

Abstract
The reduction of synovial tissue macrophage is a reliable biomarker for clinical improvement in patients with rheumatoid arthritis (RA) and macrophages are reduced in synovial tissue shortly after initiation of TNF inhibitors. The mechanism for this initial response is unclear. These studies were performed to identify the mechanisms responsible for the initial reduction of macrophages following TNF inhibition, positing that efflux to draining lymph nodes (LNs) was involved. RA synovial tissue and synovial fluid macrophages expressed CCR7, which was increased in control macrophages following incubation with TNFα. Human TNF transgenic (hTNF-Tg) mice were treated with infliximab after development of arthritis. Ankles were harvested and examined by histology, immunohistochemistry, qRT-PCR, ELISA, and flow cytometry. hTNF-Tg mice treated with infliximab demonstrated significant clinical and histologic improvement 3 days after the initiation of therapy, at which time Ly6C+ macrophages were significantly reduced in the ankles. However, no evidence was identified to support a role of macrophage efflux to draining LNs following treatment with infliximab. In contrast, apoptosis of Ly6C+ macrophages in the ankles and popliteal LNs, decreased migration of monocytes into the ankles, and a reduction of CCL2, were identified following the initiation of infliximab. These observations demonstrate that Ly6C+ macrophages apoptosis and decreased ingress of circulating monocytes into the joint are responsible for the initial reduction of macrophages following infliximab treatment in hTNF-Tg mice.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that primarily affects synovial tissue resulting in hyperplasia of the synovial lining composed of macrophages and fibroblasts and infiltration of the sublining region with a variety of cells including macrophages, fibroblasts, lymphocytes, and dendritic cells, together with abundant angiogenesis. Each of these cell types has been implicated in disease pathogenesis. If inadequately treated RA may result in cartilage loss and joint destruction. While great advances in therapy have been made including the use of non-biologic disease-modifying anti-rheumatic drugs such as methotrexate and biologic agents such as TNF inhibitors, abatacept and rituximab, the mechanism of action of effective therapy is poorly understood. However, published studies document that the extent of macrophage infiltration in the synovial tissue is a strong predictor of clinical outcome (1). Further, examination of synovial tissue biopsies before and after therapy, demonstrate that the reduction of sublining CD68+ macrophages, but not other cell types, correlates with the reduction of the DAS28, regardless of the therapy (2). Therefore, synovial tissue macrophages are a relevant biomarker for clinical response in RA.
The mechanism by which synovial tissue macrophages are reduced following effective therapy is not known. Potential mechanisms include reduced recruitment of monocytes into the tissue, increased cell death, such as apoptosis, or increased macrophage trafficking out of the tissue via the lymphatics to the lymph nodes. An understanding of the responsible mechanism is critical to identify safer, more effective therapy, especially for those who do not respond adequately to currently available treatment. A number of recent studies have employed TNF inhibitors to address the mechanism of response. Studies that examined synovial tissue apoptosis at 1, 24 and 48 hours after the initiation of therapy with infliximab in patients with RA, which resulted in significant reduction of synovial tissue macrophages, failed to demonstrate increased apoptosis (3). Clinical trials that targeted inhibition of chemokine receptors present on monocytes, CCR1, CCR2 and CCR5, in an effort to decrease monocyte migration into the joint, failed to result in significant clinical improvement in patients with RA (4-6). Further, employing a technique to directly track the migration of circulating monocytes into RA synovial tissue, no reduction of monocyte migration was observed in patients treated with adalimumab, a therapy that results in rapid reduction of synovial tissue macrophages (7). In contrast, TNF inhibition resulted in significantly reduced granulocyte migration into the joints, when measured two weeks after therapy (8). Together these observations suggest than neither macrophage apoptosis nor reduction of monocyte migration into the RA joint is responsible for the clinical response to TNF inhibitors, identifying a potential role for increased egress of macrophages, and possibly other cell types, from the RA joint as an important mechanism of action.
While CCR7 is known to be expressed on T cells and dendritic cells, our preliminary data demonstrate that CCR7 is also expressed on RA synovial tissue macrophages. Also we have shown recently that the CCR7 ligands CCL19 and CCL21 are expressed in RA synovial tissue (9). While lymph nodes highly express CCL19 and CCL21 constitutively, CCL19 and 21 are induced by inflammatory mediators including TNFα. These observations suggest that CCR7, CCL19 and CCL21 may contribute to retaining macrophages in the inflamed joint, and that effective therapy may suppress CCL19 and CCL21 in the joints, resulting in attraction of CCR7 positive cells to the lymph nodes. Additionally, CCR7 deficient mice demonstrate more chronic immune complex mediated arthritis, and the synovitis is enriched in macrophages (10). Therefore, we employed human TNF transgenic (hTNF-Tg) mice to identify the initial mechanism of response to TNF inhibition. Following treatment with infliximab joint Ly6C+ macrophages, but not other cell types, were reduced at 72 hours. In contrast to expectations increase efflux of macrophages from the joints could not be identified, nor was synovial tissue CCL19 or 21 effectively decreased prior to the reduction of macrophages. A modest increase of apoptosis of Ly6C+ macrophages was identified. Further, a significant decrease of monocytes entering the joints was identified following TNFα neutralization, which was associated with a dramatic reduction of a classical monocyte chemokine CCL2, but not Fractalkine, a non-classical monocyte chemokine. Since Ly6C+ macrophages derive from recently recruited monocytes, these observation suggest that in hTNF-Tg mice, neutralization of TNFα results in reduced trafficking of monocytes due to reduction of CCL2 and possibly other chemokines, and to a lesser degree, increased apoptosis.
Materials and Methods
Cell Culture
Human monocytes, isolated from buffy coats (Lifesource, Glenview, IL) using counter-current centrifugal elutriation (JE-6B, Beckman Coulter, Palo Alto, CA), were adhered to plastic plates for 1 hour in RPMI medium without serum and differentiated into macrophages for 7 days in RPMI-1640 medium containing 20% FBS as previously described (11-13). Macrophages were treated without or with TNFα (20ng/ml) for 16 hours and cells were harvested for immunoblot analysis.
Mouse Lines
The hTNF-Tg mouse line 196 was the generous gift of Dr. George. Kollias. CCR7-/- and CD45.1 mice were procured from Jackson Laboratory and crossed with the hTNF-Tg line to generate hTNF-Tg CCR7-/- and CD45.1 hTNF-Tg mice. All mice were bred on C57Bl/6 background. PCR was used to genotype the mice using genomic DNA extracted from tail biopsies at 10 days of age. Local ethical guidelines were followed and animal procedures approved by Northwestern University's Office of Research Safety and the Institutional Animal Care and Use Committee.
Patients and specimens
Synovial tissue was obtained from 3 patients diagnosed with RA and 3 arthritis free controls, as previously described (11). Peripheral blood was obtained from 4 healthy donors. Synovial fluid was obtained from 9 patients diagnosed with RA, according to the American College of Rheumatology classification criteria (14). Synovial fluid was obtained during routine care for active RA. Macrophages were obtained as previously described (15, 16). All participants were recruited from Northwestern Medical Faculty Foundation (now Northwestern Medicine) or the Rehabilitation Institute of Chicago. All participants provided written informed consent. These studies were approved by the Institution Review Board of Northwestern University. Although all patient specimens were obtained with informed consent, we were not able to retrieve detailed information concerning demographics, medications and disease activity, since during the study our institution initiated a policy that prevents us from retrieving clinical data from patient charts retrospectively, even though the patients consented.
Immunohistochemistry
CCR7 was detected by immunohistochemistry in formalin fixed and paraffin-embedded RA and arthritis-free control synovial tissues (16). Immunostaining was performed by the pathology core facility of Northwestern University. Slides were deparaffinized in xylene for 3 changes at 5 min each, followed by rehydration through graded alcohols. Antigen retrieval was accomplished in pH 6.0 citrate buffer solution (Dako, S1699) using a digital pressure cooker (Biocare Medical) at 125°C for 30 seconds. Endogenous peroxidase activity was quenched by 3% H2O2 for 10 min, slides were blocked with casein (Dako, X0909) for 5 minutes, and then incubated with a monoclonal mouse anti-CCR7 (R&D System, MAB197) or isotype matched mouse IgG2A control or mouse anti-CD68, followed by anti-mouse IgG (Dako #K4007) secondary antibody conjugated to HRP for 15 minutes, which was visualized with diaminobenzidine substrate, and counterstained with hematoxylin.
Quantitative RT-PCR
Total RNA was extracted from human macrophages and from homogenized mouse tissues using Trizol (Invitrogen). The reverse transcription with oligo (dT) primers was performed by using Superscript reverse transcriptase (Invitrogen) according to the manufacturer's protocol. Real-time PCR was carried out employing TaqMan Universal PCR Master Mix Kit (Applied Biosystems, CA). The primers and probes were obtained from Applied Biosystems: human CCR7 (Hs01013469_ml), mouse Ccl21 (Mm03646971-9H), mouse Ccl19 (Mn00839967_g1), and human and mouse GAPDH. The qPCR was performed with a 7300 real time PCR System (Applied Biosystems). The amplification program was: 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of 95° C for 15 seconds, and then 60° C for 1 minute. Quantitative values were derived from the threshold cycle number (Ct). The experiments were performed in triplicate and expression values were normalized to GAPDH.
Immunoblot analysis
Immunoblot analysis was performed according to an established protocol (11). Briefly, whole-cell extracts were prepared from macrophages and proteins (60μg) were electrophoresed on SDS-PAGE 12% polyacrylamide gels and transferred to Immobilon-P (Millipore, Bedford, MA) by semidry electro blotter as previously described (BioRad, Hercules, CA) (11). The membranes were then blocked in 5% non-fat milk PBS/0.2% Tween-20 (PBST) and subsequently incubated overnight at 4°C with primary antibody anti-CCR7 (mouse monoclonal, R&D System, MAB197) or anti-β-actin (Sigma, St. Louis, MO). Membranes were washed in PBST and incubated with either donkey anti-rabbit or anti-mouse secondary antibodies conjugated with horseradish peroxidase (1:3300 dilution; Amersham Pharmacia Biotech, Piscataway, NJ). The specific proteins were detected employing the Enhanced Chemiluminescent detection reagent (Amersham, Pharmacia Biotech).
Histopathologic analysis
Mouse ankles were stored in 10% neutral based formalin before being incubated in EDTA decalcification buffer for 2 weeks. Ankles were embedded in paraffin and prepared as described (17, 18), sections cut at 4μm, and stained with hematoxylin and eosin by the Mouse Histology and Phenotyping Laboratory of Northwestern University. Ankle specimens were examined by a blinded pathologist scoring the following on a scale from 1 to 5 for inflammation, erosion and pannus formation, cartilage destruction, neutrophil and lymphocyte infiltration, and the average synovial lining thickness. (15, 19, 20).
Infliximab Treatment
hTNF-Tg, hTNF-Tg CCR7-/-, or littermate control mice were administered 10 mg/kg infliximab (Janssen) intraperitoneally beginning 5-6 weeks of age. Mice were administered either 2 or 3 doses of infliximab. Mice given 2 doses were administered infliximab on day 0 and day 2 with tissue harvested on day 3, or 7 as indicated. Mice receiving 3 doses were administered infliximab on days 0, 2, and 4 with tissue harvested 7 days after the initial injection.
Clinical Evaluation of Arthritis
Mice were evaluated weekly from 3 to 10 weeks of age to determine the severity of arthritis. Joint swelling and inflammation was scored for each paw/ankle 0-3. Joint deformity was scored for each paw/ankle 0-3 including toe flexion, contraction and shortening. Grip strength was determined on 3mm diameter wires and scored 0-4 (21). The clinical score was the sum of joint inflammation, joint deformity, and grip strength, with a maximum clinical score of 28.
Mouse tissue and cell preparation
Mouse popliteal lymph nodes (pLNs), or the superficial cervical, axillary lymph nodes, as well as spleen, thymus, liver, skeletal and cardiac muscles were isolated from C57BL/6 mice and subjected to RNA extraction. For analysis by flow cytometry, single cell suspensions were obtained from ankle joints as previously described (22) with collagenase (1mg/ml) in HBSS. The pLNs were digested with collagenase (1mg/ml) and DNase (0.1mg/ml) in HBSS. Peripheral blood were collected by cardiac puncture at the time of harvest other tissues with EDTA anticoagulant.
Immunophenotyping
Cells types were identified by multi-colored flow cytometry using antibodies to F4/80, CD11b, CD45.1, CD45.2, CD64, Ly6C, CD3, CD19, Ly6G, and CD11c (eBioscience or BD Bioscience). Macrophages were defined as CD64+F4/80+CD11b+, and further identified by Ly6C+ or Ly6C- . Granulocytes were defined as CD64-CD11b+Ly6G+ and dendritic cells as CD64-MHCII+CD11c+. T cells were CD3+CD11b- and B cells CD19+CD11b-. Cell viability was determined by Live/Dead (Thermo Fisher Scientific) staining. Apoptotic and necrotic cells were evaluated with Annexin V and 7AAD (BD Bioscience) as previously described (13). Apoptotic cells were defined as Annexin V+ 7AAD-, while necrotic cells were Annexin V+ 7AAD+. CCR7 expression was assessed using an anti-CCR7 antibody (clone 4B12) or isotype control IgG (eBioscience). Cleaved caspase 3 and 8 were detected permeabilized cells according to the manufacturer's directions (Cell Signaling Technology). Data was acquired with a BD LSRII flow cytometer and analyzed with FlowJo software (Tree Star).
ELISA
Chemokine concentrations from ankles and pLNs were determined using ELISAs specific for each chemokine (R&D Systems) according to the manufacturer's protocol. Ankles and pLN were homogenized in PBS supplemented with protease inhibitor (Sigma) as previously described (18, 23). Tissue homogenates were centrifuged at 12,000g for 5 minutes at 4°C and the supernatant collected. Protein concentration was determined using a Pierce BCA protein assay kit (Thermo Fisher Scientific) (15, 19). Chemokine concentrations are displayed as pg/mg total protein.
Macrophage egress
Ankle synovial macrophages were isolated from CD45.2+ hTNF-Tg mice with arthritis. Synovial macrophages were enrichment by negative selection removing cells positive for Ly6G, CD3, CD19, CD11c and NK 1.1, sorted by flow cytometry. The donor cells (0.75×106 cells/10 μl PBS/ankle × 2 ankles) were injected into CD45.1 hTNF-Tg recipients with infliximab or PBS treated 3 hours before applying of cells. Ankles and PL LNs were harvested 16 hours after macrophage injection and macrophage egress was tracked by the presence of CD45.2 macrophages employing flow cytometry.
Monocyte Tracking
Bone marrow was collected from CD45.1 mice. CD115+ monocytes were isolated from the bone marrow using Monocyte Isolation kits (BM) (Miltenyi). hTNF-tg mice on a CD45.2 background received 2×106 CD45.1 monocytes via retro-orbital injection. Monocyte recipients received infliximab at the time of administering monocytes, and one group of monocyte recipients received an additional injection of infliximab 24 hours prior to monocytes being administered. Ankles and pLNs were collected 24 hours after monocyte injection and examined by flow cytometry for the presence of CD45.1 monocytes.
Statistical Analysis
Quantitative data were presented as mean +/- SEM. Student's 2-tailed t-test was used to analyze the significance between two groups. ANOVA was used for multiple comparisons followed by Tukey's pair wise mean comparison for multi group analysis. P values less than 0.05 were considered significant.
Results
CCR7 is expressed on RA synovial tissue macrophages
In an effort to identify the initial event in effective therapy in inflammatory arthritis, RA synovial tissue was examined. CCR7 was detected on RA synovial tissue lining and sublining CD68+ macrophages, but not on those from arthritis-free controls by immunohistochemistry (Figure 1A, B). CCR7 mRNA was increased in isolated RA synovial fluid macrophages compared with macrophages differentiated in vitro from normal monocytes (Figure 1C). CCR7 mRNA and protein was induced by in vitro differentiated macrophages following incubation with TNFα (Figure 1D).
Figure 1. CCR7 is expressed by RA synovial macrophages.
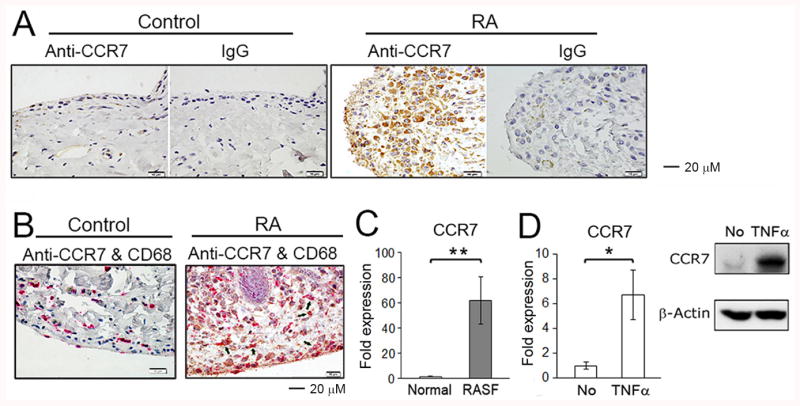
(A) CCR7 expression in arthritis-free control or RA synovial tissue was examined by immunohistochemistry using mouse monoclonal anti-CCR7 and mouse IgG isotype control antibodies. The photos shown are representative of 3 RA and 3 arthritis-free control synovial tissues. (B) The double staining for control and RA synovial tissues employing anti-CCR7 (brown) and anti-CD68 (red) antibodies. The arrows identify cells that are CCR7+CD68+. (C) CCR7 mRNA expression detected by qRT-PCR were examined employing in vitro differentiated macrophages from peripheral blood of 4 healthy donors and RA synovial fluid (SF) macrophages from 9 RA patients. * represents p<0.05 for RA SFs compared to control macrophages differentiated in vitro from normal monocytes. (D) In vitro differentiated human macrophages were incubated for 16 hours with/without TNFα (20 ng/ml). The Expression of CCR7 mRNA was determined by qRT-PCR (left panel) and CCR7 protein expression was detected by Western blot employing an anti-CCR7 antibody (right panel). * represents p<0.05 and ** p < 0.01 between the indicated groups.
CCR7 expression on murine macrophages
Wild type B6 mice were examined to identify the expression of CCR7 and the CCR7 ligands CCL19 and CCL21. Confirming prior studies, CCL19 and 21 were constitutively highly expressed in the lymph nodes, but not other tissues (Supplemental Figure 1A, B). Next, cells from the ankles were examined by flow cytometry. Total macrophages were identified as F4/80+CD11b+CD64+ cells (Supplemental Figure 1C), which were further defined as Ly6C+ or Ly6C-. For those macrophages that are also Ly6C+, most likely identify cells recently arrived in the joint derived from circulating Ly6C+ monocytes. While some authors refer to these Ly6C+ cells as monocytes, we have chosen to call them Ly6C+ macrophages because they have already emigrated from the circulation, consistent with the view that bona fide monocytes are found exclusively in the bone marrow, circulation and spleen (24, 25). In contrast to our results with human control synovial tissue by immunohistochemistry, which is less sensitive than flow cytometry, by flow CCR7 was identified on murine ankle macrophages, particularly on those that were Ly6C-, but not on neutrophils (Supplemental Figure 1D).
The arthritis of hTNF-Tg mice responds to infliximab by 72 hours
The arthritis of the hTNF-Tg mice progressed over the initial 10 weeks of observation (Figure 2A). The studies presented employed mice at 5-6 weeks of age, since arthritis was present in all mice by this time point. Following 2 treatments with infliximab, 3 days after the first injection, a significant improvement in the clinical scores was first documented (Figure 2B). Some mice receiving an additional injection at day 4, and they showed further improvement when examined 7 days after the initial injection (Figure 2B). At 5-6 weeks of age, inflammation was identified histologically (Figure 2C and Supplemental Figure 2). Compared with untreated mice, inflammation, erosion/pannus, lining thickness, and neutrophils were all significantly reduced 3 days after the initiation of treatment, and to a greater degree at 7 days (Figure 2C). Since 3 days after initiation of therapy was the earliest time point at which both clinical and histologic improvement were documented, this time point was employed for additional experiments.
Figure 2. Infliximab effectively controls the spontaneous arthritis in hTNF-Tg mice.
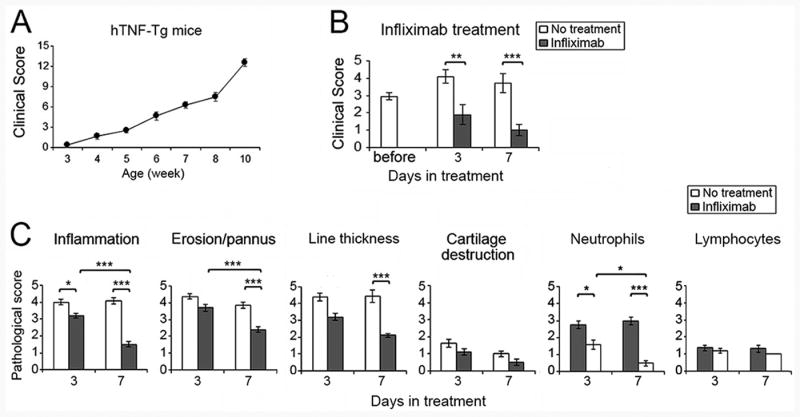
(A) Clinical evaluation of the spontaneous development of arthritis in hTNF-Tg mice. Arthritis was evaluated on a weekly basis from weeks 3 to-10. (B). Clinical evaluation of arthritis of 5-6 week old hTNF-Tg mice before or after 2 doses (3 days) or 3 doses (7 days) of infliximab (n = 8-14 mice in each group). (C). Analysis of histological assessment of ankle joints after initiating infliximab 3 or 7 days earlier in 5-6 week old mice, which are compared with hTNF-Tg mice of the same age without treatment harvested same day (n = 5 for each treatment group, and n = 4-8 mice for the controls). * represents p < 0.05, ** p <0.01 and *** p < 0.001 between the indicated groups.
Ly6C+ macrophages are reduced in ankles and draining lymph nodes at 3 days
Cells from the ankles and the draining popliteal lymph nodes (pLN) were examined by flow cytometry. While total macrophages were reduced 3 days after initiation of infliximab, this was due primarily to the reduction of ankle Ly6C+ macrophages (Figure 3A). Granulocytes, dendritic cells, and T and B lymphocytes determined by flow cytometry were not significantly reduced in the ankles of infliximab treated mice at 3 days (Figure 3A). The pLNs draining the ankles were also examined at 3 days, positing that there would be an increase of macrophages. In contrast, there was a decrease of Ly6C+ macrophages and B lymphocytes, while dendritic cells and T lymphocytes were not significantly different from the untreated controls (Figure 3B). Although there was a trend toward reduction of granulocytes, this was not significant in either the ankles or pLNs 3 days following the initiation of therapy (figure 3A, B). As in the controls, CCR7 was expressed primarily on Ly6C- macrophages and there was no reduction of the expression of CCR7 on either Ly6C+ or Ly6C- ankle macrophages following treatment with infliximab (Figure 3C, D). These observations do not support the hypothesis that the reduction of macrophages in the ankles was due to increased efflux from the joints following the initiation of therapy.
Figure 3. Ly6C+ macrophages are reduced in ankles and popliteal lymph nodes following treatment with infliximab.
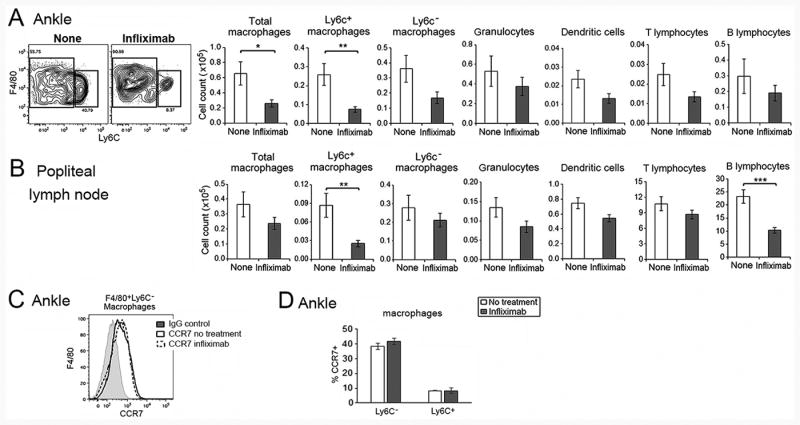
Flow cytometric analysis demonstrating the effect of infliximab on leukocytes in (A) ankles and (B) pLNs, employing hTNF-Tg mice treated with infliximab or vehicle control, harvested at day 3. The gating strategy for ankle cells is the same as presented in Supplemental Figure 1C (n = 13 each group). Panel (C) represents the CCR7 expression on Ly6C- ankle macrophages, while panel (D) presents the percent CCR7 on Ly6C+ and Ly6C- macrophages before and after infliximab (n = 3 for each group). The gating strategy was the same as presented in Supplemental Figure 1D. All data presented were obtained employing 5-6 week old hTNF-Tg mice with arthritis. * represents p < 0.05, ** p <0.01 and *** p < 0.001 between the indicated groups.
No evidence of CCR7 mediated macrophage egress
The ankles and pLNs of the hTNF-Tg mice were examined for CCR7 ligands. After treatment with infliximab, a modest decrease of CCL19 was observed in the ankles, but there was no reduction of CCL21 (Figure 4A). As anticipated, there was no change in the levels of CCL19 or 21 in the draining pLNs following treatment with infliximab. We considered the possibility that macrophage efflux was not observed because the macrophages in the ankles were not differentially labeled. Therefore, CD45.1+ macrophages were harvested from hTNF-Tg mice with arthritis and injected into the ankles of CD45.2 hTNF-Tg mice which were then treated with infliximab or not. There was a reduction of Ly6C+ CD45.1+ donor macrophages in the ankles of the treated mice following infliximab (Figure 4B). The number of donor macrophages was too low in the pLNs to discriminate Ly6C+ and Ly6C-. As expected, recipient CD45.2+Ly6C+ macrophages were reduced in both the ankles and the pLNs following treatment at 3 days (Figure 4C).
Figure 4. Macrophage egress and CCR7 were not documented following treatment with infliximab.
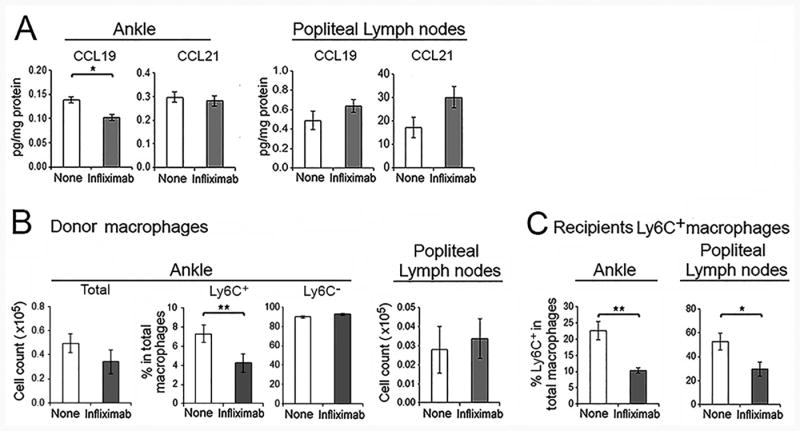
(A). CCL19 and CCL21 in ankles and pLNs of hTNF-Tg mice following treatment with infliximab at 72 hours were determined by ELISA (n = 8 for treatment and 7 for the controls). (B-C). Macrophage egress was tracked in response to infliximab. Total macrophages isolated from the ankles of CD45.1 mice were injected into the ankles of CD45.2 hTNF-Tg mice with arthritis. The recipients were then untreated or treated with infliximab. Ankles and pLNs were harvested 24 hour post-macrophage administration. (B). The CD45.1 donor macrophages in the ankles and pLNs were analyzed as the total number, and the percent of Ly6C+ and Ly6C- macrophages in the ankles. (C) Recipient CD45.2 macrophages in the same joints response to infliximab are also presented (n = 4 mice per group).
In an attempt to further examine the potential role of CCR7 and macrophage efflux, hTNF-Tg mice were crossed with CCR7-/- mice. The arthritis was modestly more severe in the hTNF-Tg CCR7-/- mice compared with the hTNF-Tg CCR7+/- mice (Figure 5A), or the hTNF-Tg mice (Figure 2A). Increased synovial lining thickness and cartilage damage were observed in the hTNF-Tg CCR7-/- mice (Figure 5B). Following treatment with infliximab, there was no difference in the reduction of Ly6C+ macrophages between the hTNF-Tg/CCR7+/- and the hTNF-Tg/CCR7-/- mice (Figure 5C). These observations do not support a role for CCR7 or macrophage efflux in response to treatment with infliximab in hTNF-Tg mice.
Figure 5. hTNF-Tg CCR7-/- mice develop more severe arthritis but respond similarly to infliximab.
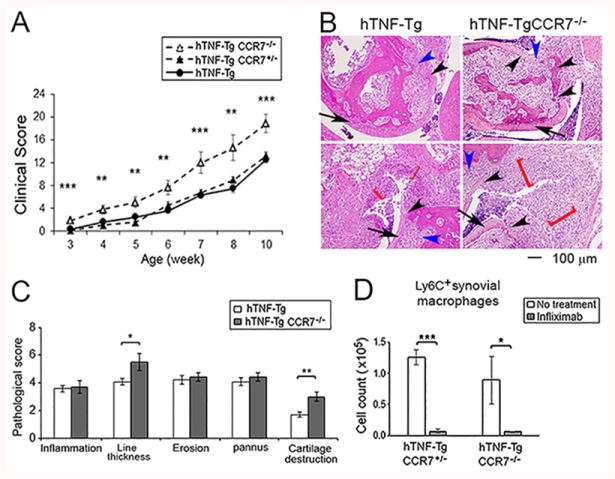
hTNF-Tg mice were crossed with CCR7-/- mice. (A) The development of arthritis in hTNF-Tg CCR7-/-, hTNF-Tg-CCR7+/- or hTNF-Tg (from figure 2A) was evaluated as in Figure 2. The significance was determined by ANOVA among the 3 groups. (B, C). Histological assessment of ankle joints of hTNF-Tg CCR7-/- and hTNF-Tg mice at the same age (n = 5 mice for hTNF-Tg CCR7-/-, and 7 mice for hTNF-Tg mice). The cartilage destruction is identified by the black arrow head and synovial lining thickness by the red bracket, and the inflammation and erosion/pannus were indicated by blue arrow head. The arrows identifies cartilage with normal morphology. (D). Ly6C+ macrophages were determined by flow cytometry and compared between hTNF-Tg-CCR7-/- and hTNF-Tg-CCR7+/- mice. Both groups of mice were treated or untreated with infliximab. The cells were harvested after 3 days following 2 dose of treatment (n = 4-6 mice for each group). * represents p < 0.05, ** p <0.01 and *** p < 0.001 between the indicated groups.
Increased apoptosis of Ly6C+ macrophages following infliximab treatment
Studies were performed to determine if there was increased cell death following treatment. The ankles of hTNF-Tg mice were isolated from untreated or infliximab treated mice at 3 days. An increase of apoptosis of Ly6C+ macrophages in both the ankles and draining pLNs was noted following treatment (Figure 6A, B). There was no difference in apoptosis of the Ly6C- macrophages, granulocytes or B or T lymphocytes in the ankles, while a slight reduction of granulocyte apoptosis was noted in the pLNs following treatment (Figure 6A, B). No increase of apoptosis of circulating Ly6C+ monocytes was noted following treatment with infliximab (data not shown). No difference in necrotic cells, defined as 7AAD+Annexin V+, was noted for any cell type in the ankles or pLNs following treatment (Figure 6A, B). Ankles were examined for the expression of cleaved caspase 8 or cleaved caspase 3 (Figure 6C, D). A significant increase of cleaved caspase 3 was observed in the Ly6C+, but not Ly6C-, ankle macrophages following infliximab (Figure 6C). There was a trend for an increase of cleaved caspase 8, which did not reach statistical significance (Figure 6D). These observations suggest that increased apoptosis may contribute to the reduction of Ly6C+ macrophages following treatment with infliximab.
Figure 6. Increased apoptosis contributes to infliximab mediated reduction of Ly6C+ macrophages in ankles and popliteal lymph nodes.
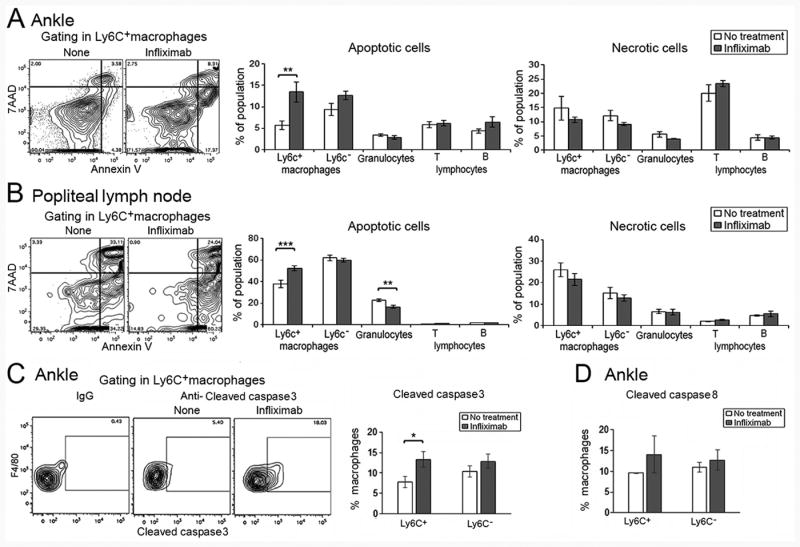
(A). Apoptosis and necrosis were defined by live/dead staining and annexin V analysis following flow cytometry. hTNF-Tg mice (5-6 weeks old) with arthritis were employed and treated with 2 doses of infliximab or vehicle control harvested at day 3. Representative staining for treated and untreated ankles for Ly6C+ macrophages is presented (left panels), while the analysis of each cell type is presented in the panels on the right. (B). The same analyses are presented for pLNs harvested at the same time. (n = 7 for no treatment and 8 for infliximab treated mice). (C) The cleaved caspase 3 and (D) cleaved caspase 8 were identified by intracellular staining followed by flow cytometry (n = 5 for no treatment and 8 for infliximab treated mice). ** represents p < 0.01 and *** p < 0.001 between the indicated groups.
Reduced chemotaxis monocytes after infliximab
In order to determine the effect of treatment on the migration of circulating monocytes into the joints, monocytes isolated from the bone marrow of CD45.1+ mice were adoptively transferred into CD45.2+ hTNF-Tg mice. Regardless of whether infliximab was injected 24 hours prior to and at the time of monocyte transfer (2 doses) or only at the time of monocyte transfer (1 dose), a significant (p < 0.001) decrease of monocyte migration into the ankles was documented (Figure 7A). No reduction of circulating monocytes was noted following infliximab treatment (data not shown). Further, the classical monocyte chemokine CCL2, but not the non-classical monocyte chemokine CX3CL1 (Fractalkine), was significantly reduced in the ankles following a single treatment with infliximab (Figure 7B). In order to identify the downstream effect of reducing macrophages or chemokines, we examined mice after 2 doses of infliximab, Ankles are harvested not at 3 days, but at 7 days. In the joints, Ly6C+ macrophages and granulocytes, but not dendritic cells or B or T lymphocytes, were significantly reduced at 7 days (Supplemental Figure 3). Examination of joint tissue on day 3, after 2 doses of infliximab, prior to the reduction of neutrophils in the ankles, demonstrated a significant reduction of the neutrophil chemokine CXCL5 (Figure 7C). These observations demonstrate that the reduction of macrophages and chemokines contributed to clinical improvement following treatment of hTNF-Tg mice with infliximab. It is likely that the reduction of macrophages was due both to increased apoptosis and reduced chemokine secretion by the remaining macrophages.
Figure 7. Reduced monocyte migration into the ankles following infliximab treatment.
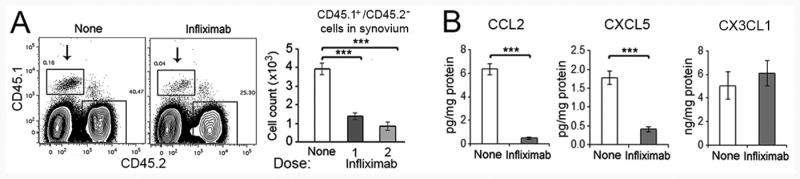
(A). Infliximab suppresses the monocyte influx into ankles in hTNF-Tg mice. Monocytes isolated from CD45.1 bone marrow were transferred into TNF-Tg (CD45.2) mice. The TNF-Tg recipients were received one dose of infliximab 24 hours before and additional dose at the same time of monocyte administrated (2 doses), or a single dose at the same time as monocytes were injected (1 dose). All mice were sacrificed 24 hours after monocyte injection, and analyzed for CD45.1 cells in the ankle joints. The panels on the left are representative gating of the CD45.1+CD45.2- donor monocytes that migrated into the ankles (indicated by arrows), with the statistical analysis presented in the panels on the right (n = 4-5 mice for each group). (B). Reduced CCL2 and CXCL5, but not CX3CL1 in the ankles of hTNF-Tg mice following treatment with infliximab (2 doses), determined by ELISA (n = 7-8 mice for each group). *** represents p < 0.001 between the indicated groups.
Discussion
This study was performed to identify the initial mechanism(s) that contribute to a reduction of macrophages in response to TNF inhibition. We hypothesized that TNFα expression would result in an increase of CCR7 on synovial tissue macrophages and an increase of the CCR7 ligands CCL19 and CCL21 would contribute to the retention of macrophages in the inflamed joint. Neutralization of TNFα would result in suppression of CCL19 and 21 which would permit CCR7+ macrophages to egress from the inflamed synovial tissue to the draining lymph nodes which constitutively express the CCR7 ligands. Employing hTNF-Tg mice, macrophages were reduced 3 days after the initiation of treatment with infliximab, which coincided with reduction of joint swelling on clinical exam and inflammation assessed histologically. This reduction was due primarily to the reduction of Ly6C+ macrophages in the ankles. At this same time point, in the ankles, neutrophils, dendritic cell and B and T lymphocytes were not significantly reduced compared with untreated controls. However, in contrast to expectations we were not able identify egress of macrophages to the draining lymph nodes, nor evidence of a role for CCR7, CCL19 or CCL21 in the initial clinical response. We did document increased apoptosis of Ly6C+ macrophages in both the ankles and the draining lymph nodes. Further a reduction of circulating monocyte migration into the inflamed joints and a marked reduction of ankle CCL2, but not CX3CL1, was documented after treatment with infliximab. These observations demonstrate that in hTNF-Tg mouse arthritis, TNFα neutralization results in the initial clinical response, at least in part, through the increase of apoptosis of Ly6C+ macrophages and reduced monocyte migration into the ankles mediated by the reduction of CCL2, at which time the neutrophil chemokine CXCL5 was also reduced.
Sublining macrophages have been shown to be a reliable biomarker for response to treatment of RA with a variety of modalities, including non-biologic DMARDs, rituximab, TNF inhibitors, and inhibitors of chemotaxis (2, 26, 27). The change in sublining CD68+ macrophages strongly correlates with the clinical response determined by change in DAS28. The reduction of macrophages is observed as early as 48 hours after the initiation of infliximab (3, 28). At this same time point T, but not B, lymphocytes were also reduced (3). However by 2 or 4 weeks after the initiation of infliximab, macrophages as well as T and B lymphocytes were significantly reduced in patients with RA (8, 29). Nevertheless, regardless of the time of biopsy after the initiation of treatment, change in sublining macrophages correlated with change in the DAS28.
Since earlier studies in RA did not identify reduced migration of monocytes into the joints, studies were performed focusing on macrophage egress to the draining lymph nodes. We previously demonstrated that CCL19 and 21 were increased in the synovial tissue of patients with RA (9). In hTNF-Tg mice, CCR7 was expressed on a subset of macrophages, CCL19 and 21 were present in the ankles and draining lymph nodes, and macrophages were reduced shortly after the introduction of infliximab. Our hypothesis was based on the fact that T cells, neutrophils and dendritic cells employ the CCR7-CCL19-CCL21 axis to migrate to afferent lymph nodes (30-32). Further, localization of macrophages in the marginal zone is mediated by CCL19 and 21 (33). Additionally, atherosclerosis regression in ApoE-deficient mice is due at least in part to macrophage egress from the aorta mediated through the CCR7-CCL19-CCL21 axis (34). In contrast, in hTNF-Tg mice we identified no role for macrophage egress or the CCR7-CCL19-CCL21 axis in the early response to infliximab.
Next we examined the role of apoptosis for the initial reduction of macrophages following therapy in hTNF-Tg mice. In patients with RA conflicting results have been published. Increased macrophage, but not T cell, apoptosis was observed at 8 weeks following the initiation of etanercept or infliximab (35). Successful treatment of RA with DMARDs was associated with increased apoptosis and reduction of macrophages (36). Infliximab resulted in increased monocyte apoptosis in patients with chronic, active Crohn's disease (37). In contrast, another study examining RA synovial tissue and peripheral blood found no evidence of increased monocyte or macrophage apoptosis examined at 1 hour and 24 hours after the initiation of infliximab (3). The reasons for these differences may include the disease activity at the time of the studies, sensitivity of the assays employed and the timing of the samples after the initiation of therapy. In the hTNF-Tg mice an increase of macrophage, but not B or T lymphocyte, apoptosis was observed following the initiation of infliximab. Increased cleaved caspase 3 was detected in ankle Ly6C+ macrophages, suggesting that in recently migrated monocyte derived macrophages, TNFα neutralization may promote apoptosis through the intrinsic pathway, possibly due to the reduction of anti-apoptotic molecules. The apoptosis occurred primarily in the Ly6C+ macrophage population. A slight reduction of neutrophil pLN apoptosis was noted following neutralization of TNFα. Consistent with our observation, spontaneous resolution of local inflammation of the peritoneum was mediated through macrophage apoptosis, and with a minor contribution of egress to the draining lymph nodes that was not mediated by CCR7 (38).
The role of reduction of monocyte chemotaxis in response to therapy in RA has been examined. Two weeks following infliximab monocyte chemokine CCL2 (MCP-1), but not CCL5 (RANTES), CCL3 (MIP1α) or CCL4 (MIP1β) was reduced when examined by immunohistochemistry (8). Serum CCL2 correlated with the swollen joint count in RA (39), and reduction of serum CCL2 was observed following treatment with infliximab or etanercept (8, 40). However, employing technetium-99m labeled autologous monocytes, no reduction of monocyte influx into the inflamed RA joints was observed 14 days after treatment with adalimumab, even though clinical benefit was observed at this time point (7). In contrast, a significant reduction in the influx of 111In labeled autologous granulocytes was observed 2 weeks after treatment with infliximab, at which point T and B lymphocytes and macrophages were reduced compared with pretreatment biopsies (8). In hTNF-Tg mice, reduced migration of monocytes into the inflamed joints was observed after a single treatment of infliximab. This was associated with a significant reduction of the CCL2, but not CX3CL1 (Fractalkine). It is possible that other monocyte chemokines, which were not examined in this study, such as CCL3 (MIP1α), 4 (MIP1β) or 5 (RANTES), may also have contributed to the reduced monocyte ingress into the ankles. The reduction of CXCL5 following infliximab, was followed by a reduction of neutrophils in the ankles at 7 days. Together these observations support the interpretation that in hTNF-Tg mice, the early effects of infliximab are at least in part due to both increased apoptosis and to reduction of monocyte migration into the joints. The reduction of granulocytes seen 7 days after the initiation of therapy, was associated with decreased CXCL5 at day 3, and not with increased granulocyte apoptosis. These studies suggest that both the reduction of macrophages by apoptosis and the suppression of chemokine secretion by the remaining macrophages contributed to the clinical benefit observed in hTNF-Tg mice treated with infliximab.
It is not clear that these observations can be directly applied to the mechanisms of response in patients with RA. Blocking of CCR2 employing a monoclonal antibody, or CCR1, which also contributes to monocyte chemotaxis mediated by a number of CCL chemokines, failed to improve RA (5, 6), although an earlier phase1b study showed clinical benefit and a reduction of macrophages in the synovial tissue following inhibition of CCR1 signaling (41). Additionally oral agonists of CCR5 were ineffective in treating patients with RA (4, 42). However, the fact that chemokine receptor blockade was not effective in RA does not mean that inhibition of monocyte chemotaxis is not a mechanism responsible for improvement in RA following neutralization of TNFα. It may be that the redundant array of chemokines expressed in the RA joint makes it difficult to block chemotaxis by blocking a single receptor. It may be that reduction of the multiple ligands within the joint, as may occur with a variety of effective therapies, is necessary to result in the reduction of the influx of monocytes into the inflamed joint.
Supplementary Material
Acknowledgments
This study was supported by the NIH (National Institute of Arthritis and Musculoskeletal and Skin Disease) grant R21AR065076.
References
- 1.Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 1996;39:115–124. doi: 10.1002/art.1780390116. [DOI] [PubMed] [Google Scholar]
- 2.Wijbrandts CA, Vergunst CE, Haringman JJ, Gerlag DM, Smeets TJ, Tak PP. Absence of changes in the number of synovial sublining macrophages after ineffective treatment for rheumatoid arthritis: Implications for use of synovial sublining macrophages as a biomarker. Arthritis Rheum. 2007;56:3869–3871. doi: 10.1002/art.22964. [DOI] [PubMed] [Google Scholar]
- 3.Wijbrandts CA, Remans PH, Klarenbeek PL, Wouters D, van den Bergh Weerman MA, Smeets TJ, Vervoordeldonk MJ, Baeten D, Tak PP. Analysis of apoptosis in peripheral blood and synovial tissue very early after initiation of infliximab treatment in rheumatoid arthritis patients. Arthritis Rheum. 2008;58:3330–3339. doi: 10.1002/art.23989. [DOI] [PubMed] [Google Scholar]
- 4.van Kuijk AW, Vergunst CE, Gerlag DM, Bresnihan B, Gomez-Reino JJ, Rouzier R, Verschueren PC, van de Leij C, Maas M, Kraan MC, Tak PP. CCR5 blockade in rheumatoid arthritis: a randomised, double-blind, placebo-controlled clinical trial. Ann Rheum Dis. 2010;69:2013–2016. doi: 10.1136/ard.2010.131235. [DOI] [PubMed] [Google Scholar]
- 5.Vergunst CE, Gerlag DM, Lopatinskaya L, Klareskog L, Smith MD, van den Bosch F, Dinant HJ, Lee Y, Wyant T, Jacobson EW, Baeten D, Tak PP. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2008;58:1931–1939. doi: 10.1002/art.23591. [DOI] [PubMed] [Google Scholar]
- 6.Vergunst CE, Gerlag DM, von Moltke L, Karol M, Wyant T, Chi X, Matzkin E, Leach T, Tak PP. MLN3897 plus methotrexate in patients with rheumatoid arthritis: safety, efficacy, pharmacokinetics, and pharmacodynamics of an oral CCR1 antagonist in a phase IIa, double-blind, placebo-controlled, randomized, proof-of-concept study. Arthritis Rheum. 2009;60:3572–3581. doi: 10.1002/art.24978. [DOI] [PubMed] [Google Scholar]
- 7.Herenius MM, Thurlings RM, Wijbrandts CA, Bennink RJ, Dohmen SE, Voermans C, Wouters D, Izmailova ES, Gerlag DM, van Eck-Smit BL, Tak PP. Monocyte migration to the synovium in rheumatoid arthritis patients treated with adalimumab. Ann Rheum Dis. 2011;70:1160–1162. doi: 10.1136/ard.2010.141549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor PC, Peters AM, Paleolog E, Chapman PT, Elliott MJ, McCloskey R, Feldmann M, Maini RN. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43:38–47. doi: 10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 9.Pickens SR, Chamberlain ND, Volin MV, Pope RM, Mandelin AM, 2nd, Shahrara S. Characterization of CCL19 and CCL21 in rheumatoid arthritis. Arthritis Rheum. 2011;63:914–922. doi: 10.1002/art.30232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wengner AM, Hopken UE, Petrow PK, Hartmann S, Schurigt U, Brauer R, Lipp M. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007;56:3271–3283. doi: 10.1002/art.22939. [DOI] [PubMed] [Google Scholar]
- 11.Huang Q, Ma Y, Adebayo A, Pope RM. Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 2007;56:2192–2201. doi: 10.1002/art.22707. [DOI] [PubMed] [Google Scholar]
- 12.Pagliari LJ, Perlman H, Liu H, Pope RM. Macrophages require constitutive NF-kappaB activation to maintain A1 expression and mitochondrial homeostasis. Mol Cell Biol. 2000;20:8855–8865. doi: 10.1128/mcb.20.23.8855-8865.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perlman H, Pagliari LJ, Georganas C, Mano T, Walsh K, Pope RM. FLICE-inhibitory protein expression during macrophage differentiation confers resistance to fas-mediated apoptosis. J Exp Med. 1999;190:1679–1688. doi: 10.1084/jem.190.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 15.Huang QQ, Koessler RE, Birkett R, Dorfleutner A, Perlman H, Haines GK, 3rd, Stehlik C, Nicchitta CV, Pope RM. Glycoprotein 96 perpetuates the persistent inflammation of rheumatoid arthritis. Arthritis Rheum. 2012;64:3638–3648. doi: 10.1002/art.34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang QQ, Sobkoviak R, Jockheck-Clark AR, Shi B, Mandelin AM, 2nd, Tak PP, Haines GK, 3rd, Nicchitta CV, Pope RM. Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol. 2009;182:4965–4973. doi: 10.4049/jimmunol.0801563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scatizzi JC, Bickel E, Hutcheson J, Haines GK, 3rd, Perlman H. Bim deficiency leads to exacerbation and prolongation of joint inflammation in experimental arthritis. Arthritis Rheum. 2006;54:3182–3193. doi: 10.1002/art.22133. [DOI] [PubMed] [Google Scholar]
- 18.Scatizzi JC, Hutcheson J, Pope RM, Firestein GS, Koch AE, Mavers M, Smason A, Agrawal H, Haines GK, 3rd, Chandel NS, Hotchkiss RS, Perlman H. Bim-Bcl-2 homology 3 mimetic therapy is effective at suppressing inflammatory arthritis through the activation of myeloid cell apoptosis. Arthritis Rheum. 2010;62:441–451. doi: 10.1002/art.27198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang QQ, Birkett R, Koessler RE, Cuda CM, Haines GK, 3rd, Jin JP, Perlman H, Pope RM. Fas signaling in macrophages promotes chronicity in K/BxN serum-induced arthritis. Arthritis Rheumatol. 2014;66:68–77. doi: 10.1002/art.38198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang QQ, Perlman H, Birkett R, Doyle R, Fang D, Haines GK, Robinson W, Datta S, Huang Z, Li QZ, Phee H, Pope RM. CD11c-mediated deletion of Flip promotes autoreactivity and inflammatory arthritis. Nat Commun. 2015;6:7086. doi: 10.1038/ncomms8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zwerina J, Hayer S, Tohidast-Akrad M, Bergmeister H, Redlich K, Feige U, Dunstan C, Kollias G, Steiner G, Smolen J, Schett G. Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: effects on synovial inflammation, bone erosion, and cartilage destruction. Arthritis Rheum. 2004;50:277–290. doi: 10.1002/art.11487. [DOI] [PubMed] [Google Scholar]
- 22.Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK, 3rd, Berdnikovs S, Filer A, Clark AR, Buckley CD, Mutlu GM, Budinger GR, Perlman H. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep. 2014;9:591–604. doi: 10.1016/j.celrep.2014.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scatizzi JC, Hutcheson J, Bickel E, Haines GK, 3rd, Perlman H. Pro-apoptotic Bid is required for the resolution of the effector phase of inflammatory arthritis. Arthritis Res Ther. 2007;9:R49. doi: 10.1186/ar2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Italiani P, Boraschi D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haringman JJ, Gerlag DM, Zwinderman AH, Smeets TJ, Kraan MC, Baeten D, McInnes IB, Bresnihan B, Tak PP. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64:834–838. doi: 10.1136/ard.2004.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bresnihan B, Pontifex E, Thurlings RM, Vinkenoog M, El-Gabalawy H, Fearon U, Fitzgerald O, Gerlag DM, Rooney T, van de Sande MG, Veale D, Vos K, Tak PP. Synovial tissue sublining CD68 expression is a biomarker of therapeutic response in rheumatoid arthritis clinical trials: consistency across centers. J Rheumatol. 2009;36:1800–1802. doi: 10.3899/jrheum.090348. [DOI] [PubMed] [Google Scholar]
- 28.Smeets TJ, Kraan MC, van Loon ME, Tak PP. Tumor necrosis factor alpha blockade reduces the synovial cell infiltrate early after initiation of treatment, but apparently not by induction of apoptosis in synovial tissue. Arthritis Rheum. 2003;48:2155–2162. doi: 10.1002/art.11098. [DOI] [PubMed] [Google Scholar]
- 29.Tak PP, Taylor PC, Breedveld FC, Smeets TJ, Daha MR, Kluin PM, Meinders AE, Maini RN. Decrease in cellularity and expression of adhesion molecules by anti-tumor necrosis factor alpha monoclonal antibody treatment in patients with rheumatoid arthritis. Arthritis Rheum. 1996;39:1077–1081. doi: 10.1002/art.1780390702. [DOI] [PubMed] [Google Scholar]
- 30.Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, Magistrelli G, Masternak K, Chevailler A, Delneste Y, Jeannin P. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood. 2011;117:1196–1204. doi: 10.1182/blood-2009-11-254490. [DOI] [PubMed] [Google Scholar]
- 31.Braun A, Worbs T, Moschovakis GL, Halle S, Hoffmann K, Bolter J, Munk A, Forster R. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat Immunol. 2011;12:879–887. doi: 10.1038/ni.2085. [DOI] [PubMed] [Google Scholar]
- 32.Russo E, Teijeira A, Vaahtomeri K, Willrodt AH, Bloch JS, Nitschke M, Santambrogio L, Kerjaschki D, Sixt M, Halin C. Intralymphatic CCL21 Promotes Tissue Egress of Dendritic Cells through Afferent Lymphatic Vessels. Cell Rep. 2016;14:1723–1734. doi: 10.1016/j.celrep.2016.01.048. [DOI] [PubMed] [Google Scholar]
- 33.Ato M, Nakano H, Kakiuchi T, Kaye PM. Localization of marginal zone macrophages is regulated by C-C chemokine ligands 21/19. J Immunol. 2004;173:4815–4820. doi: 10.4049/jimmunol.173.8.4815. [DOI] [PubMed] [Google Scholar]
- 34.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Catrina AI, Trollmo C, af Klint E, Engstrom M, Lampa J, Hermansson Y, Klareskog L, Ulfgren AK. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum. 2005;52:61–72. doi: 10.1002/art.20764. [DOI] [PubMed] [Google Scholar]
- 36.Smith MD, Weedon H, Papangelis V, Walker J, Roberts-Thomson PJ, Ahern MJ. Apoptosis in the rheumatoid arthritis synovial membrane: modulation by disease-modifying anti-rheumatic drug treatment. Rheumatology (Oxford) 2010;49:862–875. doi: 10.1093/rheumatology/kep467. [DOI] [PubMed] [Google Scholar]
- 37.Lugering A, Schmidt M, Lugering N, Pauels HG, Domschke W, Kucharzik T. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn's disease by using a caspase-dependent pathway. Gastroenterology. 2001;121:1145–1157. doi: 10.1053/gast.2001.28702. [DOI] [PubMed] [Google Scholar]
- 38.Gautier EL, Ivanov S, Lesnik P, Randolph GJ. Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood. 2013;122:2714–2722. doi: 10.1182/blood-2013-01-478206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellingsen T, Buus A, Stengaard-Pedersen K. Plasma monocyte chemoattractant protein 1 is a marker for joint inflammation in rheumatoid arthritis. J Rheumatol. 2001;28:41–46. [PubMed] [Google Scholar]
- 40.Kageyama Y, Kobayashi H, Kato N, Shimazu M. Etanercept reduces the serum levels of macrophage chemotactic protein-1 in patients with rheumatoid arthritis. Mod Rheumatol. 2009;19:372–378. doi: 10.1007/s10165-009-0175-z. [DOI] [PubMed] [Google Scholar]
- 41.Haringman JJ, Kraan MC, Smeets TJ, Zwinderman KH, Tak PP. Chemokine blockade and chronic inflammatory disease: proof of concept in patients with rheumatoid arthritis. Ann Rheum Dis. 2003;62:715–721. doi: 10.1136/ard.62.8.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerlag DM, Hollis S, Layton M, Vencovsky J, Szekanecz Z, Braddock M, Tak PP, Group ES. Preclinical and clinical investigation of a CCR5 antagonist, AZD5672, in patients with rheumatoid arthritis receiving methotrexate. Arthritis Rheum. 2010;62:3154–3160. doi: 10.1002/art.27652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.