase kinase 1 and 2 (PDK1, and PDK2) and MCT1, were significantly reduced and partially restored, and PKM1 expression significantly increased (Figure 6A–G). Moreover, expression of PTBP1, PKM1 and PKM2 was comparable with levels in untreated control BOECs, suggesting that manipulation of miR-124 alone corrects the abnormal expression of glycolytic genes observed in PAH ECs (Figure 6A–C). As expected, the expression of the precursor mRNA coding for both PKM1 and PKM2 did not change with introduction of the miR-124 mimic (Suppl. Figure 4D).
Figure 6. Upregulation of miR-124 restores the expression of glycolytic genes to control levels.
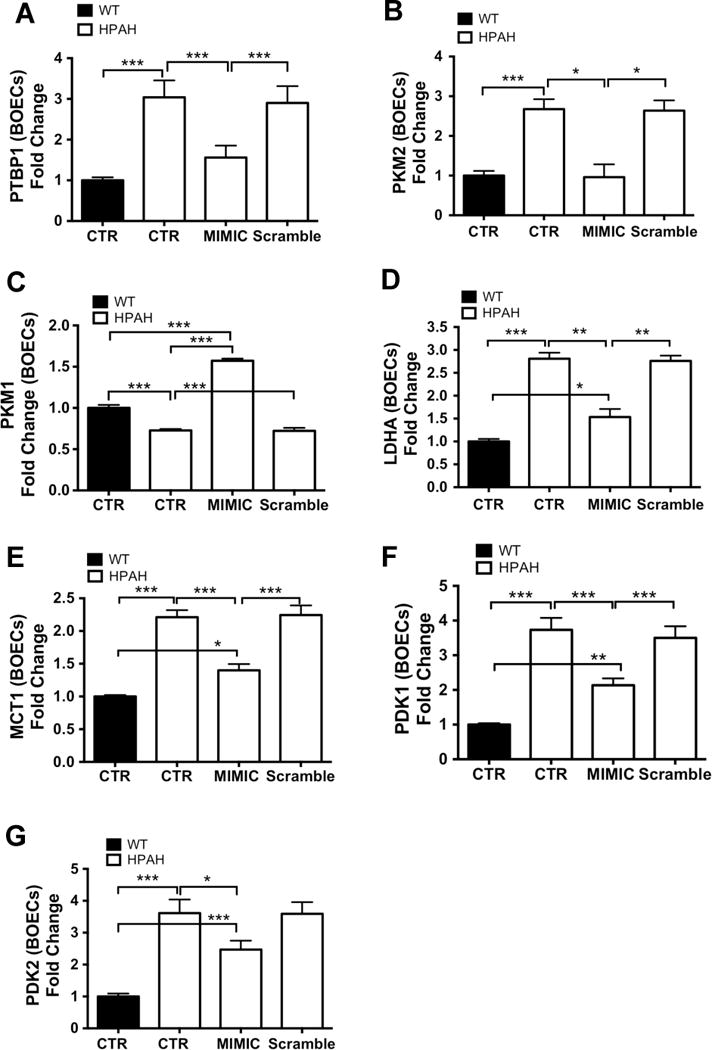
(A–G) HPAH BOECs (n=3) were transfected with a miR-124 mimic or with scrambled control. Cells treated with the transfection reagent alone were used as a negative control (CTR). Total RNA was extracted 48 hours post-transfection and analyzed by qPCR for the expression of PTBP1 (A), PKM2 (B), PKM1 (C) LDHA (D) MCT1 (E), PDK1 (F) and PDK2 (G). Gene expression was also assessed in control BOECs (n=3) to establish the basal level of expression in unaffected subjects. Data are presented as the mean ± S.E.M. Samples were tested in triplicate. Data were analyzed using a 1-way ANOVA followed by Bonferroni post hoc test. (***p<0.001, **p<0.01, *p<0.05).
miR-124 supplementation or PTBP1 silencing reverse the hyperglycolytic phenotype of PAH BOECs
To determine whether supplementation of miR-124 reverses the enhanced glycolysis observed in PAH BOECs, we measured the glycolytic flux of BMPR2 mutant HPAH, IPAH derived and control BOECs treated with a miR-124 mimic. Restoration of miR-124 levels reduced the flux of glycolysis in both PAH and control BOECs. Notably, glycolysis in mimic-treated HPAH and IPAH BOECs was comparable with the glycolytic flux of untreated and scramble-treated control cells (Figure 7A,C). Consistent with these findings, the increased lactate levels secreted from HPAH and IPAH BOECs returned to control levels after miR-124 supplementation (Figure 7B,D). miR-124 upregulation alone can therefore restore a normal glycolytic flux in PAH ECs.
Figure 7. Upregulation of miR-124 or downregulation of PTBP1 restores glycolysis, proliferation and mitochondrial activity to basal levels.
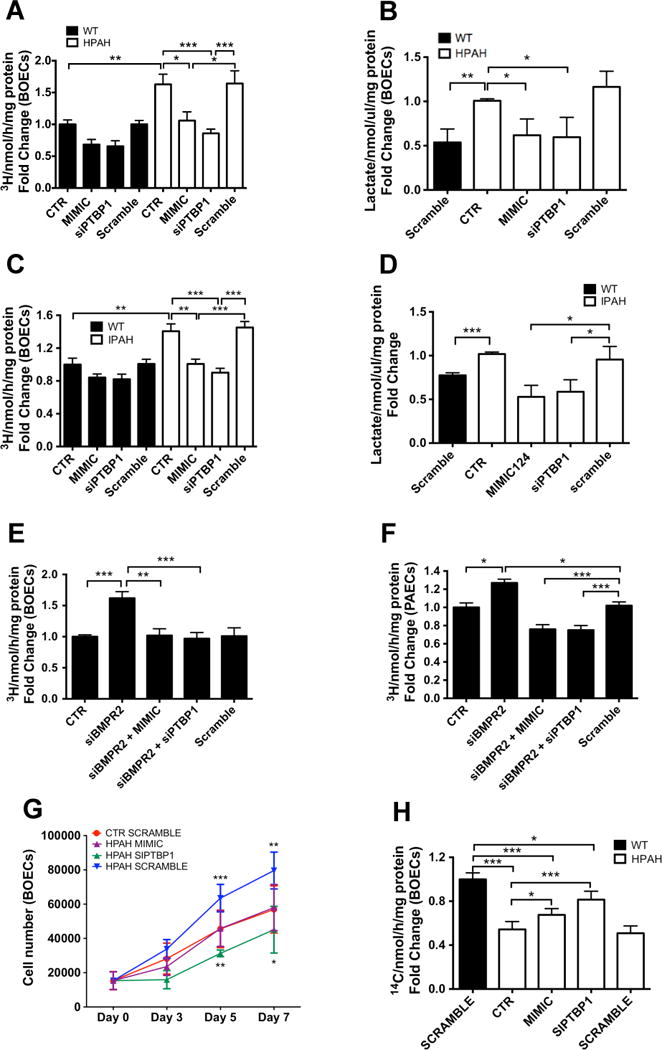
(A, C) Control (n=3; black bars) and HPAH (A, n=3; white bars) or IPAH BOECS (C, n=3; white bars) were transfected with a miR-124 mimic or a siPTBP1 and glycolytic flux was measured after 48 hours and compared with untreated (CTR) or siScramble-treated cells. (B, D) Lactate production was assessed in control and HPAH (B) or IPAH BOECs (D) treated in the same way than (A) and (C) respectively. (E–F) Glycolysis was assessed in control BOECs (E, n=3) and PAECs (F, n=3) transfected with siBMPR2, alone or in presence of miR-124 mimic or siPTBP1 and compared to untreated (CTR) or siScramble-treated cells. (G) Control BOECs (n=4) were transfected with a siScramble (red line) and HPAH cells were transfected with a miR-124 mimic (purple line), siPTBP1 (green line) or siScramble as negative control (blue line). After 48 hours, cells were serum and growth factor starved for 4 hours. Cells were counted at day 0, 3, 5 and 7. (H) TCA cycle activity was measured in HPAH BOECs untreated (CTR) or transfected with a miR-124 mimic, siPTBP1 or siScramble. Basal TCA cycle activity was also measured in control BOECs as a reference (n=4). Data are presented as the mean ± S.E.M. Every sample was tested in triplicate. Data were analyzed using a 1-way ANOVA or 2-way ANOVA (for section 7G) followed by Bonferroni post hoc test. (***p<0.001, **p<0.01, *p<0.05).
To determine whether miR-124 affects glycolysis through the regulation of PTBP1, the effect of PTBP1 manipulation on glycolysis was evaluated in HPAH BOECs. HPAH, IPAH and control BOECs were transfected with a siRNA against PTBP1 and the 3H-glucose glycolytic flux was compared with untreated and scramble-treated BOECs. Silencing of PTBP1 decreased glycolysis in PAH and control ECs, and restored a normal glycolytic flux in PAH cells (Figure 7A,C). The suppression of PTBP1 also corrected the increased lactate release from PAH BOECs (Figure 7B,D).
We next examined whether manipulation of miR-124 or PTBP1 could restore normal glycolysis following BMPR2 knockdown in control BOECs. Cells were co-transfected simultaneously with siBMPR2 and miR-124 mimic, siPTBP1 or a scrambled sequence as control. Both miR-124 upregulation and PTBP1 downregulation reversed the effect on glycolysis induced by BMPR2 silencing, and restored glycolysis to normal levels (Figure 7E). Similar results were obtained in control PAECs (Figure 7F).
PKM2 regulates LDHA expression via HIF1 activity
Having shown that miR-124 augmentation restores LDHA levels in HPAH BOECs, we questioned whether this was due to a direct effect of miR-124 on LDHA, or mediated via the effect of miR-124 on PTBP1/PKM2. Thus, we measured LDHA expression in HPAH BOECs transfected with siRNAs targeting PTBP1 or PKM237, selecting an siRNA that robustly suppressed PKM2 expression without altering PKM1 levels (Suppl. Figure 5D–E). Silencing of PTBP1 or PKM2 restored LDHA expression to control levels (Suppl. Figure 5A,C), suggesting that the effect of miR-124 on LDHA expression is mediated via PTBP1/PKM2. In addition, PKM2 can directly interact with the alpha subunit of the hypoxia inducible factor 1 (HIF1A), increasing HIF1 binding and transactivation of target genes, including LDHA, and PDK138–40. To determine the role of HIF1, we measured LDHA and PDK1 expression in HPAH BOECs 48 hours after transfection with a HIF1A siRNA (Suppl. Figure 5H). This experiment confirmed that HIF1 regulates LDHA and PDK1 expression in BOECs from PAH patients (Suppl. Figure 5F–G).
miR-124 and PTBP1 manipulation reverse the hyperproliferative phenotype of PAH BOECs
We investigated the effect of miR-124 and PTBP1 manipulation on proliferation of BOECs. HPAH BOECs were transfected with a miR-124 mimic or siPTBP1 and cultured for 7 days. Scramble-treated BMPR2 mutant and control BOECs were cultured under the same conditions for comparison. As expected, BMPR2 mutant BOECs treated with a scramble sequence demonstrated significantly higher growth rates by day 5, maintained at day 7, in comparison with control scramble-treated BOECs (Figure 7G). However, the transfection of miR-124 mimic restored cell proliferation to control levels and PTBP1 knockdown induced an even greater inhibition of cell proliferation (Figure 7G). In addition to previously reported abnormalities in ECs derived from PAH patients, we identified increased migratory capacity as a further phenotype of BMPR2 mutant ECs (Suppl. Figure 6).
miR-124 and PTBP1 manipulation partially restore mitochondrial activity in PAH BOECs
Having shown that manipulation of miR-124 and PTBP1 reduces lactate production (Figure 7B,D) and partially corrects the expression level of PDK1 and PDK2 (Figure 6E–F), we evaluated the effect of miR-124 and PTBP1 manipulation on mitochondrial activity. TCA cycle flux was measured in BMPR2 mutant BOECs treated with 14C-glucose and measuring 14C–CO2 production. Experiments were conducted in cells transfected with miR-124 mimic, siPTBP1, or scramble sequence. Scramble-treated control BOECs were also assessed to determine the basal flux of healthy cells. The experiment confirmed the reduced mitochondrial activity of untreated and scramble-treated BMPR2 mutant HPAH BOECs versus control cells, and proved that the manipulation of miR-124 and PTBP1 can partially correct the reduced mitochondrial activity (Figure 7H).
BMPR-II regulation of miR-124 does not involve SMAD1/5 or LIMK1
Since BMPR-II regulates miR-124 expression, we explored potential mechanisms. siRNA silencing of the canonical downstream mediators of BMP signalling, SMAD1 and SMAD5, did not affect expression of miR-124 or its downstream targets in control PAECs (Suppl. Figure 7A–D). Alternative, non-canonical signalling downstream of BMPR-II includes interaction with a specific lim kinase (LIMK), LIMK141. We therefore assessed the expression of miR-124 and its targets in control PAECs after the downregulation of LIMK1 with a specific siRNA and in control BOECs treated with a LIMK inhibitor, as previously reported42. These experiments failed to demonstrate an involvement of LIMK1 in the observed metabolic changes (Suppl. Figure 7A–H).
miR-124 and its glycolytic targets are dysregulated in experimental PAH
We next questioned whether dysregulation of the miR-124/PTBP1/PKM2 axis occurs in the lungs of rats with experimental PAH. We measured the expression levels of miR-124 and glycolytic genes in the SUGEN-hypoxia rat model of PAH, characterised by severely elevated RVSP and RV/LV+S43 (Figure 8A–B), and associated with increased glycolysis in pulmonary vascular cells44. Analysis of miR-124 expression in whole lung mRNA from SUGEN-hypoxia rats revealed downregulation of miR-124 compared to control animals (Figure 8C). In addition, the analysis of Ptbp1 gene and protein expression revealed upregulation in SUGEN-hypoxia rats, and Pkm2 gene expression was also increased (Figure 8D–F). Immunostaining for PTBP1 in lung tissue confirmed the expression of PTBP1 in the pulmonary endothelium and adventitia of hypertensive pulmonary arteries (Figure 8G). Mct1 and Ldha mRNA expression were also increased in the lungs of SUGEN-hypoxia rats, confirming the presence of a hyperglycolytic program of gene expression in this model (Figure 8H–I). SUGEN-hypoxia rats also demonstrated markedly reduced expression of lung Bmpr2 (Figure 8J).
Figure 8. Dysregulation of miR-124 and glycolytic genes is conserved in SUGEN-hypoxia rats.
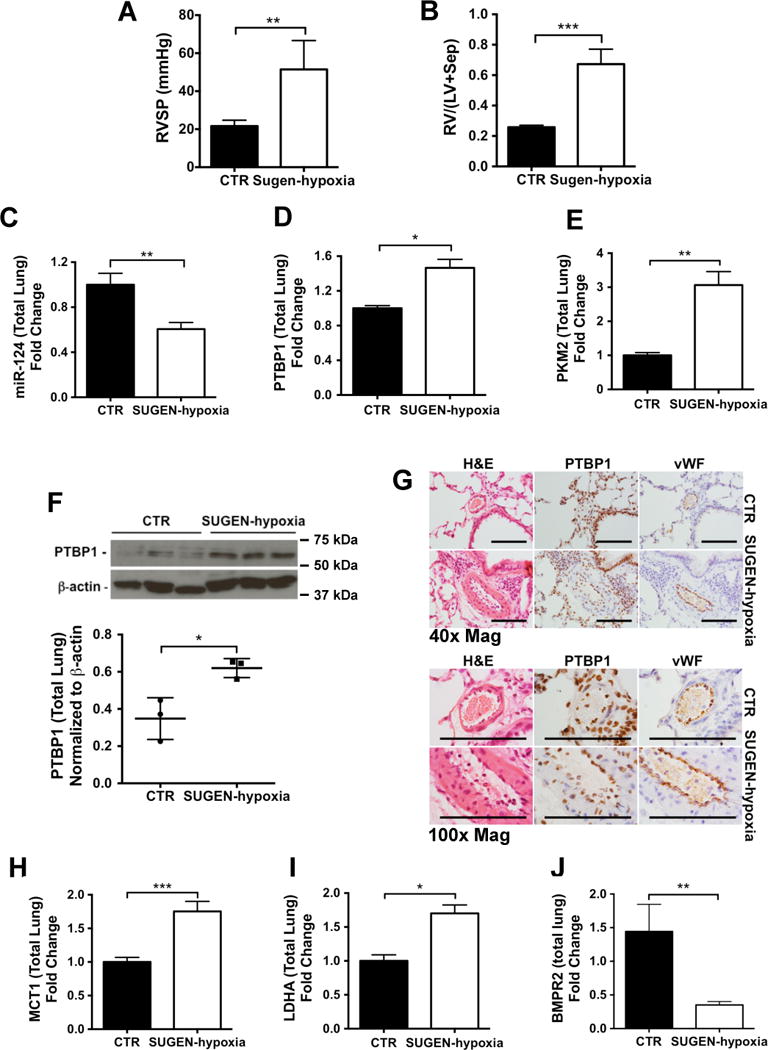
To induce the development of PAH, 12 weeks old Sprague–Dawley rats received a single injection of SUGEN-5416 on day 1 (20mg/kg) and were maintained in 10% O2 for 3 weeks. Thereafter, rats were returned to normoxia for 5 weeks. At the 8-week time point, right ventricular systolic pressure (A, RVSP) was measured and right ventricular hypertrophy was assessed (B, Fulton index, RV/(LV+Sep)) to confirm the development of PAH. (C–E) miR-124 (C), Ptbp1 (D) Pkm2 (E) gene expression was analysed by qPCR. Data were analyzed using an unpaired t-test (**p<0.01, *p<0.05). (F) Immunoblot analysis of PTBP1 protein expression in whole lung lysates extracted from 3 SUGEN-hypoxia and 3 control rats and quantitative densitometry analysis of the immunoblots. Data were analyzed using an unpaired t-test (*p<0.05). (E) PTBP1 localisation was confirmed by PTBP1 immunostaining in lung tissue from SUGEN-hypoxia and control rats. Scale bars: 100 μm. (H–J) The same samples used in C, D and E were analysed by qPCR to assess the expression levels of Mct1 (H), Ldha (I) and Bmpr2 (J). Data are presented as the mean ± S.E.M. Every sample was tested in triplicate. Data were analyzed using an unpaired t-test (***p<0.001, **p<0.01, *p<0.05).
Discussion
Understanding the role played by altered cellular metabolism in the development of PAH is an area of intense interest. Systems biology approaches have revealed evidence for a profound alteration in energy production in cells isolated from the pulmonary vasculature of PAH patients6, 7. Furthermore, expression arrays from Bmpr2 mutant mice indicate that nearly 50% of the significantly altered genes cluster in metabolic gene ontology groups, although specific metabolic pathways remain to be identified45. In PAH, ECs exhibit profound dysfunction2. Enhanced endothelial apoptosis and hyperproliferation together contribute to loss of small pulmonary arterioles and vascular occlusion. The presence of plexiform lesions in severe PAH represents an extreme example of disordered angiogenesis2. A central role of EC dysfunction in PAH is therefore well established. In addition, proliferation of pulmonary artery fibroblasts and smooth muscle cells also contributes to sustained vascular remodelling in established PAH2.
Cellular metabolism is an important determinant of endothelial phenotype and function9. Despite the close proximity of ECs to circulating oxygen, these cells do not rely on oxidative metabolism but instead rely on glycolysis for more than 80% of their ATP requirements8. Moreover, glycolytic flux in ECs is further increased when switching from a quiescent state to a proliferative and migratory phenotype. This phenomenon has been reported previously in hyperproliferative ECs isolated from IPAH patients6, 7. Of note, the introduction of BMPR-II mutations in pulmonary microvascular cells results in altered gene expression, 40% of which are related to metabolic pathways46, suggesting a mechanistic link between BMPR2 expression and metabolic dysfunction. Here, we confirm the presence of increased nucleotide levels in BOECs derived from HPAH and IPAH patients, as well as increases in steady state levels of glycolysis and glycolytic flux associated with increased proliferation. BOECs have been reported to be a readily accessible surrogate for PAECs23, 47, recapitulating the same abnormalities observed in PAECs derived from PAH patients in terms of increased cell proliferation, impaired network formation and susceptibility to apoptosis. Such cells have the advantage of easy isolation from peripheral blood, and the possibility to resample the same subject on multiple occasions. Here, we further demonstrate that BOECs isolated from PAH patients with or without BMPR2 mutations also exhibit increased glycolysis and dysregulation of glycolytic genes comparable with PAECs isolated from patients. The main focus of this study was to identify potential mechanisms responsible for this hyperglycolytic phenotype, and the potential involvement of BMPR-II in these changes, which has not been elucidated previously.
As part of an unbiased screen, we identified miR-124 as markedly suppressed in BOECs from HPAH and IPAH patients. Interestingly, previous studies reported dysregulation of miR-124 in fibroblasts and smooth muscle cells from the hypertensive lung vasculature4, 20, which suggests a conserved and central role for this pathway in PAH. Our study is the first to link this finding with the metabolic alterations in ECs from patients with PAH. Abnormal miR-124 expression is documented in cancer cells, where it contributes to proliferation and invasion21. This miRNA also plays an essential role in neuronal development, since overexpression of miR-124 promotes neuronal differentiation48. Here we show that a direct target of miR-124, the splicing factor PTBP1, is responsible for the effect of this miRNA on the heightened glycolytic flux and hyperproliferation of ECs derived from PAH patients.
PTBP1 is a ubiquitous RNA-binding protein, consisting of four RNA recognition motifs29. By binding to widely separated sequences on the same RNA molecule, PTBP1 induces a substantial restructuring of the target RNA and the introduction of RNA loops. Through this mechanism, PTBP1 is known to regulate not only RNA splicing but also RNA polyadenylation, translation and stability29. PTBP1 is downregulated by the activity of miR-124, as previously described during neurogenesis28. Our findings further support a direct link between miR-124 and PTBP1 expression in the pulmonary vasculature, and demonstrate how these changes perturb the glycolytic state of ECs.
PTBP1 is one of the main regulators of the enzyme pyruvate kinase (PK), which plays a central role in glycolysis by catalysing the dephosphorylation of phosphoenolpyruvate (PEP) into pyruvate. Recently, it was reported in cancer cells that PTBP1 promotes the switching in expression of two forms of pyruvate kinase, PKM1 and PKM2, by suppressing PKM1-specific exon 9 inclusion, and promoting PKM2-specific exon 10 inclusion. This results in increased aerobic glycolysis, consequently increasing cell proliferation28, 30. Pyruvate kinase has four isoforms, produced from two distinct genes, which are expressed in an isoform specific manner in tissues with different metabolic requirements. Among these, PKM1 is normally expressed in tissues where energy needs to be provided rapidly, such as muscle and brain. In contrast, PKM2 is mainly expressed in the embryo and is progressively replaced by PKM1 in adult tissues22. However, when quiescent cells re-enter the cell cycle, the tissue specific isoenzymes are replaced by PKM2. Most tumours mainly express PKM231. PKM2 is the preferential form expressed by hyperproliferative cells, due to its ability to shift between a tetrameric highly active form, that produces ATP efficiently, and a dimeric form, which is much less active and less ATP-efficient but which regulates the accumulation of glycolytic intermediates required for the synthesis of cell components needed by rapidly dividing cells31 (Suppl. Figure 8). PKM2-specific exon 10 can also interact with HIF1A in the nucleus and function as a transcriptional coactivator for the expression of HIF targets (LDHA and PDK1 among others), promoting the shift from oxidative phosphorylation to glycolytic metabolism39. LDHA catalyses the conversion of pyruvate into L-lactate, while the 4 PDK isoforms (PDK1–4) are responsible for the phosphorylation/inactivation of pyruvate dehydrogenase (PDH) involved in the conversion of pyruvate into acetyl-CoA. Both these enzymes can reduce the production of Acetyl-CoA, the main fuel for glucose oxidation, thus affecting mitochondrial respiration. Increased expression of PTBP1 shifts the balance of PKM isoforms towards PKM2,28, 30 therefore inducing the downstream effects on growth and metabolism previously described. Only one study has previously demonstrated a direct link between miR-124, PTBP1 and PKM2 in the regulation of aerobic glycolysis49.
We found that PKM2, together with PTBP1, were increased in the lungs of rats with PAH induced by exposure to SUGEN-hypoxia, one of the most severe rodent models of PAH, associated with reduced expression of BMPR2 and miR-124. In vitro, miR-124 enhancement or PTBP1 silencing restored normal levels of PKM2 in PAH ECs. This was associated with restoration of normal glycolytic flux and inhibition of cell hyperproliferation. miR-124 dysregulation in PAH cells could therefore explain the sustained hyperproliferative phenotype of PAECs in culture. We also show that reduced expression of miR-124 is linked to widespread alterations in the expression of glycolytic enzymes including MCT1, LDHA, PDK1 and PDK2, which were restored to basal levels by miR-124 enhancement. PTBP1, PKM2 and HIF1A siRNA silencing also restored basal expression levels of PDK1 and LDHA, suggesting that the shift from glucose oxidation to glycolysis in PAH BOECs is partly promoted by the recognized interaction between PKM2 and HIF1, responsible for HIF1 activation and associated with a state of pseudo-hypoxia in these cells38, 39.
Further studies are required to elucidate the precise mechanistic link between BMPR2 expression and glycolysis. We have established that the downregulation of BMPR2 leads to reduced expression of miR-124, increased expression of miR-124 targets, and increased glycolysis. However, the mechanism appears to be independent of canonical BMPR-II signalling via SMAD1/5, or non-canonical signalling via LIMK1. Further experiments will be necessary to identify specific mechanisms by which BMPR2 regulates expression of miR-124. Given that the penetrance of BMPR2 mutations is only 20–30% within families with PAH, it is possible that BMPR-II deficiency lowers the threshold for dysregulation of the miR124/PTBP1/PKM axis, which can then be further critically reduced by additional environmental or genetic triggers for disease. More in depth experiments are also required to determine if the increase of mitochondrial respiration induced by miR-124 and PTBP1 manipulation is caused by increased production of acetyl-CoA following restoration of PDK1 and PDK2 expression, or is due to mechanisms involving different miR-124/PTBP1/HIF1 targets.
Although we provide compelling evidence that the miR-124/PTBP1/PKM2 axis is dysregulated in the pulmonary hypertensive lungs of SUGEN-hypoxia rats, a limitation of this study is the lack of an in vivo intervention to confirm that manipulation of this pathway is causally linked to the development of PAH. Such approaches would include the delivery of a miR-124 mimic to the pulmonary vasculature, or the inhibition of PKM2 activity with agents such as Shikonin50. In a linked manuscript, Zhang et al. provide preliminary evidence that Shikonin inhibits the metabolic reprogramming in the chronically hypoxic mouse lung, with a trend towards reduced right ventricular systolic pressures51. Further optimisation will be required in preclinical models to determine whether manipulation of this pathway represents a viable approach for the clinic.
A further limitation of this study is the small sample of subjects used in the miRNA PCR array. The design of this experiment was constrained by the rarity of patient specimens and high costs. The miRNA candidates identified in the initial screen did not pass typical false discovery rate-corrected thresholds, and therefore were prioritized based on p-values unadjusted for multiple comparisons. Thus, some of these candidates should be interpreted cautiously, as we cannot exclude the possibility of false positives. Nevertheless, we independently confirmed significant changes in the levels of selected miRNAs in PAH cells by quantitative PCR analysis using a larger sample size (Suppl. Figure 2). Moreover, we have demonstrated through extensive molecular, cellular, animal and human data that miR-124 dysregulation plays a critical role in the pathobiology of PAH.
In summary, we provide evidence for a central role for reduced miR-124 in the heightened glycolysis of ECs derived from PAH patients. Restoration of miR-124 levels in vitro restored normal expression of PTBP1, PKM2 and other glycolytic genes, which in turn reversed EC hyperproliferation, one of the most well described phenotypes of PAH ECs. Similar dysregulation of the miR-124/PTBP1/PKM2 axis has been shown to play a central role in the abnormal metabolic, proliferative and inflammatory phenotype of pulmonary artery fibroblasts derived from patients with PAH and from calves with severe hypoxia-induced pulmonary hypertension51. Consistent with our observations, these authors confirm that manipulation of miR-124 or PTBP1 alone corrects the hyperglycolytic phenotype and increases mitochondrial respiration in pulmonary hypertensive fibroblasts. Taken together, these observations strongly suggest that the manipulation of the miR-124/PTBP1/PKM2 pathway represent a novel strategy to achieve additional benefits in the treatment of PAH, by correcting the conserved metabolic phenotype of key cell types contributing to pulmonary vasculature remodeling.
Supplementary Material
CLINICAL PERSPECTIVE.
What is New?
Alterations in energy production have been critically involved in the abnormal cellular responses observed in pulmonary vascular cells from patients with pulmonary arterial hypertension (PAH).
These include increased glucose uptake and utilization via glycolysis, accompanied by reduced mitochondrial oxidative phosphorylation.
Mutations in the bone morphogenetic protein type 2 receptor (BMPR2), the commonest genetic cause of PAH, alter endothelial function but the mechanisms are unclear.
Here, we demonstrate that reduced expression of miR-124 in PAH endothelial cells is responsible for the dysregulation of the splicing factor polypyrimidine-tract-binding protein (PTBP1) and its target, pyruvate kinase M2 (PKM2), a major regulator of glycolysis, which contributes to abnormal cell proliferation.
Reduced BMPR2 levels are associated with reduced miR-124 expression.
Clinical Implications
Correction of the dysregulated pathway linking miR-124, PTBP1 and PKM2 is sufficient to restore the normal glycolytic flux in PAH endothelial cells and partially correct reduced mitochondrial activity, with a subsequent inhibition of the hyperproliferative phenotype characteristic of pulmonary vascular endothelial cells from PAH patients.
These findings provide new avenues for the treatment of PAH.
Acknowledgments
We thank the Cambridge National Institute of Health Research Biomedical Research Centre and Cell Phenotyping Hub for providing infrastructure and core facilities. Additional support was provided by the BHF Cambridge Centre for Cardiovascular Research Excellence. We also thank Baraa Kwieder and Christopher Huang for their technical assistance.
Sources of Funding
This work was supported by a British Heart Foundation (BHF) Immediate Post-doctoral Fellowship (Dr. Paola Caruso, FS/13/19/29931). Professor Morrell is funded by a BHF Personal Chair Award (CH/09/001/25945) and BHF Programme Grant (RG/13/4/30107). Professor Stewart is funded by the Canadian Institutes of Health Research (Grant #57726 DJS).
Footnotes
CARUSO, Role of miR-124 in endothelial glycolysis
Disclosures
None
References
- 1.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 2.Eddahibi S, Morrell N, d’Ortho MP, Naeije R, Adnot S. Pathobiology of pulmonary arterial hypertension. Eur Respir J. 2002;20:1559–72. doi: 10.1183/09031936.02.00081302. [DOI] [PubMed] [Google Scholar]
- 3.Dewachter L, Adnot S, Guignabert C, Tu L, Marcos E, Fadel E, Humbert M, Dartevelle P, Simonneau G, Naeije R, Eddahibi S. Bone morphogenetic protein signalling in heritable versus idiopathic pulmonary hypertension. Eur Respir J. 2009;34:1100–10. doi: 10.1183/09031936.00183008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang K, Peng X, Zhang X, Wang Y, Zhang L, Gao L, Weng T, Zhang H, Ramchandran R, Raj JU, Gou D, Liu L. MicroRNA-124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. J Biol Chem. 2013;288:25414–27. doi: 10.1074/jbc.M113.460287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plecita-Hlavata L, Tauber J, Li M, Zhang H, Flockton AR, Pullamsetti SS, Chelladurai P, D’Alessandro A, El Kasmi KC, Jezek P, Stenmark KR. Constitutive Reprogramming of Fibroblast Mitochondrial Metabolism in Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2016;55:47–57. doi: 10.1165/rcmb.2015-0142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu W, Erzurum SC. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Comprehensive Physiology. 2011;1:357–72. doi: 10.1002/cphy.c090005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1342–7. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verdegem D, Moens S, Stapor P, Carmeliet P. Endothelial cell metabolism: parallels and divergences with cancer cell metabolism. Cancer & metabolism. 2014;2:19. doi: 10.1186/2049-3002-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eelen G, Cruys B, Welti J, De Bock K, Carmeliet P. Control of vessel sprouting by genetic and metabolic determinants. Trends in endocrinology and metabolism: TEM. 2013;24:589–96. doi: 10.1016/j.tem.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature reviews Cancer. 2011;11:325–37. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 11.Galie N, Ghofrani AH. New horizons in pulmonary arterial hypertension therapies. European respiratory review : an official journal of the European Respiratory Society. 2013;22:503–14. doi: 10.1183/09059180.00006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, Rehman J, Archer SL. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. Journal of molecular medicine. 2012;90:31–43. doi: 10.1007/s00109-011-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–50. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- 14.Arunachalam G, Upadhyay R, Ding H, Triggle CR. MicroRNA Signature and Cardiovascular Dysfunction. Journal of cardiovascular pharmacology. 2015;65:419–29. doi: 10.1097/FJC.0000000000000178. [DOI] [PubMed] [Google Scholar]
- 15.Gao P, Sun L, He X, Cao Y, Zhang H. MicroRNAs and the Warburg Effect: new players in an old arena. Current gene therapy. 2012;12:285–91. doi: 10.2174/156652312802083620. [DOI] [PubMed] [Google Scholar]
- 16.Bienertova-Vasku J, Novak J, Vasku A. MicroRNAs in pulmonary arterial hypertension: pathogenesis, diagnosis and treatment. Journal of the American Society of Hypertension : JASH. 2015;9:221–34. doi: 10.1016/j.jash.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Courboulin A, Paulin R, Giguere NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Cote J, Simard MJ, Bonnet S. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med. 2011;208:535–48. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White K, Dempsie Y, Caruso P, Wallace E, McDonald RA, Stevens H, Hatley ME, Van Rooij E, Morrell NW, MacLean MR, Baker AH. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension. 2014;64:185–94. doi: 10.1161/HYPERTENSIONAHA.113.03037. [DOI] [PubMed] [Google Scholar]
- 19.White K, Lu Y, Annis S, Hale AE, Chau BN, Dahlman JE, Hemann C, Opotowsky AR, Vargas SO, Rosas I, Perrella MA, Osorio JC, Haley KJ, Graham BB, Kumar R, Saggar R, Saggar R, Wallace WD, Ross DJ, Khan OF, Bader A, Gochuico BR, Matar M, Polach K, Johannessen NM, Prosser HM, Anderson DG, Langer R, Zweier JL, Bindoff LA, Systrom D, Waxman AB, Jin RC, Chan SY. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO molecular medicine. 2015;7:695–713. doi: 10.15252/emmm.201404511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang D, Zhang H, Li M, Frid MG, Flockton AR, McKeon BA, Yeager ME, Fini MA, Morrell NW, Pullamsetti SS, Velegala S, Seeger W, McKinsey TA, Sucharov CC, Stenmark KR. MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ Res. 2014;114:67–78. doi: 10.1161/CIRCRESAHA.114.301633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papagiannakopoulos T, Kosik KS. MicroRNAs: regulators of oncogenesis and stemness. BMC medicine. 2008;6:15. doi: 10.1186/1741-7015-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M, Zhang J, Manley JL. Turning on a fuel switch of cancer: hnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010;70:8977–80. doi: 10.1158/0008-5472.CAN-10-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LS, Marchesan D, Yang J, Suntharalingam J, Soon E, Exley A, Stewart S, Hecker M, Zhu Z, Gehling U, Seeger W, Pepke-Zaba J, Morrell NW. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009;180:780–7. doi: 10.1164/rccm.200810-1662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, Aldred MA, Yu PB, Upton PD, Morrell NW. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nature medicine. 2015;21:777–85. doi: 10.1038/nm.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D’Alessandro A, Gray AD, Szczepiorkowski ZM, Hansen K, Herschel LH, Dumont LJ. Red blood cell metabolic responses to refrigerated storage, rejuvenation, and frozen storage. Transfusion. 2017;57:1019–1030. doi: 10.1111/trf.14034. [DOI] [PubMed] [Google Scholar]
- 26.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–8. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 27.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res. 2014;115:189–202. doi: 10.1161/CIRCRESAHA.115.303404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell. 2007;27:435–48. doi: 10.1016/j.molcel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawicka K, Bushell M, Spriggs KA, Willis AE. Polypyrimidine-tract-binding protein: a multifunctional RNA-binding protein. Biochemical Society transactions. 2008;36:641–7. doi: 10.1042/BST0360641. [DOI] [PubMed] [Google Scholar]
- 30.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–8. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Seminars in cancer biology. 2005;15:300–8. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Halestrap AP. Monocarboxylic acid transport. Comprehensive Physiology. 2013;3:1611–43. doi: 10.1002/cphy.c130008. [DOI] [PubMed] [Google Scholar]
- 33.Halestrap AP. The SLC16 gene family - structure, role and regulation in health and disease. Molecular aspects of medicine. 2013;34:337–49. doi: 10.1016/j.mam.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 34.Pullen TJ, da Silva Xavier G, Kelsey G, Rutter GA. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1) Mol Cell Biol. 2011;31:3182–94. doi: 10.1128/MCB.01433-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ormiston ML, Toshner MR, Kiskin FN, Huang CJ, Groves E, Morrell NW, Rana AA. Generation and Culture of Blood Outgrowth Endothelial Cells from Human Peripheral Blood. Journal of visualized experiments : JoVE. 2015:e53384. doi: 10.3791/53384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toshner M, Morrell NW. Endothelial progenitor cells in pulmonary hypertension - dawn of cell-based therapy? Int J Clin Pract Suppl. 2010:7–12. doi: 10.1111/j.1742-1241.2009.02232.x. [DOI] [PubMed] [Google Scholar]
- 37.Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med. 2012;209:217–24. doi: 10.1084/jem.20111487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–44. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget. 2011;2:551–6. doi: 10.18632/oncotarget.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011;76:347–53. doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- 41.Johnson JA, Hemnes AR, Perrien DS, Schuster M, Robinson LJ, Gladson S, Loibner H, Bai S, Blackwell TR, Tada Y, Harral JW, Talati M, Lane KB, Fagan KA, West J. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2011;302:L474–84. doi: 10.1152/ajplung.00202.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott RW, Hooper S, Crighton D, Li A, Konig I, Munro J, Trivier E, Wickman G, Morin P, Croft DR, Dawson J, Machesky L, Anderson KI, Sahai EA, Olson MF. LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J Cell Biol. 2010;191:169–85. doi: 10.1083/jcb.201002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15:427–38. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 44.Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, Thenappan T, Bache-Wiig P, Piao L, Paul J, Chen CT, Archer SL. Lung (1)(8)F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:670–9. doi: 10.1164/rccm.201108-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, Tada Y, Tanabe N, Tatsumi K, Hemnes AR, West JD. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ. 2012;2:201–13. doi: 10.4103/2045-8932.97606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Majka S, Hagen M, Blackwell T, Harral J, Johnson JA, Gendron R, Paradis H, Crona D, Loyd JE, Nozik-Grayck E, Stenmark KR, West J. Physiologic and molecular consequences of endothelial Bmpr2 mutation. Respir Res. 2011;12:84. doi: 10.1186/1465-9921-12-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lavoie JR, Ormiston ML, Perez-Iratxeta C, Courtman DW, Jiang B, Ferrer E, Caruso P, Southwood M, Foster WS, Morrell NW, Stewart DJ. Proteomic analysis implicates translationally controlled tumor protein as a novel mediator of occlusive vascular remodeling in pulmonary arterial hypertension. Circulation. 2014;129:2125–35. doi: 10.1161/CIRCULATIONAHA.114.008777. [DOI] [PubMed] [Google Scholar]
- 48.Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci U S A. 2006;103:2422–7. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Y, Zhao X, Zhou Y, Hu Y. miR-124, miR-137 and miR-340 regulate colorectal cancer growth via inhibition of the Warburg effect. Oncology reports. 2012;28:1346–52. doi: 10.3892/or.2012.1958. [DOI] [PubMed] [Google Scholar]
- 50.Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, Sun X, Yang M, Billiar TR, Wang H, Cao L, Jiang J, Tang D. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nature communications. 2016;7:13280. doi: 10.1038/ncomms13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang H, Wang D, Li M, Plecitá-Hlavatá L, D’Alessandro A, Tauber J, Riddle S, Kumar S, Flockton A, McKeon BA, Frid MG, Reisz JA, Caruso P, El Kasmi KC, Ježek P, Morrell NW, Hu C, Stenmark KR. The metabolic and proliferative state of vascular adventitial fibroblasts in pulmonary hypertension is regulated through a miR-124/PTBP1/PKM axis. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.117.028069. Linked manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.