

Abstract
A new series of oxadiazolylbiphenylthiazoles was prepared with the objective of improving the limited solubility of first-generation derivatives while maintaining antibacterial activity against drug-resistant Staphylococcus aureus. Studying the structure-activity relationship at the cationic part provided the piperazine-1-carboximidamide derivative 27 with a MIC (MRSA) value of 1.1 μg/mL, bactericidal mode of action, and a 50-fold improvement in aqueous solubility. Additionally, 27 exhibited a wider safety margin against mammalian cells, and most importantly, a significant improvement in oral bioavailability.
Keywords: antibiotic drug resistance, Caenorhabditis elegans, Methicillin-resistant Staphylococcus aureus, MRSA, pharmacokinetics
Graphical Abstract

1. INTRODUCTION
The notable rise in bacterial resistance to antibiotics poses a serious threat to the entire structure of modern medicine. Patients receiving an organ transplant, cancer chemotherapy, artificial-joint replacements, or undergoing surgery or dental procedures are at significant risk of acquiring a bacterial infection.[1] Though prophylactic antibiotics are utilized to try to circumvent infection, patients still contract infections that are often resistant to treatment with one or more antibiotic classes. Staphylococcus aureus remains a leading source of antibiotic-resistant hospital-acquired infections in the United States,[2] Europe,[3] and Asia. Amongst drug-resistant staphylococci, strains of methicillin-resistant S. aureus (MRSA) are responsible for nearly half of all documented fatalities due to antibiotic-resistant infections in the United States alone.[2] Furthermore, several clinical isolates of MRSA have developed resistance to multiple antibiotic classes including β-lactam antibiotics,[4] macrolides,[5] and fluoroquinolones.[5] Additionally, strains of S. aureus have been isolated that exhibit resistance to antibiotics deemed agents of last resort including vancomycin[6] and linezolid.[7]
Remarkably, in the past 30 years no new antibiotic class effective against MRSA has been discovered[8] and received regulatory approval. This necessitates the medicinal chemistry community to respond to the need for developing new antibiotics with novel chemical scaffolds. The phenylthiazoles developed by our group represent a new antibiotic class that demonstrates potent antibacterial activity against multidrug-resistant staphylococcal strains.[9, 10] Though the first-generation phenylthiazoles possess many desirable qualities for an antibacterial agent including rapid bactericidal activity and activity against a broad range of MRSA strains, one substantial limitation is their limited stability to hepatic metabolism.[9] The initial structure-activity relationship (SAR) study identified two essential structural elements; a nitrogenous head connected to the phenylthiazole ring from one side and a lipophilic tail from the other end.[11] Incorporating the hydrolysable Schiff’s base moiety (C=N) within a pyrimidine nucleus resulted in second-generation derivatives that were more stable to hepatic metabolism (Figure 1).[2] On the other hand, rigorous SAR study of the lipophilic side chain including substitution of the alkane with a phenyl ring resulted in the most potent lipophilic moiety identified thus far (Figure 1).[9] The main disadvantage of adding an extra phenyl group to the phenylthiazole scaffold is deterioration of aqueous solubility (Figure 1). For example, the biphenyl derivative 1b was 24-times less soluble than the lead compound 1a in phosphate-buffered saline (Figure 1).[9] The issue of aqueous solubility worsened with the 2nd generation biphenyl analogue 1c (Figure 1)[12]. The inclusion of an extra pyrimidine ring in 1c significantly decreased its aqueous solubility (Figure 1). Drugs with such low solubility (e.g. telaprevir) exhibit limited oral bioavailability that does not exceed 3%, even when administrated as a nanosuspension.[13] Thus addressing the solubility issue of the phenylthiazole compounds is critical to their development as viable therapeutic agents.
Figure 1.

Developmental progress of phenylthiazole antibiotics and the new approach to improve physicochemical properties.
Designing compounds with balanced physicochemical properties is crucial since these properties are inextricable from their biochemical characteristics and are directly connected with other ADME parameters. In this regard previous attempts to improve the aqueous solubility of phenylthiazole antibiotics focused on increasing the tPSA/decreasing lipophilicity via replacing the phenyl ring with more polar/less hydrophobic groups including azoles, oxazole or thiophen; unfortunately, such efforts furnished less active analogues.[2] In the present work, a new strategy to tackle the poor aqueous solubility of biphenylthiazole antibiotics was implemented, and the pyrimidine ring was replaced with a more polar oxadiazole isostere that has fewer carbons and a more polar surface area (Figure 1). Additionally, the proper nitrogenous element at oxadiazole position-5 has been deeply explored via synthesizing a large set of 5-oxadiazolylbiphenylthiazoles with different amines, hydrazines, guanidines and carboximidines substituents.
2. RESEULTS AND DISCUSSION
2.1. CHEMISTRY
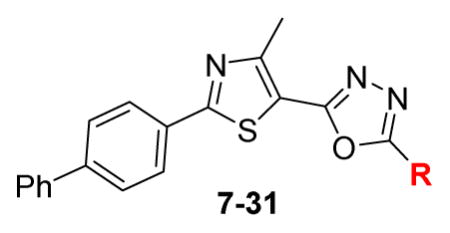
Synthesis of the starting material 3 was previously reported via C-C cross-coupling of 2-bromo4-methyl-thiazole-5-carboxylic acid ethyl ester with 4-biphenyl boronic acid in a moderate yield.[14] However, we prepared this compound in higher yield using thioamide 2 and α-chloroacetylacetone (Scheme 1). The key intermediate, acid hydrazide 4, was obtained via treatment of the corresponding ethyl ester 3 with hydrazine hydrate (Scheme 1). Adding carbon disulfide to 4, in the presence of potassium hydroxide, followed by methylation of the free thiol group afforded the 5-methylmercapto derivative 5. The latter derivative was oxidized by mCPBA to yield methylsulfone analogue 6. Analogue 6 was treated with different nitrogenous nucleophiles to afford the final products 7–30 (Scheme 1).
Scheme 1. Reagents and conditions.

(a) Absolute EtOH, α-chloroacetylacetone, heat at reflux, 4 h, 95%; (b) Absolute EtOH, NH2NH2·H2O, heat at reflux, 8 h; 94%; c) CS2, KOH, EtOH, heat at reflux, 12 h; (d) dimethyl sulfate, KOH, H2O, stirring at 23°C, 2 h; 89% (2 steps); (e) MCPBA, dry DCM, 23°C, 16 h; 93; (f) appropriate amine, hydrazine, guanidine or carboximidate; K2CO3, DMF, heat at 80°C for 0.5 – 12 h, 54 – 97%.
2.2. BIOLOGICAL RESULTS AND DISCUSSION
Anti-MRSA activity
At the outset of this study, fourteen derivatives carrying different amines (compounds 7–20) were prepared. In general, compounds with secondary and tertiary amine side chains, 7–17, were inactive against MRSA (Table 1). Using piperazine, hydroxylazetidine or hydrazine as the nitrogenous head provided three compounds (19 – 21) with better MIC values (Table 1). By extending the nitrogenous head, nine more derivatives (compounds 22–30) with guanidine and guanidine-like head groups were obtained. The guanidine-containing analogue 22 inhibited the growth of MRSA (2658 RCMB) at a concentration almost one-fold better than the MIC for the lead compound 1b and was as potent as vancomycin (MIC = 1.56 μg/mL, Table 1). Parallel to this, the aqueous solubility of compound 22 was assessed using a turbidity assay in phosphate buffered saline; remarkably 22 exhibited a marked improvement in aqueous solubility that was 43-times better than the solubility obtained for the corresponding pyrimidine analogue 1c (Table 2).
Table 1.
Antimicrobial activity (μg/mL) of tested phenylthiazoles against MRSA (2658 RCMB).
| |||||
|---|---|---|---|---|---|
| R | MIC (μg/mL) | cpd | R | MIC (μg/mL) | |
| 1b | NA | 2.75 | 19 |
|
1.56 |
| 7 | HN-Me | 12.5 | 20 |
|
3.12 |
| 8 | HN-Et | > 25 | 21 |
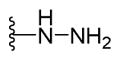
|
3.12 |
| 9 | HN-Pr | > 25 | 22 |
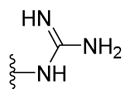
|
1.56 |
| 10 | HN-Bu | > 25 | 23 |
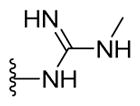
|
1.56 |
| 11 |
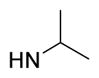
|
> 25 | 24 |
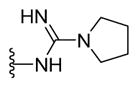
|
12.5 |
| 12 |
|
> 25 | 25 |
|
> 25 |
| 13 |
|
> 25 | 26 |
|
1.56 |
| 14 |
|
> 25 | 27 |
|
1.17 |
| 15 |
|
> 25 | 28 |
|
> 25 |
| 16 |
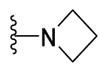
|
> 25 | 29 |
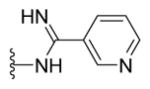
|
> 25 |
| 17 |
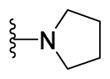
|
> 25 | 30 |
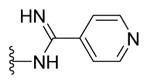
|
> 25 |
| 18 |
|
12.5 | Vancomycin | N.A. | 1.56 |
N.A. = Not applicable
Table 2.
Solubility Evaluation in phosphate buffered saline (PBS).
| Compound Tested | Solubility limit (μM) | CLogP |
|---|---|---|
| 1a | 62.5 | 4.3 |
| 1b | 2.7 | 4.2 |
| 1c | 2.2 | 4.3 |
| 19 | 45 | 4.4 |
| 20 | 150 | 3.5 |
| 21 | 77 | 4.2 |
| 22 | 128 | 3.4 |
| 23 | 89 | 2.1 |
| 27 | 104 | 3.3 |
| Tamoxifen | 15.6 | |
| Verapamil | > 500 |
Solubility limit corresponds to the highest concentration of test compound where no precipitate was detected (OD540). All tested compounds meet Lipinski’s rule of five.
Adding a methyl group to the terminal guanidine afforded compound 23 with no change in the anti-MRSA activity (Table 1). Further increase in the hydrocarbon size around the peripheral nitrogen provided compounds 24 and 25 with weaker antibacterial activity against tested MRSA strain (Table 1). On the other hand, analogues containing an additional polar atom on the nitrogenous side chain (compounds 26 and 27) resulted in more potent compounds. For example, the MIC of the morpholine analogue 26 is on par with the first compound in this series, 22, and vancomycin, while the piperazine-containing derivative 27 showed slight improvement in anti-MRSA potency with a MIC value of 1.1 μg/mL (Table 1). This value is slightly more potent than that of vancomycin and two-fold better than the biphenylthiazole lead compound 1b (MIC = 2.7 μg/mL) (Table 1).
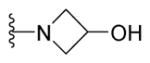
Next, incorporating the terminal nitrogen within the pyridine ring system (compounds 28–30) completely obliterated the antibacterial activity of these analogues (Table 1). As demonstrated earlier, replacement of the terminal guanidine with amines furnished inactive or weakly active analogues (compounds 7–18). The only compounds with amine-containing side chains that showed acceptable MIC values were 19 and 20 (Table 1). Interestingly, both compounds carry more polar nitrogenous side chains (i.e. piperazine and hydroxyazetidine). Comparing the MIC values of the piperazinyl derivative 19 (MIC = 1.56 μg/mL) with its corresponding aminocyclohexyl analogue 13 (MIC > 25 μg/mL), and the hydroxyazetidine 20 (MIC = 3.12 μg/mL) with its closely related azetidine analogue 16 (MIC > 25 μg/mL) highlights the crucial role of the terminal heteroatom as a HBD and/or acceptor.
The most potent compound 27 in this series was selected for further assays. First, it was tested for activity against additional strains of drug-resistant S. aureus (Table 1S). Compound 27 inhibited growth of strains of S. aureus exhibiting high-level resistance to mupirocin (NRS107, MIC = 1024 μg/mL),[15] linezolid (NRS119, MIC >128 μg/mL),[15] macrolides (NRS384), and tetracyclines (NRS384). Additionally, compound 27 (MIC = 16 μg/mL) inhibited growth of three vancomycin-resistant S. aureus clinical isolates (VRS10, VRS11a and VRS12). This represents a 30-fold improvement in activity when compared with vancomycin (MIC >512 μg/mL).
In order to examine the antibacterial spectrum of activity of 27, its MIC was determined against key Gram-positive (vancomycin –resistant E. faecium and E. faecalis) and Gram-negative (A. baumannii, E. cloacae, K. pneumoniae, and P. aeruginosa) bacterial pathogens (Table 1S). The first-generation phenylthiazoles exhibited potent antibacterial activity against strains of vancomycin-resistant enterococci (VRE). Interestingly, 27 lacks activity against both strains of VRE (MIC >64 μg/mL). Additionally, the compound is ineffective at inhibiting growth of Gram-negative bacteria. Similar to linezolid, the MIC of 27 against A. baumannii, E. cloacae, K. pneumoniae, and P. aeruginosa exceeded 64 μg/mL. This indicates 27 has more selective anti-MRSA activity compared to the first-generation phenylthiazoles.
With this data in hand, we moved next to evaluate if 27 was bacteriostatic or bactericidal against MRSA, using a time-kill assay. This assay was performed against MRSA USA300, a strain that is responsible for the majority of community-acquired MRSA (CA-MRSA) infections in the United States.[16] Compound 27 produced a 3-log10 reduction in bacterial CFU after 12 h and completely eradicated MRSA CFU after 24 h, similar to vancomycin (Figure 2). This confirms 27 is bactericidal, at the concentration tested (Figure 2).
Figure 2. Time-kill analysis of biphenylthiazole 27 and vancomycin against methicillin-resistant Staphylococcus aureus (MRSA USA300) over a 24-hour incubation period at 37 °C.

DMSO served as a negative control. The error bars represent standard deviation values obtained from triplicate samples used for each compound/antibiotic studied.
Structure-solubility-relationship
Early in drug discovery, profiling of drug properties has always been a prominent component of the development phase, since ADME, toxicity and pharmacodynamic properties are dependent upon aqueous solubility and the partition coefficient (logP). In this regard, the “brick-like” properties of biphenylthiazole lead 1b (Saq = 2.2 μM) was immensely improved by replacing the pyrimidine linker with the more polar oxadiazole ring. In brief, the six most potent derivatives showed noticeable improvement in aqueous solubility. The hydroxylazetidine 20 is the most soluble derivative developed thus far (Saq = 150 μM). In contrast, the structurally-related piperazinyl analogue 19 was the least soluble derivative synthesized (Saq = 45 μM, Table 2). Adding a carboximidamide (with two HBD’s) to the structure of 19 yielded compound 27. This compound exhibited a noticeable one-fold improvement in aqueous solubility (Saq = 104 μM, Table 2).
Preliminary toxicity assessment using human colorectal cells (HRT-18)
Improvement in the aqueous solubility of the compounds also led to expanding the safety margin from 32×MIC value, in the case of the lead biphenylthiazole 1a, to more than 400×MIC (relative to MRSA (2658 RCMB)) for the oxadiazole-containing derivative 27 (not toxic at 512 μg/mL). Additionally, 26 was not toxic to HRT-18 cells up to 256 μg/mL (Figure 3). Additionally, both compounds were highly tolerable when tested against non-cancerous cell line BHK-21 (Table 2S).
Figure 3. Toxicity analysis of active compounds against human colorectal cells (HRT-18).

Percent viable mammalian cells (measured as average absorbance ratio (test agent relative to DMSO)) for cytotoxicity analysis of active compounds (tested in triplicate) at different concentrations against HRT-18 cells using the MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay. Dimethyl sulfoxide (DMSO) was used as a negative control to determine a baseline measurement for the cytotoxic impact of each compound. The absorbance values represent an average of a minimum of three samples analyzed for each compound. Error bars represent standard deviation values for the absorbance values. A two-way ANOVA, with post hoc Dunnet’s multiple comparisons test, determined statistical difference between the values obtained for compound 26 and DMSO at 512 μg/mL (denoted by the asterisk) (P < 0.05).
Multi-step resistance selection against MRSA
To examine the probability of rapid emergence of resistance to compounds 26 and 27, a multi-step resistance selection experiment against MRSA was conducted. Initially the MICs of compounds 26 and 27 and the antibiotic rifampicin were determined against MRSA USA300, using the broth microdilution method. Bacteria were then subcultured for fourteen passages over two weeks, to determine if a shift in the MIC of each agent was observed. No increase in MIC was observed for either compound 26 or 27 over fourteen passages (Figure 4), indicating MRSA mutants exhibiting resistant to these compounds could not be isolated. In contrast, the MIC of rifampicin increased rapidly from 0.006 μg/mL (initial day) to 1.6 μg/mL after the fourth passage, indicating the formation of resistance. The MIC for rifampicin against MRSA continued to rise, increasing more than 40,000-fold after the fourteenth passage. Rifampicin is a bactericidal antibiotic that exerts its antibacterial effect by targeting RNA polymerase. The rapid development of MRSA resistance to rifampicin has been noted in previous reports.[17, 18] This is one of the primary reasons for its limited use as a single agent for treatment of MRSA infections.
Figure 4.

Multi-step resistance selection of compounds 26, 27, and rifampicin against methicillin-resistant S. aureus USA300. MRSA was serially passaged over a 14-day period and the broth microdilution assay was utilized to determine the minimum inhibitory concentration of each compound against MRSA after each successive passage. A four-fold shift in MIC would be indicative of bacterial resistance to the test agent.
In vivo PK profiling
Encouraged by the promising solubility and preliminary toxicological properties of 27, the key PK parameters were determined in rats. Remarkably, the maximum plasma concentration (Cmax) after a 25 mg/kg oral dose of 27 exceeded its MIC value against MRSA (2658 RCMB) by nearly six-fold (Table 3). More importantly, the proper aqueous solubility criteria reflected positively on the absorption of 27 from the rat GIT. In comparison with 1a, which is not bioavailable orally, compound 27 revealed oral bioavailability of 42.5% (Table 3). This value can be further enhanced by reducing the particle size, selecting the correct polymorphic type, and using suitable solid dosage form excipients.
Table 3.
Pre-clinical in vivo PK for compound 27 after 25 mg/kg oral dose was administered to a rat.
| Cmax (ng/mL) | tmax (h) | AUClast ((h)*(ng/mL)) | thalf (h) | %F | |
|---|---|---|---|---|---|
|
|
|||||
| 27 | 6222.1 | 1.7 | 83173.60 | 2.04 | 42.5 |
| 1aa | ND | ND | ND | ND | 0% |
Plasma concentrations of 1a were under the lowest detectable limit, so all PK parameters after 25 mg/kg oral dose could not be calculated
3. CONCLUSION
In conclusion, incorporating the Schiff’s bond of 1st generation anti-MRSA compounds with the oxadiazole ring produced a new biphenylthiazole series with enhanced physicochemical properties and oral bioavailability. In addition, the new generation exhibited significant improvement in their toxicity profile with low incidence of MRSA to generate resistant mutant against this new class of biphenylthiazoles, while maintaining potent antibacterial activity against MRSA cells. These attributes collectively suggest byphenylthiazoles as potential candidates for clinical applications.
4. EXPERIMENTAL SECTION
4.1. General
1H NMR spectra were run at 400 MHz and 13C spectra were determined at 100 MHz in deuterated chloroform (CDCl3), or dimethyl sulfoxide (DMSO-d6) on a Varian Mercury VX-400 NMR spectrometer. Chemical shifts are given in parts per million (ppm) on the delta (δ) scale. Chemical shifts were calibrated relative to those of the solvents. Flash chromatography was performed on 230–400 mesh silica. The progress of reactions was monitored with Merck silica gel IB2-F plates (0.25 mm thickness). The infrared spectra were recorded in potassium bromide disks on pye Unicam SP 3300 and Shimadzu FT IR 8101 PC infrared spectrophotometer. Mass spectra were recorded at 70 eV. High resolution mass spectra for all ionization techniques were obtained from a FinniganMAT XL95. Melting points were determined using capillary tubes with a Stuart SMP30 apparatus and are uncorrected. All yields reported refer to isolated yields.
Ethyl 2-([1,1′-biphenyl]-4-yl)-4-methylthiazole-5-carboxylate (3)
[14] Compound 2 (3 g, 14 mmol) and ethyl 2-chloro-3-oxobutanoate (3.88 mL, 4.62 g, 28 mmol) were added to absolute ethanol (30 mL). The reaction mixture was heated at reflux for 4 h. After removal of solvent under reduced pressure, the solid residue was purified by crystallization from ethanol to provide the desired product as white crystals (4.3 g, 95%) mp = 105 °C; MS (m/z) 323; 1H NMR (DMSO-d6) d: 8.04 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 7.6 Hz, 2H), 7.45 (t, J = 7.2 Hz, 2H), 7.37 (t, J = 7.2 Hz, 1H), 4.28 (q, J = 4.8 Hz, 2H), 2.67 (s, 3H), 1.33 (t, J = 4.8 Hz, 3H); 13C NMR (DMSO-d6) d: 168.97, 161.80, 160.80, 143.24, 139.28, 131.52, 129.51, 128.64, 127.88, 127.57, 127.15, 121.52, 61.62, 17.70, 14.58; MS (m/z) 323; Anal. Calc. for: (C19H17NO2S): C, 70.56; H, 5.30; N, 4.33 %; Found: C, 70.66; H, 5.31; N, 4.34%.
2-([1,1′-Biphenyl]-4-yl)-4-methylthiazole-5-carbohydr-azide (4)
To a solution of 3 (1.29 g, 4 mmol) in ethanol (15 mL), hydrazine hydrate (99%, 1 mL, 20 mmol) was added dropwise. The reaction mixture was heated at reflux for 6 h then allowed to cool down to room temperature. The formed solid was separated by filtration and crystallized from ethanol to provide the desired product as white crystals (1.1 g, 93%) mp = 204–205 °C; 1H NMR (DMSO-d6) d: 9.56 (brs, 1H), 8.00 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 7.6 Hz, 2H), 7.48 (t, J = 7.2 Hz, 2H), 7.39 (t, J = 7.2 Hz, 1H), 4.53 (brs, 2H), 2.59 (s, 3H); 13C NMR (DMSO-d6) d: 165.96, 161.51, 155.38, 142.72, 139.42, 131.88, 129.53, 128.57, 127.95, 127.28, 127.15, 125.10, 17.46; MS (m/z) 309; Anal. Calc. for: (C17H15N3OS): C, 66.00; H, 4.89; N, 13.58 %; Found: C, 66.01; H, 4.90; N, 13.58%.
2-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-5-(me-thylthio)-1,3,4-oxadiazole (5)
Potassium hydroxide (0.4 g, 10 mmol) was added to a solution of 4 (3 g, 10 mmol) in ethanol (15 mL), followed by drop-wise addition of carbon disulphide (3 mL, 110 mmol) over 30 min. The reaction mixture was stirred at room temperature for an additional 15 min and then heated to reflux until the evolution of hydrogen sulfide gas ceased. After completion of the reaction, as monitored by TLC, the obtained intermediate was poured on cold water (50 mL), filtered, washed with water, dried and crystallized from ethanol to provide yellow crystals (3.3 g, 89%) mp > 300 °C; 1H NMR (DMSO-d6) d: 8.05 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 7.2 Hz, 2H), 7.48 (t, J = 6.8 Hz, 2H), 7.40 (t, J = 6.8 Hz, 1H), 2.64 (s, 3H). The obtained yellow solid and potassium hydroxide (0.25 g, 4.2 mmol) were dissolved in water (15 mL). Then, dimethyl sulfate (0.5 mL, 4 mmol) was added dropwise with vigorous stirring. After 2 h, the formed solid was filtered and washed with copious amounts of water to yield the titled compound as a yellowish white solid (0.68 g, 91%); mp = 173 °C; 1H NMR (DMSO-d6) d: 8.08 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 7.2 Hz, 2H), 7.49 (t, J = 7.2 Hz, 2H), 7.42 (t, J = 7.2 Hz, 1H), 2.75 (s, 3H), 2.69 (s, 3H); 13C NMR (DMSO-d6) d: 168.01, 164.86, 160.59, 156.52, 143.24, 139.29, 131.34, 129.56, 128.69, 128.01, 127.58, 127.20, 114.24, 17.64, 14.87; MS (m/z) 365; HRMS (EI) m/z 365.0669 M+, calcd for C19H15N3OS2 365.0657; Anal. Calc. for: (C19H15N3OS2): C, 62.44; H, 4.14; N, 11.50%; Found: C, 62.44; H, 4.15; N, 11.51%.
2-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-5-(methylsulfonyl)-1,3,4-oxadiazole (6)
To a solution of 5 (0.5 g, 1.3 mmol) in dry DCM (5 mL), m-CPBA (0.514 g, 2.9 mmol) diluted with DCM (5 mL) was added portion-wise with continuous stirring. Afterward, the reaction mixture was kept at 23 °C for 16 h. Additional DCM (10 mL) was added and the reaction mixture was washed with 25 mL of 5% aqueous solution of sodium metabisulfite and 25 mL of 5% aqueous sodium carbonate. The organic layer was separated, dried and concentrated under reduced pressure to give the desired product as yellow crystals (0.5 g, 93%) mp = 144 °C; 1H NMR (DMSO-d6) d: 8.09 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 7.6 Hz, 2H), 7.58 (t, J = 6.8 Hz, 2H), 7.52 (t, J = 6.8 Hz, 1H), 3.83 (s, 3H), 2.89 (s, 3H); 13C NMR (DMSO-d6) d: 169.08, 167.02, 161.13, 158.26, 143.70, 139.21, 131.28, 129.48, 128.67, 127.93, 127.65, 127.12, 113.58, 43.40, 17.78; MS (m/z) 397; HRMS (EI) m/z 397.0551 M+, calcd for C19H15N3O3S2 397.0555; Anal. Calc. for: (C19H15N3O3S2): C, 57.42; H, 3.80; N, 10.57%; Found: C, 57.44; H, 3.81; N, 10.57%.
Compounds 7–31
To a solution of 6 (0.1 g, 0.25 mmol) in dry DMF (5 mL), appropriate amine, hydrazine, guanidine or carboximidate (0.4 mmol) was added. The reaction mixture was heated at 80 °C for 30 min to 12 h, and then poured over ice water (50 mL). The formed solid was extracted with ethyl acetate (10 mL). The organic layer was evaporated under reduced pressure. The obtained crude material was then purified by crystallization or column chromatography. Physical properties and spectral analysis of isolated products are listed below:
5-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-N-methyl-1,3,4-oxadiazol-2-amine (7)
Yellow solid (0.08 g, 88%) mp = 223 °C; 1H NMR (DMSO-d6) d: 8.03 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.74 (brs, 1H), 7.71 (d, J = 7.6 Hz, 2H), 7.48 (t, J = 6.8 Hz, 2H), 7.41 (t, J = 6.8 Hz, 1H), 2.84 (s, 3H), 2.67 (s, 3H); 13C NMR (DMSO-d6) d: 166.07, 164.25, 153.99, 153.03, 142.85, 139.35, 131.58, 129.54, 128.60, 127.92, 127.35, 127.15, 115.43, 29.48, 17.40; MS (m/z) 348; HRMS (EI) m/z 348.1060 M+, calcd for C19H16N4OS 348.1045; Anal. Calc. for: (C19H16N4OS): C, 65.50; H, 4.63; N, 16.08%; Found: C, 65.51; H, 4.63; N, 16.08%.
5-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-N-ethyl-1,3,4-oxadiazol-2-amine (8)
Yellow solid (0.05 g, 55%) mp = 200 °C; 1H NMR (DMSO-d6) d: 8.04 (d, J = 8.4 Hz, 2H), 7.88 (brs, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 7.6 Hz, 2H), 7.50 (t, J = 7.2 Hz, 2H), 7.38 (t, J = 7.2 Hz, 1H), 3.22 (q, J = 7.2 Hz, 2H), 2.67 (s, 3H), 1.17 (t, J = 7.2 Hz, 3H); 13C NMR (DMSO-d6) d: 166.02, 163.54, 153.94, 152.88, 142.84, 139.36, 131.60, 129.53, 128.60, 127.92, 127.36, 127.15, 115.48, 37.95, 17.40, 14.93; MS (m/z) 362; HRMS (EI) m/z 362.1208 M+, calcd for C20H18N4OS 362.1201; Anal. Calc. for: (C20H18N4OS): C, 66.28; H, 5.01; N, 15.46%; Found: C, 66.29; H, 5.03; N, 15.47%.
5-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-N-propyl-1,3,4-oxadiazol-2-amine (9)
Yellow solid (0.076 g, 80%) mp = 184 °C ; 1H NMR (DMSO-d6) d: 8.04 (d, J = 8 Hz, 2H), 7.90 (brs, 1H), 7.82 (d, J = 8 Hz, 2H), 7.72 (d, J = 7.6 Hz, 2H), 7.50 (t, J = 7.2 Hz, 2H), 7.38 (t, J = 7.2 Hz, 1H), 3.17 (t, J = 7.2 Hz, 2H), 2.68 (s, 3H), 1.58 (sixt, J = 7.6 Hz, 2H), 0.90 (t, J = 7.6 Hz, 3H); 13C NMR (DMSO-d6) d: 166.01, 163.73, 153.92, 152.81, 142.86, 139.37, 131.61, 129.55, 128.61, 127.94, 127.37, 127.16, 115.50, 44.87, 22.49, 17.41, 1168; MS (m/z) 376; HRMS (EI) m/z 376.1348 M+, calcd for C21H20N4OS 376.1358; Anal. Calc. for: (C21H20N4OS): C, 67.00; H, 5.35; N, 14.88%; Found: C, 67.01; H, 5.36; N, 14.88%.
5-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-N-butyl-1,3,4-oxadiazol-2-amine (10)
Yellow solid (0.09 g, 93%) mp = 145 °C; 1H NMR (DMSO-d6) d: 8.03 (d, J = 8 Hz, 2H), 7.86 (brs, 1H), 7.79 (d, J = 8 Hz, 2H), 7.73 (d, J = 7.6 Hz, 2H), 7.50 (t, J = 7.2 Hz, 2H), 7.39 (t, J = 7.2 Hz, 1H), 3.21 (t, J = 7.2 Hz, 2H), 2.67 (s, 3H), 1.56 (pent, J = 6.8 Hz, 2H), 1.33 (sixt, J = 7.2 Hz, 2H), 0.89 (t, J = 7.2 Hz, 3H); 13C NMR (DMSO-d6) d: 165.99, 163.70, 153.91, 152.81, 142.84, 139.36, 131.60, 129.53, 128.60, 127.92, 127.35, 127.15, 115.49, 42.76, 31.27, 19.84, 17.40, 14.06; MS (m/z) 390; HRMS (EI) m/z 390.1499 M+, calcd for C22H22N4OS 390.1514; Anal. Calc. for: (C22H22N4OS): C, 67.67; H, 5.68; N, 14.35%; Found: C, 67.67; H, 5.69; N, 14.37%.
5-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-N-isopropyl-1,3,4-oxadiazol-2-amine (11)
Yellow solid (0.06 g, 81%) mp = 220°C; 1H NMR (DMSO-d6) d: 8.05 (d, J = 8.4 Hz, 2H), 7.83 (brs, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 7.6 Hz, 2H), 7.50 (t, J = 7.2 Hz, 2H), 7.42 (t, J = 7.2 Hz, 1H), 3.73 (sept, J = 6.8 Hz, 1H), 2.68 (s, 3H), 1.21 (d, J = 6.8 Hz, 6H); 13C NMR (DMSO-d6) d: 166.01, 162.96, 153.92, 152.78, 142.86, 139.37, 131.61, 129.55, 128.62, 127.95, 127.37, 127.17, 115.51, 45.44, 22.70, 17.40; MS (m/z) 376; HRMS (EI) m/z X M+, calcd for C21H20N4OS 376.1358; Anal. Calc. for: (C21H20N4OS): C, 67.00; H, 5.35; N, 14.88%; Found: C, 67.01; H, 5.35; N, 14.90%.
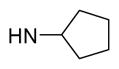
5-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-N-cyclopentyl-1,3,4-oxadiazol-2-amine (12)
Yellow solid (0.06 g, 60%) mp = 195 °C; 1H NMR (DMSO-d6) d: 8.04 (d, J = 8 Hz, 2H), 7.82 (brs, 1H), 7.80 (d, J = 8 Hz, 2H), 7.72 (d, J = 7.6 Hz, 2H), 7.50 (t, J = 7.2 Hz, 2H), 7.38 (t, J = 7.2 Hz, 1H), 3.89 (m, 1H), 2.68 (s, 3H), 1.88 (m, 2H), 1.60 (m, 4H), 1.57 (m, 2H); 13C NMR (DMSO-d6) d: 166.01, 163.25, 153.92, 152.85, 142.86, 139.37, 131.61, 129.55, 128.62, 127.95, 127.37, 127.16, 115.52, 54.88, 32.65, 23.69, 17.41; MS (m/z) 402; HRMS (EI) m/z 402.1496 M+, calcd for C23H22N4OS 402.1514; Anal. Calc. for: (C23H22N4OS): C, 68.63; H, 5.51; N, 13.92%; Found: C, 68.64; H, 5.53; N, 13.94%.
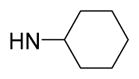
5-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-N-cyclohexyl-1,3,4-oxadiazol-2-amine (13)
Light brown solid (0.08 g, 76%) mp = 186 °C; 1H NMR (DMSO-d6) d: 8.04 (d, J = 8.4 Hz, 2H), 7.86 (brs, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 6.8 Hz, 2H), 7.48 (t, J = 6.8 Hz, 2H), 7.38 (t, J = 6.8 Hz, 1H), 3.38 (m, 1H), 2.68 (s, 3H), 1.95 (m, 2H), 1.72 (m, 2H), 1.57 (m, 2H), 1.28 (m, 2H), 0.98 (m, 2H); 13C NMR (DMSO-d6) d: 165.98, 162.97, 153.88, 152.71, 142.85, 139.36, 131.61, 129.54, 128.61, 127.93, 127.36, 127.16, 115.52, 52.36, 32.67, 25.60, 24.78, 17.40; MS (m/z) 416; HRMS (EI) m/z 416.1677 M+, calcd for C24H24N4OS 416.1671; Anal. Calc. for: (C24H24N4OS): C, 69.20; H, 5.81; N, 13.45%; Found: C, 69.22; H, 5.82; N, 13.46%.
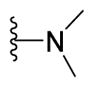
5-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-N,N-dimethyl-1,3,4-oxadiazol-2-amine (14)
Yellow solid (0.07 g, 80%) mp = 185 °C; 1H NMR (DMSO-d6) d: 8.03 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.4 Hz, 2H), 7.71(d, J = 7.2 Hz, 2H), 7.50 (t, J = 7.6 Hz, 2H), 7.37 (t, J = 7.6 Hz, 1H), 3.04 (s, 6H), 2.67 (s, 3H); 13C NMR (DMSO-d6) d: 166.13, 164.63, 154.00, 153.46, 142.83, 139.33, 131.57, 129.53, 128.60, 127.90, 127.34, 127.14, 115.35, 38.12, 17.40; MS (m/z) 362; HRMS (EI) m/z 362.1212 M+, calcd for C20H18N4OS 362.1201; Anal. Calc. for: (C20H18N4OS): C, 66.28; H, 5.01; N, 15.46%; Found: C, 66.28; H, 5.03; N, 15.47%.
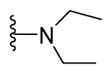
5-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-N,N-diethyl-1,3,4-oxadiazol-2-amine (15)
Brown solid (0.06 g, 66%) mp = 178 °C; 1H NMR (DMSO-d6) d: 8.04 (d, J = 8 Hz, 2H), 7.79 (d, J = 8 Hz, 2H), 7.72 (d, J = 7.2 Hz, 2H), 7.49 (t, J = 6.8 Hz, 2H), 7.40 (t, J = 6.8 Hz, 1H), 3.46 (q, J = 6.8 Hz, 4H), 2.66 (s, 3H), 1.61 (t, J = 6.8 Hz, 6H); 13C NMR (DMSO-d6) d: 166.01, 163.44, 153.81, 153.17, 142.80, 139.34, 131.58, 129.58, 128.59, 127.89, 127.34, 127.13, 115.47, 43.57, 17.35, 13.48; MS (m/z) 390; HRMS (EI) m/z 390.1528 M+, calcd for C22H22N4OS 390.1514; Anal. Calc. for: (C22H22N4OS): C, 67.67; H, 5.68; N, 14.35%; Found: C, 67.69; H, 5.69; N, 14.36%.
2-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-5-(azetidin-1-yl)-1,3,4-oxadiazole (16)
Yellow solid (0.07 g, 70%) mp = 228 °C; 1H NMR (DMSO-d6) d: 8.06 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 7.2 Hz, 2H), 7.50 (t, J = 7.6 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 4.17 (t, J = 7.6 Hz, 4H), 2.76 (s, 3H), 2.42 (p, J = 7.6 Hz, 2H); 13C NMR (DMSO-d6) d: 166.49, 164.79, 154.53, 154.31, 142.95, 139.34, 131.55, 129.56, 128.64, 127.96, 127.42, 127.18, 115.13, 56.46, 52.55, 17.82; MS (m/z) 374; HRMS (EI) m/z 374.1215 M+, calcd for C21H18N4OS 374.1201; Anal. Calc. for: (C21H18N4OS): C, 67.36; H, 4.85; N, 14.96%; Found: C, 67.36; H, 4.86; N, 14.97%.
2-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-5-(pyrrolidin-1-yl)-1,3,4-oxadiazole (17)
Yellow solid (0.089 g, 91%) mp = 226 °C; 1H NMR (DMSO-d6) d: 8.05 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 7.2 Hz, 2H), 7.50 (t, J = 7.2 Hz, 2H), 7.40 (t, J = 7.2 Hz, 1H), 3.48 (t, J = 6.4 Hz, 4H), 2.69 (s, 3H), 1.96 (t, J = 6.4 Hz, 4H); 13C NMR (DMSO-d6) d: 166.45, 164.33, 154.47, 153.24, 142.93, 139.32, 131.65, 129.55, 128.63, 127.96, 127.38, 127.17, 115.17, 48.03, 25.56, 17.40; MS (m/z) 388; HRMS (EI) m/z 388.1340 M+, calcd for C22H20N4OS 388.1358; Anal. Calc. for: (C22H20N4OS) (M.W. = 388): C, 68.02; H, 5.19; N, 14.42%; Found: C, 68.02; H, 5.21; N, 14.43%.
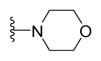
4-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}morpholine (18)
Yellow solid (0.098 g, 97%) mp = 285 °C; 1H NMR (DMSO-d6) d: 8.07 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 7.2 Hz, 2H), 7.50 (t, J = 7.2 Hz, 2H), 7.41 (t, J = 7.2 Hz, 1H), 3.72 (t, J = 4.4 Hz, 4H), 3.46 (t, J = 4.4 Hz, 4H), 2.68 (s, 3H); 13C NMR (DMSO-d6) d: 166.50, 163.99, 154.50, 153.92, 142.92, 139.32, 131.52, 129.55, 128.63, 127.95, 127.39, 127.15, 115.14, 65.58, 46.24, 17.46; MS (m/z) 404; HRMS (EI) m/z 404.1292 M+, calcd for C22H20N4O2S 404.1307; Anal. Calc. for: (C22H20N4O2S): C, 65.33; H, 4.98; N, 13.85%; Found: C, 65.35; H, 4.99; N, 13.87%.
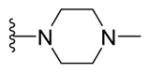
2-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-5-(4-methylpiperazin-1-yl)-1,3,4-oxadiazole (19)
Yellow solid (0.099 g, 97%) mp = 177 °C; 1H NMR (DMSO-d6) d: 8.04 (d, J = 8 Hz, 2H), 7.82 (d, J = 8 Hz, 2H), 7.74 (d, J = 7.6 Hz, 2H), 7.50 (t, J = 7.6 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 3.48 (t, J = 4.8 Hz, 4H), 2.68 (s, 3H) 2.43 (t, J = 4.8 Hz, 4H), 2.21 (s, 3H); 13C NMR (DMSO-d6) d: 166.41, 164.02, 154.41, 153.79, 142.92, 139.33, 131.55, 129.55, 128.63, 127.95, 127.39, 127.16, 115.20, 53.79, 46.13, 18.99, 17.47; MS (m/z) 417; HRMS (EI) m/z 417.1625 M+, calcd for C23H23N5OS 417.1623; Anal. Calc. for: (C23H23N5OS): C, 66.16; H, 5.55; N, 16.77%; Found: C, 66.17; H, 5.57; N, 16.79%.
1-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}azetidin-3-ol (20)
Yellow solid (0.05 g, 54%) mp = 210 °C; 1H NMR (DMSO-d6) d: 8.06 (d, J = 8.4 Hz, 2H), 7.87 (d, J = 8.4 Hz, 2H), 7.83 (d, J = 7.6 Hz, 2H), 7.50 (t, J = 7.6 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 5.80 (brs, 1H), 4.82 (pent, J = 4.8 Hz, 1H), 4.36 (dd, J = 4.6 Hz, J = 9.2 Hz, 2H), 3.95 (dd, J = 4.8 Hz, J = 9.2 Hz, 2H), 2.70 (s, 3H); 13C NMR (DMSO-d6) d: 166.55, 164.85, 154.61, 154.45, 142.83, 139.33, 131.52, 129.56, 128.64, 127.96, 127.43, 127.17, 115.92, 62.39, 62.20, 17.44; MS (m/z) 390; HRMS (EI) m/z 390.1164 M+, calcd for C21H18N4O2S 390.1150; Anal. Calc. for: (C21H18N4O2S): C, 64.60; H, 4.65; N, 14.35%; Found: C, 64.61; H, 4.67; N, 14.36%.
2-{2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl}-5-hydrazinyl-1,3,4-oxadiazole (21)
Yellow fluffy powder (0.06 g, 75%) mp = 260 °C; 1H NMR (DMSO-d6) d: 8.74 (brs, 1H), 8.03 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 7.2 Hz, 2H), 7.50 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.6 Hz, 1H), 4.55 (brs, 2H), 2.69 (s, 3H); 13C NMR (DMSO-d6) d: 165.98, 163.97, 153.90, 152.73, 142.86, 139.37, 131.61, 129.55, 128.01, 127.95, 127.28, 127.16, 115.55, 17.43; MS (m/z) 349; HRMS (EI) m/z 349.1010 M+, calcd for C18H15N5OS 349.0997; Anal. Calc. for: (C18H15N5OS): C, 61.87; H, 4.33; N, 20.04%; Found: C, 61.88; H, 4.32; N, 20.06%.
1-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}guanidine (22)
Grayish solid (0.07 g, 76%) mp = 198 °C; 1H NMR (DMSO-d6) d: 9.22 (brs, 1H), 8.32 (brs, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 7.2 Hz, 2H), 7.48 (t, J = 7.6 Hz, 2H), 7.39 (t, J = 7.6 Hz, 1H), 6.95 (brs, 2H), 2.68 (s, 3H); 13C NMR (DMSO-d6) d: 167.32, 166.08, 159.43, 153.94, 152.71, 142.82, 139.39, 131.67, 129.54, 128.59, 127.93, 127.24, 127.16, 116.13, 17.41; MS (m/z) 376; HRMS (EI) m/z 376.1110 M+, calcd for C19H16N6OS 376.1106; Anal. Calc. for: (C19H16N6OS): C, 60.62; H, 4.28; N, 22.33%; Found: C, 60.63; H, 4.29; N, 22.25%.
1-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}-3-methylguanidine (23)
Yellowish brown solid (0.07 g, 75%) mp = 194 °C; 1H NMR (DMSO-d6) d: 9.19 (brs, 1H), 8.56 (brs, 1H), 8.05 (d, J = 8 Hz, 2H), 7.81 (d, J = 8 Hz, 2H), 7.74 (d, J = 7.6 Hz, 2H), 7.51 (t, J = 7.6 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 2.85 (s, 3H), 2.75 (brs, 1H), 2.70 (s, 3H); 13C NMR (DMSO-d6) d: 167.33, 166.10, 159.44, 154.00, 153.72, 142.83, 139.37, 131.62, 129.51, 128.62, 127.96, 127.38, 127.17, 116.13, 29.50, 17.42; MS (m/z) 390; HRMS (EI) m/z 390.1254 M+, calcd for C20H18N6OS 390.1263; Anal. Calc. for: (C20H18N6OS): C, 61.52; H, 4.65; N, 21.52%; Found: C, 61.53; H, 4.66; N, 21.52%.
N-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}pyrrolidine-1-carboximidamide (24)
Brown solid (0.08 g, 79%) mp = 238 °C; 1H NMR (DMSO-d6) d: 8.63 (brs, 1H), 8.04 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 7.2 Hz, 2H), 7.62 (brs, 1H), 7.48 (t, J = 7.2 Hz, 2H), 7.38 (t, J = 7.2 Hz, 1H), 3.31 (m, 4H), 2.70 (s, 3H), 1.87 (m, 4H); 13C NMR (DMSO-d6) d: 166.93, 166.05, 155.77, 154.02, 152.69, 142.80, 139.39, 131.68, 129.53, 128.59, 127.78, 127.34, 127.15, 116.07, 31.14, 25.33, 17.56; MS (m/z) 430; HRMS (EI) m/z 430.1570 M+, calcd for C23H22N6OS 430.1576; Anal. Calc. for: (C23H22N6OS): C, 64.17; H, 5.15; N, 19.52%; Found: C, 64.18; H, 5.16; N, 19.53%.
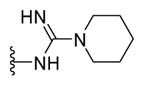
N-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}piperidine-1-carboximidamide (25)
Brown solid (0.09 g, 85%) mp = 240 °C; 1H NMR (DMSO-d6) d: 8.30 (brs, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.90 (brs, 1H), 7.86 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 7.6 Hz, 2H), 7.51 (t, J = 7.6Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 3.55 (m, 4H), 2.71 (s, 3H), 1.59 (m, 4H), 1.51 (m, 2H); 13C NMR (DMSO-d6) d: 167.15, 166.17, 156.63, 154.15, 152.88, 142.85, 139.38, 131.65, 129.55, 128.61, 127.96, 127.37, 127.16, 116.02, 45.53, 25.72, 24.27, 17.54; MS (m/z) 444; HRMS (EI) m/z 444.1719 M+, calcd for C24H24N6OS 444.1732; Anal. Calc. for: (C24H24N6OS): C, 64.84; H, 5.44; N, 18.90%; Found: C, 64.85; H, 5.46; N, 18.91%.
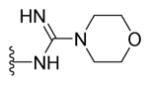
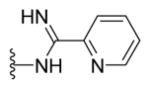
N-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}morpholine-4-carboximidamide (26)
Yellow solid (0.07 g, 65%) mp = 260 °C; 1H NMR (DMSO-d6) d: 8.32 (brs, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.95 (brs, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 6.8 Hz, 2H), 7.51 (t, J = 7.6 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 3.63 (t, J = 4.4 Hz, 4H), 3.54 (t, J = 4.4 Hz, 4H), 2.71 (s, 3H); 13C NMR (DMSO-d6) d: 167.34, 166.94, 164.81, 157.21, 154.32, 142.93, 139.38, 131.64, 129.56, 128.63, 127.97, 127.40, 127.18, 115.91, 66.14, 44.87, 17.54; MS (m/z) 446; HRMS (EI) m/z 446.1533 M+, calcd for C23H22N6O2S 446.1525; Anal. Calc. for: (C23H22N6O2S): C, 61.87; H, 4.97; N, 18.82%; Found: C, 61.88; H, 4.97; N, 18.83%.
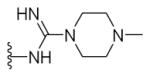
N-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}-4-methylpiperazine-1-carboximid-amide (27)
Yellow solid (0.07 g, 63%) mp = 262 °C; 1H NMR (DMSO-d6) d: 8.31 (brs, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.91 (brs, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 7.6 Hz, 2H), 7.51 (t, J = 7.6 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 3.54 (t, J = 4.8 Hz, 4H), 2.70 (s, 3H), 2.32 (t, J = 4.8 Hz, 4H), 2.18 (s, 3H); 13C NMR (DMSO-d6) d: 166.98, 166.26, 156.96, 154.25, 153.04, 142.87, 139.37, 131.64, 129.55, 128.61, 127.95, 127.38, 127.16, 115.95, 54.57, 46.02, 44.40, 17.53; MS (m/z) 459; HRMS (EI) m/z 459.1822 M+, calcd for C24H25N7OS 459.1841; Anal. Calc. for: (C24H25N7OS): C, 62.72; H, 5.48; N, 21.33%; Found: C, 62.74; H, 5.49; N, 21.34%.
N-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}picolinimidamide (28)
Yellow solid (0.1 g, 91%) mp = 225 °C; 1H NMR (DMSO-d6) d: 9.51 (brs, 1H), 8.78 (d, J = 5.2 Hz, 1H), 8.71 (brs, 1H), 8.45 (d, J = 4.8 Hz, 1H), 8.09 (d, J = 8.4 Hz, 2H), 8.00 (t, J = 4.8 Hz, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.75 (d, J = 6.8 Hz, 2H), 7.63 (t, J = 4.8 Hz, 1H), 7.51 (t, J = 7.6 Hz, 2H), 7.38 (t, J = 7.6 Hz, 1H), 2.79 (s, 3H); 13C NMR (DMSO-d6) d: 167.11, 166.01, 160.55, 155.72, 155.37, 152.81, 148.99, 142.98, 139.27, 135.90, 131.48, 129.55, 128.61, 127.96, 127.37, 127.16, 126.71, 122.53, 115.93, 17.66; MS (m/z) 438; HRMS (EI) m/z 438.1277 M+, calcd for C24H18N6OS 438.1263; Anal. Calc. for: (C24H18N6OS): C, 65.74; H, 4.14; N, 19.17%; Found: C, 65.75; H, 4.15; N, 19.19%.
N-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}nicotinimidamide (29)
Yellow solid (0.1 g, 91%) mp = 289 °C; 1H NMR (DMSO-d6) d: 9.55 (brs, 1H), 9.21 (s, 1H), 9.03 (brs, 1H), 8.76 (d, J = 6.4 Hz, 1H), 8.41 (t, J = 8 Hz, 1H), 8.07 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 7.6 Hz, 2H), 7.54 (d, J = 8 Hz, 1H), 7.46 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.6 Hz, 1H), 2.76 (s, 3H); 13C NMR (DMSO-d6) d: 167.25, 166.44, 160.62, 155.64, 155.13, 152.90, 149.14, 143.05, 139.32, 135.91, 131.49, 129.75, 129.54, 128.64, 127.95, 127.49, 127.17, 124.00, 115.28, 17.65; MS (m/z) 438; HRMS (EI) m/z 438.1268 M+, calcd for C24H18N6OS 438.1263; Anal. Calc. for: (C24H18N6OS): C, 65.74; H, 4.14; N, 19.17%; Found: C, 65.75; H, 4.16; N, 19.18%.
N-{5-(2-([1,1′-Biphenyl]-4-yl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl}isonicotinimidamide (30)
Yellow solid (0.07 g, 64%) mp = 257 °C; 1H NMR (DMSO-d6) d: 9.56 (brs, 1H), 9.12 (brs, 1H), 8.77 (d, J = 6 Hz, 2H), 8.07 (d, J = 8.4 Hz, 2H), 7.98 (d, J = 6 Hz, 2H), 7.83 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 7.6 Hz, 2H), 7.50 (t, J = 7.6 Hz, 2H), 7.42 (t, J = 7.6 Hz, 1H), 2.76 (s, 3H); 13C NMR (DMSO-d6) d: 167.33, 166.39, 160.37, 155.74, 155.34, 150.81, 143.08, 141.19, 139.33, 131.49, 129.55, 128.65, 127.97, 127.50, 127.18, 121.18, 115.25, 17.67; MS (m/z) 438; HRMS (EI) m/z 438.1249 M+, calcd for C24H18N6OS 438.1263; Anal. Calc. for: (C24H18N6OS): C, 65.74; H, 4.14; N, 19.17%; Found: C, 65.76; H, 4.15; N, 19.18%.
4.2.Microbiological assays
Determination of Minimum Inhibitory Concentration (MIC)
MRSA clinical isolate 2658 RCMB was obtained from the Regional Center of Mycology & Biotechnology, Cairo, Egypt. S. aureus clinical isolates (NRS107, NRS119, NRS123, NRS384, VRS10, VRS11a, and VRS12), vancomycin-resistant E. faecium ATCC 700221, vancomycin-resistant E. faecalis ATCC 51299, Acinetobacter baumannii ATCC 19606, Enterobacter cloacae BAA-1154, Klebsiella pneumoniae BAA-1706 and P. aeruginosa ATCC 15442 were obtained through the Network of Antimicrobial Resistance in Staphylococcus aureus (NARSA) program, BEI Resources, and the American Type Culture Collection.
The MICs of the newly synthesized compounds, tested against the bacterial strains noted above, were determined using the broth microdilution method in accordance with the recommendations contained in the Clinical and Laboratory Standards Institute guidelines. Bacteria were cultured in cation-adjusted Mueller Hinton broth (for S. aureus), brain heart infusion broth (for E. faecium), or Tryptic soy broth (for E. faecalis, A. baumannii, E. cloacae, K. pneumoniae, and P. aeruginosa) in a 96-well plate. Compounds, using triplicate samples, were added to the plate and serially diluted. Plates were incubated at 37 °C for at least 20 hours prior to determining the MIC. Plates were visually inspected and the MIC was categorized as the concentration at which no visible growth of bacteria was observed.
Time-kill assay of biphenylthiazole compounds against MRSA
MRSA USA400 cells in logarithmic growth phase (OD600 = 0.796) were diluted to 9.20 × 105 colony-forming units (CFU/mL) and exposed to concentrations equivalent to 4 × MIC (in triplicate) of compound 27 and vancomycin in Tryptic soy broth. Aliquots (100 μL) were collected from each treatment after 0, 2, 4, 6, 8, 10, 12, and 24 hours of incubation at 37 °C and subsequently serially diluted in PBS. Bacteria were then transferred to Tryptic soy agar plates and incubated at 37 °C for 18–20 hours before viable CFU/mL was determined.
In vitro cytotoxicity analysis of diphenylurea compounds against HRT-18 cells
Compounds 26 and 27 (at 64, 128, 256, and 512 μg/mL) were assayed against a human colorectal (HRT-18) cell line to determine the potential toxic effect to mammalian cells in vitro. Cells were cultured in RPMI-1640 medium supplemented with 10% fetal horse serum at 37 °C with CO2 (5%). Control cells received DMSO alone at a concentration equal to that in drug-treated cell samples. Cells were incubated with compounds (in triplicate) in a 96-well plate at 37 °C with CO2 (5%) for two hours. The assay reagent MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (Promega, Madison, WI, USA) was subsequently added and the plate was incubated for four hours. Absorbance readings (at OD490) were taken using a kinetic microplate reader (Molecular Devices, Sunnyvale, CA, USA). The quantity of viable cells after treatment with each compound was expressed as a percentage of the viability of DMSO-treated control cells (average of triplicate wells ± standard deviation). The toxicity data was analyzed via a one-way ANOVA, with post hoc Dunnet’s multiple comparisons test (P < 0.05), utilizing GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA).
In vitro cytotoxicity analysis of biphenylthiazole compounds against BHK-21
Cell culture
Baby hamster kidney fibroblasts (BHK-21) was originally purchased from American type culture collection (ATCC, Wesel, Germany) and grown in the tissue culture lab of the Egyptian company for production of vaccines, sera and drugs (Vacsera, Giza, Egypt). The cells were transferred to our laboratory and maintained in Dulbecco Modified Eagle’s medium (DMEM) supplemented with 1% of 100 mg/ mL of streptomycin, 100 units/ mL of penicillin and 10% of heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA) in a humidified, 5% (v/v) CO2 atmosphere at 37 °C.
Exponentially growing cells were trypsinized, counted and seeded at the appropriate densities (1000 cells/0.33 cm2 well) into 96-well microtiter plates. Cells were incubated in a humidified atmosphere at 37°C for 24 hours. Then, cells were exposed to different compounds at the desired concentrations (0,16,32,64, 128, 256, 512 & 1024 μg/ml) for 6 or 24 hours. At the end of the treatment period, media were removed; cells were incubated with 200 μl of 5% 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide (MTT) solution/well (Sigma Aldrich, MO) and allowed to metabolize the dye into a colored-insoluble formazan complex for 2 hours. Medium was discarded from the wells and the formazan crystals were dissolved in 200 μl/well acidified isopropanol for 30 min, covered with aluminum foil and with continuous shaking using a MaxQ 2000 plate shaker (Thermo Fisher Scientific Inc, MI) at room temperature. Absorbance was measured at 570 nm using a Epoch-2C Microplate Reader (Bio Tek, VT). Cell viability was expressed relative to the untreated control cells and the IC50s were calculated using Graph pad prism 5 software (GraphPad Software, Inc., CA, USA).
Multi-step resistance selection against MRSA
To assess the ability of MRSA to develop resistance to compounds 26 and 27, a multi-step resistance selection experiment was performed, as described elsewhere.[19] The broth microdilution method was utilized to determine the MIC of 27 and ciprofloxacin exposed to MRSA USA300 (NRS384), at subinhibitory concentrations, for 14 passages over a period of two weeks. Resistance was classified as a greater than four-fold increase in the initial MIC, as reported elsewhere.[20] Results presented in Figure 4 are from two independent experiments.
4.3.Pharmacokinetics and Solubility Assays
PBS Solubility Screen
Serial dilutions of the tested compounds, reserpine, tamoxifen, and verapamil were prepared in phosphate buffered saline (PBS) at 100 × the final concentration. The solutions were diluted 100-fold into PBS in a 96-well plate and mixed. The absorbance of the PBS-containing plate was measured prior to addition of the test agents to determine the background absorbance. After 2 h, the presence of precipitate was detected by turbidity (absorbance at 540 nm). An absorbance value of greater than (mean + 3x standard deviation of the blank), after subtracting the pre-experiment background, is indicative of turbidity. The solubility limit is reported as the highest experimental concentration with no evidence of turbidity.
In vivo Pharmacokinetics
This assay has been conducted at a credited bioequivalence center (http://www.grc-me.com/pk_pd.html). Pharmacokinetic studies were performed in male naïve Sprague Dawley (SD) rats, (three animals) following Institutional Animal Care and Use Committee guidelines. IV bolus of 1 μM solution or oral dosing (25 mg/kg) was administered by gavage in a vehicle containing 5% ethanol, 45% PEG 400, and 50% water. Blood samples were collected over a 24-hour period post dose into Vacutainer tubes containing EDTA-K2. Plasma was isolated, and the concentration of compound 27 in plasma was determined with LC/MS/MS after protein precipitation with acetonitrile.
Non-compartmental pharmacokinetic analysis was performed on plasma concentration data to calculate pharmacokinetic parameters using Kinetica® 2000 (release 4.4.1)
Supplementary Material
Oxadiazolylbiphenylthiazoles is a promising class of antibiotics with anti-MRSA activity
Compound 27 exhibited 50-fold improvement in aqueous solubility and oral bioavailability.
MRSA mutants exhibiting resistance to these class of compounds could not be isolated.
Compounds 26 and 27 have balanced PD/PK and toxicological properties
Acknowledgments
This work was supported by Science & Technology Development Funds (STDF), Grant#5503.
Abbreviations
- CFU
colony forming unit
- GIT
gastrointestinal tract
- HBD
hydrogen bond donor
- HRT-18
human colorectal cells
Footnotes
Supporting Information. Spectral data of all new compounds.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Enzler MJ, Berbari E, Osmon DR. Antimicrobial prophylaxis in adults. Mayo Clinic proceedings. 2011;86:686–701. doi: 10.4065/mcp.2011.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2013. 2013. [Google Scholar]
- 3.World Health Organization. Report on the Burden of Endemic Health Care-Associated Infection Worldwide. 2011:1–40. [Google Scholar]
- 4.Chambers HF. Community-associated MRSA--resistance and virulence converge. N Engl J Med. 2005;352:1485–1487. doi: 10.1056/NEJMe058023. [DOI] [PubMed] [Google Scholar]
- 5.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355:666–674. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 6.Hiramitsu K. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect Dis. 2001;1:147–155. doi: 10.1016/S1473-3099(01)00091-3. [DOI] [PubMed] [Google Scholar]
- 7.Peter Wilson J, Andrews A, Charlesworth R, Walesby R, Singer M, Farrell DJ, Robbins M. Linezolid resistance in clinical isolates of Staphylococcus aureus. J Antimicrob Chemother. 2003;51:186–188. doi: 10.1093/jac/dkg104. [DOI] [PubMed] [Google Scholar]
- 8.The Pew Charitable Trusts. [Accessed on February 21, 2017];A Scientific Roadmap for Antibiotic Discovery. 2016 May 11; URL: http://www.pewtrusts.org/en/research-and-analysis/reports/2016/05/a-scientific-roadmap-for-antibiotic-discovery, in.
- 9.Mohammad H, Mayhoub AS, Ghafoor A, Soofi M, Alajlouni RA, Cushman M, Seleem MN. Discovery and characterization of potent thiazoles versus methicillin- and vancomycin-resistant Staphylococcus aureus. J Med Chem. 2014;57:1609–1615. doi: 10.1021/jm401905m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seleem MA, Disouky AM, Mohammad H, Abdelghany TM, Mancy AS, Bayoumi SA, Elshafeey A, El-Morsy A, Seleem MN, Mayhoub AS. Second-Generation Phenylthiazole Antibiotics with Enhanced Pharmacokinetic Properties. J Med Chem. 2016;59:4900–4912. doi: 10.1021/acs.jmedchem.6b00233. [DOI] [PubMed] [Google Scholar]
- 11.Mohammad H, Reddy PV, Monteleone D, Mayhoub AS, Cushman M, Hammac GK, Seleem MN. Antibacterial Characterization of Novel Synthetic Thiazole Compounds against Methicillin-Resistant Staphylococcus pseudintermedius. PloS One. 2015;10:e0130385. doi: 10.1371/journal.pone.0130385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagras M, Mohammad H, Mandour MS, Hegazy HA, Ghaiaty A, Seleem MN, Mayhoub AS. Investigating the Antibacterial Activity of Biphenylthiazoles against Methicillin- and Vancomycin-Resistant Staphylococcus aureus (MRSA and VRSA) J Med Chem. 2017 doi: 10.1021/acs.jmedchem.7b00392. (under reviewing) [DOI] [PubMed] [Google Scholar]
- 13.Kwong AD, Kauffman RS, Hurter P, Mueller P. Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nature Biotechnology. 2011;29:993–1003. doi: 10.1038/nbt.2020. [DOI] [PubMed] [Google Scholar]
- 14.Kessler U, Ranadheera C, Joubert M, Giethlen B, Garrido F. Small molecule inhibitors of influenza a and b virus and respiratory syncytial virus replication. 2010128163. Pike Pharma Gmbh; PCT Int Appl. 2010
- 15.Mohammad H, Cushman M, Seleem MN. Antibacterial Evaluation of Synthetic Thiazole Compounds In Vitro and In Vivo in a Methicillin-Resistant Staphylococcus aureus (MRSA) Skin Infection Mouse Model. PloS One. 2015;10:e0142321. doi: 10.1371/journal.pone.0142321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chylek P, Robinson S, Dubey MK, King MD, Fu Q, Clodius WB. Comparison of near-infrared and thermal infrared cloud phase detections. J Geophys Res-Atmos. 2006;111 [Google Scholar]
- 17.Forrest GN, Tamura K. Rifampin combination therapy for nonmycobacterial infections. Clin Microbiol Rev. 2010;23:14–34. doi: 10.1128/CMR.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitz FJ, Fluit AC, Hafner D, Beeck A, Perdikouli M, Boos M, Scheuring S, Verhoef J, Kohrer K, Von Eiff C. Development of resistance to ciprofloxacin, rifampin, and mupirocin in methicillin-susceptible and -resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother. 2000;44:3229–3231. doi: 10.1128/aac.44.11.3229-3231.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh MP, Petersen PJ, Weiss WJ, Janso JE, Luckman SW, Lenoy EB, Bradford PA, Testa RT, Greenstein M. Mannopeptimycins, new cyclic glycopeptide antibiotics produced by Streptomyces hygroscopicus LL-AC98: antibacterial and mechanistic activities. Antimicrob Agents Chemother. 2003;47:62–69. doi: 10.1128/AAC.47.1.62-69.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farrell DJ, Robbins M, Rhys-Williams W, Love WG. Investigation of the potential for mutational resistance to XF-73, retapamulin, mupirocin, fusidic acid, daptomycin, and vancomycin in methicillin-resistant Staphylococcus aureus isolates during a 55-passage study. Antimicrob Agents Chemother. 2011;55:1177–1181. doi: 10.1128/AAC.01285-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.