

Abstract
The mitochondrial Bit1 protein exerts tumor-suppressive function in NSCLC through induction of anoikis and inhibition of EMT. Having this dual tumor suppressive effect, its downregulation in the established human lung adenocarcinoma A549 cell line resulted in potentiation of tumorigenicity and metastasis in vivo. However, the exact role of Bit1 in regulating malignant growth and transformation of human lung epithelial cells, which are origin of most forms of human lung cancers, has not been examined. To this end, we have downregulated the endogenous Bit1 expression in the immortalized non-tumorigenic human bronchial epithelial BEAS-2B cells. Knockdown of Bit1 enhanced the growth and anoikis insensitivity of BEAS-2B cells. In line with their acquired anoikis resistance, the Bit1 knockdown BEAS-2B cells exhibited enhanced anchorage-independent growth in vitro but failed to form tumors in vivo. The loss of Bit1-induced transformed phenotypes was in part attributable to the repression of E-cadherin expression since forced exogenous E-cadherin expression attenuated the malignant phenotypes of the Bit1 knockdown cells. Importantly, we show that the loss of Bit1 expression in BEAS-2B cells resulted in increased Erk activation, which functions upstream to promote TLE1-mediated transcriptional repression of E-cadherin. These collective findings indicate that loss of Bit1 expression contributes to the acquisition of malignant phenotype of human lung epithelial cells via Erk activation-induced suppression of E-cadherin expression.
1. Introduction
Bcl2 inhibitor of transcription 1 (Bit1) is a 179-residue mitochondrial protein which is part of an apoptosis pathway that is uniquely regulated by integrin-mediated cell attachment [1-8]. Following of loss of cell attachment, Bit1 is released to the cytosol, forms a complex with the transcriptional regulator Amino-terminal Enhancer of Split (AES) protein, and induces a caspase-independent form of apoptosis. While the Bit1 apoptosis function is unresponsive to the effects of several key anti-apoptotic factors such as Bcl-2, Bcl-xL, phosphatidylinositol 3-kinase, the integrin-mediated cell attachment is the only upstream treatment that can suppress apoptosis induced by cytosolic Bit1 pointing to the possibility that Bit1 may play a fundamental role in the anchorage dependence of normal adherent cells [1]. As an effector of anoikis (detachment-induced apoptosis), the Bit1 pathway is likely to be bypassed by cancer cells to acquire anchorage independence and malignant transformation, and hence may function as a tumor suppressor.
To date, several lines of evidence exist to support the role of Bit1 as a putative tumor suppressor in Non-small cell lung cancer (NSCLC), which is the most common form of human lung cancer. First, Bit1 is selectively downregulated in advanced stages of human NSCLC tumors as compared to normal lung tissues [6]. Second, the sole inhibitor of the Bit1 apoptosis function, the TLE1 corepressor, is upregulated in human lung cancer tissues and functions as a lung-specific oncogene in a transgenic mouse model [9]. In line with the tumor suppressive function of the Bit1 pathway, cytoplasmic targeting of the Bit1 protein or its cell death domain (CDD) effectively triggered apoptosis in caspase-resistant NSCLC cells while disruption of the Bit1 pathway in these cells potentiated their anoikis resistance and anchorage-independent growth in vitro and tumorigenicity in vivo [6]. Consistent with the Bit1 apoptosis and anoikis function, the enhanced tumorigenicity of Bit1 knockdown cells is in part due to reduced basal apoptosis.
In addition to its role in apoptosis, Bit1 exerts an inhibitory effect on Epithelial-to-Mesenchymal Transition (EMT), a key event and determinant of cancer progression and aggressiveness, in NSCLC [7]. As an inhibitor of EMT, suppression of endogenous Bit1 expression in A549 NSCLC cell line via siRNA and shRNA strategies induced mesenchymal phenotypes, including enhanced fibroblastoid morphology and migratory cell potential with concomitant repression of the epithelial marker E-cadherin expression. On the other hand, exogenous Bit1 expression in NSCLC cells promoted epithelial transition characterized by cuboidal-like epithelial cell phenotype, reduced cell motility, and upregulated E-cadherin expression. Underscoring the importance of the Bit1 EMT inhibitory function, ectopic Bit1 was shown to be effective in blocking the metastatic potential of NSCLC cells in vivo [7].
The molecular basis underlying the tumor suppressor function of Bit1 has begun to be unraveled. Our collective data indicate that the oncogenic TLE1 corepressor pathway is an important molecular target of Bit1 function [6-8]. To induce anoikis and inhibit EMT, Bit1 turns off the TLE1 corepressor function, particularly TLE1-mediated repression of the epithelial marker E-cadherin. Through genetic analysis, we have shown that the Bit1 induction of E-cadherin expression is a necessary molecular event for Bit1-dependent anoikis and EMT inhibitory function [7-8]. Although the molecular details of how Bit1 inhibits the oncogenic TLE1 transcriptional machinery remain under active investigation, the inhibition of TLE1 corepressor function by Bit1 occurs in part through AES [7]. It is noteworthy that Bit1 is tethered on the outer mitochondrial membrane facing the cytoplasm [10] and has recently been found to interact with Focal Adhesion Kinase (FAK) in the plasma membrane [11], hence raising a possibility that Bit1 may regulate oncogenic signaling pathways that are upstream of the TLE1 protein. Indeed, Bit1 has been found to inhibit the Extracellular regulated kinase (ERK) pathway in mouse embryonic fibroblasts (MEF) and cancer cells, and such inhibition of the Erk pathway contributes to Bit1 anoikis function [3,4]. The effect of Bit1 regulation of the Erk pathway on TLE1 corepressor function particularly in NSCLC has not been elucidated.
Since most previous studies in support of the lung tumor suppressive function of Bit1 were done in established NSCLC cell lines, here we investigated the role of Bit1 in malignant transformation of the immortalized non-tumorigenic human bronchial epithelial BEAS-2B cells. Our results showed that downregulation of endogenous Bit1 expression in BEAS-2B cells potentiates their malignant potential characterized by increased growth, anoikis resistance, and anchorage-independent growth in vitro but is insufficient to promote their tumor growth in vivo. The observed loss of Bit1-induced malignant phenotypes was in part due to the Erk-activation dependent TLE1 mediated transcriptional repression of the E-cadherin gene.
2. Materials and methods
2.1. Cell culture and transfection assays
The human bronchial epithelial cell line BEAS-2B was purchased from ATCC and cultured in BEGM Bronchial Epithelial Cell Growth Medium (Lonza). As described previously [7], the stable BEAS-2B Bit1 shRNA and control shRNA pool of cells were generated via transfection with pRS vector containing the short hairpin RNA against Bit1 (Origene) or the non-targeting scrambled shRNA (Origene) and selection with 0.5 μg/ml puromycin (Invitrogen). Individual puromycin-resistant clones were screened for Bit1 downregulation by immunoblotting using a specific antibody to Bit1. Two Bit1 shRNA knockdown-positive clones and two control shRNA clones were pooled for further characterization. To create stable exogenous E-cadherin expressing Bit1 knockdown cells, the Bit1 shRNA pool of BEAS-2B cells were transfected with the full-length E-cadherin or empty vector construct (Origene). In parallel, the control shRNA pool of BEAS-2B cells were transfected with the empty vector. Subsequently, transfected cells were grown in the presence of 0.5 μg/ml puromycin and 450 ug/ml G418 for selection. Several resistant clones were isolated by cloning rings, and the level of Bit1 knockdown and E-cadherin expression was confirmed by immunoblotting with a specific Bit1 and E-cadherin antibody. Several control shRNA/vector, Bit1 shRNA/vector, Bit1 shRNA/E-cadherin clones were pooled together to generate the control shRNA/vector, Bit1 shRNA/vector, Bit1 shRNA/E-cadherin pools, respectively.
2.2. Chemical reagents, antibodies, and plasmids
The antibodies against PTRH2 (Bit1), total Erk1/2, phosphorylated, Erk1/2 (Thr-202/Tyr-204), total Akt, and activated Akt were from Cell Signaling Technology (Danvers, MA). The mouse monoclonal anti-B-actin antibodies were purchased from Sigma (St. Louis, MO) while the anti-E-cadherin antibodies were purchased from BD Biosciences (San Diego, CA). The MEK/ERK inhibitor U0126 was purchased from Calbiochem (La Jolla, CA). The full-length E-cadherin and the corresponding control constructs were obtained from Origene.
2.3. Cell proliferation, anoikis, and soft agar assays
Anchorage-dependent growth was quantified using the alamar blue staining (Invitrogen) and subsequent fluorescence reading (485 nm excitation wavelength and 520 nm emission wavelength) using the microplate reader (BioTek Instruments) as described previously [8]. In addition, cell growth was determined by clonogenic assay. Briefly, cells were plated at 2,500 cells/per well in a 6-well plate in maintenance medium. After a 2–3-week incubation at 37 °C, colonies were stained with methylene blue and visible colonies were counted. The anchorage-independent growth of cells was assessed using the 96-well plate format as described previously [8]. Briefly, 5,000 cells in 0.3% agar solution were plated onto wells precoated with 0.6% agar in the culture medium. The growth of the resulting colonies was quantified by counting the number of visible colonies (with a diameter greater than 30 uM) and by alamar fluorescence assay [8]. To quantitate for anoikis, cells were cultured onto Polyhema coated 96 well plates in complete growth medium containing 0.5% methylcellulose at a density of 1.0 ×104/well at the indicated time points. Detached cells were then collected and subjected to the Cell Death ELISA apoptosis assay as previously described [8].
2.4. Protein preparation and western blotting assays
Protein preparation and immunoblotting were performed as described previously [7,8]. Briefly, cells were harvested by adding ice-cold NP-40 lysis buffer (1% NP-40; 20mM Tris-HCL [pH7.4]; 150 mM NaCl; 10% glycerol, 2 mM sodium vanadate; 1 mM henylmethylsulfonyl fluoride; 10 μg/ml leupeptin; and 5 μg/ml aprotinin) and incubating at 4°C for 20 min. For immunoblot analysis, equal amounts of proteins were resolved on 4-20% gradient Tris-glycine gels (Invitrogen) and then electrophoretically transferred to nitrocellulose membrane. The membranes were incubated with primary antibodies overnight at 4°C followed by secondary antibodies conjugated with horseradish peroxidase. Membranes were developed using the ECL detection system.
2.5. Total RNA extraction and quantitative Real-Time PCR
As described previously [8], total RNA was extracted from 1.0×106 cultured cells using the RNeasy kit (Qiagen) and quantified by spectrophotometry (NanoDrop 8000, Thermo Scientific). Total RNA was reversed transcribed using the Superscript First-Strand Synthesis Kit for RT-PCR (Invitrogen) as prescribed by the supplier. cDNA was quantified by real-time PCR on the ABI Prism 7900 Sequence Detection System (Applied Biosystems) with the following human E-cadherin (forward primer: AGGCTAGAGGGTCACCGCGTC and reverse primer: GCTTTGCAGTTCCGACGCCAC) using the Sybr Green PCR Core reagents (Applied Biosystems, Foster City, CA). Amplification of the same cDNAs with human GAPDH primers (forward primer: CCCACTCCTCCACCTTTGAC and reverse primer: TTGCTGTAGCCAAATTCGTTGT) was used for internal normalization.
2.6. Promoter luciferase analysis
Control shRNA and Bit1 shRNA pool of BEAS-2B cells were cotransfected with the E-cadherin luciferase promoter-reporter construct (SwitchGear Genomics) and GAPDH luciferase promoter-reporter vector (SwitchGear Genomics) as an internal control for luciferase activity as described previously [8]. Following 24h of incubation, the cells were subjected to a luciferase assay (SwitchGear Genomics' LightSwitch Luciferase Assay System) following the manufacturer's protocol. Luciferase activity was normalized to GAPDH luciferase activity. To determine the effect of Erk inhibition on E-cadherin promoter activity in Bit1 knockdown cells, the Bit1 shRNA cells were treated with 10 μM UO126, and 24 h later cells were cotransfected with the E-cadherin luciferase promoter-reporter construct and GAPDH luciferase promoter-reporter vector (SwitchGear Genomics). Following 24h of incubation, the cells were subjected to a luciferase assay (SwitchGear Genomics' LightSwitch Luciferase Assay System) as indicated above. Each of the above experiment was performed in triplicate and repeated at least three times.
2.7. ChIP assay
The control shRNA and Bit1 shRNA pool of BEAS-2B cells treated or untreated with 10 μM UO126 were grown to 70% to 80 % confluence, crosslinked with 1% formaldehyde, and then processed using the EpiSeeker ChIP Kit – One Step (Abcam) as described previously [8]. The resulting chromatin fragments were immunoprecipitated with the anti-TLE1 antibody-ChIP grade (Abcam), anti-Acetyl-Histone H3-ChIP (anti-Ac-H3) grade (EMD MILLIPORE), or non-specific IgG (Santa Cruz Biotechnology). Subsequent downstream steps were conducted following the protocol from the EpiSeeker ChIP Kit (Abcam). The PCRs were conducted using the E-cadherin primers (5′-CCCACCACGTACAAGGGTC-3′(sense), 5′-ATGCCATCGTTGTTCACTGGA-3′ (antisense)) with the following program: 45 cycles at 95°C for 30 s, 56°C for 30s, and 72°C for 30s [8]. The amplified E-cadherin DNA promoter fragment was separated on 1.5% agarose gel and visualized with ethidium bromide. The purified DNA samples were used as an input for PCR reactions. The PCR product was then subjected to densitometric quantification using QuantityOne software (Bio-Rad) and fold enrichment was determined and expressed as a +TLE1-specific antibody or +Acetyl-Histone H3-specific antibody over non-specific IgG as described previously [8]. The ChIP experiments were repeated at least three times.
2.8. In vivo tumorigenesis assay
All procedures were done according to protocols approved by the Institutional Committee for Use and Care of Laboratory Animals of Xavier University of Louisiana Institutional Animal Care and Use Committee (IACUC, Approval Number 060911-001BI). Eight-week-old female athymic nude mice (BALB/c) were used for the tumorigenesis assay [8]. The control shRNA/vector, Bit1 shRNA/vector, Bit1 shRNA/E-cadherin pool of BEAS-2B cells as well as A549 cells (1.0 ×106) were injected subcutaneously (8 animals/group), and the tumor sizes were measured periodically with a caliper at the indicated time points. Tumor volume was determined by the formula (d1×d22)/2 where d1 represents the larger diameter and d2 the smaller diameter.
2.9. Statistical analysis
Data are presented as means (±S.D.). For western blots and ChIP assays, experiments were performed at least three times. Statistical differences between groups were established at a P value < 0.05 using the Student's t-test (two-tailed). All calculations were done using the NCSS statistical software (NCSS, Kaysville, UT).
3. Results
3.1. Downregulation of Bit1 expression enhances growth and anoikis insensitivity of BEAS-2B cells
To define the tumor suppressive role of Bit1 in lung cancer, we previously silenced endogenous Bit1 expression in the immortalized non-tumorigenic human bronchial epithelial BEAS-2B cell line via the shRNA strategy [7]. In contrast to the stable control shRNA pool of BEAS-2B cells, the stable Bit1 shRNA pool of BEAS-2B cells was shown to exhibit EMT phenotypes including enhanced spindle-shaped morphology, increased motility, and reduced E-cadherin expression [7]. Here, we examined the effects of loss of Bit1 expression on other malignant phenotypes including alteration in growth kinetics and anoikis resistance. As shown in Figs. 1A-1B, stable downregulation of Bit1 expression resulted in enhanced growth of BEAS-2B in monolayer cell culture. Importantly, the minimal clonogenic ability of BEAS-2B cells was significantly enhanced based on the increased number of larger colonies in Bit1 shRNA cells as compared to control shRNA cells (Figs. 1C-1D). Considering that normal human epithelial cells are generally considered sensitive to anoikis, which is a deterrent to malignant transformation, we then examined if Bit1 downregulation alters the anoikis sensitivity of BEAS-2B cells. As shown in Fig. 1E, while the control shRNA and Bit1 shRNA cells exhibited the same level of spontaneous apoptosis when grown attached to a culture dish, the Bit1 shRNA cells showed a significantly reduced level of cell death in suspension as compared to control shRNA cells. Taken together, these findings indicate that loss of Bit1 expression conferred enhanced anchorage-dependent growth and anoikis insensitivity in immortalized non-tumorigenic BEAS-2B cells.
Fig.1.
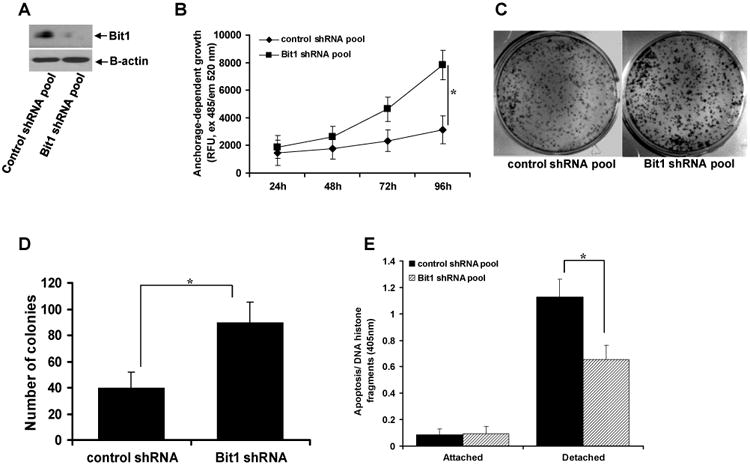
Knockdown of endogenous Bit1 expression in BEAS-2B cells promotes growth and anoikis resistance. A. Stable control shRNA and Bit1 shRNA pool of BEAS-2B cells were subjected to total cell lysate isolation, SDS-PAGE, and immunoblotting against specific antibodies to Bit1 and B-actin. B. The growth of control shRNA and Bit1 shRNA cells was quantified by examining the number of metabolically active cells at the indicated time points with the use of alamar blue assay as described in materials and methods. C. and D. The growth of control shRNA and Bit1 shRNA cells was also examined by clonogenic assay. The representative colonies were visualized by methylene blue staining (C) and quantified (D). E. Control shRNA and Bit1 shRNA cells were cultured in attached and detached conditions for 24h and then harvested for Cell Death Elisa as described in materials and methods. In B, D, and E, three independent experiments were performed in triplicates, * indicates p<0.05 by Student's t test.
3.2. Knockdown of Bit1 promotes anchorage-independent growth in vitro but fails to induce tumor growth in vivo of BEAS-2B cells
Considering that anchorage-independent growth potential is a critical determinant of malignant transformation of human epithelial cells, we then investigated the effect of loss of Bit1 expression in the anchorage-independent growth of BEAS-2B cells. As shown in Fig. 2A, the Bit1 shRNA BEAS-2B cells showed enhanced growth as compared to control shRNA cells in suspension. Consistent with this result, the Bit1 shRNA cells exhibited increased growth in a semisolid agar medium relative to control shRNA cells as quantified by counting the number of sizable colonies (Fig. 2B) and by alamar blue staining (Fig. 2C). In line with their increased anchorage-independent growth potential, the Bit1 knockdown cells exhibited a much a larger colony size as compared to that of the slow-growing control shRNA cells (Fig. 2D).
Fig. 2.
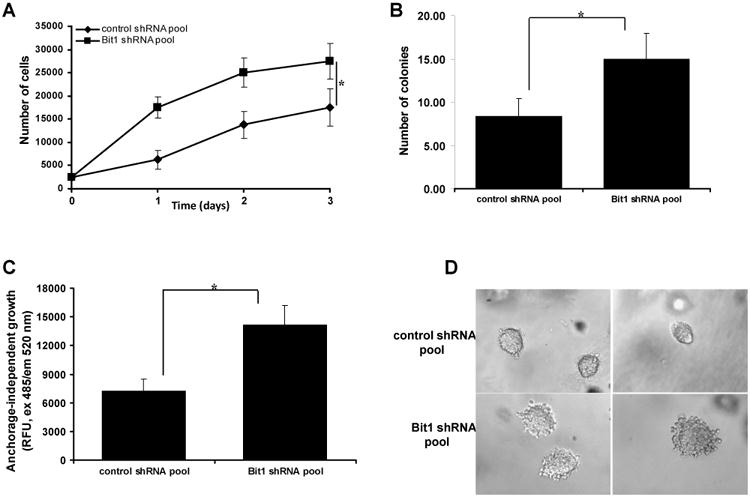

Downregulation of endogenous Bit1 expression in BEAS-2B cells promotes anchorage-independent growth in vitro but is not sufficient to induce tumorigenicity in vivo. A. Control shRNA and Bit1 shRNA pool of cells were plated onto poly-hema coated wells and then subjected to alamar blue assay at various time points. B., C., and D. Control shRNA and Bit1 shRNA cells were cultured in soft agar, and the growth of cells was quantified by counting the number of sizable colonies (B) and alamar blue assay (C). The representative colonies are shown in D. E. Control shRNA and Bit1 shRNA BEAS-2B cells as well as A549 cells were injected subcutaneously in 8-week old BALB/c nude mice. Tumors were measured periodically with a caliper on the days after injection. Shown are the mean tumor volumes as indicated in E. In A, B, and C, * indicates p<0.05 by Student's t test.
To examine whether the enhanced anchorage-independent growth potential of the Bit1 knockdown cells translates to tumorigenicity in vivo, we then injected the control shRNA and Bit1 shRNA cells subcutaneously in nude mice. As a positive control, we also injected the human adenocarcinoma A549 cell line subcutaneously. As indicated in Fig. 2E, the mice receiving the control shRNA BEAS-2B cells nor the Bit1 shRNA BEAS-2B cells formed tumors following implantation into nude mice. In contrast, the A549 cells formed large tumors and grew robustly as we have described previously [6]. Hence, while the loss of endogenous Bit1 expression confers the enhanced anchorage-independent growth of BEAS-2B cells in vitro, downregulation of Bit1 alone is insufficient for these cells to form tumors in vivo.
3.3. E-cadherin repression contributes to loss of Bit1-induced lung epithelial cell growth, anoikis resistance, and transformation
We previously showed that Bit1 induces the epithelial marker E-cadherin gene expression` to induce anoikis and inhibit EMT in the human adenocarcinoma A549 cell line [7,8]. Here, we examined the possibility that E-cadherin is a downstream target of Bit1 in regulating the malignant phenotype of BEAS-2B cells. To this end, we first examined whether E-cadherin expression is altered upon knockdown of endogenous Bit1 expression. Consistent with our previous findings demonstrating that Bit1 functions as an inducer of E-cadherin expression [7,8], the Bit1 shRNA cells exhibited decreased levels of E-cadherin protein as compared to control shRNA cells (Fig. 3A). In addition, the Bit1 knockdown cells showed reduced E-cadherin mRNA transcript (Fig. 3B) and reporter activity (Fig. 3C), indicating loss of Bit1 induces suppression of E-cadherin transcription in BEAS-2B cells. To examine whether E-cadherin repression contributes to the enhanced malignant phenotypes of the Bit1 knockdown cells, we stably expressed exogenous E-cadherin in the Bit1 shRNA cells (Fig. 3D) and then subjected the cells to various transformation assays. As shown in Figs. 3E and 3F, exogenous E-cadherin significantly attenuated the induced anchorage-dependent growth and anoikis resistance of Bit1 knockdown cells. Importantly, forced ectopic E-cadherin expression inhibited the enhanced anchorage-independent growth of BEAS-2B cells as imposed by the loss of Bit1 expression (Fig. 3G). Taken together, these findings indicate that repression of E-cadherin expression is a necessary molecular event contributing to the loss of Bit1-induced malignant phenotype in BEAS-2B cells, and further suggest that E-cadherin is a downstream target of Bit1 in its tumor suppressive function.
Fig. 3.
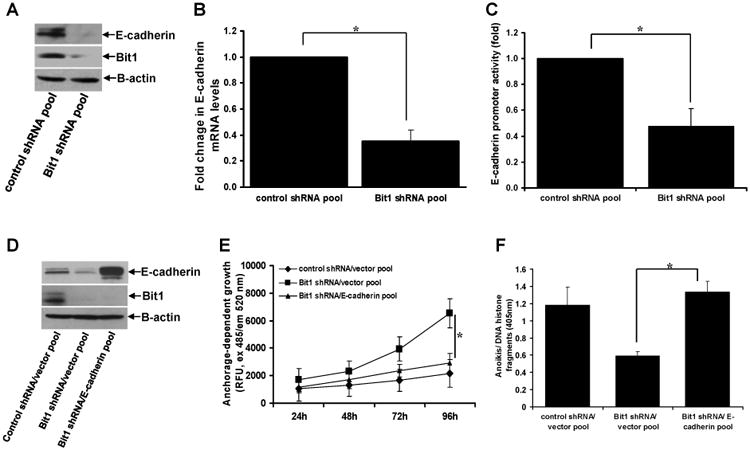

Loss of Bit1-induced E-cadherin repression contributes to the enhanced malignant phenotypes of the Bit1 knockdown BEAS-2B cells. A. Control shRNA and Bit1 shRNA cells were subjected to total cell lysate isolation, SDS-PAGE, and immunoblotting against specific antibodies to E-cadherin, Bit1, and B-actin. B. and C. Control shRNA and Bit1 shRNA cells were subjected to RNA isolation and Real time PCR analysis to quantify for E-cadherin mRNA levels (B) and E-cadherin promoter activity (C). D. Stable control shRNA/vector, Bit1 shRNA/vector, and Bit1shRNA/E-cadherin pool of cells were generated as described in the materials and method and subjected to immunoblotting against specific antibodies to E-cadherin, Bit1, and B-actin. E. The control shRNA/vector, Bit1 shRNA/vector, and Bit1shRNA/E-cadherin pool of cells were cultured in normal culture conditions and subjected to alamar blue assay. F. The control shRNA/vector, Bit1 shRNA/vector, and Bit1shRNA/E-cadherin pool of cells were cultured in detached conditions for 24h and then harvested for Cell Death Elisa assay. G. The control shRNA/vector, Bit1 shRNA/vector, and Bit1shRNA/E-cadherin pool of cells were cultured in soft agar, and the number of sizable colonies was quantified. In B, C, E, F, and G, * indicates p<0.05 by Student's t test.
3.4. Loss of Bit1 expression induces Erk activation to promote TLE1-mediated repression of E-cadherin
It has been reported that activation of survival pathways including the MAPK/Extracellular regulated kinase (Erk) pathway promotes E-cadherin suppression [12-14]. Based on our previous data indicating that Bit1 functions as an inhibitor of the Erk activity [3,4], we then examined the possibility that the observed loss of Bit1 induced E-cadherin suppression in BEAS-2B cells may result from Erk activation. First, we determined the level of Erk activation in control shRNA and Bit1 shRNA cells using immunoblotting assay with a specific antibody against phosphorylated Erk. Indeed, consistent with our previous findings, downregulation of endogenous Bit1 expression resulted in increased levels of active phosphorylated Erk (Fig. 4A). In contrast, loss of Bit1 expression had no significant effect on the activation of the PI3K/Akt survival pathway, indicating the specific effect of Bit1 on Erk. To address whether the Erk pathway impinges on E-cadherin expression in Bit1 knockdown cells, we treated the Bit1 shRNA cells with U0126, a pharmacological inhibitor of the MAPK/Erk pathway. As shown in Fig. 4B, the Erk inhibitor markedly diminished the level of Erk activation in Bit1 knockdown cells to the level seen in control shRNA cells. Importantly, treatment of Bit1 shRNA cells with U0126 resulted in a significant increase in the level of E-cadherin protein (Fig. 4B) and reporter activity (Fig. 4C) comparable to that observed in control shRNA cells, indicating that inhibition of the Erk pathway attenuates the E-cadherin suppression in Bit1 knockdown cells. In line with our findings that E-cadherin repression contributes to the malignant phenotype of Bit1 knockdown cells (Figs. 3D-3G), treatment of Bit1 shRNA cells with the Erk inhibitor also reduced the enhanced anoikis resistance (Fig. 4D) and anchorage-independent growth (Fig. 4E) of the Bit1 knockdown cells. These collective findings indicate that Erk activation at least in part mediates the E-cadherin repression in BEAS-2B cells following loss of Bit1 expression.
Fig. 4.
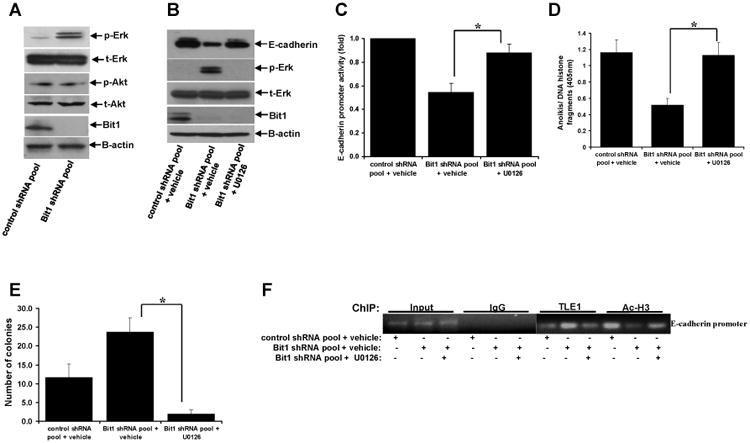

Knockdown of Bit1-induced Erk activation promotes TLE1-mediated transcriptional repression of E-cadherin. A. Control shRNA and Bit1 shRNA pool of BEAS-2B cells were subjected to immunoblotting with the indicated antibodies. B. Control shRNA and Bit1 shRNA cells were treated with U0126 or vehicle as indicated, and 24 h later cells were harvested and cell extracts were prepared and subjected to immunoblotting with the indicated antibodies. C. Control shRNA and Bit1 shRNA cells were treated with U0126 or vehicle as indicated, and 24 h later cells were subjected to E-cadherin promoter luciferase assay as described in the materials and methods. D. Control shRNA and Bit1 shRNA cells treated with U0126 or vehicle were cultured in suspension for 24 h and then harvested and subjected to Cell Death Elisa assay. E. Control shRNA and Bit1 shRNA cells treated with U0126 or vehicle were subjected to a soft agar assay as described in materials and methods, and the resulting number of colonies was quantified. F. and G. Control shRNA and Bit1 shRNA cells treated with U0126 or vehicle were subjected to ChIP assay. Chromatin was precipitated using ChIP-validated antibodies against TLE1, acetyl-histone H3 (Ac-H3), or control IgG as detailed in the materials and methods. The E-cadherin promoter sequence was amplified by PCR and subjected to agarose gel electrophoresis. The ChIP experiments were repeated at least three times and a representative experiment is shown F. Enrichment of the E-cadherin promoter fragment in TLE1-ChIP and acetyl-histone 3-ChIP over IgG-antibody is shown in G. In C, D, E, and G, three independent experiments were performed in triplicates, *indicates p<0.05 by Student's t test.
Since we previously found that the corepressor TLE1 in conjunction with the transcription factor Zeb1 induces transcriptional repression of E-cadherin in lung cancer cells [8], we then examined if Erk activation targets the TLE1 corepressor activity to regulate E-cadherin expression. In line with our previous findings [7], knockdown of endogenous Bit1 expression resulted in an increase of TLE1 occupancy with a concomitant decrease in the histone acetylation on the E-cadherin promoter (Figs. 4F and 4G). Notably, treatment of Bit1 shRNA cells with the Erk inhibitor U0126 attenuated the observed increase in TLE1 occupancy and decrease in acetylated histone on the E-cadherin promoter (Figs. 4F and 4G). Taken together, these findings suggest that the loss of Bit1-induced Erk activation promotes E-cadherin downregulation at least in part through the TLE1 corepressor function.
4. Discussion
The mitochondrial Bit1 protein was originally identified as part of an apoptosis pathway that is regulated by integrin-mediated cell attachment and which may play a fundamental role in the anchorage dependence of normal adherent cells [1]. As an anoikis effector, Bit1 is released to the cytosol following loss of cell attachment and interacts with the transcriptional regulator Amino-terminal Enhancer of Split (AES) to induce a caspase-independent form of apoptosis. Considering that anoikis resistance is a determinant of transformation and tumorigenicity, the Bit1 anoikis pathway is likely to be bypassed by malignant cells to become anchorage independent, and hence may exert tumor suppressive function. Indeed, several lines of evidence exist in support of Bit1 as a putative tumor suppressor in NSCLC: i) Bit1 was selectively downregulated in advanced stages of human NSCLC tumors as compared to normal lung tissues in a human lung cancer tissue array studies [6], ii) the sole inhibitor of Bit1 apoptosis function, the TLE1 corepressor, is upregulated in human lung cancer tissues and functions as a lung-specific oncogene [9], and iii) disruption of the Bit1 pathway in the human lung adenocarcinoma A549 cells potentiated their anoikis resistance and anchorage-independent growth in vitro and tumorigenicity and metastasis in vivo [6,7]. Here, we report that sole downregulation of Bit1 expression in the non-tumorigenic human bronchial epithelial BEAS-2B cells augmented their anoikis resistance and anchorage-independent growth in vitro but failed to induce tumor growth in vivo. The latter finding suggests that additional oncogenic events such as activation of pro-oncogenes are needed to induce tumorigenicity of BEAS-2B cells in vivo. Mechanistically, we have found that the enhanced malignant phenotype of the Bit1 knockdown cells is in part attributable to the Erk activation-induced repression of E-cadherin expression, which underscores E-cadherin as an important molecular target of the Bit1 tumor suppressive function.
E-cadherin is an epithelial cell adhesion protein widely known for its function in keeping epithelial cells tightly bound to each other. As a cell adhesive molecule, its loss of expression in cancer cells promotes their motility and invasiveness, hence downregulation of E-cadherin is a determinant of EMT and metastasis [15]. Recently, a novel role for the E-cadherin transmembrane protein in regulating epithelial cell survival and apoptosis has emerged. In particular, forced knockdown of E-cadherin expression in human breast epithelial cells as well as mouse mammary tumor cells induces resistance to anoikis [16,17]. In NSCLC cells in particular, E-cadherin expression exerts an inhibitory effect on anchorage-independent growth [18]. Having this dual role in regulating cell-cell adhesion and cell survival, the multifunction E-cadherin protein is, therefore, a target of tumor-promoting pathways. For example, activation of oncogenes and silencing of tumor suppressor genes may result in inhibition of E-cadherin expression. Here, we show that repression of E-cadherin is a necessary molecular event for the promotion of malignant phenotypes in the Bit1 knockdown BEAS-2B cells. This finding is consistent with our previous results demonstrating that forced ectopic E-cadherin expression in the Bit1 knockdown human lung adenocarcinoma A549 cells which exhibit suppressed E-cadherin expression attenuated their enhanced migratory potential [7] and anoikis resistance [8]. Furthermore, silencing of E-cadherin expression in exogenous Bit1 expressing A549 cells attenuated their reduced anoikis sensitivity and motility [7,8]. Although our collective data point to E-cadherin as an important molecular target of the Bit1 tumor suppressive function, the downstream signaling pathway(s) impacted by the loss of E-cadherin expression following downregulation of Bit1 remains to be determined. It is noteworthy that E-cadherin inhibits the Rho family proteins, RhoA and Cdc42, to exert its inhibitory effect on the transformed phenotypes in NSCLC cell lines [19]. Hence, it will be interesting to determine whether the loss of Bit1-induced E-cadherin repression and transformation in lung epithelial cells are associated with activation of the Rho family of GTPases.
The silencing of E-cadherin gene expression in various human epithelial tumors has been attributed primarily via transcriptional and epigenetic mechanisms [15]. To this end, we have recently identified the Zeb1/TLE1 transcriptional machinery as an important determinant of E-cadherin repression in NSCLC cells [8]. In particular, the transcription factor Zeb1, a key EMT mediator whose expression is negatively correlated with E-cadherin protein and mRNA levels in lung cancer cells [20], recruits the TLE1 corepressor in association with the chromatin remodelling enzyme Histone deacetylase 1 (HDAC1) to the E-cadherin promoter to impose histone deacetylation and gene silencing. Importantly, such Zeb1/TLE1-mediated E-cadherin transcription repression is negatively regulated by Bit1 to induce E-cadherin expression in established NSCLC cells [7,8]. In line with this notion, we show here that loss of Bit1 expression in human lung epithelial cells results in enhanced TLE1-mediated transcriptional silencing of E-cadherin, as evidenced by the increase in TLE1 occupancy with a concomitant decrease in histone acetylation on the E-cadherin promoter. As a critical downstream target inhibited by lung tumor suppressor Bit1, the ZEB1/TLE1/E-cadherin transcriptional mechanism may regulate and dictate the acquisition of tumorigenic phenotypes by the lung epithelial cells. Indeed, we recently showed that the Zeb1/TLE1-mediated suppression of E-cadherin expression promotes tumorigenicity in NSCLC cells [8].
The exact mechanism underlying the negative regulation of TLE1 corepressor function by Bit1 remains to be fully delineated. One such possible mechanism involves the transcriptional regulator protein AES, a Groucho related binding partner of nuclear TLE1. Acting as a competitor for AES binding with TLE1, Bit1, which is tethered on the outer mitochondrial membrane facing the cytoplasm [10], may interact with cytosolic AES and such functional interaction may result in channelling of nuclear AES away to the cytoplasm and subsequent impingement of TLE1 nuclear function. Here, we present findings in support of a novel mechanism by which Bit1 negatively regulates the TLE1 corepressor function via inhibition of the Erk pathway. Loss of endogenous Bit1 expression in BEAS-2B resulted in increased Erk activation whose inhibition led to transcriptional induction of E-cadherin expression. Importantly, inhibition of Erk activity impinges on TLE1 corepressor function on the E-cadherin promoter. While the exact mechanism by which the Erk pathway affects TLE1 nuclear function needs to be delineated, it remains a possibility that active Erk may directly phosphorylate TLE1, and such Erk-mediated phosphorylation will enhance its corepressor activity. In support of this notion, the nuclear TLE1 protein undergoes cofactor-dependent phosphorylation which promotes its interaction with chromatin and anti-neurogenic function [21]. The molecular details of how the Erk pathway regulates TLE1 nuclear function as well as the mode by which Bit1 negates Erk activation will be the focus of our future studies.
In summary, we provide evidence that loss of the tumor suppressor Bit1 expression is an important molecular event contributing to the malignant transformation of the human bronchial epithelial cells by suppressing E-cadherin expression. Mechanistically, the loss of Bit1-induced E-cadherin repression is via activation of the Erk pathway which functions upstream to potentiate the TLE1-mediated transcriptional repression of E-cadherin. Considering that currently available targeted therapies for lung cancer are limited, it will be important to determine if the enhanced Erk activation upon downregulation of Bit1 as observed in BEAS-2B and cancer cells [3-4] may result in acquired tumor vulnerability and addiction that can be exploited and targeted with pharmacological approaches.
Highlights.
Downregulation of Bit1 expression potentiates cell growth, anoikis resistance, and anchorage-independent potential of BEAS-2B cells.
Loss of Bit1 induced E-cadherin repression contributes to enhanced malignant phenotypes of the Bit1 knockdown cells.
The increased Erk activation in Bit1 knockdown cells promotes TLE1-mediated transcriptional repression of E-cadherin.
Acknowledgments
This work was supported by the Louisiana Cancer Research Consortium start up grant (to HB), NIH 2R15 CA158677-02 Grant (to HB), NIH RCMI grant #8G12MD007595 (Xavier University of Louisiana), and NIH R25GM060926 (Xavier University of Louisiana).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jan Y, Matter M, Pai JT, Chen YL, Pilch J, Komatsu M, Ong E, Fukuda M, Ruoslahti E. A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell. 2004;116:751–762. doi: 10.1016/s0092-8674(04)00204-1. [DOI] [PubMed] [Google Scholar]
- 2.Biliran H, Jan Y, Chen R, Pasquale EB, Ruoslahti E. Protein kinase D is a positive regulator of Bit1 apoptotic function. J Biol Chem. 2008;283:28029–28037. doi: 10.1074/jbc.M803139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kairouz-Wahbe R, Biliran H, Luo X, Khor I, Wankell M, Besch-Williford C, Pascual J, Oshima R, Ruoslahti E. Anoikis effector Bit1 negatively regulates Erk activity. Proc Natl Acad Sci U S A. 2008;105:1528–1532. doi: 10.1073/pnas.0711357105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karmali PP, Brunquell C, Tram H, Ireland SK, Ruoslahti E, Biliran H. Metastasis of tumor cells is enhanced by downregulation of Bit1. PLoS One. 6:e23840. doi: 10.1371/journal.pone.0023840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunquell C, Biliran H, Jennings S, Ireland SK, Chen R, Ruoslahti E. TLE1 is an anoikis regulator and is downregulated by Bit1 in breast cancer cells. Mol Cancer Res. 10:1482–1495. doi: 10.1158/1541-7786.MCR-12-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao X, Jennings S, Ireland SK, Pham T, Temple B, Davis M, Chen R, Davenport I, Biliran H. The anoikis effector Bit1 displays tumor suppressive function in lung cancer cells. PLoS One. 9:e101564. doi: 10.1371/journal.pone.0101564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao X, Pham T, Temple B, Gray S, Cannon C, Chen R, Abdel-Mageed AB, Biliran H. The Anoikis Effector Bit1 Inhibits EMT through Attenuation of TLE1-Mediated Repression of E-Cadherin in Lung Cancer Cells. PLoS One. 11:e0163228. doi: 10.1371/journal.pone.0163228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao X, et al. TLE1 inhibits anoikis and promotes tumorigenicity in human lung cancer cells through ZEB1-mediated E-cadherin repression. Oncotarget. 2017:1–15. doi: 10.18632/oncotarget.19703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen T, van Tuyl M, Iyengar P, Jothy S, Post M, Tsao MS, Lobe CG. Grg1 acts as a lung-specific oncogene in a transgenic mouse model. Cancer Res. 2006;66:1294–1301. doi: 10.1158/0008-5472.CAN-05-1634. [DOI] [PubMed] [Google Scholar]
- 10.Chen R, Braun GB, Luo X, Sugahara KN, Teesalu T, Ruoslahti E. Application of a proapoptotic peptide to intratumorally spreading cancer therapy. Cancer Res. 73:1352–1361. doi: 10.1158/0008-5472.CAN-12-1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffiths GS, Grundl M, Leychenko A, Reiter S, Young-Robbins SS, Sulzmaier FJ, Caliva MJ, Ramos JW, Matter ML. Bit-1 mediates integrin-dependent cell survival through activation of the NFkappaB pathway. J Biol Chem. 286:14713–14723. doi: 10.1074/jbc.M111.228387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lau MT, So WK, Leung PC. Fibroblast growth factor 2 induces E-cadherin down-regulation via PI3K/Akt/mTOR and MAPK/ERK signaling in ovarian cancer cells. PLoS One. 8:e59083. doi: 10.1371/journal.pone.0059083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW, O'Regan RM. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–2488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- 14.Saegusa M, Hashimura M, Kuwata T, Okayasu I. Requirement of the Akt/beta-catenin pathway for uterine carcinosarcoma genesis, modulating E-cadherin expression through the transactivation of slug. Am J Pathol. 2009;174:2107–2115. doi: 10.2353/ajpath.2009.081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nollet F, Berx G, van Roy F. The role of the E-cadherin/catenin adhesion complex in the development and progression of cancer. Mol Cell Biol Res Commun. 1999;2:77–85. doi: 10.1006/mcbr.1999.0155. [DOI] [PubMed] [Google Scholar]
- 16.Derksen PW, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10:437–449. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 18.Takeyama Y, et al. Knockdown of ZEB1, a master epithelial-to-mesenchymal transition (EMT) gene, suppresses anchorage-independent cell growth of lung cancer cells. Cancer Lett. 296:216–224. doi: 10.1016/j.canlet.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asnaghi L, Vass WC, Quadri R, Day PM, Qian X, Braverman R, Papageorge AG, Lowy DR. E-cadherin negatively regulates neoplastic growth in non-small cell lung cancer: role of Rho GTPases. Oncogene. 29:2760–2771. doi: 10.1038/onc.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohira T, et al. WNT7a induces E-cadherin in lung cancer cells. Proc Natl Acad Sci U S A. 2003;100:10429–10434. doi: 10.1073/pnas.1734137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buscarlet M, Hermann R, Lo R, Tang Y, Joachim K, Stifani S. Cofactor-activated phosphorylation is required for inhibition of cortical neuron differentiation by Groucho/TLE1. PLoS One. 2009;4:e8107. doi: 10.1371/journal.pone.0008107. [DOI] [PMC free article] [PubMed] [Google Scholar]